

Abstract
The role of caspase-8 and its adaptor Fas-associated death domain (FADD) in lymphocyte apoptosis is well defined, but their functions in other hemopoietic lineages are not clear. We were unable to generate transgenic mice expressing dominant inhibitors of FADD or caspase-8 in hemopoietic cells, possibly because their expression may have precluded production of vital hemopoietic cells. When using a retroviral gene delivery system, fetal liver stem cells expressing a dominant-negative mutant of FADD (FADD-DN) were unable to generate myeloid or lymphoid cells upon transplantation into lethally irradiated mice. However, fetal liver stem cells expressing very low levels of the caspase-8 inhibitor cytokine response modifier A (CrmA) could reconstitute the hemopoietic system. This level of CrmA expression provided some protection against Fas ligand (FasL)–induced apoptosis and promoted accumulation of myeloid cells in the bone marrow, but it did not inhibit mitogen-induced proliferation of B or T lymphocytes. Using an in vitro colony formation assay, we found that fetal liver stem cells expressing FADD-DN, CrmA, or a dominant-negative mutant of caspase-8 could not proliferate in response to cytokine stimulation. These data demonstrate that the enzymatic activity of caspase-8 and its adaptor FADD are required for cytokine-induced proliferation of hemopoietic progenitor cells.
Introduction
Cell death in mammals can be induced via 2 distinct pathways1: one regulated by the B-cell lymphoma 2 (Bcl-2) protein family (often referred to as the “mitochondrial” or “intrinsic” pathway) and the other activated by so-called “death receptors,” a subgroup of the tumor necrosis factor receptor (TNF-R) family.2 “Death ligands,” such as Fas ligand (FasL), bind and cluster their cognate death receptors, which in turn recruit and cluster, via a homotypic interaction involving “death domains” (DDs), the adaptor protein Fas-associated DD (FADD) with or without the help of the adaptor TNF-R–associated DD (TRADD).2 When FADD binds to Fas or other death receptors, it is able to recruit, via the homotypic interaction of death effector domains (DEDs), pro–caspase-8 (and in humans also pro–caspase-10). Pro–caspase-8 has low inherent enzymatic activity, but, when it is aggregated in the DISC (death-inducing signaling complex) by ligated death receptors, a critical level of activity is achieved, and the zymogens are able to activate each other.2 The activated caspase-8 can then proteolytically activate downstream so-called effector caspases, which cleave vital cellular proteins and thereby cause cell demolition.
The role of death receptors in hemopoietic progenitors and myeloid cells has not yet been studied in detail. Fas-deficient mutant Lpr mice have normal numbers of granulocytes and macrophages, although a small increase in numbers of myeloid colony-forming cells in the bone marrow has been reported.3 In contrast, transgenic mice overexpressing Bcl-2 in the myeloid lineage under the control of the hMRP8 promoter develop progressive monocytosis and die by 1 year from neutropenia because granulopoiesis favors formation of immature cell types.4 Of interest, hMRP8-Bcl2/Lpr double-mutant mice are predisposed to acute myeloblastic leukemia.3 These results demonstrate that the Fas death receptor–signaling and the Bcl-2–regulated apoptosis pathways are distinct in myeloid cells and that defects in both can synergize to cause leukemia.
To assess the function of all death receptors in the control of programmed death of myeloid cells, we attempted to generate transgenic mice expressing a dominant-interfering mutant of FADD, FADD-DN, or an inhibitor of caspase-8 enzymatic activity, cytokine response modifier A (CrmA), throughout the hemopoietic compartment using the Vav gene promoter. We were unable to generate such mice and speculate that this could be due to embryonic lethality caused by defects in hemopoiesis.
Mice deficient in FADD or caspase-8 die during embryogenesis, and their cells are resistant to death receptor–induced apoptosis.5-9 Transgenic expression of a dominant-interfering mutant of FADD (FADD-DN) does not only block death receptor–induced apoptosis but also inhibits mitogen- or antigen-induced activation and proliferation of mature T cells.10,11 Similar defects were found in FADD–/– T cells in chimeric mice generated by injection of FADD–/– embryonic stem (ES) cells into rag-deficient blastocysts.6 Defects in T-cell proliferation were also found in a small subset of patients with autoimmune lymphoproliferative syndrome with a mutation in the caspase-8 gene12 and in gene-targeted mice in which the caspase-8 gene was inactivated only in T lymphocytes.13 Hence, both FADD and caspase-8 are required for cell activation and proliferation, at least in the T-lymphoid lineage. T cells from mice lacking Fas, TNF-R1, or both receptors proliferate normally in response to mitogens or antigens.10,14 This may indicate that other death receptors act upstream of FADD and caspase-8 in T-cell proliferation. Alternatively, mitogens and antigens may activate T-cell proliferation via FADD and caspase-8 through a mechanism that is independent of death receptors.
To investigate the role of FADD and caspase-8 in proliferation of hemopoietic progenitor cells, we infected fetal liver cells in vitro with retroviruses encoding dominant inhibitors of FADD or caspase-8 function. Our analysis demonstrates that both FADD and caspase-8 are required for cytokine-induced proliferation of hemopoietic progenitor cells.
Materials and methods
Transgenic mice
The VavP transgene vector15 was used to generate VavP–FLAG–FADD-DN and VavP-FLAG-CrmA mice. The vector was digested with SfiI and NotI, and the relevant cDNA was subcloned into the 9.9-kb vector. The inserts encoding FLAG-tagged FADD-DN or FLAG-tagged CrmA were prepared from pEF–FLAG–FADD-DN10 or pEF-FLAG-CrmA,1 respectively. The pIC19H backbone (2.7 kb) was removed from the vector construct by digestion with HindIII and electrophoresis of digests through low melting point agarose. Vectors were then purified using Elutip-d (Schleicher and Schuell, Keene, NH), precipitated with ethanol and dissolved in sterile 10 mM Tris (tris(hydroxymethyl)aminomethane)–HCl (pH 7.4), 0.1 mM EDTA (ethylenediaminetetraacetic acid). The constructs containing FLAG–FADD-DN or FLAG-CrmA with elements of the SRα expression vector and the simian virus 40 (SV40) late region polyadenylation signal were injected into the pronucleus of C57BL/6J mouse zygotes and transferred into pseudo-pregnant females. Transgenic pups were identified by dot blot hybridization or by polymerase chain reaction (PCR) amplification of DNA from whole blood or tail biopsies.
PCR analysis
Heparinized blood (2 μL) was diluted in 38 μL water and heated to 95°C in a thermal cycler for 15 minutes. Primers to amplify the SV40 late-region polyadenylation signal present in the transgene were 5′-GCC GCA GAC ATG ATA AGA TAC ATT GAT G and 5′-AAA ACC TCC CAC ACC TCC CCC TGA A. Tail DNA for PCR was prepared from 1-cm biopsies by digestion with proteinase K (Roche, Indianapolis, IN), precipitation with isopropanol, washing with 70% ethanol, and resuspension in 10 mM Tris-HCl, 1 mM EDTA, pH 8.0 (TE).
