

Abstract
Phosphatase and tensin homolog deleted on chromosome 10 (PTEN), a multifunctional tumor suppressor, has been shown to play a regulatory role in cell migration. Dictyostelium discoideum cells lacking PTEN exhibited impaired migration toward chemoattractant gradients. In the present study, we investigated the involvement of PTEN in chemotaxis of mammalian cells by examining PTEN-null Jurkat T cells. We observed that, in contrast to observations made in D discoideum, PTEN-null Jurkat T cells exhibited potent chemotactic responses to the chemokine stromal cell–derived factor 1α (SDF-1α), indicating that PTEN was not requisite for CXC chemokine receptor 4 (CXCR4)–mediated chemotaxis of Jurkat cells. Conversely, reconstitution of PTEN in Jurkat cells by using a tetracycline (Tet-on)–inducible expression system down-regulated CXCR4-mediated chemotaxis. Furthermore, we established the lipid phosphatase activity of PTEN as essential for its inhibitory effect on chemotaxis. In addition, using PTEN-expressing T-cell lines and primary T cells, we demonstrated that down-regulation of PTEN expression with vector-based small interfering RNAs (siRNAs) enhanced CXCR4-mediated chemotaxis. Based on these results, we conclude that PTEN expression negatively regulates chemotaxis of lymphoid mammalian cells via its lipid phosphatase activity. Our findings may account for the reported increase in metastatic activity of PTEN-null tumor cells.
Introduction
Chemotaxis is a property of many motile eukaryotic cells that results from a cascade of intracellular events initiated in cells with the capacity to detect an extracellular chemoattractant gradient.1,2 The initiation of cell chemotaxis requires the binding of a chemoattractant to its receptor, usually a 7 transmembrane G protein–coupled receptor (GPCR), and subsequent activation of pertussis toxin (PTX)–sensitive G protein (Gi), which results in a chain of signaling events, from activation of the phosphatidylinositol 3-kinase (PI3K) pathway and downstream small G proteins Regulator of adenylyl cyclase/cell division control protein-42 (Rac/cdc42), polymerization of F actin at the leading edge of the cells, to the formation of a pseudopod and cell polarization.3-5 Mounting evidence has established the extensive involvement of chemoattractants, chemokines in particular, in homeostatic homing of leukocytes, inflammation, development, wound healing, angiogenesis, and immune responses.6-8 Chemokines and their receptors also play a critical role in metastasis of malignant tumor cells.9-11
Phosphatase and tensin homolog deleted on chromosome 10 (PTEN), one of the most frequently mutated or deleted tumor suppressor genes in human cancers,12-14 has been shown to play an important role in regulating cell migration. Previous studies showed that PTEN expression inhibited migration of certain human tumor cell lines, although the driving forces behind such migration and its underlying mechanisms remained obscure.15,16 The involvement of PTEN in the chemotactic movement of cells was initially evidenced in Dictyostelium discoideum, as cells lacking PTEN exhibited impaired migration toward chemoattractants.17-19 Although the evidence obtained from D discoideum study was considered conclusive, whether the observations could be extrapolated to mammalian cells remained to be determined. To date, the reports directly addressing the involvement of PTEN in chemotactic movement of mammalian cells have been confusing. While Fox et al20 demonstrated that PTEN+/– mouse B lymphocytes exhibited enhanced chemotactic responses to chemokine stromal cell–derived factor 1α (SDF-1α), Anzelon et al21 reported that the conditional deletion of PTEN from mouse B cells resulted in a general impairment in chemotaxis induced by SDF-1α and B-lymphocyte chemoattractant (BLC). These discrepancies point to the necessity to further elucidate the role of PTEN in regulating the chemotaxis of mammalian cells.
A longstanding challenge in tumor biology is to decipher how metastasis, the chief cause of morbidity and mortality in most cancers, occurs in human malignancy. Although accumulating evidence suggested organ-specific metastasis is governed, in part, by interactions between chemokine receptors on cancer cells and chemokine ligands in target organs,9-11 the underlying regulatory mechanisms have remained largely unknown. Thus, we reasoned that it was of significance to address directly the role of PTEN, a multifunctional tumor suppressor, in modulating chemotaxis of mammalian cells, including human cancer cells. To this end, we used Jurkat cells, a naturally occurring PTEN-null and CXC chemokine receptor 4–positive (CXCR4+) human leukemia T-cell line, to investigate the role of PTEN in chemotaxis. We found that PTEN-null Jurkat cells and small interfering RNA (siRNA)– transfected primary T cells exhibited potent chemotactic responses to SDF-1α, indicating that PTEN was not indispensable for chemotactic movement of lymphoid cells, a sharp contrast to early observations made in D discoideum. By using a tetracycline (Tet-on)–inducible system, we then demonstrated that reconstitution of PTEN expression inhibited CXCR4-mediated chemotaxis. Importantly, we further established that the lipid phosphatase activity of PTEN was essential for its role as a suppressor of chemotaxis. Our results may serve to invite more extensive and straightforward approaches to investigate the antimetastatic function of PTEN in human malignancy.
Materials and methods
Reagents
Recombinant human chemokines (SDF-1α, interferon-inducible protein-10 [IP-10], RANTES [regulated on activation, normal T expressed and secreted], macrophage-derived chemokine [MDC], liver and activation-regulated chemokine [LARC], I-309, macrophage inflammatory protein 3β [MIP-3β], and Eotaxin3) and cytokines (human interleukin-2 [IL-2] and mouse IL-12) were purchased from Pepro Tech (Rocky Hill, NJ). Recombinant human insulin-like growth factor 1 (IGF-1) was obtained from R&D (Minneapolis, MN). Anti–human PTEN (clone 6H2.1) was obtained from Cascade Bioscience (Winchester, MA). Anti–phosphorylated (phospho)–Akt (Ser473) and anti–extracellular signal–related kinase 1 (ERK1)/ERK2 were purchased from Cell Signaling Technology (Beverly, MD). Phycoerythrin (PE)–conjugated anti–human CXCR4 was obtained from R&D. PE-conjugated mouse isotype–matched immunoglobulin G2a κ (IgG2a κ) was obtained from BD Bioscience Pharmingen (Palo Alto, CA). Pertussis toxin (PTX), wortmannin, and LY294002 were obtained from Sigma-Aldrich Chemicals (St Louis, MO). Doxycycline was purchased from BD Bioscience Pharmingen.
