

Abstract
Resistance to the Ableson protein tyrosine (Abl) kinase inhibitor imatinib mesylate has become a critical issue for patients in advanced phases of chronic myelogenous leukemia. Imatinib-resistant tumor cells develop, in part, as a result of point mutations within the Abl kinase domain. As protein kinase B (Akt) plays a pivotal role in Abl oncogene-mediated cell survival, we hypothesize that concurrent inhibition of Akt will sensitize resistant cells to the residual apoptotic activity of imatinib mesylate, thereby overcoming the resistance. Here, we examined the effect of OSU-03012, a celecoxib-derived phosphoinositide-dependent kinase-1 (PDK-1) inhibitor, on imatinib mesylate-induced apoptosis in 2 clinically relevant breakpoint cluster region (Bcr)-Abl mutant cell lines, Ba/F3p210E255K and Ba/F3p210T315I. The 50% inhibitory concentration (IC50) values of imatinib mesylate to inhibit the proliferation of Ba/F3p210E255K and Ba/F3p210T315I were 14 ± 4 and 30 ± 2 μM, respectively. There was no cross-resistance to OSU-03012 in these mutant cells with an IC50 of 5 μM irrespective of mutations. Nevertheless, in the presence of OSU-03012 the susceptibility of these mutant cells to imatinib-induced apoptosis was significantly enhanced. This synergistic action was, at least in part, mediated through the concerted effect on phospho-Akt. Together these data provide a novel therapeutic strategy to overcome imatinib mesylate resistance, especially with the Abl mutant T315I.
Introduction
It has been well established that breakpoint cluster region/Ableson protein tyrosine (Bcr-Abl) kinase, the product of the Philadelphia chromosome, plays an obligatory role in the pathogenesis of chronic myelogenous leukemia (CML).1 This causal relationship underlies the clinical success of using the Bcr-Abl tyrosine kinase inhibitor imatinib mesylate (ST1571; Gleevec) to target this molecular defect in CML, as evidenced by the complete remission and remarkably few associated side effects in patients with first chronic-phase CML.2-5 However, patients in more advanced phases of CML either fail to respond or quickly relapse following an initial response to imatinib mesylate.6,7 Acquisition of the imatinib mesylate-resistant phenotype is attributable to at least 2 major cellular mechanisms: amplification of the Bcr-Abl gene and mutations in the Abl catalytic domain.8 Mutations within the kinase domain represent the more commonly identified mechanism associated with relapse,9,10 among which Y253F/H, E255K/V, T315I, and M351T are characterized as the most clinically relevant mutants.11-19 Especially, E255K and T315I exhibit nearly 2-orders-of-magnitude lower biochemical and cellular sensitivity to imatinib mesylate,20 with the resulting 50% inhibitory concentration (IC50) values greatly exceeding the therapeutically attainable concentration of the drug. Three-dimensional structural data indicate that the mechanisms by which E255K and T315I affect imatinib mesylate's interactions with the catalytic domain vary.8 For example, conversion of T315 to an Ile residue in the catalytic domain results in the loss of a hydrogen bonding with imatinib mesylate, thereby restricting imatinib mesylate's access to its binding site.9,12 In contrast, the structural effect of the E225K on imatinib mesylate binding is subtle since this residue is located in the nucleotide-binding loop for adenosine triphosphate (ATP). Conceivably, this mutation affects imatinib mesylate binding by altering the conformational flexibility of the neighboring nucleotide-binding and activation loops.8 As a result of significantly reduced sensitivity to imatinib mesylate, both mutations exhibit high degrees of cellular resistance to imatinib mesylate.20 Thus, it is of urgency to develop an alternative strategy to overcome this imatinib mesylate resistance.
From a mechanistic perspective, expression of the Bcr-Abl oncogene up-regulates multiple downstream signaling pathways, including those mediated by phosphatidylinositol 3-kinase (PI3K)/Akt, Ras/mitogen-activated protein kinase (MAPK), and signal transducer and activator of transcription (STAT).1,21 Of these pathways, the PI3K/Akt signaling cascade plays an especially pivotal role in Abl oncogene-mediated proliferation, survival, and transformation.22-25 For example, recent evidence indicates that CML cells were susceptible to the growth-inhibitory effects of the PI3K inhibitor LY294002 but not the MAPK inhibitor PD98059.26 In addition, PI3K inhibitors have been shown to synergize with imatinib mesylate in inhibiting CML cell growth.27 Together these findings suggest the clinical relevance of targeting Akt signaling in imatinib-resistant patients.28
Recently, based on our finding that the cyclooxygenase-2 (COX-2) inhibitor celecoxib mediates apoptosis by blocking phosphoinositide-dependent kinase-1 (PDK-1)/Akt signaling independently of COX-2 inhibition, we have used celecoxib as a scaffold to develop a novel class of PDK-1 inhibitors with high potency in deactivating Akt and inducing apoptosis in cancer cells.29 These celecoxib-derived PDK-1 inhibitors, however, are devoid of COX-2 inhibitory activity. Here, we examined the effect of an optimal inhibitor (OSU-03012), alone or in combination with imatinib mesylate, in Bcr-Abl-expressing Ba/F3 cells (Ba/F3p210Bcr-Abl) vis-à-vis 2 mutant cell lines (Ba/F3p210E255K and Ba/F3p210T315I), both of which are highly resistant to high doses of imatinib mesylate.30,31 Previous studies have shown that arsenic oxide (As2O3), 5-aza-2-deoxycytidine (decitabine), or the farnesyl transferase inhibitor SCH66336 could not cooperate with imatinib mesylate in enhancing its in vitro efficacy in Ba/F3p210T315I cells.30,32 Here we hypothesize that concurrent inhibition of PDK-1/Akt signaling would sensitize imatinib mesylate-resistant cells to the residual apoptotic effects of imatinib mesylate, thereby overcoming the resistance. This premise is corroborated by the ability of OSU-03012 to restore the sensitivity of Ba/F3p210E255K and Ba/F3p210T315I to imatinib mesylate by shifting the dose-response curve to the left by more than 1 log unit.