Dot blot hybridization
Tail DNA was phenol/chloroform extracted and washed, and duplicate 6-μg samples were spotted onto Hybond N+ membranes (Amersham Pharmacia Biosciences [APB], Piscataway, NJ). Membranes were probed with an α-32P–deoxyadenosine triphosphate (dATP)–labeled fragment of SV40 sequence present in the transgene constructs. After probing, membranes were washed in 2 × standard saline citrate/0.1% sodium dodecyl sulfate (SDS) at 65°C for 20 minutes. Bound probe was detected by exposing membranes to AR Hyperfilm (APB).
Tissue culture
Jurkat cells and primary mouse cells were cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 13 μM folic acid, 250 μM l-asparagine, 50 μM 2-mercaptoethanol, and 10% fetal calf serum (Trace Bioscience, Castle Hill, NSW, Australia) (FCS). Stably transfected Neuro-2A cells expressing mouse FasL16 and parental Neuro-2A cells were grown in DMEM containing 10% fetal calf serum. Both cell lines possessed the selectable neo marker and were maintained in culture with 0.5 mg/mL geneticin (Life Technologies, Gaithersburg, MD). For cell death assays, Neuro-2A cells were plated 24 hours prior to addition of target cells in DMEM + 10% FCS without addition of geneticin.
Immunofluorescence staining, FACS analysis, and cell sorting
Single-cell suspensions from thymus, lymph nodes (LNs), spleen, or bone marrow (BM) were prepared in balanced salt solution containing 2% fetal calf serum. Cells were stained with one or a combination of monoclonal antibodies to B220 (RA3-6B2), CD4 (H129.19), CD8 (YTS 169), Gr-1 (RB6-8C5), Ly5.1 (A2D1.7), Ly5.2 (5.450.15.2), macrophage antigen-1 (Mac-1; MI/70), Ter119 (TER-119), or thymocyte 1 antigen (Thy-1; T24.31.2) conjugated with fluorescein isothiocyanate (FITC), R-phycoerythrin (R-PE), cyanine 5 (Cy5) (Molecular Probes, Eugene, OR) or biotin (Sigma, St Louis, MO), washed and stained with propidium iodide (PI) to distinguish live and dead cells. Streptavidin conjugated with FITC, R-PE, or Tricolor (Caltag, San Francisco, CA) was used to reveal biotinylated antibodies. Cells were analyzed in a fluorescence-activated cell scan (FACScan; Becton Dickinson, Franklin Lakes, NJ) or sorted in a DIVA (Becton Dickinson) or MoFlo (Cytomation, Denver, CO) cell sorter.
For intracellular staining, cells were fixed in 1% paraformaldehyde for 10 minutes on ice, washed twice, and stained with anti-FLAG M2 (Sigma) or anti–human FADD (Transduction Laboratories, Lexington, KY) monoclonal antibodies (mAbs) in balanced salt solution with 2% FCS and 0.3% saponin (Sigma) for 30 minutes on ice. Cells were then washed and incubated with FITC-conjugated goat anti–mouse immunoglobulin G (IgG) antibodies (Silenus Laboratories, Melbourne, Australia).
Cell-death assays
Cell-death assays were performed in 96-well flat-bottom tissue culture plates (Falcon, Franklin Lakes, NJ). FACS-sorted CD4+CD8+ thymocytes, B cells (Thy-1–Gr-1–Mac-1–Ter119– cells from LNs), T cells (B220– Gr-1–Mac-1–Ter119– cells from LNs), and granulocytes (Gr-1+ Mac-1+ from bone marrow) were cultured in FMA at a concentration of 2.5 × 105/mL. Activated T cells were generated by culturing spleen cells for 3 days with 2 μg/mL concanavalin A (Sigma) and then for 2 days in interleukin 2 (IL-2). Activated B cells were generated by culturing spleen cells for 3 days with 20 μg/mL lipopolysaccharide (LPS; Difco Laboratories, Detroit, MI) in the presence of IL-2, IL-4, and IL-5. These cytokines were produced by X63/0 hybridoma cells transfected with expression vectors for these cytokines.17 Cytokine withdrawal assays were performed on activated B or T cells that had been repeatedly washed through an FCS cushion. Cell death stimuli included coculture with Neuro-2A cells expressing FasL,16 recombinant FLAG-tagged human FasL (Alexis, Lausen, Switzerland) oligomerized with 1 μg/mL M2 anti-FLAG mAb, etoposide (Pharmacia Upjohn, Uppsala, Sweden) and dexamethasone (Sigma) at the specified concentrations. For γ-irradiation, a 60Co source was used. Cell viability was assessed by staining with propidium iodide with or without additional staining with Annexin V–FITC, followed by analysis in a FACScan.
Hemopoietic reconstitution of lethally irradiated mice
All experiments with animals were performed according to the guidelines of the Melbourne Health Research Directorate Animal Ethics Committee. Six- to 12-week-old C57BL/6.SJL-Ptprca(Ly5.1) mice were irradiated twice with 5.5 Gy 2 to 3 hours apart and then injected into the tail vein with 1 × 106 fetal liver cells. Mice were given 1.67 mg/mL neomycin (Sigma) in the drinking water for 2 weeks after irradiation. The extent of hemopoietic reconstitution was assessed 8 weeks after fetal liver cell transplantation by immunofluorescent staining and FACS analysis of blood leukocytes.
Retroviral caspase-8, CrmA, and FADD expression constructs
The murine stem cell virus–internal ribosome entry site–green fluorescent protein (MSCV-IRES-GFP) retroviral construct was based on the Moloney murine leukemia virus.18 The retroviral vector polylinker sequence contains BglII, XhoI, and EcoRI restriction sites. Inserts were amplified or cut directly from pEF-based vectors with BglII and EcoRI or XhoI, and EcoRI-compatible restriction sites. Inserts were ligated into the retroviral vector polylinker sequence. The inserts and original vectors were FLAG-tagged CrmA from pEF-FLAG-CrmA,1 FLAG-CrmA with an active site mutation (mutation T291R) from pEF-FLAG-CrmA, caspase-8 from pCR3.V78–caspase-8–FLAG–C-terminal, caspase-8 active site mutant (mutation C360G) from pCR3.V78–caspase-8 mutant–FLAG–C-terminal (gifts of Prof J. Tschopp, Institute for Biochemistry, University of Lausanne, Lausanne, Switzerland), FADD from pEF-EE-FADD, FADD-DN from pEF–FLAG–FADD-DN,10 FADD-DN with a mutation that abolishes binding to Fas from point mutagenesis (V121N) of MSCV–FLAG–FADD-DN–IRES–GFP using a Quick Change mutagenesis kit (Qiagen, Valencia, CA), caspase-9 dominant negative (C287S) from pEF–FLAG–caspase-9–DN and cytoplasmic region of Fas from pEFBos-FLAG-cytFas. The Phoenix cell line was used for retroviral packaging (a gift of Dr G. Nolan, Stanford University, Stanford, CA) and transfected with retroviral constructs as described.18 Viral supernatant was harvested and filtered through a 0.8-μm Millex syringe filter unit (Millipore, Bedford, MA).