Cell lines and culture conditions
The Tet-on–inducible Jurkat cell lines have been described previously.22 2D6 T cells and 2B4 T cells, kind gifts from Dr Hiromi Fujiwara of Osaka University in Japan, were also described in our previous publications.23-25 While all the cell lines were maintained in RPMI 1640 medium containing 10% (vol/vol) fetal bovine serum (FBS; HyClone, Logan, UT), 10 mM glutamine, 100 international units (IU)/mL penicillin, and 100 μg/mL streptomycin (Quality Biologicals, Gaithersburg, MD), and additional 800 μg/mL geneticin (GIBCO, Carlsbad, CA) was added into the culture medium to maintain Tet-on Jurkat cells, and 250 pg/mL of IL-12 was added to maintain 2D6 T cells.23 The treatment conditions for doxycycline or other chemical reagents were specified in the figure legends. HEK293 and HEK293/CCR5 cells were described previously.26 Human peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll-Hypaque density gradient centrifugation from leukopacks supplied by the Department of Transfusion Medicine under an approved human subjects protocol (Clinical Center, National Institutes of Health, Bethesda, MD).
Purification of human CD4 T cells
PBMCs were isolated by Ficoll-Paque density gradient centrifugation from leukopacks supplied by the Department of Transfusion Medicine under approved human subjects protocol institutional review board (IRB) no. 99-CC-0168. Approval was obtained from the National Cancer Institute (NCI) IRB for these studies. Informed consent was provided according to the Declaration of Helsinki. Human peripheral blood CD4 T cells were purified from PBMCs by the use of magnetic-activated cell sorting (MACS) human CD4 T-cell isolation kit (Miltenyi Biotech, Auburn, CA) following the manufacturer's recommendation. The purity of CD4 T cells (> 95%) was confirmed by flow cytometry.
Plasmids and transfection
Tet-on response plasmid pTRE-HA-PTEN (PTEN/wt) has been described previously.22 The naturally occurring PTEN mutants G129E (lipid phosphatase–dead, protein phosphatase–live) and G129R (phosphatase-dead) were introduced into pTRE-HA-PTEN by Quickchange (Stratagene, La Jolla, CA) site-directed mutagenesis. The cDNA plasmids were transiently transfected into Tet-on Jurkat cells by utilizing Fugene 6 Tranfection Reagents (Roche Diagnostic, Indianapolis, IN), which was performed according to the manufacturer's protocol. Six hours after transfection, doxycycline (1 μg/mL) was added to the culture to induce PTEN expression. After an additional 48 hours, cells were harvested for Western blotting and chemotaxis assay.
The DNA vectors for siRNA constructs, pRNAT-U6.1/Hygro/green fluorescent protein (GFP), were from GenScript (Piscataway, NJ). Three siRNA insert sequences for PTEN targeting, which were selected by using the software siRNA Construct Builder (GenScript), were as follows: GGATCCCGTTCCGCCACTGAACATTGGAATTGATATCCGTTCCAATGTTCAGTGGCGGAATTTTTTCCAAAAGCTT (siPTEN construct no.1), GGATCCCGCACGCTCTATACTGCAAATGTTGATATCCGCATTTGCAGTATAGAGCGTGCTTTTTTCCAAAAGCTT (siPTEN construct no. 2), GGATCCCGTATAGGTCAAGTCTAAGTCGATTGATATCCGTCGACTTAGACTTGACCTATATTTTTTCCAAAAGCTT (siPTEN construct no. 3). As a negative control, a mock vector was also designed, whose inserted sequence was GGATCCCGCGAGTTAATTACACGCGATCCTTGATATCCGGGATCGCGTGTAATTAACTCGTTTTTTCCAAAAGCTT (scrambled). The 3 siRNA constructs were combined into 1 pool and transfected into 2D6 T cells or 2B4 T cells using Fugene 6 Tranfection Reagents according to the manufacturer's protocol (Roche Diagnostic). Stable cell clones were obtained with hygromycin selection, based on GFP expression. PTEN expression following the transfection was measured by Western blotting.
The transfection of primary CD4 T cells with vector-based siRNA constructs were carried out by using Nucleofect II Device and Human T cell Nucleofector Kit from Amaxa Biosystems (Amaxa, Cologne, Germany) according to the manufacture's protocol for unstimulated human T cells. 27 We used 5 × 106 purified unstimulated CD4 T cells, 3 μg DNA, and Program U-014 (Amara) for each transfection because our pilot studies revealed that these conditions yielded a higher proportion of surviving transfected cells than when we used phytohemagglutinin (PHA)–stimulated T cells. The cells were cultured in RPMI 1640 supplemented with 10% (vol/vol) FBS for 4 hours after transfection, then human IL-2 (20 U/mL) was added to the culture. After additional 20 hours of culture, we pooled multiple wells of cells transfected with the same vector and purified the viable and positive cells by cell sorting using the DakoCytomation MoFlo System (Fort Collins, CO), based on GFP expression. The purified cells were 93% positive for GFP and 98% of them excluded trypan blue. PTEN expression was measure by Western blotting.
Chemotaxis
Chemotaxis assays were performed using 48-well microchemotaxis chambers.26 Jurkat cells, other T-cell lines, or peripheral blood T cells were resuspended in binding medium (RPMI 1640 medium containing 1% bovine serum albumin [Sigma-Aldrich A4503], 25 mM HEPES [N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid], pH 8.0) at 2.0 × 106. In some experiments, cells were pretreated with chemical inhibitors under conditions specified in the figure legends. Chemokines, diluted in binding medium, were placed in the lower wells of the microchemotaxis chambers (Neuroprobe, Rockville, MD). 5-μm polyvinyl-free polycarbonate membranes (Neuroprobe) were treated overnight at 4°C in RPMI containing 10 μg/mL fibronectin (Sigma-Aldrich), air-dried and placed over the chemoattractants to separate the lower and upper wells of the chamber. After the microchemotaxis chamber was assembled, 50 μL cells were added in the upper wells. After incubation at 37°C for 3 hours in humidified air with 5% CO2, the membranes were removed, scraped, and stained. The cells migrated across the membranes were counted with the use of a Bioquant semiautomatic counting system (R&M Biometrics, Nashville, TN), which objectively determines the average number of cells in 6 defined microscopic fields of each condition on the filter membrane. The results were reported as the mean number of cells per high-powered field (HPF).