Materials and methods
Reagents and cell culture
Imatinib mesylate, also known as STI571, was obtained from commercial imatinib mesylate capsules (Novartis Pharmaceuticals, East Hanover, NJ) by solvent extraction followed by recrystallization. The PDK-1 inhibitor OSU-03012 was synthesized as described.29 Rabbit polyclonal anti-Akt and rabbit monoclonal anti-PARP (anti-poly-adenosine diphosphate [ADP]-ribose polymerase) were purchased from Cell Signaling Technology (Beverly, MA). Rabbit antibodies against phospo-Thr308-Akt were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse monoclonal anti-cytochrome c, anti-Bcr, and antiactin were from BD Pharmingen (San Diego, CA), Oncogene (Boston, MA), and ICN Biomedicals (Costa Mesa, CA), respectively. Goat anti-rabbit and goat anti-mouse immunoglobulin G (IgG) horseradish peroxidase conjugates were from Jackson ImmunoResearch Laboratories (West Grove, PA). A murine myeloid hematopoietic cell line (32D) and a lymphoid cell line (Ba/F3) were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS; Gibco, Carlsbad, CA); 15% Walter and Eliza Hall Institute (WEHI)-conditioned media as an interleukin 3 (IL-3) source; and 50 units/mL penicillin G, 50 μg/mL streptomycin, and 10 μg/mL gentamicin (Sigma, St Louis, MO). Ba/F3p210Bcr-Abl and 2 imatinib-resistant Ba/F3p210 mutant cell lines, Ba/F3p210E255K and Ba/F3p210T315I, were generated as previously reported.31 These cells were cultured in RPMI 1640 medium containing 10% FBS, 50 units/mL penicillin G, 50 μg/mL streptomycin, and 10 μg/mL gentamicin (Sigma) at 37°C in 5% CO2.
Cell proliferation assay (MTS assay)
Cell proliferation was analyzed by the MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assay (Promega, Madison, WI) in 6 replicates. Cells (5000/well) were grown in 10% FBS-supplemented RPMI 1640 medium in 96-well flat-bottomed plates and exposed to various concentrations of individual agents or combination of drugs dissolved in dimethyl sulfoxide (DMSO; final concentration ≤ 0.1%) in the same medium. Control groups received DMSO vehicle at a concentration equal to that in drug-treated cells. After 48-hour treatment, MTS and the phenazine methosulfate (PMS) detection reagent were mixed at a ratio of 20:1 (MTS/PMS) and immediately added to the culture medium at a ratio of 1:5. Cells were incubated in the CO2 incubator at 37°C for 3 hours and the production of formazan was analyzed by measuring the absorbance at 492 nm in a plate reader.
Immunoblotting
The general procedure for the Western blot analysis of Bcr-Abl, Akt, phospho-Akt, and actin was performed as follows. Cells were collected by centrifugation at 2000g and resuspended in radioimmunoprecipitation assay (RIPA) lysis buffer consisting of 50 mM tris(hydroxymethyl)aminomethane (Tris)-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA (ethylenediaminetetraacetic acid), 1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), and a mixture of protease inhibitor cocktail (100 μM 4-(2-aminoethyl)benzenesulfonyl fluoride, 80 nM aprotinin, 5 μM bestatin, 1.5 μM E-64 protease inhibitor, 2 μM leupeptin, 1 μM pepstatin A [Calbiochem, La Jolla, CA]) and phosphatase inhibitors (10 μM sodium fluoride, 5 μM sodium vanadate, and 10 μM β-glycerol phosphate). The mixture was sonicated for 5 seconds and protein contents were analyzed by using the Bradford assay kit (Bio-Rad, Hercules, CA). Twenty-five micrograms total protein was resolved in SDS-polyacrylamide gels on a Minigel apparatus and transferred to a nitrocellulose membrane using a semidry transfer cell. The transblotted membrane was washed 3 times with Tris-buffered saline (TBS) containing 0.05% Tween 20 (TBST). After blocking with TBST containing 5% nonfat milk for 60 minutes, the membrane was incubated with the appropriate primary antibody at 1:1000 dilution in TBST-5% nonfat milk at 4°C overnight and then washed 3 times with TBST. The membrane was probed with horseradish peroxidase-conjugated secondary antibody at 1:3000 for 1 hour at room temperature and was then washed with TBST 3 times. The immunoblots were visualized by enhanced chemiluminescence. The density of the blot was further analyzed by densitometry using the Gel-Pro Analyzer (Media Cybernetics, San Diego, CA).
Assessment of apoptosis
Flow cytometric analysis. Fluorescein-conjugated annexin V (annexin V-FITC) and propidium iodide (PI; BD Pharmingen) were used to quantify the percentage of cells undergoing apoptosis by following the protocol provided by the vender. In short, after drug treatment the cells were collected and resuspended in 1 mL binding buffer (10 mM Hepes [N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid], pH 7.4; 140 mM NaCl; 2.5 mM CaCl2) at a concentration of 5 × 106 cells/mL. A 200-μL solution (1 × 106) was transferred to a culture tube, to which were added annexin V-FITC and PI. The cells were gently vortexed and incubated for 15 minutes at room temperature in the dark. An additional 800 μL of binding buffer was added to each tube and the samples were analyzed by flow cytometry.
Cytochrome c release analysis. Cytosol-specific mitochondria-free lysates were prepared as previously described.33 Drug-treated cells were collected by centrifugation at 1000g for 5 minutes. The pellet fraction was recovered, placed on ice, and resuspended in 100 μL of a chilled hypotonic lysis solution (220 mM mannitol; 68 mM sucrose; 50 mM PIPES [piperazine diethanesulfonic acid]-KOH, pH 7.4; 50 mM KCl; 5 mM EDTA; 2 mM MgCl2; 1 mM dithiothreitol; and the aforementioned protease inhibitors cocktails). After a 20-minute incubation on ice, the mixture was centrifuged at 600g for 10 minutes. The supernatant was collected in a microcentrifuge tube and centrifuged at 14 000g for 30 minutes. An equivalent amount of protein (25 μg) from each supernatant was resolved by 15% SDS-polyacrylamide gel electrophoresis and blotted with anti-cytochrome c antibody by following the procedure described under “Immunoblotting.”
Western blot analysis of PARP cleavage. Cells that were drug treated for 48 hours were collected, washed with ice-cold phosphate-buffered saline (PBS), and resuspended in the aforementioned lysis buffer. Soluble cell lysates were collected after centrifugation at 10 000g for 5 minutes. Equivalent amounts of proteins (50 μg) from each lysate were resolved in 10% SDS-polyacrylamide gels. Bands were transferred to nitrocellulose membranes and analyzed by immunoblotting with monoclonal anti-PARP antibody.