Retroviral infection
Fetal liver cells were harvested from E13 C57BL/6 embryos and cultured in DMEM containing 20% FCS and 100 ng/mL stem cell factor (purified from culture supernatants of transfected Pichia pastoris [a gift of Dr H. Martin of Walter and Eliza Hall Institute of Medical Research]), 50 ng/mL thrombopoietin (Kirin Brewery, Tokyo, Japan), 500 ng/mL Flk ligand (purified from supernatant of transfected Chinese hamster ovary [CHO] DHFR– cells), and 100 U/ml IL-6 (both gifts of Dr H. Martin) for 24 hours at 37° prior to infection. Wells of a 6-well plate (nontreated; Falcon) were coated with 4 μg/cm2 RetroNectin (Takara Bio, Otsu, Japan) in 2 mL phosphate-buffered saline (PBS), and cells were infected according to the manufacturer's instructions. Infected cells were harvested from plates using cell dissociation buffer (Invitrogen, Carlsbad, CA), washed, and resuspended in PBS for injection into irradiated mice or resuspended in balanced salt solution containing 10% FCS and 4 μg/mL propidium iodide for FACS sorting. Cells for agar assays were sorted using a MoFlo sorter (Cytomation) to collect viable GFP+PI– cells.
Jurkat cells expressing the ecotropic retroviral receptor (a gift of Prof G. Nolan) were infected as described earlier in this section and sorted in a MoFlo cell sorter. GFP+PI– cells were collected and then used in cell death assays or for preparation of cell lysates for Western blot analysis.
Western blot analysis
Cells (1 × 106 cells/20 μL) were lysed in RIPA buffer (PBS with 1% NP-40 [Nonidet P-40, [Octylphenoxy]polyethoxyethanol], 0.5% sodium deoxycholate, and 0.1% SDS) containing 2 μM pefabloc, 1 μg/mL aprotinin, and 1 μg/mL leupeptin. Samples were then prepared in an equal volume of 2 × loading buffer, (125 mM Tris-HCl pH 6.8, 2% SDS, 20% glycerol, 2% 2-mercaptoethanol, and 0.02% bromophenol blue) and resolved by electrophoresis on 4% to 20% gradient Tris-glycine polyacrylamide gels (Invitrogen) in buffer containing SDS. Resolved proteins were transferred to nitrocellulose membranes (Hybond C-extra; APB) by electroblotting. Membranes were blocked for 2 hours in PBS containing 5% skim milk and 0.1% Tween-20 (Sigma) and probed with 3 μg/mL M2 anti-FLAG (Sigma), N6 anti-Hsp70 (a gift of Dr R. Anderson, Peter MacCallum Cancer Institute, Melbourne, Australia), or 1 μg/mL clone 1 anti–human FADD mAb (Transduction Laboratories). Peroxidase-conjugated sheep anti–mouse Ig antibodies (Silenus Laboratories) and electrochemiluminescence (ECL) reagent were used for detection.
Hemopoietic colony assays
Fetal liver cells infected with retrovirus and expressing GFP (sorted by FACS) were plated at 1.25 × 104 cells/mL in DMEM containing 10% bovine calf serum (HyClone, Logan, UT), 100 ng/mL stem cell factor (SCF), 2500 U/mL IL-3, 2 U/mL erythropoietin (EPO), and 0.3% Bacto agar (Difco Laboratories) and incubated at 37°C in a humidified 10% CO2 incubator. After 7 days, colonies were counted, and agar cultures were fixed with 2.5% glutaraldehyde in PBS.
Results
Failure to generate transgenic mice expressing FADD-DN or CrmA throughout the hemopoietic system
The promoter elements of the Vav gene were used in an attempt to drive expression of FADD-DN or CrmA in all hemopoietic cell lineages. Both transgene-encoded proteins were FLAG-tagged to facilitate detection of expression. PCR and Southern blot analysis showed that 20 primary mice of a total of 115 pups, generated by pronuclear injection into C57BL/6 oocytes, contained the FLAG–FADD-DN transgene (Figure 1A-B) and 20 of 131 contained the FLAG-CrmA transgene (Figure S1A-B, available on the Blood website; see the Supplemental Figures link at the top of the online article). Breeding strains were established from 4 FLAG–FADD-DN and 4 FLAG-CrmA primary transgenic mice. PCR screening of tail DNA showed that the transmission frequency of the transgene was as expected, with a rate of 46 of 97 FLAG–FADD-DN–positive and 49 of 102 FLAG-CrmA–positive pups, respectively (Figure 1B; Figure S1B). However, neither FLAG–FADD-DN nor FLAG-CrmA protein expression could be detected in lymphoid or myeloid cells of any transgene-positive mice of any of these lines by Western blotting or flow cytometric analysis using antibodies against FADD (which bind FADD-DN) or FLAG (Figure 1C-D; Figure S1C-D). Moreover, these transgenic mice all had normal numbers of myeloid and lymphoid cells in their hemopoietic organs (not shown).
Figure 1.

Generation and analysis of VavP–FLAG–FADD-DN transgenic mice. (A) Tail DNA was extracted from primary and first generation VavP–FLAG–FADD-DN transgenic mice. Dot hybridization to a 32P-labeled SV40 probe was used to detect the presence of the transgene. DNA was spotted in duplicate, and transgene copy number can be estimated by comparing intensity of hybridization to that of control SV40 DNA of known concentration. These are typical results with 2 examples highlighting a transgene positive (solid line) and a negative (broken line) mouse. (B) PCR using SV40-specific primers was used to detect transgene sequences in primary transgenic mice or first-generation descendants. Results are representative of multiple screens. (C) Western blots on extracts of 106 cells from spleen or thymus of VavP–FLAG–FADD-DN transgene-positive mice were probed with either anti–human FADD mAb (which recognizes FLAG–FADD-DN) or anti-FLAG mAb. (D) In an alternative method for detecting transgene expression, cells from lymph nodes (LNs), thymus, or bone marrow of VavP–FLAG–FADD-DN transgenic mice were stained with antibodies to Thy1, CD8, CD4, Gr-1, and/or Mac-1, sorted in a FACS machine, fixed, permeabilized, stained with anti-FLAG mAb, and analyzed in a FACScan. Intracellular staining of thymocytes from Lck–FLAG–FADD-DN transgenic mice10 with anti-FLAG mAb was the positive control. Results are representative of multiple screens (> 12 PCR-positive mice). (E) Granulocytes (Mac-1+Gr-1+) were FACS-sorted from bone marrow of VavP–FLAG–FADD-DN transgene-positive progeny and control mice and cultured for 24 hours in medium alone or in the presence of oligomerized recombinant FasL (100 ng/mL). Cell viability was determined by staining with PI and FACS analysis. Numbers in quadrants represent percentages of cells within each staining profile. FSC indicates forward light scatter. Results shown are representative of more than 12 assays.
Immature thymocytes (CD4+CD8+), granulocytes (Gr-1+Mac-++), monocytes (Gr-1–Mac-1+), mature T cells (Thy1+), or B cells (B220+), activated B cells or monocytes from transgene-positive mice were cultured in the presence of oligomerized FasL. Cell viability was assessed by staining with PI and FACS analysis and compared with viability of cells from wild-type (wt) and Fasdeficient mutant Lpr mice (Figure 1E; Figures S1E, S2-S3). Cells from VavP–FLAG–FADD-DN and VavP-FLAG-CrmA mice showed similar extent of FasL-induced cell death as did wt cells. These results are consistent with the failure to detect transgenic protein in these mice. At the same time, we were readily able to generate transgenic mice expressing Bcl215 under the control of the Vav promoter, indicating that selection against transgenic mice expressing the VavP–FLAG–FADD-DN or VavP-FLAG-CrmA constructs may have occurred. This may indicate that interfering with FADD or caspase-8 function causes embryonic lethality resulting from hemopoietic failure.