Flow cytometry
The surface expression of chemokine receptor CXCR4 on Jurkat cells was monitored by fluorescence-activated cell sorter (FACS) analysis. Jurkat cells (1 × 106 with/without doxycycline treatment) were washed with FACS buffer (1 × Dulbecco phosphate-buffered saline [DPBS] without calcium and magnesium, 1% fetal calf serum [FCS] and 0.02% sodium azide), and stained with PE-conjugated mouse anti–human CXCR4 mAb (20 μL/sample), or PE-conjugated mouse isotype–matched IgG2a. Stained cells were washed and analyzed on a FACScan (BD Biosciences, Mountain View, CA) using CellQuest software (BD Biosciences).
Cell sorting of GFP-positive T cells after siRNA transfection was carried out using the DakoCytomation MoFlo System.
Western blotting
The cultured cells (1 × 106) were harvested and lysed by adding 100 μLof 1 × sodium dodecyl sulfate (SDS) sample buffer (62.5 nM Tris [tris(hydroxymethyl)aminomethane]–HCl, pH 6.8 at 25°C, 2% wt/vol SDS, 10% glycerol, 50 mM dithiothretol, 0.01% bromophenol blue). The samples were then sonicated for 10 seconds on ice, boiled for 5 minutes, cooled on ice, and centrifuged at 17/500g for 5 minutes. The protein concentration of the extracts was estimated with a BCA Protein Assay kit (PIERCE, Rockford, IL) and 25 μg of proteins were loaded and separated on a 4% to 12% NuPAGE Bis-Tris Gel (Invitrogen, Carsbad, CA). The proteins in the gel were then electrotransferred onto Immobilon membrane (Millipore, Bedford, MA) with an Xcell blot Module (Invitrogen) using 1 × NuPAGE transfer buffer. The polyvinylidene difluoride (PVDF) membrane with the transferred proteins was sequentially washed with 1 × TBS/T (1 × Tris-buffered saline, 0.1% Tween 20) for 5 minutes, incubated for 1 hour in blocking buffer (1 × TBS, 0.1% Tween 20, 5% wt/vol nonfat milk), and incubated at 4°C overnight in blocking buffer containing 1:1000 dilution of the primary antibodies. The membrane was then washed 3 times for 5 minutes each with TBS/T and incubated with 10 mL of 1:2000 dilution of horseradish peroxidase (HPR)–conjugated second antibodies (Cell Signaling Technology) for 1 hour. After washing 3 times with TBS/T, the membrane was incubated with working solution of ECLplus Detection Reagents (Amersham, Piscataway, NJ) for 5 minutes and exposed to an X-ray film, which was then developed using an automatic processor (X-OMAT 2000A; Kodak, Eastman, NY). The same membrane was incubated in stripping solution (62.5 mM Tris-HCl, pH6.7, 2% SDS, 100mM 2 mercaptoethanol [2-ME]) at 50°C for 30 minutes and then reprobed as before.
Results
Several previous studies have reported that Jurkat cells were PTEN deficient;22,28-30 however, there were also reports showing that Jurkat cells expressed wild-type PTEN.31 Thus, we started our experiments by examining PTEN protein expression in Jurkat cells. Several Jurkat cell lines, obtained from different laboratories, were tested by Western blotting and none was found to be PTEN positive (Figure 1A and data not shown).
Figure 1.

PTEN-null Jurkat T cells exhibited potent chemotactic response to SDF-1α. (A) Jurkat cells were PTEN deficient. Lanes 1 to 6 contained lysates of wild-type Jurkat cells obtained from the laboratories of Drs Ji-Ming Wang, William L. Farrar, and Daniel W. McVicar, as well as our lab (all at NCI-Frederick, Frederick, MD), respectively, and the Tet-on and Tet-off Jurkat T-cell lines purchased from Clontech (Palo Alto, CA). Lanes 7 to 9 showed the lysates of HEK293 cells and PBMCs, used as positive controls. Cultured cells were lysed and electrophoresed on an SDS–polyacrylamide gel electrophoresis (PAGE) gel, transferred to nitrocellulose membranes, and probed with anti-PTEN or anti-ERK1/2 as a loading control. (B) Jurkat cells express CXCR4. The surface expression of chemokine receptor CXCR4 in Jurkat cells was measured by flow cytometry. Cells were stained with anti-hCXCR4 (solid line) or isotype-matched human IgG2a as a negative control (dotted line). (C) Jurkat cells exhibit dose-dependent chemotactic responses to SDF-1α. Chemotaxis was performed using 48-well chemotaxis chambers as described in “Materials and methods.” Different concentrations (0-1000 ng/mL) of SDF-1α were used for chemotaxis. (D) SDF-1α–induced chemotaxis of Jurkat cells was PTX sensitive. Jurkat cells were pretreated with different doses of PTX for 30 minutes at room temperature (RT) and chemotaxis assay was carried out as described in “Materials and methods”. All the results in this figure were representative of at least 3 independent experiments. (C-D) Data shown are means ± SD.
Jurkat cells are human acute leukemia T-cell lines.32 As expected, FACS analysis confirmed surface expression of CXC chemokine receptor CXCR4 in these cells (Figure 1B and data not shown). Thus, PTEN-null Jurkat cells represented a useful model to study the role of PTEN in chemotaxis of mammalian cells.
When stimulated with the CXCR4 ligand, SDF-1α (CXCL12), Jurkat cells exhibited potent migratory responses in chemotaxis assays. Since earlier reports showed that D discoideum lacking PTEN were defective in directional movements toward chemoattractant sources (chemotaxis), but exhibited enhanced random movements (chemokinesis),17 we further characterized SDF-1α–stimulated Jurkat cell migration in chemotaxis assays to determine whether it was due to chemotaxis or chemokinesis. As shown in Figure 1, SDF-1α stimulated typical bell-shaped dose-dependent chemotactic responses (Figure 1C) that were inhibited by pertussis toxin (PTX) (Figure 1D). When the chemokine concentration gradient was eliminated by adding equal concentrations of SDF-1α to the upper wells of chemotaxis chambers, Jurkat cell migration was abolished (data not shown). Furthermore, stimulation with various other chemokines did not elicit chemotactic response in Jurkat cells. Taken together, our results indicated that, in contrast to previous findings in D discoideum, PTEN-null Jurkat cells exhibited potent chemotactic responses to SDF-1α.