Statistical analysis and determination of synergism
The medium-effect method was used to analyze dose-response data for single drug or multiple drugs. The synergistic effect of multiple drugs was determined by the definition of Chou and Talalay.34 A combination index (CI) less than 1 was defined as synergism. By using the software package Calcusyn (Biosoft, Cambridge, United Kingdom), the values of IC50, the drug concentration required for 50% growth inhibition, and CI were calculated. These drugs were assumed as totally independent modes of action and are therefore mutually nonexclusive. The sign test, a nonparametric test,35 was used to test the hypothesis that combination indices for drugs were less than 1. The Jonckheere-Terpstra test, a nonparametric test for trends in ordered groups,35 was used to evaluate the decreasing trend in the relationship between drug concentrations and the densitometry values of phospho-Akt.
Results
Differential inhibitory effects of imatinib mesylate on Akt activation
In line with the earlier reports,30,31 Ba/F3p210Bcr-Abl, Ba/F3p210E255K, and Ba/F3p210T315I exhibited differential sensitivity to imatinib mesylate's antiproliferative effects. MTS assays indicate that the IC50 values for Ba/F3p210E255K and Ba/F3p210T315I were 14 ± 4 and 30 ± 2 μM, respectively, 2 orders of magnitude greater than the wild-type counterpart (0.13 ± 0.01 μM; Figure 1A). Imatinib mesylate, however, was ineffective against the cell lines 32D and untransfected Ba/F3 (with IC50 > 50 μM; Figure 1A), indicating the pivotal role of Bcr-Abl in the drug action. Furthermore, characterizations of cytochrome c release and phosphatidylserine externalization indicate that the thresholds for imatinib mesylate to trigger apoptosis in Ba/F3p210E255K and Ba/F3p210T315I cells were approximately 10 and 20 μM, respectively, vis-à-vis 0.1 μM for Ba/F3p210Bcr-Abl (Figure 1B-C). Flow analysis indicates that at 10 μM the extents of imatinib mesylate-induced apoptotic death (annexin V-positive cells) were 97%, 35%, and 6% in Ba/F3p210Bcr-Abl, Ba/F3p210E255K, and Ba/F3p210T315I, respectively (Figure 1C both quadrants B2 and B4).
Figure 1.

Differential susceptibility of Ba/F3p210Bcr-Abl, Ba/F3p210E255K, and Ba/F3p210T315I cells to imatinib mesylate. (A) Dose-response curves obtained by MTS assays after 48-hour exposure of Ba/F3p210Bcr-Abl (wild-type [WT]; •), Ba/F3p210E255K (E225K; ♦), and Ba/F3p210T315I (T315I; ▴) cells versus the control 32D (▪) and untransfected Ba/F3 cells (×) to imatinib mesylate. Each data point represents means ± SD (n = 6). (B) Dose-dependent effect of imatinib mesylate on cytochrome c (Cyt. c) release. The immunoblots are representative of 3 independent experiments. (C) Flow cytometric analysis of apoptotic death in the 3 cell lines overexpressing wild-type or mutant Bcr-Abl after treatment with DMSO vehicle or 1 or 10 μM imatinib mesylate for 48 hours. Results are representative of at least 3 independent experiments. B1, B2, B3, and B4 represent annexin V-/PI+, annexin V+/PI+Z (late apoptosis), annexin V-/PI-, and annexin V+/PI- (early apoptosis), respectively; FL1-H indicates annexin V-FITC; FL3-H, propidium iodide. The percentages in the graphs represent the percent of cell numbers in the respective quadrants.
As Akt signaling represents a major pathway through which Bcr-Abl mediates oncogenic effects in CML cells, we further assessed the effect of imatinib mesylate on Akt activation in wild-type versus mutant Bcr-Abl-overexpressing Ba/F3 cells. Because overexpression of Bcr-Abl up-regulates PI3K/Akt signaling,36,37 Ba/F3p210Bcr-Abl, Ba/F3p210E255K, and Ba/F3p210T315I exhibited substantially higher levels of Akt phosphorylation, irrespective of mutations, compared with that of 32D and untransfected Ba/F3 cells (Figure 2A). However, the respective susceptibility of these 3 cell lines to the inhibitory effect of imatinib mesylate on Akt varied to a great extent.
Figure 2.

Dose-dependent effect of imatinib mesylate on Akt dephosphorylation in Ba/F3p210Bcr-Abl, Ba/F3p210E255K, and Ba/F3p210T315I cells. (A) Bcl-Abl expression versus phospho-Thr308 Akt levels in 32D, Ba/F3, Ba/F3p210Bcr-Abl (WT), Ba/F3p210E255K (E225K), and Ba/F3p210T315I (T315I) cells. As shown, Akt phosphorylation is up-regulated by Bcr-Abl irrespective of mutations. (B, left) Dose-dependent effect of imatinib mesylate on Akt Thr308 phosphorylation (p-Thr308-Akt) in the 3 cell lines after 36-hour exposure. All immunoblots are representative of 3 independent experiments. (B, right) Bars represent the means of relative p-Akt level compared with the DMSO control of 3 independent determinations ± SD. According to the Jonckheere-Terpstra test, the trend P values were .004 and .022 for E225K and T315I, respectively.
Densitometry analysis of the immunoblots shows that exposure of Ba/F3p210Bcr-Abl to imatinib mesylate, even at concentrations as low as 0.5 μM, led to complete Akt deactivation (Figure 2B). A decreasing trend of phospho-Akt was noted with increasing imatinib mesylate concentrations in Ba/F3p210E255K and Ba/F3p210T315I (trend P values were .004 and .022, respectively, according to the Jonckheere-Terpstra test). The estimated IC50 values for imatinib-mediated Akt dephosphorylation were approximately 10 and 20 μM for Ba/F3p210E255K and Ba/F3p210T315I, respectively. Together these data suggested a putative link between imatinib mesylate resistance and Akt signaling. Accordingly, we hypothesized that concurrent inhibition of Akt could lower the threshold of imatinib-mediated apoptosis, thereby overcoming imatinib mesylate resistance.