Low levels of CrmA but not FADD-DN can be expressed broadly in hemopoietic cells using retroviral vectors
In an attempt to overcome potential embryonic lethality and to generate animals expressing FLAG–FADD-DN or FLAG-CrmA throughout the hemopoietic compartment, embryonic day 13 (E13) C57BL/6-(Ly5.2) fetal liver stem cells were infected in vitro with replication-incompetent retroviruses encoding GFP alone (control), FLAG–FADD-DN plus GFP or FLAG-CrmA plus GFP (GFP expression linked by an IRES). Infected fetal liver cells were then injected into lethally irradiated C57BL/6-Ly5.1 recipient mice, which were killed for analysis 8 to 10 weeks later. Approximately 85% of the mice survived reconstitution with GFP or FLAG-CrmA-GFP–expressing fetal liver stem cells and 30% to 90% of their leukocytes expressed GFP, with no significant differences in frequency of GFP+ cells between these 2 groups (Figure 2A). The level of GFP expression was, however, higher in cells infected with the control GFP virus than in those infected with the FLAG-CrmA/IRES-GFP virus. This may have been due to a selection bias favoring low expression of FLAG-CrmA (and hence GFP), or, given the nature of the vector, it may have been due to translational drop-off after the IRES. The FLAG-CrmA-GFP construct gave rise to FLAG-CrmA protein expression (relatively low levels) in thymocytes, granulocytes, macrophages, and splenocytes (Figure 2B). The levels of FLAG-CrmA appeared even lower in mature Thy1+ T cells sorted from lymph nodes or spleen than in thymocytes. In mice reconstituted with fetal liver cells infected with the FLAG–FADD-DN–GFP retrovirus no leukocytes expressing FADD-DN could be found (not shown). These results indicate that it is possible to achieve long-term pan-hemopoietic expression of low levels of CrmA but not FADD-DN.
Figure 2.
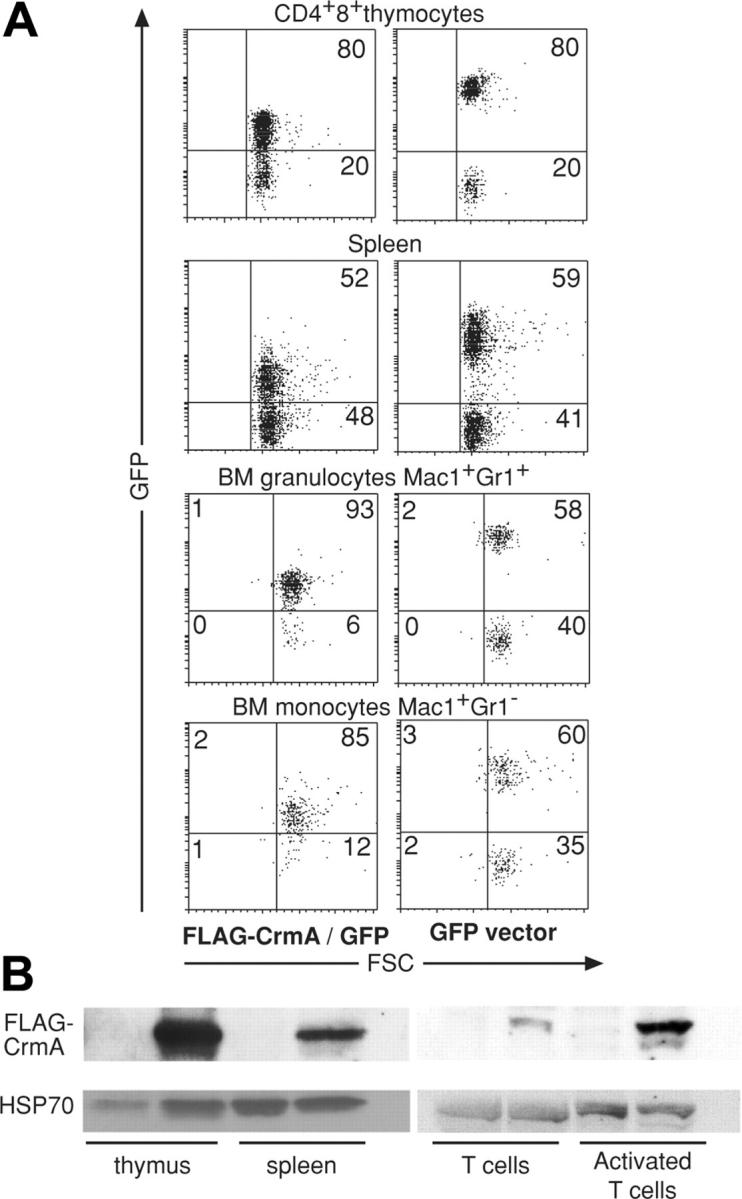
Analysis of mice reconstituted with FLAG-CrmA-GFP retrovirus-infected (E14) fetal liver cells. (A) Cells were harvested from mice 8 weeks after reconstitution and stained with mAbs to cell lineage-specific markers and analyzed by FACS. GFP expression was compared between mice reconstituted with fetal liver cells infected with FLAG-CrmA-GFP–expressing virus or control (GFP alone) virus. Numbers in quadrants represent percentages of cells within each staining profile. (B) Lysates from cells of hemopoietic organs were used for Western blot analysis using anti-FLAG mAb to assess levels of FLAG-CrmA protein expression. Results are representative of more than 6 analyses.
CrmA expression protects cells from FasL-induced apoptosis but not from stimuli activating the Bcl-2–regulated apoptotic pathway
As expected from experiments with transgenic mice expressing FLAG-CrmA under control of a T-cell–specific promoter,19 thymocytes expressing FLAG-CrmA-GFP were resistant to apoptosis induced by oligomerized-soluble FasL or membrane-bound FasL (expressed on the surface of Neuro2A cells stably transfected with a FasL expression vector) (Figure 3A). The extent of protection was similar to that observed in Fas-deficient thymocytes from Lpr mutant mice. Granulocytes and monocytes expressing FLAG-CrmA-GFP were also resistant to FasL, but to a lesser degree than cells from Lpr mice (Figure 3A). Mature T cells and activated B cells showed only modest resistance to FasL, probably reflecting the lower level of FLAG-CrmA expression in these cells.
Figure 3.

FLAG-CrmA expression in hemopoietic cells from reconstituted mice inhibits FasL-induced apoptosis but does not protect against apoptosis induced by Bcl-2–regulated death stimuli. (A) Immature thymocytes (CD4+CD8+), BM granulocytes (Mac-1+Gr-1+), BM monocytes (Mac-1+Gr-1–), naive LN B cells (B220+), and naive LN T cells (Thy1+) were FACS sorted from mice reconstituted with fetal liver cells infected with a FLAG-CrmA-GFP or a control GFP retrovirus and cultured for 24 hours in medium alone or in the presence of oligomerized recombinant FasL (100 or 10 ng/mL). B cells and monocytes were activated in vitro with LPS plus IL-2, IL-4, and IL-5 or with interferon-γ (IFNγ), respectively, prior to death assays. Cells, isolated and prepared as described in panel A, were also cocultured with Neuro2A cells expressing membrane-bound FasL. (B) Bcl-2–regulated apoptosis was induced by γ-irradiation or treatment with etoposide or dexamethasone. Comparisons are made with cells from Fas-deficient Lpr mutant mice. (C) Cells were also cultured in medium alone, and viability was assessed by PI staining and FACS analysis at the specified time points. Data represent mean ± SD from 3 mice of each type.