To more directly address the role of PTEN in chemotaxis, PTEN expression was reconstituted in Jurkat cells by using a tetracycline-inducible expression system as previously described.22 PTEN expression upon doxycycline (dox) treatment was confirmed in a PIJ (PTEN-inducible Jurkat)–17 clone but not in a non–PTEN-expressing control clone, Con18, by Western blotting (Figure 2A). PTEN expression in PIJ-17 Jurkat cells was detectable as early as 3 hours after dox induction, reaching maximum expression between 24 and 48 hours, and was persistent at the maximal level 120 hours after induction (Figure 2C). PTEN expression inversely correlated with phosphorylation of Akt, indicating that the expressed PTEN was functional (Figure 2A,C).
Figure 2.

Reconstitution of PTEN in Jurkat cells down-regulated CXCR4-mediated chemotaxis. (A) Doxycycline (dox) induced PTEN expression in PIJ-17 but not in Col-18 or Tet-on Jurkat cells. PIJ-17, Con-18, and Tet-on Jurkat cells were treated with 1 μg/mL dox for 48 hours and the expression of PTEN (top panel) and Ser473-phosphorylated AKT (middle panel) was measured by Western blotting. The samples were also blotted with anti-ERK1/2 as loading control (bottom panel). (B) Expression of PTEN down-regulated SDF-1α–mediated chemotaxis. PIJ-17 and Con-18 Jurkat cells were cultured with or without the presence of dox (1 μg/mL) for 48 hours and chemotaxis was carried out as in Figure 1. The results were representative of at least 5 independent experiments. Data shown are means ± SD. (C-D) Time course of PTEN expression under dox induction and its effects on chemotaxis. PIJ-17 Jurkat cells were treated with 1 μg/mL dox for the indicated periods of time, and the levels of PTEN and Ser473-phosphorylated AKT were detected by Western blotting (C). The same cells were also assayed for chemotactic responses (D). Data shown are means ± SD. The results were representative of 3 independent experiments. (E) PTEN expression had no effects on CXCR4 surface expression. PIJ-17 Jurkat T cells were cultured with or without dox (1 μg/mL) for 48 hours and CXCR4 expression were detected by flow cytometry. The results were representative of 2 independent experiments. The histograms show cell-surface staining with anti-hCXCR4 (solid line) or isotype-matched human IgG2a (dashed line).
Chemotaxis assays showed that expression of PTEN substantially inhibited SDF-1α–stimulated chemotaxis of PIJ-17 cells (Figure 2B). The effect on chemotaxis, which was detectable 6 hours after dox induction and persistent for at least 5 days, correlated with the level of PTEN expression and phosphorylation of Akt (Figure 2C-D). However, expression of PTEN did not affect surface expression of CXCR4 on Jurkat cells (Figure 2E), suggesting that PTEN negatively modulated SDF-1α–stimulated chemotaxis, presumably by inhibiting chemotactic signaling pathway downstream of CXCR4. In agreement with the previous report,22 in the timeframe of our experiments, dox treatment did not affect the viability of PIJ-17 Jurkat cells (data not shown).
PTEN is a dual-functional protein and lipid phosphatase. The major known physiologically relevant substrate of PTEN is the lipid second messenger phosphatidylinositol 3,4,5-trisphosphate (PIP3).33 However, both lipid and protein phosphatase activities have been suggested to be involved in regulating cellular migration.16,34 To investigate which of the phosphatase activities of PTEN, lipid or protein, were responsible for the inhibitory effect of PTEN on chemotaxis, Tet-on–inducible vectors expressing PTEN/wt, PTEN/G129E, and PTEN/G129R were transiently transfected to Tet-on Jurkat cells. G129E and G129R are 2 naturally occurring PTEN mutants found in human malignancy. G129E is a mutant lacking lipid phosphatase activity while retaining protein phosphatase activity, whereas G129R lacks both lipid and protein phosphatase activities.15 After 6 hours following transfection, dox (1 μg/mL) was added to the medium to induce PTEN expression. After an additional 48 hours, PTEN expression was detected in PTEN vector–transfected cells but not in the mock transfection group. Phosphorylation of Akt was decreased in the PTEN/wt group, but not in mutant groups (PTEN/G129R or PTEN/G129E), indicating that the expressed mutant PTEN indeed lacked lipid phosphatase activity (Figure 3A). Chemotaxis assays were performed 48 hours after transfection. As shown in Figure 3B, the effect of PTEN on chemotaxis was abolished by a single point mutation, which inactivates PTEN's lipid phosphatase activity. Of note, PTEN/G129E, a mutant lacking lipid phosphatase activity but retaining protein phosphatase activity, was also defective in modulating chemotaxis, indicating that the lipid, but not protein, phosphatase activity of PTEN was essential.
Figure 3.

The lipid phosphatase activity was essential for PTEN's role as a negative regulator of chemotaxis. Tet-on Jurkat cells were transiently transfected with Tet-inducible vectors expressing PTEN/wt, PTEN/G129E, PTEN/G129R, or a mock vector. After 6 hours, dox (1 μg/mL) was added to the culture medium. Cells were cultured for an additional 48 hours and then harvested for Western blotting (A) and chemotaxis (B). The results were representative of 3 independent experiments. Data shown are means ± SD.
Since the lipid phosphatase activity was essential for the effect of PTEN on chemotaxis, we postulated that, by antagonizing the PI3K-PIP3 pathway, PTEN negatively modulated CXCR4-mediated chemotaxis in Jurkat T cells. Although PI3K-PIP3 have been proposed as central players in chemotaxis in a number of mammalian cells, its effect varies, depending on the cell types and chemokine receptors involved.35,36 PI3K-independent pathways have also been proposed.35,36 Therefore, we examined whether blocking the PI3K-PIP3 pathway impaired CXCR4-mediated chemotaxis. PIJ-17 Jurkat cells, with or without dox treatment, were preincubated with 2 chemical inhibitors of PI3K, wortmannin or LY294002, and chemotaxis assays were performed. As shown in Figure 4, both wortmannin and LY294002 inhibited SDF-1α–stimulated chemotaxis in a dose-dependent manner. Interestingly, although chemotaxis was markedly inhibited with PTEN expression, it was further decreased by the pretreatment with PI3K inhibitors. However, chemotaxis was never decreased to basal levels, even at higher doses of chemical inhibitors (Figure 4 and data not shown), suggesting that pathways independent of PI3K may also regulate SDF-1α–stimulated chemotaxis of Jurkat cells.
Figure 4.