The PDK-1/Akt signaling inhibitor OSU-03012 induces apoptosis irrespective of Bcr-Abl mutation
To test this hypothesis we examined the antiproliferative effects of the PDK-1 inhibitor OSU-03012, a structurally optimized derivative of celecoxib, which exhibits IC50 in PDK-1 inhibition of 5 μM.29 However, the consequent effect on intracellular Akt is more pronounced presumably due to the concurrent action of protein phosphatase 2A in Akt dephosphorylation. As a result, this agent could affect Akt phosphorylation levels as low as 1 μM.29
As shown in Figure 3, 32D, Ba/F3, Ba/F3p210Bcr-Abl, Ba/F3p210E255K, and Ba/F3p210T315I were equally susceptible to the antiproliferative effects of OSU-03012, with the respective IC50 values of 4.4 ± 0.1 μM, 4.8 ± 0.1 μM, 4.9 ± 1.0 μM, 4.8 ± 0.1 μM, and 4.5 ± 0.3 μM, respectively (Figure 3A). Analyses of cytochrome c release, PARP cleavage, and annexin V/PI staining in Ba/F3p210Bcr-Abl, Ba/F3p210E255K, and Ba/F3p210T315I cells demonstrate that the OSU-03012-mediated cell death was mainly attributable to apoptosis (Figure 3B-C).
Figure 3.
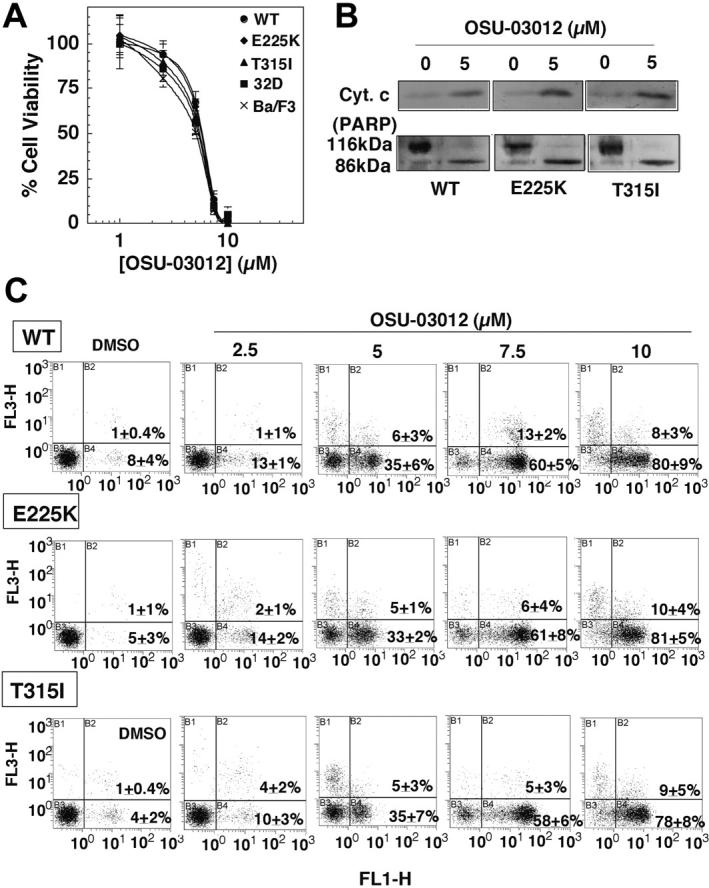
Ba/F3p210Bcr-Abl, Ba/F3p210E255K, and Ba/F3p210T315I cells are equally susceptible to OSU-03012 irrespective of Bcr-Abl mutations. (A) Dose-response curves obtained by MTS assays after 48-hour exposure of the control 32D and untransfected Ba/F3 cells versus Ba/F3p210Bcr-Abl (WT), Ba/F3p210E255K (E225K), and Ba/F3p210T315I (T315I) cells to OSU-03012. Each data point represents means ± SD (n = 6). (B) Effect of 5 μM OSU-03012 on cytochrome c release. The immunoblots are representative of 3 independent experiments. (C) Flow cytometric analysis of apoptotic death in the 3 cell lines treated with DMSO vehicle or OSU-03012 at 2.5, 5, 7.5, or 10 μM for 48 hours. Results are representative of at least 3 independent experiments. The percentages in the graphs represent the percent of cell numbers in each quadrant.
In addition, Western blot analysis indicates that OSU-03012 was able to diminish the phospho-Akt level at a concentration as low as 1 μM in these 3 cell lines irrespective of Bcr-Abl mutation (data not shown). Exposure to OSU-03012 over the range of 5 to 7.5 μM resulted in complete Akt dephosphorylation, which corresponds to the precipitous drop in cell viability between 5 and 7.5 μM (from 60% to 10%) in the dose-response curves (Figure 3A). Together these data clearly demonstrate the lack of cross-resistance to OSU-03012 in these imatinib mesylate-resistant cells.
OSU-03012 sensitizes imatinib mesylate-resistant cells to imatinib-induced apoptosis
To explore the effect of OSU-03012 on imatinib mesylate resistance, Ba/F3p210E255K and Ba/F3p210T315I cells, both of which exhibited IC50 greater than 10 μM against imatinib mesylate, were treated with varying concentrations of imatinib mesylate in the presence of 5 μM OSU-03012 or vice versa. As shown by the dose-response curves in Figure 4A, OSU-03012 sensitized, to a great extent, Ba/F3p210E255K and Ba/F3p210T315I cells to imatinib-induced cell death. For example, imatinib mesylate alone was ineffective in preventing cell proliferation within therapeutically attainable concentrations (≤ 5 μM; Figure 4A left panel dotted lines). However, in the presence of 5 μM OSU-03012, the susceptibility of these mutant cells to imatinib-induced apoptosis increased by more than one order of magnitude (Figure 4A left panel solid lines) with an apparent IC50 of 1 μM. Annexin V analysis indicates that in combination with 5 μM OSU-03012, imatinib mesylate at 2.5 and 5 μM caused 70% and greater than 95% cell death, respectively (Figure 4B). Similarly, imatinib mesylate at 5 μM also sensitized Ba/F3p210E255K and Ba/F3p210T315I cells to OSU-03012-mediated apoptosis (Figure 4A right panel) with a reduction of IC50 from 5 μM to less than 3 μM. Medium dose analysis of apoptosis induction in Ba/F3p210E255K and Ba/F3p210T315I cells was carried out over a range of OSU-03012 and imatinib mesylate concentrations at a fixed ratio (1:1) for 48 hours, after which CI values for apoptosis were determined in relation to the fraction affected. As shown in Figure 4C, the resulting CI values were significantly less than 1, which are considered as a synergistic interaction. A combination of imatinib mesylate and OSU-03012 at 5 μM each represented an effective treatment to completely eliminate these imatinib-resistant cells.