As expected,1,19 cells expressing FLAG-CrmA-GFP showed normal sensitivity to stimuli that induce apoptosis through the Bcl-2–regulated pathway, such as γ-irradiation, treatment with dexamethasone or etoposide (Figure 3B), or cytokine deprivation (Figure 3C). These results demonstrate that the retroviral delivery system allows sufficient CrmA expression to inhibit death receptor–induced apoptosis in cells of multiple hemopoietic lineages.
Effect of FLAG-CrmA-GFP expression on numbers of hemopoietic cells
Because CrmA inhibits apoptosis induced by all known death receptors (reviewed in Ashkenazi and Dexit2 and Strasser et al20), and because we were able to express functional levels of FLAG-CrmA in many hemopoietic lineages (Figures 2, 3), we investigated the role of death receptor–mediated apoptosis in the homeostasis of myeloid cells. Leukocytes in the hemopoietic organs of mice reconstituted with GFP-expressing or FLAG-CrmA-GFP–expressing retroviruses were quantified by cell counting in a hemocytometer and by determining numbers of GFP+ cells by flow cytometry. Figure 4 shows that there was no significant difference in total cellularity or percentage of GFP+ cells in spleen, lymph nodes, thymus, and bone marrow of mice reconstituted with GFP or FLAG-CrmA-GFP retrovirus-infected fetal liver cells. There was a tendency toward accumulation of FLAG-CrmA-GFP–expressing monocytes and granulocytes, but because of the wide variability in reconstitution efficiency these differences were not statistically significant.
Figure 4.

Organ cellularity and numbers of retroviral construct–expressing cells in reconstituted mice. (A) The total number of cells per organ in mice reconstituted with fetal liver stem cells infected with FLAG-CrmA-GFP or GFP control retrovirus was quantified by preparing single-cell suspensions and counting cells in a hemocytometer. (B) The proportion of GFP-expressing cells in each organ was quantified by FACS analysis. (C) The total number of GFP+ granulocytes and monocytes in bone marrow (2 femurs and 2 tibias) of reconstituted mice was determined directly by FACS analysis. Data represent mean ± SD from organs of 3 mice of each type. ▪ indicates FLAG-CrmA/GFP; and  , GFP vector.
, GFP vector.
FADD-DN and CrmA inhibit colony formation by hemopoietic progenitor cells
The failure to generate hemopoietic cells expressing FADD-DN in irradiated mice reconstituted with fetal liver cells infected with the FLAG–FADD-DN–GFP retrovirus may indicate that FADD-DN, even at low expression levels, leads to a failure in progenitor cell proliferation. To investigate specifically the role of FADD and caspase-8 in hemopoietic progenitor cell proliferation, in vitro colony-forming assays were performed. Fetal (E13) liver cells were infected with a GFP control retrovirus or retroviruses encoding GFP plus FLAG-tagged CrmA (FLAG-CrmA-GFP) or FLAG-tagged FADD-DN (FLAG–FADD-DN–GFP). Infected cells were purified by cell sorting (GFP+), plated in soft agar containing SCF, EPO plus IL-3, and colonies were counted after 7 days of incubation. Expression of FLAG-CrmA-GFP or FLAG–FADD-DN–GFP strongly inhibited proliferation and colony formation by hemopoietic progenitor cells. Compared with control GFP virus–expressing cells, which produced 280 ± 20 colonies per 104 cells plated, only 24 ± 3 or 20 ± 1 colonies were formed by FLAG-CrmA-GFP or FLAG–FADD-DN–GFP virus–infected cells, respectively (Figure 5A-B). These results indicate that FADD and caspase-8 are essential for cytokine-induced proliferation of hemopoietic progenitor cells.
Figure 5.

Expression of CrmA or FADD-DN inhibits colony formation by hemopoietic progenitor cells. Fetal liver cells from E14 C57BL/6 embryos were infected with retroviruses encoding FLAG-CrmA-GFP, FLAG–FADD-DN–GFP, or GFP alone (control vector) for 48 hours in the presence of IL-6, mSCF, TPO, and Flk ligand (Flk)L. After infection, cells were sorted for GFP expression and cultured for 7 days in agar with SCF, IL-3, and EPO. (A) Compared with GFP vector–expressing cells, hemopoietic progenitor cells expressing FLAG-CrmA or FLAG–FADD-DN formed very few colonies. Colonies were viewed under a Leica DMLIL microscope (Leica, Wetzlar, Germany) using a 20× objective (0.0-0.4 NA) and photos were taken with an Axiocam camera and processed using Axiovision software (both from Zeiss, Hallbergmoos, Germany). (B) The numbers of colonies are shown graphically with data representing mean ± SD from 6 to 9 independent experiments for each vector. CFU indicates colony-forming units. (C) Similar experiments were performed with fetal liver cells infected with retroviruses encoding GFP alone (vector), wt FLAG-CrmA, mutated FLAG-CrmA (CrmAmt), which does not inhibit caspase-8, dominant-interfering catalytically inactive caspase-8 (FLAG–caspase-8–DN) or dominant-interfering catalytically inactive caspase-9 (FLAG–caspase-9–DN). (D) Retroviruses encoding GFP alone (vector), FLAG–FADD-DN, a mutant of FLAG–FADD-DN that is unable to bind to the intracellular region of Fas26 or the cytoplasmic region of Fas (FLAG-cFas), were also used. The numbers of colonies are shown graphically with data representing arithmetic mean ± SD from 6 to 9 independent experiments. (E) Freshly isolated fetal liver cells, not cultured and not enriched for progenitor cells, were plated in agar culture in the presence of Z-Val-Ala-Asp-(ome)-fluoromethyl-ketone (Z-VAD-fmk) at the specified concentrations together with optimal cytokines. Colonies were counted after 7 days in culture, and numbers represent means ± SDs from 3 separate experiments.
Catalytic activity of caspase-8 appears to be required for hemopoietic progenitor cell proliferation
The inhibition of proliferation and colony formation by FLAG-CrmA is most likely due to inhibition of caspase-8 function. To investigate this further, we used several retroviral constructs encoding mutated forms of CrmA or caspase-8. Expression of a CrmA mutant (T291R), which is unable to block the enzymatic activity of caspase-8,21 did not inhibit colony formation by hemopoietic progenitor cells (Figure 5C). This indicates that CrmA inhibits proliferation by interfering with the enzymatic activity of caspases. The notion that caspase catalytic activity is required for progenitor cell proliferation is further supported by the finding that Z-VAD-fmk (a chemical pan-caspase inhibitor) substantially reduced myeloid colony formation in agar (Figure 5E).
CrmA shows selective binding to and inhibition of different caspases, preferring caspase-1 followed by caspase-8 over the other caspases.22 To specifically address the role of caspase-8, we constructed a retrovirus-encoding caspase-8 with a point mutation at the catalytic site (FLAG–caspase-8–DN–GFP), which acts as a dominant-interfering mutant.23 As a control, we used a retrovirus encoding a dominant-interfering mutant of caspase-9 (FLAG–caspase-9–DN–GFP).24 Hemopoietic colony formation was significantly inhibited by FLAG–caspase-8–DN whereas FLAG–caspase-9–DN had no effect (Figure 5C).