PI3K inhibitors reproduced the inhibitory effect of PTEN on CXCR4-mediated chemotaxis. PIJ-17 Jurkat cells were cultured with or without dox (1 μg/mL) and were then pretreated with wortmannin (A) or LY294002 (B) for 1 hour at RT at the indicated concentrations. The cells were then subjected to chemotaxis assays with SDF-1α (100 ng/mL). The basal migration in the absence of SDF-1α was indicated as medium only. The results were representative of 4 independent experiments. Data shown are means ± SD.
Next, we addressed whether the effect of PTEN was restricted to CXCR4-mediated chemotaxis. We tested Jurkat cell responses to other chemokines whose receptors are reportedly expressed on different subsets of T lymphocytes. Of all the chemokines tested, Jurkat cells responded only to SDF-1α (Figure 5A). However, Jurkat cells demonstrated substantial chemotactic responses to IGF-1, whose receptor IGFR, a receptor tyrosine kinase, was reportedly expressed on Jurkat cells.37 Interestingly, expression of PTEN in PIJ-17 Jurkat cells substantially inhibited IGF-1–stimulated chemotaxis as well (Figure 5B).
Figure 5.
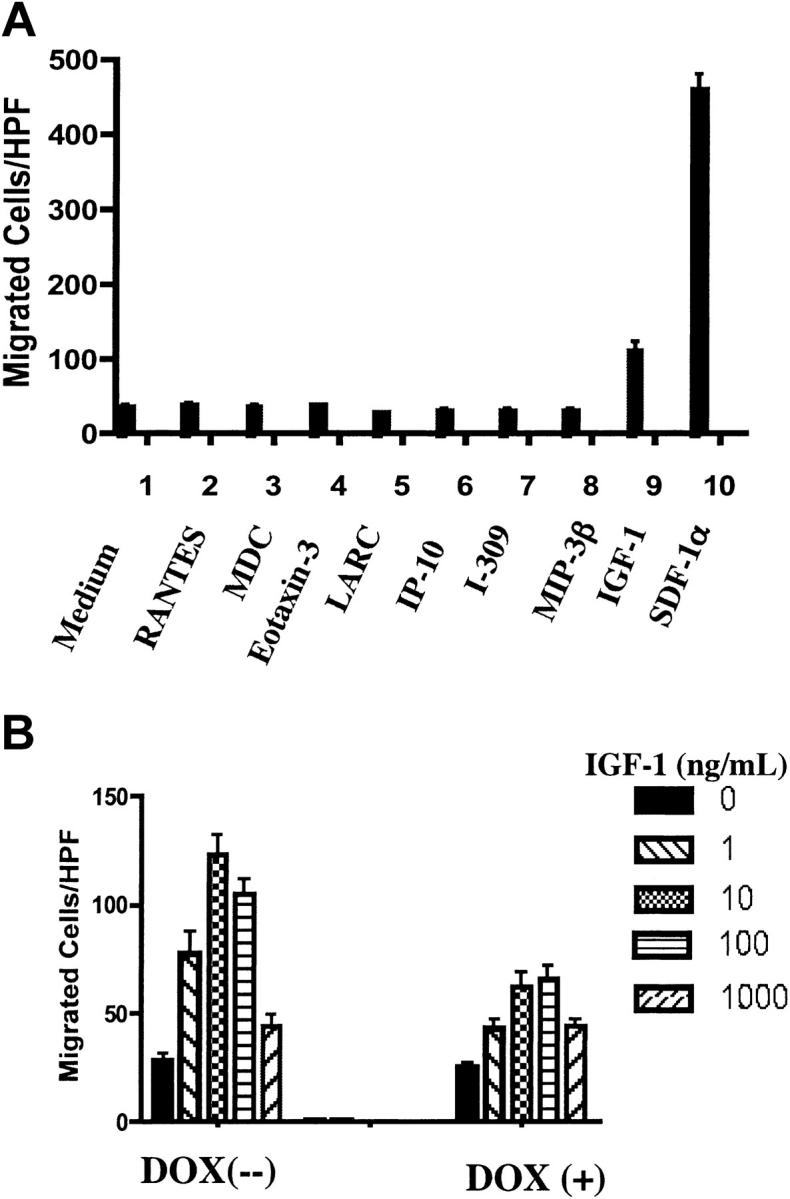
Restoration of PTEN expression in Jurkat cells down-regulated IGF1-mediated chemotaxis. (A) The chemotactic responses of cultured PIJ-17 Jurkat cells (without dox) to different chemoattractants were tested. The concentrations used were determined previously as optimal for chemotaxis of different T lymphocyte subsets: RANTES (50 ng/mL), MDC (100 ng/mL), eotaxin3 (100 ng/mL), LARC (1000 ng/mL), IP-10 (100 ng/mL), I-309 (10 ng/mL), MIP-3β (100 ng/mL), IGF-1 (10 ng/mL), SDF-1α (100 ng/mL). The results were representative of 3 independent experiments. (B) PIJ-17 Jurkat cells were cultured with or without dox (1 μg/mL) for 48 hours and chemotaxis assays were performed as in panel A. The results were representative of 3 independent experiments. Data shown are means ± SD.
To address whether our observation that PTEN expression inhibited Jurkat T-cell migration could be extrapolated to other mammalian cells, we used vector-based siRNAs to inhibit PTEN expression in 2 additional T-cell lines, 2D6 T cells and 2B4 T cells. 2D6 cells were established from a terminally differentiated T-helper 1 (Th1) cell38 clone that is IL-12 responsive. Our previous observations demonstrated that 2D6 T cells expressed chemokine receptors including CXCR4 and showed chemotactic responses to chemokine ligands (P.G. and Hiromi Fujiwara, unpublished observations, August 2002).24 The chemotactic ability of 2B4/CXCR4 transfectant T cells were described in our previous publication.25 Following transfection with vector-based PTEN-targeting siRNAs, stable clones were obtained with hygromycin selection, based on GFP expression (data not shown). Western blotting confirmed a substantial reduction in PTEN expression in siRNA/PTEN-transfected cells, but not in mock vector–transfected cells (Figure 6A). Consistent with results in Jurkat T cells, inhibition of PTEN expression significantly enhanced SDF-1α–induced chemotaxis of 2D6 T cells (Figure 6B) and 2B4 T cells (Figure 6C), indicating that PTEN negatively regulated CXCR4-mediated chemotaxis in 2 additional T-cell lines, 2D6 and 2B4 T cells.
Figure 6.