Figure 4.
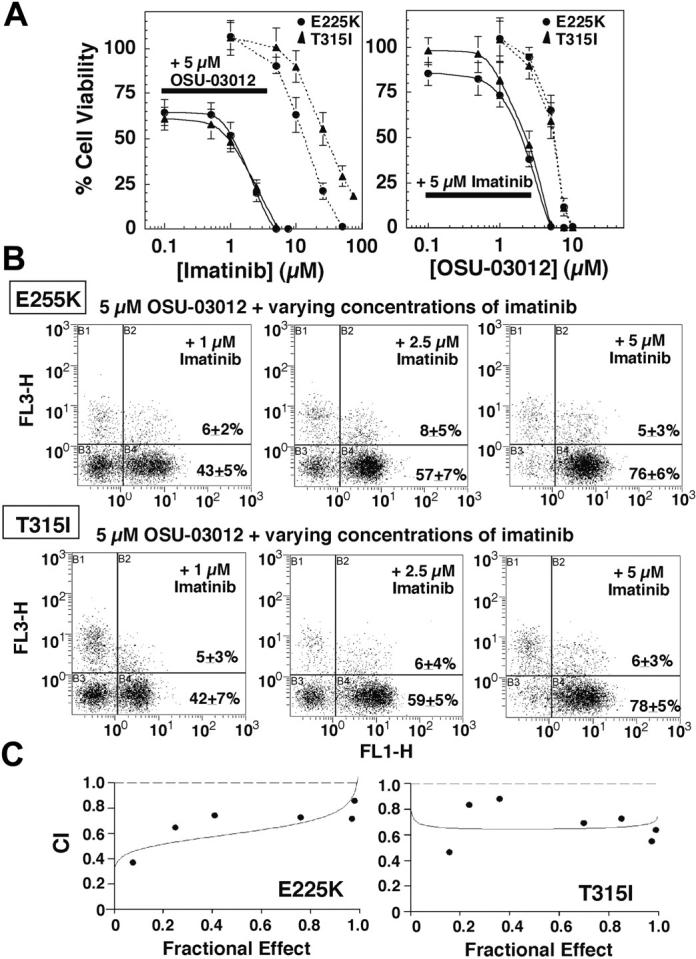
OSU-03012 sensitizes Ba/F3p210E255K and Ba/F3p210T315I cells to imatinib-induced apoptosis. (A, left) Dose-response curves obtained by MTS assays after 48-hour exposure of Ba/F3p210E255K (E225K) and Ba/F3p210T315I (T315I) cells to a combination of 5 μM OSU-03012 and varying concentration of imatinib mesylate (solid curves) or varying concentrations of imatinib mesylate alone (dotted curves). (Right) Dose-response curves obtained by MTS assays after 48-hour exposure to a combination of 5 μM imatinib mesylate and varying concentration of OSU-03012 (solid curves) or varying concentrations of OSU-03012 alone (dotted curves). Each data point represents means ± SD (n = 6). (B) Flow cytometric analysis of apoptotic death in Ba/F3p210E255K (top row) and Ba/F3p210T315I (bottom row) cells treated with 1, 2.5, and 5 μM imatinib mesylate in combination with 5 μM OSU-03012 for 48 hours. Results are representative of at least 3 independent experiments. (C) Ba/F3p210E255K and Ba/F3p210T315I cells were exposed to varying concentrations of OSU-03012 and imatinib mesylate at a fixed ratio (1:1) for 48 hours, after which CI values for apoptosis were determined in relation to the fraction affected using the medium dose effect analysis. CI values less than 1 are considered as a synergistic interaction. Mutually nonexclusive CI for combination at the IC50 was 0.602 for Ba/F3p210E255K and 0.649 for Ba/F3p210T315I. The P values for sign test comparing CI equal to 1 versus CI less than 1 are .016 for Ba/F3p210E255K and .008 for Ba/F3p210T315I. The curves depict the simulations of mutually nonexclusive CI values by Calcusyn.
Such a synergy, however, was not noted with 32D and Ba/F3 cells that lacked Bcr-Abl expression (Figure 5A), even though OSU-03012 was able to facilitate Akt dephosphorylation in both cell systems (Figure 5B). This synergistic action was, at least in part, mediated through the concerted effect of OSU-03012 and imatinib mesylate on phospho-Akt. Exposure of Ba/F3p210E255K and Ba/F3p210T315I cells to the drug combination did not affect the Bcr-Abl expression level (Figure 6A). However, imatinib mesylate at varying concentrations could augment the effect of 5 μM OSU-03012 on phospho-Akt in both cell lines (Figure 6B). Complete Akt dephosphorylation was achieved with a combination of 5 μM imatinib mesylate and 5 μM OSU-03012, in line with the optimal combination to elicit complete apoptotic death in these mutant cells. Similar augmenting effects were also noted with the combination of varying concentrations of OSU-03012 with 5 μM imatinib mesylate (Figure 6B).
Figure 5.

OSU-03012-mediated Akt dephosphorylation does not sensitize 32D and untransfected Ba/F3 cells to imatinib-induced apoptotic death. (A) Dose-response curves obtained by MTS assays after 48-hour exposure to a combination of 5 μM OSU-03012 and varying concentrations of imatinib mesylate. Each data point represents mean ± SD. (B) Effect of 5 μM OSU-03012 on Thr308 phosphorylation of Akt in 32D (•) and Ba/F3 (▴) cells after 36-hour exposure. The immunoblots are representative of 3 independent experiments.
Figure 6.