To ensure that all the expression constructs were producing functional protein, we infected Jurkat cells expressing the mouse ecotropic virus receptor (Jurkat-EcoR cells) and analyzed infected cells by Western blotting and apoptosis assays. All proteins were expressed at readily detectable levels (Figure S4A). As expected from previous analyses of transgenic mice19 or transfected cells,1,23 FLAG-CrmA and FLAG–caspase-8–DN inhibited FasL-induced apoptosis in these cells but had no effect on etoposide-induced cell death (Figure S4B). Although mutant FLAG-CrmA was expressed in infected Jurkat-EcoR cells (Figure S4A), it had no effect on FasL-induced apoptosis (Figure S4B). In agreement with studies of hemopoietic cells lacking caspase-9,25 FLAG–caspase-9–DN had a small inhibitory effect on etoposide-induced apoptosis but no effect on Fas-mediated cell death (Figure S4B). These results demonstrate that all our retroviral vectors expressed functional proteins and indicate that the enzymatic activity of caspase-8 but not that of caspase-9 is critical for proliferation of hemopoietic progenitor cells.
Interaction with Fas is not required for FADD-DN to inhibit proliferation of hemopoietic progenitor cells
It is not clear whether FADD is activated in hemopoietic progenitor cell proliferation by death receptors or by a different mechanism. In an attempt to investigate the role of death receptors in this process, a point mutation that abrogates the ability of FADD to bind to Fas, but does not affect its other protein-protein interactions, was introduced into the FLAG–FADD-DN–GFP construct.26 Expression of this mutated FLAG–FADD-DN protein in fetal liver cells inhibited hemopoietic colony formation, indicating that interaction with death receptors is not required for FADD-DN to inhibit cell proliferation (Figure 5D). This suggests that growth stimuli may activate proliferation via FADD and caspase-8 through a mechanism that does not involve death receptor ligation.
Enforced expression of death domains other than that of FADD does not inhibit hemopoietic colony formation
To ensure that the consequences of FLAG–FADD-DN and mutated FLAG–FADD-DN were not due to a nonspecific effect of overexpression of any death domain, we generated a retroviral expression construct encoding the cytoplasmic (death domain containing) region of Fas (FLAG-cFas). Hemopoietic progenitor cells expressing this protein were able to proliferate and form colonies normally in agar (Figure 5D). Therefore, the effects seen with FLAG–FADD-DN and mutated FLAG–FADD-DN are unlikely due to nonspecific effects of overexpression of any death domain but rather a consequence of their ability to interfere, by acting as true dominant negatives, with endogenous FADD function.
Jurkat cells infected with FLAG–FADD-DN, mutated FLAG–FADD-DN, or cFas retroviral constructs produce protein
Jurkat cells expressing the murine ecotropic retrovirus receptor were infected with retroviral constructs encoding FLAG–FADD-DN, mutated FLAG–FADD-DN or FLAG-cFas (all containing IRES-GFP), sorted by FACS for GFP expression and lysed for Western blot analysis or cultured for apoptosis assays. All retroviral expression constructs gave rise to proteins of the expected size (Figure S4A). As expected,10 FLAG–FADD-DN but not mutated FLAG–FADD-DN or FLAG-cFas protected Jurkat cells from Fas-mediated apoptosis, but none had any effect on etoposide-induced cell death (Figure S4B).
Inhibition of hemopoietic colony formation by FLAG–FADD-DN, FLAG-CrmA, and FLAG–caspase-8–DN appears not to be associated with increased cell death
Reduced colony formation by hemopoietic progenitor cells expressing FLAG–FADD-DN, FLAG-CrmA, or FLAG–caspase-8–DN could theoretically be due to slower cell cycling, induction of cell death, or inhibition of entry into the cell cycle. To investigate this, we analyzed fetal liver cells expressing the various constructs by FACS to determine their viability and cell-cycle distribution. As controls, we used retroviral vectors encoding GFP alone, FADD, or caspase-8 (both also encoding GFP). Expression of FLAG–FADD-DN (76% ± 8% viability), mutant FLAG-cFas (76% ± 6%), FLAG-CrmA (77% ± 8%), or FLAG–caspase-8–DN (75% ± 7%) did not increase the numbers of dead cells compared with fetal liver cells infected with control (GFP) vector alone (74% ± 5%; Figure 6A-B). In contrast, expression of wt FADD (52% ± 9% viability) or caspase-8 (46% ± 3%) did induce significant cell killing (Figure 6A-B). In addition, DNA staining demonstrated that there was no significant difference in cell-cycle distribution between cells expressing FLAG–FADD-DN, FLAG-CrmA, FLAG–caspase-8–DN, or control GFP-expressing cells (Figure 6C). These results indicate that the inhibition of hemopoietic colony formation by FADD-DN, FADD-DN mutant, CrmA, and caspase-8–DN expression is not primarily due to effects on cell viability or progression of cells through the cycle after they have entered the first S phase, but it may rather be due to defects in entering the cell cycle from the G0 state.
Figure 6.

Inhibition of colony formation by FLAG-CrmA, FLAG–caspase-8–DN, and FLAG–FADD-DN does not appear to be due to induction of cell death. Fetal liver cells from E14 C57BL/6 embryos were infected with retroviruses encoding GFP alone, FLAG-CrmA, FLAG–caspase-8–DN, FLAG–caspase-9–DN, FLAG–FADD-DN, or FLAG-cFas for 48 hours in the presence of IL-6, mSCF, TPO, and FlkL. (A) After 2 days, viability of transfected (GFP+) cells was assessed by PI staining and FACS analysis. As a control to document that induction of apoptosis can be detected using this procedure, cells were infected with retroviruses encoding wt FLAG-FADD (52% ± 9% viability) or FLAG–caspase-8 (46% ± 3% viability). Numbers in quadrants represent percentages of cells within each staining profile. (B) Summary of cell-viability data representing means ± SDs from 3 to 6 separate experiments for each of the expression vectors. *P < .05. (C) Cell-cycle distribution of transfected (GFP+) cells was determined by staining fixed and permeabilized cells with PI. Data represent means ± SDs from 3 to 6 separate experiments for each of the expression vectors.
Discussion
Experiments with gene knock-out and transgenic mice have demonstrated that caspase-8 and its adaptor FADD are essential for the apoptosis of lymphocytes and fibroblasts that is induced by activation of death receptors, such as Fas.5-7,10,13 Surprisingly, it has been found that caspase-8 and FADD also play a critical role in mitogen- and antigen-induced proliferation of T lymphocytes.6,9-13,27 In the case of caspase-8 it is, however, not yet clear whether its enzymatic activity or some other function are required for T-cell proliferation. To investigate the role of caspase-8 enzymatic activity and FADD in death receptor–induced apoptosis and cytokine-induced proliferation of myeloid cells and hemopoietic progenitors, we attempted to generate transgenic mice expressing dominant inhibitors of caspase-8 (CrmA) or FADD (FADD-DN) in all hemopoietic cell types under control of the Vav gene promoter. We generated lines of mice that bore germ line integrations of a VavP–FLAG–FADD-DN or VavP-FLAG-CrmA construct, but none expressed either transgene (Figures 1, 2; Figures S1-S2). We suspect that this might be due to an inhibitory effect of FLAG–FADD-DN and FLAG-CrmA on stem cell and/or progenitor cell proliferation, causing failure of hemopoiesis and embryonic lethality. Viable chimeric mice containing apparently functional erythroid and myeloid compartments were produced when FADD–/– ES cells were injected into rag-deficient blastocysts.6 Although it is clear that in these chimaeras B and T cells were derived from the FADD–/– ES cells, it was never established whether the FADD-deficient cells could successfully contribute to erythropoiesis and/or myelopoiesis. It will be highly interesting to use this chimeric mouse system to assess the role of FADD in hemopoiesis.