Inhibition of PTEN expression by vector-based siRNA enhanced SDF-1α–induced chemotaxis of T cells. (A) Western blotting confirmed down-regulation of PTEN expression in siRNA/PTEN-transfected cells. The nontransfected cells and the cells transfected with siRNA/PTEN constructs or mock vectors (stable transfection for 2D6 and 2B4 T-cell lines and transient transfection for primary T cells) were lysed and PTEN protein expression detected by Western blotting as described in “Materials and methods”. The samples were also blotted with anti-ERK1/2 as loading control (bottom panel). The results were representative of 3 independent experiments for T-cell lines and 2 independent experiments for primary T cells. (B-D) CXCR4-mediated chemotaxis of 2D6 T cells (B), 2B4 T cells (C), and primary CD4 T cells (D) was enhanced by inhibition of PTEN expression. The chemotaxis assays were performed as described in “Materials and methods.” The results were representative of 3 independent experiments for T-cell lines and 2 independent experiments for primary T cells. Data shown are means ± SD.
Finally, we tested the effect of PTEN down-regulation on chemotaxis of primary T cells. CD4+ T cells were purified from human peripheral blood, with a purity of more than 95%, followed by transfection with vector-based siRNAs using the Amaxa System (Amara). After 24 hours of culture, we purified GFP+ viable cells by cell sorting. As shown in Figure 6A, transfection of vector-based siRNAs substantially inhibited PTEN expression in primary CD4 T cells. Consistent with results in 2B4 and 2D6 T cell lines, inhibition of PTEN expression markedly enhanced SDF-1α–stimulated chemotaxis of human primary T cells (Figure 6D).
Discussion
In this report, we investigated the naturally occurring PTEN-null human Jurkat T-cell line to characterize the involvement of PTEN in chemotaxis of mammalian cells. Our findings are as follows: (1) in contrast to early observations made in D discoideum, PTEN-null Jurkat cells exhibited potent CXCR4-mediated chemotaxis, indicating that PTEN was not indispensable for proper chemotactic movement of Jurkat cells; (2) using a Tet-on–inducible system, we found that reconstitution of PTEN expression in Jurkat cells down-regulated CXCR4-mediated chemotaxis; and (3) using 2 naturally occurring PTEN mutants, G129E and G129R, we further identified the lipid phosphatase activity as essential for PTEN's role as a negative modulator of CXCR4-mediated chemotaxis. This was further supported by the observations that PI3K inhibitors mimicked PTEN's effect of negatively regulating CXCR4-mediated chemotaxis. The inhibitory effect of PTEN on chemotaxis was not restricted to CXCR4-mediated chemotaxis of Jurkat cells since restoration of PTEN expression down-regulated IGF-1–stimulated chemotaxis as well. Furthermore, suppression of PTEN expression by siRNA also enhanced CXCR4-mediated chemotaxis by PTEN-expressing T-cell lines and primary T cells. These findings indicate that PTEN expressed by mammalian cells performs a critical inhibitory function in the signal cascade that leads to cell migration.
Considering that the single-cell D discoideum that lacked PTEN showed defective directional movement toward chemoattractant sources but enhanced random movement,17-19 our contrasting observations in PTEN-null Jurkat cells are of interest. Although single-cell organisms such as D discoideum share many mechanisms in common with leukocytes of mammalian species for sensing and responding to chemoattractant gradients, it is highly probable that the complexity of the signaling pathways elicited by chemoattractants may vary with the species of migrating cells or types of the chemoattractants. Interestingly, in D discoideum, results from Iijima and Devreotes17, Fumamoto et al18 elegantly demonstrated that, following stimulation by chemoattractants, PI3K was relocated to the leading edge while PTEN moved to the lateral sides and the posterior of the chemotaxing cells, thus increasing the intracellular PIP3 gradient and sharpening the subsequent localization of Pleckstrin homology (PH) domain–containing proteins to the leading edge of the cells. However, Xu et al39 did not observe PTEN exclusion from the leading edge of the HL-60 myeloid cells following chemoattractants' stimulation. These earlier results together with the present study suggest that the observations made in single-cell organisms such as D discoideum are not recapitulated in certain mammalian cell lines. At present, we have no specific answers to the fundamental question as to how lymphoid cells, in the absence of PTEN, can generate the intracellular molecular gradient that is essential for cellular polarization and chemotaxis. Presumably, prolonged phosphorylation of components in this pathway, which occurs in the absence of PTEN, favors chemotaxis and the actual chemotactic mechanisms in mammalian cells are distinct from those of prokaryotic cells.
There have been few earlier studies of the role of PTEN in mammalian cell chemotaxis. Two groups have directly observed chemotactic behaviors of PTEN deficient B lymphocytes, but the results were divergent.20,21 Fox et al20 demonstrated that PTEN+/– mouse B lymphocytes exhibited enhanced chemotactic responses to SDF-1α stimulation; however, Anzelon et al21 reported just the opposite: the conditional deletion of PTEN in mouse B cells resulted in an overall impairment in chemotaxis. The reason for this discrepancy remains unknown. In PTEN+/– cells used by Fox et al, the expression level of PTEN never went below 50% of the wild-type cells, whereas the PTENloxp/loxP B cells used by Anzelon et al were PTEN deficient. Since the level of PTEN has been proposed as a decisive factor in determining its multiple functions,40 it is possible that cells might behave differently, or in the opposite manner, as PTEN level is further reduced. However, our study does not support such a possibility. In PTEN-null Jurkat cells, enhanced, instead of impaired, chemotaxis was observed. Restoration of PTEN expression substantially down-regulated CXCR4-mediated chemotaxis, which was correlated with PTEN expression levels (Figure 2 C-D). On the other hand, although Anzelon et al reported impaired chemotaxis of the PTENloxp/loxP B cells in the in vitro assays, it is interesting to notice that, in those conditional knockout mice, as indicated by the preferential generation of marginal zone B cells, B1 cells, and germinal center formation,21 the general homeostatic homing of the PTEN-deficient B cells to specific secondary lymphoid compartments was not impaired, as has been observed in the knockout phenotypes of certain chemokine receptors, such as CCR7 or CXCR5.41-43 The reported increase in B-cell cellularity in specific second lymphoid compartments suggest that PTEN deficiency did not actually impair the specific homing of lymphocytes to secondary lymphoid organs in vivo, a process in which chemotaxis plays an indispensable role.44
The present study demonstrated that Jurkat cells with complete loss of PTEN exhibited enhanced directional movement toward SDF-1α as well as IGF-1 and restoration of PTEN expression inhibited such migration. Although SDF-1α and IGF1 use receptors of different families, their intracellular signaling pathways converge at the PI3K-PIP3 level.37 Indeed, we established that the lipid phosphatase activity was essential for the role of PTEN as a negative regulator of chemotaxis (Figures 3 and 4), suggesting that, by antagonizing the PI3K-PIP3 pathway, PTEN inhibited chemotaxis. Of note, we never observed complete abolition of chemotaxis by PTEN expression in Jurkat cells, indicating a delicate balance existing between PTEN and PI3K, and perhaps other pathways, that favor cell migration. Although the lipid substrates of PTEN and its implications in tumorigenesis have been intensively studied,13,45,46 the present results reveal a previously unappreciated aspect of the multifunctional nature of PTEN in tumor biology. Since chemokines, and perhaps other chemoattractants of the growth factor family, have been proposed as promoters of cancer cell metastasis to specific target organs,9-11 the present results prompted us to hypothesize that tumor cells with “loss of function” of PTEN may develop enhanced chemotactic ability, resulting in increased metastatic spread. Indeed, accumulating evidence demonstrated that “loss of function” of PTEN has been associated with late-stage, more aggressive, and metastatic cancers.12,14,47-49 Experiments using vector-based siRNA constructs to knockdown PTEN or Tet-inducible systems to restore PTEN expression in several human tumor cell models are currently in progress and their in vitro and in vivo behaviors in directional migration will be examined.