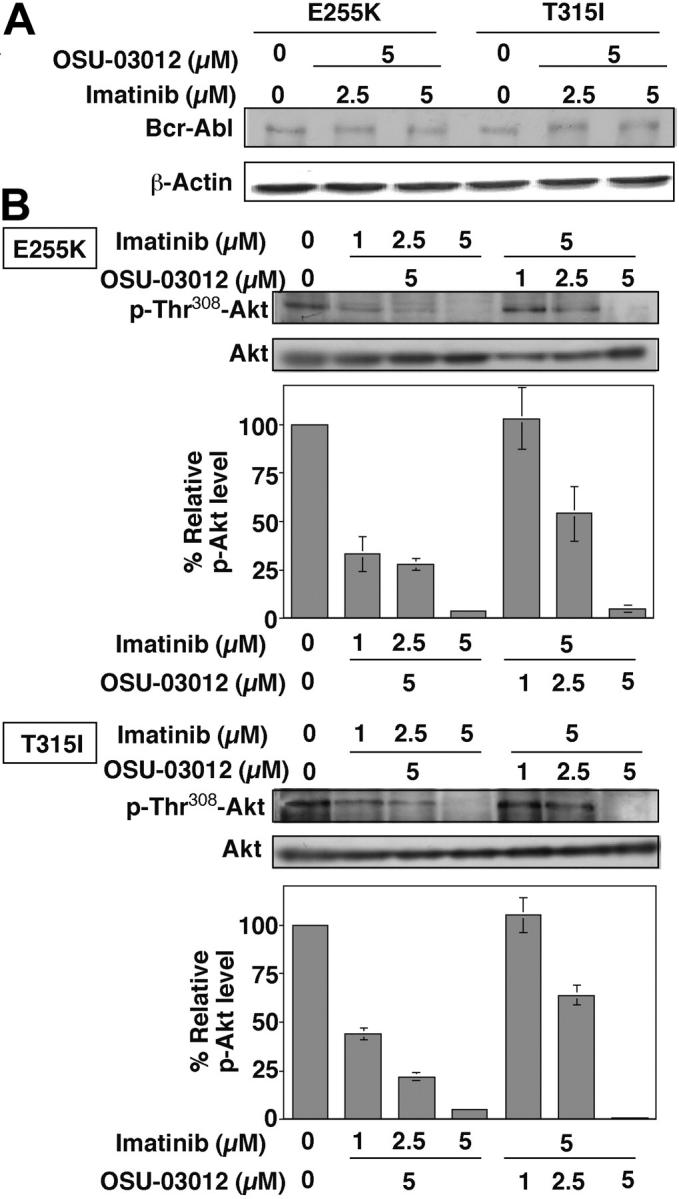
Combination effect of OSU-03012 and imatinib mesylate at different concentrations. Combination effect of OSU-03012 and imatinib mesylate at different concentrations on the state of Bcr-Abl expression (A) and Akt phosphorylation (B) in Ba/F3p210E255K and Ba/F3p210T315I cells. The immunoblots are representative of 3 independent experiments. Bars represent means ± SD (n = 3). No appreciable changes in Bcr-Abl expression levels were noted. However, a decreasing trend of p-Akt with increasing doses of imatinib mesylate and OSU-03012 in combination with 5 μM OSU-03012 and 5 μM imatinib mesylate, respectively.
Discussion
Development of new therapeutic strategies to overcome imatinib mesylate resistance in accelerated CML has been the focus of many recent investigations. In the literature, at least 3 distinct approaches have been reported. First, recent efforts have led to the identification of several novel Abl inhibitors capable of inhibiting some or all of mutant Abl kinases, which include PD180970,31 BMS-354825,38 and AP23464.39 In addition, the Bcr-Abl chaperone heat shock protein 90 inhibitors geldanamycin and 17-allyaminogelanamycin have also been shown to inhibit the growth of imatinib-resistant hematopoietic cells found in patients with T315I and E255K mutation.40 Second, cotreatment of imatinib-resistant cells with antileukemic agents such as As2O3, decitabine, the farnesyl transferase inhibitor SCH66336, and the histone deacetylase inhibitors suberoylanilide hydroxamic acid (SAHA) and butyrate could enhance the antiproliferative activity of imatinib mesylate.30,32,41 Third, the combination of different target-directed therapeutic agents such as the proteasome inhibitor bortezomib in conjunction with the cyclin-dependent kinase inhibitor flavopiridol or with SAHA has also been shown to effectively induce apoptosis in imatinib-resistant cells.42,43 As many of these strategies remain ineffective against the T315I mutant cells, this study is aimed at developing an alternative strategy to overcome imatinib mesylate resistance.
Despite reduced binding affinity, imatinib mesylate still displays differential residual activity against these mutations. However, its ability to activate apoptotic signaling in mutant cell lines diminishes. In light of the pivotal role in Akt in regulating apoptosis threshold, we hypothesize that concurrent inhibition of Akt would achieve a mechanistic synergy by sensitizing imatinib-resistant cells to the residual apoptotic efficacy of imatinib mesylate. To examine this premise, we evaluate the effect of OSU-03012, a celecoxib-derived PDK-1 inhibitor lacking COX-2 inhibitory activity, on imatinib mesylate resistance in 2 clinically relevant cell lines, Ba/F3p210E255K and Ba/F3p210T315I, compared with untransfected Ba/F3 and 32D cells. Despite low levels of phospho-Akt, both Ba/F3 and 32D cells were sensitive to the apoptosis-inducing effect of OSU-03012, which has also been observed in many types of cancer cells with normal phosphatase with tensin homology (PTEN) function.29 The effect of OSU-03012 on apoptosis in Ba/F3 and 32D might involve both Akt-dependent and -independent mechanisms. First, Akt, not even constitutively activated, still plays a role in the survival of these proliferating cells. Second, PDK-1 has non-Akt targets such as p70S6K that are also involved in cell survival and proliferation.