Using a retroviral gene delivery system to infect fetal liver stem cells prior to reconstitution of lethally irradiated mice, we were able to generate hemopoietic cells expressing FLAG-CrmA. The levels of CrmA expression achieved were rather low, and consequently FasL-induced apoptosis was only partially inhibited (Figure 3A). Notably, this retroviral gene delivery/hemopoietic reconstitution protocol did not allow generation of myeloid or lymphoid cells expressing detectable levels of FLAG–FADD-DN. These findings indicate that there was selection against cells expressing high levels of FLAG-CrmA (which might inhibit proliferation) and even those expressing low levels of FLAG–FADD-DN. This is consistent with the idea that caspase-8 and FADD may be required for normal hemopoiesis. These observations may also indicate that the level of caspase-8 enzymatic activity and its mechanism differ between death receptor–induced apoptosis and cytokine-induced proliferation. Even small changes in the function of FADD appear to disrupt proliferation signaling, but higher levels of CrmA are required to inhibit proliferation compared with what is needed to inhibit death receptor–induced apoptosis. This may be due to differences in caspase-8 sequestration, magnitude of activation, or threshold of activation.
Although only low levels of FLAG-CrmA expression were achieved in hemopoietic cells of reconstituted animals, this allowed us to examine the role of caspase-8 in myeloid cells. Granulocytes and IFNγ-stimulated macrophages expressing FLAG-CrmA were found to be resistant to FasL, demonstrating that caspase-8 is required for this pathway to apoptosis, not only in lymphocytes and fibroblasts, but also in myeloid cells. This observation is consistent with the finding that caspase-8 gene deletion rendered myeloid cells resistant to Fas-induced apoptosis.9 There was a trend toward accumulation of myeloid cells in the bone marrow of mice reconstituted with FLAG-CrmA–expressing fetal liver stem cells, indicating that death receptors may play a role in myeloid cell homeostasis.
To specifically investigate the role of FADD and caspase-8 enzymatic activity in hemopoietic progenitor cell proliferation, we performed agar culture assays for hemopoietic colony formation by fetal liver cells. We found that expression of FLAG–FADD-DN, FLAG-CrmA, or FLAG–caspase-8–DN inhibited cell proliferation (Figure 5). The inhibitory effect of FLAG–FADD-DN was independent of its ability to bind Fas and was specific, since no inhibition was seen with overexpression of another death domain, namely that of the cytoplasmic region of Fas (FLAG-cFas). The inhibitory effect of CrmA appears to be due to inhibition of caspase-8 because FLAG–caspase-8–DN also inhibited colony formation, whereas FLAG–caspase-9–DN did not (Figure 5). The studies with the caspase-8 inhibitors are consistent with the observation that deletion of the caspase-8 gene in myeloid progenitors inhibited their proliferation in response to cytokines.9 They also show that it is the enzymatic activity of caspase-8 and not some other function that is required for myeloid cell proliferation. Collectively, these results indicate that both FADD and caspase-8 are required not only for T-cell proliferation but also for proliferation of hemopoietic progenitor cells. These results are the first to show that FADD and caspase-8 are required not only for proliferation induced by antigen receptor stimulation but also that induced by cytokines.
Recently, a domain in FADD has been implicated as playing a critical role in the proliferation-inducing activity of this adaptor.28 Mice expressing FADD mutated at the serine 191 phosphorylation site were runted, anemic, and had abnormalities in their immune system consistent with a defect in cell proliferation, but Fas-mediated apoptosis was not impaired. T cells expressing this FADD mutant showed defects in cell-cycle progression. This indicates that separate domains within FADD allow this protein to have distinct functions in 2 signaling pathways and thus balance cell proliferation and apoptosis.
It is not clear what lies upstream of FADD or downstream of caspase-8 in signaling for cell proliferation, but death receptors may not be involved, because no abnormalities in cell proliferation have been found so far in mice lacking 1 or 2 death receptors (for review see Ashkenazi and Dixit2 and Newton and Strasser29) and because a mutant of FADD-DN unable to interact with Fas could still inhibit hemopoietic colony formation (Figure 5D).
Of interest, mice deficient in FLIP (FLICE [FADD-like interleukin-1β–converting enzyme]–inhibitory protein), which resembles catalytically inactive caspase-8, die during embryogenesis, and this phenotype mimics that of mice lacking either caspase-8 or FADD.30 FLIP molecules are known regulators of death receptor–induced apoptosis, which can interfere with caspase-8 activation in the DISC.31,32 The gene encoding mammalian FLIP (called Cflar) is located adjacent to the Casp8 and Casp10 genes. Alternative splicing of Cflar transcripts results in short (FLIPs) and long (FLIPL) protein products. FLIPs consists of 2 DEDs (related to the DEDs in caspase-8), whereas FLIPL additionally contains (catalytically inactive) domains related to p20 and p10 of caspase-8. FLIPs and its viral homologs, vFLIPs, are thought to function exclusively as inhibitors of death receptor–mediated apoptosis.31,33 The function of FLIPL may vary according to its expression levels. It has been reported that high levels of FLIPL inhibit death receptor–induced apoptosis, whereas low levels may promote activation of caspase-8.34 The embryonic lethality seen in FLIP-deficient mice may indicate that, like caspase-8 and FADD, FLIP has a dual role in apoptosis and cell proliferation.29 One possibility is that FLIP plays a role in formation of an essential FADD/caspase-8 scaffold required for proliferation signaling. According to a recent study in T lymphocytes,35 this process may be critical for optimal activation of Rel/nuclear factor-κB transcription factors, but it still needs to be determined whether this applies to the role of FADD and caspase-8 in cytokine-induced proliferation of hemopoietic progenitors.
In conclusion, our data identify a cytokine-induced cell proliferation–signaling pathway in hemopoietic progenitor cells that uses 2 components of the death receptor–signaling pathway. Both FADD and caspase-8 have roles in 2 distinct processes that place them at a strategic point in the homeostatic control of the hemopoietic system. Now that these dichotomous roles have been uncovered, it will be important to identify the proteins that mediate the proliferative effects of FADD and caspase-8.
Supplementary Material
Acknowledgments
We thank J. Morrow and A. Naughton for animal care and Dr F. Battye, V. Lapatis, C. Tarlinton, and C. Clark for cell sorting. We thank Profs J. Adams, S. Cory, and D. Vaux for insightful discussions and comment on this manuscript.
Prepublished online as Blood First Edition Paper, May 19, 2005; DOI 10.1182/blood-2005-01-0284.