Acknowledgments
The authors are grateful to Drs Paul Hu, Gareth Davies, and Xin Chen for advice. We thank Drs Ji-Ming Wang, William L. Farrar, and Xia Zhang for critical review of this manuscript. We thank Dr Helene Tonoli for kindly providing us with IGF-1, Dr Esta Sterneck for sharing Amaxa Nucleofector device, and Dr Hiromi Fujiwara for 2D6 and 2B4 T cells.
Prepublished online as Blood First Edition Paper, June 30, 2005; DOI 10.1182/blood-2004-08-3362.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Locati M, Murphy PM. Chemokines and chemokine receptors: biology and clinical relevance in inflammation and AIDS. Annu Rev Med. 1999;50: 425-440. [DOI] [PubMed] [Google Scholar]
- 2.Meili R, Firtel RA. Two poles and a compass. Cell. 2003;114: 153-156. [DOI] [PubMed] [Google Scholar]
- 3.Weiner OD, Neilsen PO, Prestwich GD, Kirschner MW, Cantley LC, Bourne HR. A PtdInsP(3)- and Rho GTPase-mediated positive feedback loop regulates neutrophil polarity. Nat Cell Biol. 2002;4: 509-513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laudanna C, Kim JY, Constantin G, Butcher E. Rapid leukocyte integrin activation by chemokines. Immunol Rev. 2002;186: 37-46. [DOI] [PubMed] [Google Scholar]
- 5.Mellado M, Rodriguez-Frade JM, Manes S, Martinez AC. Chemokine signaling and functional responses: the role of receptor dimerization and TK pathway activation. Annu Rev Immunol. 2001;19: 397-421. [DOI] [PubMed] [Google Scholar]
- 6.Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12: 121-127. [DOI] [PubMed] [Google Scholar]
- 7.Oppenheim JJ, Yang D, Biragyn A, Howard OM, Plotz P. Chemokine receptors on dendritic cells promote autoimmune reactions. Arthritis Res. 2002;4: S183-S188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Richmond A. Nf-kappa B, chemokine gene transcription and tumour growth. Nat Rev Immunol. 2002;2: 664-674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muller A, Homey B, Soto H, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410: 50-56. [DOI] [PubMed] [Google Scholar]
- 10.Wang JM, Deng X, Gong W, Su S. Chemokines and their role in tumor growth and metastasis. J Immunol Methods. 1998;220: 1-17. [DOI] [PubMed] [Google Scholar]
- 11.Murphy PM. Chemokines and the molecular basis of cancer metastasis. N Engl J Med. 2001;345: 833-835. [DOI] [PubMed] [Google Scholar]
- 12.Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275: 1943-1947. [DOI] [PubMed] [Google Scholar]
- 13.Steck PA, Pershouse MA, Jasser SA, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15: 356-362. [DOI] [PubMed] [Google Scholar]
- 14.Di Cristofano A, Pandolfi PP. The multiple roles of PTEN in tumor suppression. Cell. 2000;100: 387-390. [DOI] [PubMed] [Google Scholar]
- 15.Raftopoulou M, Etienne-Manneville S, Self A, Nicholls S, Hall A. Regulation of cell migration by the C2 domain of the tumor suppressor PTEN. Science. 2004;303: 1179-1181. [DOI] [PubMed] [Google Scholar]
- 16.Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. 1998;280: 1614-1617. [DOI] [PubMed] [Google Scholar]
- 17.Iijima M, Devreotes P. Tumor suppressor PTEN mediates sensing of chemoattractant gradients. Cell. 2002;109: 599-610. [DOI] [PubMed] [Google Scholar]
- 18.Funamoto S, Meili R, Lee S, Parry L, Firtel RA. Spatial and temporal regulation of 3-phosphoinositides by PI 3-kinase and PTEN mediates chemotaxis. Cell. 2002;109: 611-623. [DOI] [PubMed] [Google Scholar]
- 19.Comer FI, Parent CA. PI 3-kinases and PTEN: how opposites chemoattract. Cell. 2002;109: 541-544. [DOI] [PubMed] [Google Scholar]
- 20.Fox JA, Ung K, Tanlimco SG, Jirik FR. Disruption of a single Pten allele augments the chemotactic response of B lymphocytes to stromal cell-derived factor-1. J Immunol. 2002;169: 49-54. [DOI] [PubMed] [Google Scholar]
- 21.Anzelon AN, Wu H, Rickert RC. Pten inactivation alters peripheral B lymphocyte fate and reconstitutes CD19 function. Nat Immunol. 2003;4: 287-294. [DOI] [PubMed] [Google Scholar]
- 22.Seminario MC, Precht P, Wersto RP, Gorospe M, Wange RL. PTEN expression in PTEN-null leukaemic T cell lines leads to reduced proliferation via slowed cell cycle progression. Oncogene. 2003;22: 8195-8204. [DOI] [PubMed] [Google Scholar]
- 23.Nakahira M, Ahn HJ, Park WR, et al. Synergy of IL-12 and IL-18 for IFN-gamma gene expression: IL-12-induced STAT4 contributes to IFN-gamma promoter activation by up-regulating the binding activity of IL-18-induced activator protein 1. J Immunol. 2002;168: 1146-1153. [DOI] [PubMed] [Google Scholar]
- 24.Mukai T, Iwasaki M, Gao P, et al. IL-12 plays a pivotal role in LFA-1-mediated T cell adhesiveness by up-regulation of CCR5 expression. J Leukoc Biol. 2001;70: 422-430. [PubMed] [Google Scholar]
- 25.Gao P, Zhou XY, Yashiro-Ohtani Y, et al. The unique target specificity of a nonpeptide chemokine receptor antagonist: selective blockade of two Th1 chemokine receptors CCR5 and CXCR3. J Leukoc Biol. 2003;73: 273-280. [DOI] [PubMed] [Google Scholar]