Both mutant cell lines exhibited the same susceptibility to OSU-03012 as their wild-type counterpart Ba/F3p210Bcr-Abl irrespective of the Bcr-Abl mutations. The lack of cross-resistance of OSU-03012 and imatinib mesylate underscores the functional relevance of targeting PDK-1/Akt signaling in imatinib mesylate-resistant mutant cells. It is especially noteworthy that OSU-03012 showed augmenting effects with imatinib mesylate on apoptosis in mutant imatinib mesylate-resistant cells at therapeutically attainable concentrations (≤ 5 μM), shifting the dose-response curves by more than 1 log unit. For example, the IC50 and IC80 values for imatinib mesylate in the presence of 5 μM OSU-03012 in Ba/F3p210T315I cells were 1 and 2.5 μM, respectively, vis-à-vis 30 and 65 μM for imatinib mesylate alone. This synergy is in sharp contrast to earlier reports that many antileukemic agents such as As2O3, decitabine, and SCH66336 could not synergize with imatinib mesylate in inhibiting the growth of Ba/F3p210T315I cells.30,32
Our data suggest that the ability of OSU-03012 to facilitate imatinib mesylate-mediated antiproliferative effects in these imatinib-resistant cells could be rationalized by the mechanistic synergy at the phospho-Akt level (ie, OSU-03012 was able to suppress Akt phosphorylation at low μM levels independent of Bcr-Abl mutations). As Akt contributes to enhanced survival of Bcr-Abl-expressing cells, the reduced apoptosis threshold renders imatinib mesylate-resistant cells sensitive to the residual antiproliferative activity of imatinib mesylate. Consequently, the molecular basis underlying this augmenting effect of OSU-03012 differs from that of other antileukemic agents such as As2O3 (Bcr-Abl expression),30 decitabine (DNA hypomethylation),30 SCH66336 (protein farnesylation),32 and flavopiridol (cyclin-dependent kinase).44 It is noteworthy that OSU-03012 is currently undergoing preclinical testing under the Rapid Access to Intervention Development (RAID) program at the National Cancer Institute. Our data indicate that oral administration of this agent in tumor-bearing nude mice even at 200 mg/kg for 1 month gave rise to no weight loss or apparent toxicity at necropsy (C.S.C., unpublished data, August 2003). Thus, this combination represents a viable strategy for clinical testing in imatinib-resistant CML.
Prepublished online as Blood First Edition Paper, January 21, 2005; DOI 10.1182/blood-2004-07-2967.
Supported by grant CA-94829 from the National Cancer Institute, grant DAMD17-02-1-0117 from the Department of Defense, and the D. Warren Brown Foundation.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Goldman JM, Melo JV. Chronic myeloid leukemia: advances in biology and new approaches to treatment. N Engl J Med. 2003;349: 1451-1464. [DOI] [PubMed] [Google Scholar]
- 2.Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001; 344: 1038-1042 [DOI] [PubMed] [Google Scholar]
- 3.le Coutre P, Mologni L, Cleris L, et al. In vivo eradication of human BCR/ABL-positive leukemia cells with an ABL kinase inhibitor. J Natl Cancer Inst. 1999;91: 163-168. [DOI] [PubMed] [Google Scholar]
- 4.May TS. Gleevec: tailoring to fit. Drug Discov Today. 2003;8: 188-189. [DOI] [PubMed] [Google Scholar]
- 5.Kantarjian H, Sawyers C, Hochhaus A, et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med. 2002;346: 645-652. [DOI] [PubMed] [Google Scholar]
- 6.Talpaz M, Silver RT, Druker BJ, et al. Imatinib induces durable hematologic and cytogenetic responses in patients with accelerated phase chronic myeloid leukemia: results of a phase 2 study. Blood. 2002;99: 1928-1937. [DOI] [PubMed] [Google Scholar]
- 7.Sawyers CL, Hochhaus A, Feldman E, et al. Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II study. Blood. 2002;99: 3530-3539. [DOI] [PubMed] [Google Scholar]
- 8.Gambacorti-Passerini CB, Gunby RH, Piazza R, Galietta A, Rostagno R, Scapozza L. Molecular mechanisms of resistance to imatinib in Philadelphia-chromosome-positive leukaemias. Lancet Oncol. 2003;4: 75-85. [DOI] [PubMed] [Google Scholar]
- 9.Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293: 876-880. [DOI] [PubMed] [Google Scholar]
- 10.Hochhaus A, Kreil S, Corbin AS, et al. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia. 2002;16: 2190-2196. [DOI] [PubMed] [Google Scholar]
- 11.Corbin AS, Buchdunger E, Pascal F, Druker BJ. Analysis of the structural basis of specificity of inhibition of the Abl kinase by STI571. J Biol Chem. 2002;277: 32214-32219. [DOI] [PubMed] [Google Scholar]
- 12.Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289: 1938-1942. [DOI] [PubMed] [Google Scholar]
- 13.Hochhaus A, Kreil S, Corbin A, et al. Roots of clinical resistance to STI-571 cancer therapy. Science. 2001;293: 2163. [PubMed] [Google Scholar]
- 14.Barthe C, Cony-Makhoul P, Melo JV, Mahon JR. Roots of clinical resistance to STI-571 cancer therapy. Science. 2001;293: 2163. [DOI] [PubMed] [Google Scholar]
- 15.Hofmann WK, Jones LC, Lemp NA, et al. Ph(+) acute lymphoblastic leukemia resistant to the tyrosine kinase inhibitor STI571 has a unique BCR-ABL gene mutation. Blood. 2002;99: 1860-1862. [DOI] [PubMed] [Google Scholar]
- 16.Branford S, Rudzki Z, Walsh S, et al. High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood. 2002;99: 3472-3475. [DOI] [PubMed] [Google Scholar]
- 17.Roche-Lestienne C, Soenen-Cornu V, Grardel-Duflos N, et al. Several types of mutations of the Abl gene can be found in chronic myeloid leukemia patients resistant to STI571, and they can pre-exist to the onset of treatment. Blood. 2002; 100: 1014-1018. [DOI] [PubMed] [Google Scholar]
- 18.Shah NP, Nicoll JM, Nagar B, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2: 117-125. [DOI] [PubMed] [Google Scholar]