Supported by fellowships and grants from the National Health and Medical Research Council (NHMRC; Canberra, Australia), the Leukemia and Lymphoma Society of America (Specialized Center of Research; SCOR), and the National Institutes of Health (NIH).
The online version of the article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Strasser A, Harris AW, Huang DCS, Krammer PH, Cory S. Bcl-2 and Fas/APO-1 regulate distinct pathways to lymphocyte apoptosis. EMBO J. 1995;14: 6136-6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281: 1305-1308. [DOI] [PubMed] [Google Scholar]
- 3.Traver D, Akashi K, Weissman IL, Lagasse E. Mice defective in two apoptosis pathways in the myeloid lineage develop acute myeloblastic leukemia. Immunity. 1998;9: 47-57. [DOI] [PubMed] [Google Scholar]
- 4.Lagasse E, Weissman IL. bcl-2 inhibits apoptosis of neutrophils but not their engulfment by macrophages. J Exp Med. 1994;179: 1047-1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yeh WC, Pompa JL, McCurrach ME, et al. FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science. 1998;279: 1954-1958. [DOI] [PubMed] [Google Scholar]
- 6.Zhang J, Cado D, Chen A, Kabra NH, Winoto A. Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature. 1998;392: 296-300. [DOI] [PubMed] [Google Scholar]
- 7.Varfolomeev EE, Schuchmann M, Luria V, et al. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998;9: 267-276. [DOI] [PubMed] [Google Scholar]
- 8.Sakamaki K, Inoue T, Asano M, et al. Ex vivo whole-embryo culture of caspase-8–deficient embryos normalize their aberrant phenotypes in the developing neural tube and heart. Cell Death Differ. 2002;9: 1196-1206. [DOI] [PubMed] [Google Scholar]
- 9.Kang TB, Ben-Moshe T, Varfolomeev EE, et al. Caspase-8 serves both apoptotic and nonapoptotic roles. J Immunol. 2004;173: 2976-2984. [DOI] [PubMed] [Google Scholar]
- 10.Newton K, Harris AW, Bath ML, Smith KGC, Strasser A. A dominant interfering mutant of FADD/Mort1 enhances deletion of autoreactive thymocytes and inhibits proliferation of mature T lymphocytes. EMBO J. 1998;17: 706-718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Newton K, Kurts C, Harris AW, Strasser A. Effects of a dominant interfering mutant of FADD on signal transduction in activated T cells. Curr Biol. 2001;11: 273-276. [DOI] [PubMed] [Google Scholar]
- 12.Chun HJ, Zheng L, Ahmad M, et al. Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature. 2002;419: 395-399. [DOI] [PubMed] [Google Scholar]
- 13.Salmena L, Lemmers B, Hakem A, et al. Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev. 2003;17: 883-895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hildeman DA, Zhu Y, Mitchell TC, et al. Activated T cell death in vivo mediated by pro-apoptotic Bcl-2 family member, Bim. Immunity. 2002;16: 759-767. [DOI] [PubMed] [Google Scholar]
- 15.Ogilvy S, Metcalf D, Print CG, Bath ML, Harris AW, Adams JM. Constitutive bcl-2 expression throughout the hematopoietic compartment affects multiple lineages and enhances progenitor cell survival. Proc Natl Acad Sci U S A. 1999;96: 14943-14948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang DC, Hahne M, Schroeter M, et al. Activation of Fas by FasL induces apoptosis by a mechanism that cannot be blocked by Bcl-2 or Bcl-xL. Proc Natl Acad Sci U S A. 1999;96: 14871-14876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karasuyama H, Melchers F. Establishment of mouse cell lines which constitutively secrete large quantities of interleukin 2, 3, 4 or 5, using modified cDNA expression vectors. Eur J Immunol. 1988;18: 97-104. [DOI] [PubMed] [Google Scholar]
- 18.Pear WS, Nolan GP, Scott ML, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci U S A. 1993;90: 8392-8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith KGC, Strasser A, Vaux DL. CrmA expression in T lymphocytes of transgenic mice inhibits CD95 (Fas/APO-1)–transduced apoptosis, but does not cause lymphadenopathy or autoimmune disease. EMBO J. 1996;15: 5167-5176. [PMC free article] [PubMed] [Google Scholar]
- 20.Strasser A, O'Connor L, Dixit VM. Apoptosis signaling. Annu Rev Biochem. 2000;69: 217-245. [DOI] [PubMed] [Google Scholar]
- 21.Tewari M, Telford WG, Miller RA, Dixit VM. CrmA, a poxvirus-encoded Serpin, inhibits cytotoxic T-lymphocyte-mediated apoptosis. J Biol Chem. 1995;270: 22705-22708. [DOI] [PubMed] [Google Scholar]
- 22.Zhou Q, Snipas S, Orth K, Muzio M, Dixit VM, Salvesen GS. Target protease specificity of the viral serpin CrmA. J Biol Chem. 1997;272: 7797-7800. [DOI] [PubMed] [Google Scholar]
- 23.Muzio M, Stockwell BR, Stennicke HR, Salvesen GS, Dixit VM. An induced proximity model for caspase-8 activation. J Biol Chem. 1998;273: 2926-2930. [DOI] [PubMed] [Google Scholar]
- 24.Fearnhead HO, Rodriguez J, Govek E-E, et al. Oncogene-dependent apoptosis is mediated by caspase-9. Proc Natl Acad Sci U S A..1998;95: 13664-13669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marsden V, O'Connor L, O'Reilly LA, et al. Apoptosis initiated by Bcl-2–regulated caspase activation independently of the cytochrome c/Apaf-1/caspase-9 apoptosome. Nature. 2002;419: 634-637. [DOI] [PubMed] [Google Scholar]
- 26.Chinnaiyan AM, O'Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81: 505-512. [DOI] [PubMed] [Google Scholar]
- 27.Newton K, Harris AW, Strasser A. FADD/MORT1 regulates the pre-TCR checkpoint and can function as a tumour suppressor. EMBO J. 2000;19: 931-941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hua ZC, Sohn SJ, Kang C, Cado D, Winoto A. A function of Fas-associated death domain protein in cell cycle progression localized to a single amino acid at its C-terminal region. Immunity. 2003;18: 513-521. [DOI] [PubMed] [Google Scholar]
- 29.Newton K, Strasser A. Caspases signal not only apoptosis but also antigen-induced activation in cells of the immune system. Genes Dev. 2003;17: 819-825. [DOI] [PubMed] [Google Scholar]
- 30.Yeh WC, Itie A, Elia AJ, et al. Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity. 2000;12: 633-642. [DOI] [PubMed] [Google Scholar]
- 31.Irmler M, Thome M, Hahne M, et al. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388: 190-194. [DOI] [PubMed] [Google Scholar]
- 32.Scaffidi C, Schmitz I, Krammer PH, Peter ME. The role of c-FLIP in modulation of CD95-induced apoptosis. J Biol Chem. 1999;274: 1541-1548. [DOI] [PubMed] [Google Scholar]
- 33.Thome M, Schneider P, Hofmann K, et al. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 1997; 386: 517-521. [DOI] [PubMed] [Google Scholar]
- 34.Chang DW, Xing Z, Pan Y, et al. c-FLIPL is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J. 2002;21: 3704-3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Su H, Bidere N, Zheng L, et al. Requirement for caspase-8 in NF-kappaB activation by antigen receptor. Science. 2005;307: 1465-1468. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.