- 26.Howard OM, Dong HF, Yang D, et al. Histidyl-tRNA synthetase and asparaginyl-tRNA synthetase, autoantigens in myositis, activate chemokine receptors on T lymphocytes and immature dendritic cells. J Exp Med. 2002;196: 781-791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maasho K, Marusina A, Reynolds NM, Coligan JE, Borrego F. Efficient gene transfer into the human natural killer cell line, NKL, using the Amaxa nucleofection system. J Immunol Methods. 2004;284: 133-140. [DOI] [PubMed] [Google Scholar]
- 28.Xu Z, Stokoe D, Kane LP, Weiss A. The inducible expression of the tumor suppressor gene PTEN promotes apoptosis and decreases cell size by inhibiting the PI3K/Akt pathway in Jurkat T cells. Cell Growth Differ. 2002;13: 285-296. [PubMed] [Google Scholar]
- 29.Shan X, Czar MJ, Bunnell SC, et al. Deficiency of PTEN in Jurkat T cells causes constitutive localization of Itk to the plasma membrane and hyper-responsiveness to CD3 stimulation. Mol Cell Biol. 2000;20: 6945-6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sakai A, Thieblemont C, Wellmann A, Jaffe ES, Raffeld M. PTEN gene alterations in lymphoid neoplasms. Blood. 1998;92: 3410-3415. [PubMed] [Google Scholar]
- 31.Wang X, Gjorloff-Wingren A, Saxena M, Pathan N, Reed JC, Mustelin T. The tumor suppressor PTEN regulates T cell survival and antigen receptor signaling by acting as a phosphatidylinositol 3-phosphatase. J Immunol. 2000;164: 1934-1939. [DOI] [PubMed] [Google Scholar]
- 32.Schneider U, Schwenk HU, Bornkamm G. Characterization of EBV-genome negative “null” and “T” cell lines derived from children with acute lymphoblastic leukemia and leukemic transformed non-Hodgkin lymphoma. Int J Cancer. 1977;19: 621-626. [DOI] [PubMed] [Google Scholar]
- 33.Maehama T, Dixon JE. PTEN: a tumour suppressor that functions as a phospholipid phosphatase. Trends Cell Biol. 1999;9: 125-128. [DOI] [PubMed] [Google Scholar]
- 34.Maier D, Jones G, Li X, et al. The PTEN lipid phosphatase domain is not required to inhibit invasion of glioma cells. Cancer Res. 1999;59: 5479-5482. [PubMed] [Google Scholar]
- 35.Ward SG. Do phosphoinositide 3-kinases direct lymphocyte navigation? Trends Immunol. 2004;25: 67-74. [DOI] [PubMed] [Google Scholar]
- 36.Cronshaw DG, Owen C, Brown Z, Ward SG. Activation of phosphoinositide 3-kinases by the CCR4 ligand macrophage-derived chemokine is a dispensable signal for T lymphocyte chemotaxis. J Immunol. 2004;172: 7761-7770. [DOI] [PubMed] [Google Scholar]
- 37.Curnock AP, Sotsios Y, Wright KL, Ward SG. Optimal chemotactic responses of leukemic T cells to stromal cell-derived factor-1 requires the activation of both class IA and IB phosphoinositide 3-kinases. J Immunol. 2003;170: 4021-4030. [DOI] [PubMed] [Google Scholar]
- 38.Maruo S, Ahn HJ, Yu WG, et al. Establishment of an IL-12-responsive T cell clone: its characterization and utilization in the quantitation of IL-12 activity. J Leukoc Biol. 1997;61: 346-352. [DOI] [PubMed] [Google Scholar]
- 39.Xu J, Wang F, Van Keymeulen A, et al. Divergent signals and cytoskeletal assemblies regulate self-organizing polarity in neutrophils. Cell. 2003;114: 201-214. [DOI] [PubMed] [Google Scholar]
- 40.Trotman LC, Niki M, Dotan ZA, et al. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003;1: E59[AUXX]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Forster R, Schubel A, Breitfeld D, et al. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99: 23-33. [DOI] [PubMed] [Google Scholar]
- 42.Forster R, Mattis AE, Kremmer E, Wolf E, Brem G, Lipp M. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell. 1996;87: 1037-1047. [DOI] [PubMed] [Google Scholar]
- 43.Reif K, Ekland EH, Ohl L, et al. Balanced responsiveness to chemoattractants from adjacent zones determines B-cell position. Nature. 2002;416: 94-99. [DOI] [PubMed] [Google Scholar]
- 44.Cyster JG. Chemokines and cell migration in secondary lymphoid organs. Science. 1999;286: 2098-2102. [DOI] [PubMed] [Google Scholar]
- 45.Myers MP, Pass I, Batty IH, et al. The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc Natl Acad Sci U S A. 1998;95: 13513-13518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ward SG, Cantrell DA. Phosphoinositide 3-kinases in T lymphocyte activation. Curr Opin Immunol. 2001;13: 332-338. [DOI] [PubMed] [Google Scholar]
- 47.Bandyopadhyay S, Pai SK, Hirota S, et al. PTEN up-regulates the tumor metastasis suppressor gene Drg-1 in prostate and breast cancer. Cancer Res. 2004;64: 7655-7660. [DOI] [PubMed] [Google Scholar]
- 48.Liu TC, Lin PM, Chang JG, Lee JP, Chen TP, Lin SF. Mutation analysis of PTEN/MMAC1 in acute myeloid leukemia. Am J Hematol. 2000;63: 170-175. [DOI] [PubMed] [Google Scholar]
- 49.Wang S, Gao J, Lei Q, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4: 209-221. [DOI] [PubMed] [Google Scholar]