- 19.von Bubnoff N, Schneller F, Peschel C, Duyster J. BCR-ABL gene mutations in relation to clinical resistance of Philadelphia-chromosome-positive leukaemia to STI571: a prospective study. Lancet. 2002;359: 487-491. [DOI] [PubMed] [Google Scholar]
- 20.Corbin AS, La Rosee P, Stoffregen EP, Druker BJ, Deininger MW. Several Bcr-Abl kinase domain mutants associated with imatinib mesylate resistance remain sensitive to imatinib. Blood. 2003; 101: 4611-4614. [DOI] [PubMed] [Google Scholar]
- 21.Steelman LS, Pohnert SC, Shelton JG, Franklin RA, Bertrand FE, McCubrey JA. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia. 2004;18: 189-218. [DOI] [PubMed] [Google Scholar]
- 22.Skorski T, Kanakaraj P, Nieborowska-Skorska M, et al. Phosphatidylinositol-3 kinase activity is regulated by BCR/ABL and is required for the growth of Philadelphia chromosome-positive cells. Blood. 1995;86: 726-736. [PubMed] [Google Scholar]
- 23.Skorski T, Bellacosa A, Nieborowska-Skorska M, et al. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathway. EMBO J. 1997;16: 6151-6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neshat MS, Raitano AB, Wang HG, Reed JC, Sawyers CL. The survival function of the Bcr-Abl oncogene is mediated by Bad-dependent and -independent pathways: roles for phosphatidylinositol 3-kinase and Raf. Mol Cell Biol. 2000;20: 1179-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kharas MG, Deane JA, Wong S, et al. Phosphoinositide 3-kinase signaling is essential for ABL oncogene-mediated transformation of B-lineage cells. Blood. 2004;103: 4268-4275. [DOI] [PubMed] [Google Scholar]
- 26.Kawauchi K, Ogasawara T, Yasuyama M, Ohkawa S. Involvement of Akt kinase in the action of STI571 on chronic myelogenous leukemia cells. Blood Cells Mol Dis. 2003;31: 11-17. [DOI] [PubMed] [Google Scholar]
- 27.Klejman A, Rushen L, Morrione A, Slupianek A, Skorski T. Phosphatidylinositol-3 kinase inhibitors enhance the anti-leukemia effect of STI571. Oncogene. 2002;21: 5868-5876. [DOI] [PubMed] [Google Scholar]
- 28.Marley SB, Lewis JL, Schneider H, Rudd CE, Gordon MY. Phosphatidylinositol-3 kinase inhibitors reproduce the selective antiproliferative effects of imatinib on chronic myeloid leukaemia progenitor cells. Br J Haematol. 2004;125: 500-511. [DOI] [PubMed] [Google Scholar]
- 29.Zhu J, Huang JW, Tseng PH, et al. From the cyclooxygenase-2 inhibitor celecoxib to a novel class of 3-phosphoinositide-dependent protein kinase-1 inhibitors. Cancer Res. 2004;64: 4309-4318. [DOI] [PubMed] [Google Scholar]
- 30.La Rosee P, Johnson K, Corbin AS, et al. In vitro efficacy of combined treatment depends on the underlying mechanism of resistance in imatinib-resistant Bcr-Abl-positive cell lines. Blood. 2004; 103: 208-215. [DOI] [PubMed] [Google Scholar]
- 31.La Rosee P, Corbin AS, Stoffregen EP, Deininger MW, Druker BJ. Activity of the Bcr-Abl kinase inhibitor PD180970 against clinically relevant Bcr-Abl isoforms that cause resistance to imatinib mesylate (Gleevec, STI571). Cancer Res. 2002; 62: 7149-7153. [PubMed] [Google Scholar]
- 32.Hoover RR, Mahon FX, Melo JV, Daley GQ. Overcoming STI571 resistance with the farnesyl transferase inhibitor SCH66336. Blood. 2002; 100: 1068-1071. [DOI] [PubMed] [Google Scholar]
- 33.Yang CC, Lin HP, Chen CS, Yang YT, Tseng PH, Rangnekar VM. Bcl-xL mediates a survival mechanism independent of the phosphoinositide 3-kinase/Akt pathway in prostate cancer cells. J Biol Chem. 2003;278: 25872-25878. [DOI] [PubMed] [Google Scholar]
- 34.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22: 27-55. [DOI] [PubMed] [Google Scholar]
- 35.Hollander M, Wolfe D. Nonparametric Statistical Methods. 2nd ed. New York, NY: John Wiley and Sons; 1999.
- 36.Calabretta B, Skorski T. BCR/ABL regulation of PI-3 kinase activity. Leuk Lymphoma. 1996;23: 473-476. [DOI] [PubMed] [Google Scholar]
- 37.Zou X, Calame K. Signaling pathways activated by oncogenic forms of Abl tyrosine kinase. J Biol Chem. 1999;274: 18141-18144. [DOI] [PubMed] [Google Scholar]
- 38.Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305: 399-401. [DOI] [PubMed] [Google Scholar]
- 39.O'Hare T, Pollock R, Stoffregen EP, et al. Inhibition of wild-type and mutant Bcr-Abl by AP23464, a potent ATP-based oncogenic protein kinase inhibitor: implications for CML. Blood. 2004;104: 2532-2539. [DOI] [PubMed] [Google Scholar]
- 40.Gorre ME, Ellwood-Yen K, Chiosis G, Rosen N, Sawyers CL. BCR-ABL point mutants isolated from patients with imatinib mesylate-resistant chronic myeloid leukemia remain sensitive to inhibitors of the BCR-ABL chaperone heat shock protein 90. Blood. 2002;100: 3041-3044. [DOI] [PubMed] [Google Scholar]
- 41.Yu C, Rahmani M, Almenara J, et al. Histone deacetylase inhibitors promote STI571-mediated apoptosis in STI571-sensitive and -resistant Bcr/Abl+ human myeloid leukemia cells. Cancer Res. 2003;63: 2118-2126. [PubMed] [Google Scholar]
- 42.Dai Y, Rahmani M, Pei XY, Dent P, Grant S. Bortezomib and flavopiridol interact synergistically to induce apoptosis in chronic myeloid leukemia cells resistant to imatinib mesylate through both Bcr/Abl-dependent and -independent mechanisms. Blood. 2004;104: 509-518. [DOI] [PubMed] [Google Scholar]
- 43.Yu C, Rahmani M, Conrad D, Subler M, Dent P, Grant S. The proteasome inhibitor bortezomib interacts synergistically with histone deacetylase inhibitors to induce apoptosis in Bcr/Abl+ cells sensitive and resistant to STI571. Blood. 2003; 102: 3765-3774. [DOI] [PubMed] [Google Scholar]
- 44.Yu C, Krystal G, Dent P, Grant S. Flavopiridol potentiates STI571-induced mitochondrial damage and apoptosis in BCR-ABL-positive human leukemia cells. Clin Cancer Res. 2002;8: 2976-2984. [PubMed] [Google Scholar]