

Abstract
Cytokines are critical in regulating the development and function of diverse cells. Janus kinase 3 (Jak3) is a tyrosine kinase expressed in hematopoietic cells that associates with the common gamma chain (γc) and is required for signaling for a family of cytokines including interleukin-2 (IL-2), IL-4, IL-7, IL-9, IL-15, and IL-21; deficiency of either Jak3 or γc results in severe combined immunodeficiency (SCID). While Jak3 is essential for lymphoid-cell development, the potential roles for Jak3 in regulating dendritic cells (DCs) were unclear. Herein, we show that although CD8+CD11c+ splenic DCs are absent in Jak3-/- mice, bone marrow–derived DCs developed normally in vitro from Jak3-/- precursor cells. In fact, the survival of Jak3-/- DCs was enhanced, and they expressed lower levels of proapoptotic proteins. Jak3-/- DCs exhibited normal antigen uptake and up-regulation of costimulatory molecules. However, Jak3-/- DCs produced more IL-12 and IL-10 in response to Toll-like receptor ligands, which correlated with enhanced T helper 1 (Th1) differentiation in vivo. In summary, Jak3 is not essential for DC development but unexpectedly appears to be an important negative regulator. These results may be relevant clinically for patients with SCID who have undergone hematopoietic stem cell transplantation and for patients who might be treated with a Jak3 inhibitor.
Introduction
The development and differentiation of all lineages of hematopoietic cells is exquisitely regulated by multiple cytokines.1 Interleukin-7 (IL-7), for example, has critical functions in regulating the development of T and B cells, whereas IL-15 is an important regulator of natural killer cells.2,3 Moreover, the differentiation of T cells to helper cells, effector cells, and memory cells is also controlled by cytokines.2,4-6 Accordingly, defects in expression of these cytokines or their receptors profoundly affect the development and differentiation of lymphocytes. In humans, this is evidenced by the disorder severe combined immunodeficiency (SCID), which can be due to mutation of the common gamma chain (γc) or mutation of IL-7 receptor (IL-7R) itself.7-10
The first step in cytokine signal transduction is the activation of Janus family kinases, which comprise 4 members—Jak1, Jak2, Jak3, and Tyk2—that associate with different cytokine receptors.11-13 Jak3 associates with γc and is critical for cytokines that use this receptor subunit. Humans and mice deficient in Jak3 also suffer from SCID.13-18 Together, mutations of IL-7R, γc, and Jak3 account for almost two thirds of SCID, consistent with the critical roles of cytokine-mediated signaling in lymphoid development and function.
Dendritic cells (DCs) are also key players in the immune response, linking innate and adaptive responses. As immature cells, DCs are scattered throughout the body in nonlymphoid organs where they exert a sentinel function by taking up antigen and initiating inflammatory and immune responses. After encountering antigen, DCs mature and migrate to regional lymph nodes. Activated DCs express high levels of costimulatory molecules (CD40, CD80, and CD86) and major histocompatibility complex (MHC) molecules. Inflammatory stimuli, microbial products, and cognate T-cell signals such as CD40 ligand (CD40L) can all induce a program of maturation in DCs and induce the secretion of cytokines such as IL-12. Consequently, DCs stimulated in this manner are very efficient activators of T cells, promoting the differentiation of naive T cells into T helper 1 (Th1) cells.4
In terms of development, the origin of DCs remains murky. While it is clear that they can be generated from myeloid cells, it is also clear that lymphoid precursors contain cells that can differentiate into DCs.19,20 Given the profound effect of Jak3 and its cognate cytokines on lymphoid development, it was of interest to examine DC development and function in Jak3-deficient mice. In this regard, it is known that Jak3 is expressed in myeloid cells, where it is inducible in response to cytokines and Toll-like receptor (TLR) ligands, whereas γc is constitutively expressed.21,22 In contrast to the dramatic defects in lymphocyte maturation, however, no intrinsic defects have been found in myeloid cells lacking Jak3.21,23-25 Moreover, after hematopoietic stem cell transplantation, myeloid lineage cells are still predominantly of recipient origin, suggesting that Jak3-dependent signals are dispensable in myeloid cells.26 Jak3-/- mice show abnormal expansion and infiltration of organs by myeloid lineage cells; however, this is thought to be a consequence of activated T cells that accumulate in aging Jak3-/- mice.27 Therefore, one would predict that, to the extent that DCs are related to myeloid cells, their development should not be impaired in Jak3- and γc-deficient mice.
We were therefore interested in investigating the role of Jak3 in DC function and the ability of Jak3-/- DCs to regulate T-cell differentiation. We also sought to compare and contrast Jak3 and γc deficiency to determine if γc-independent functions of Jak3 were apparent. Herein, we report that Jak3-/- bone marrow–derived DCs (BMDCs) developed and matured normally. Surprisingly, Jak3-/- and γc-/- DCs exhibited a higher survival rate upon cytokine removal. They produced higher levels of IL-12 and IL-10 upon stimulation with TLR ligands, and normal T cells responded to Jak3-/- DCs by producing relatively higher levels of interferon-γ (IFN-γ). Together these data indicate that Jak3 and γc are not required for DC development or signaling through TLRs. However, they do serve to negatively regulate DC survival and cytokine production.
Materials and methods
Mice
Jak3-/- (B6;129S4-Jak3tm1Ljb/J), γc-/- (B6.129S4-Il2rgtm1Wjl), and wild-type C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME). IL-15-/- (C57BL/6-IL15tm1Imx), OTII TCR transgenic mice (CD45.2+), and C57BL/6 (CD45.1+) congenic mice were purchased from Taconic (Germantown, NY). Jak3-/- and γc-/- mice were bred under specific pathogen-free conditions at the National Institutes of Health (NIH) and were used between 6 to 12 weeks of age in accordance with institutional animal care guidelines.
Reagents
The following items were purchased: lipopolysaccharide (LPS) from Escherichia coli serotype 055:B5, Zymosan A from Saccharomyces cerevisiae (Sigma, St Louis, MO), cytosine-phosphate-guanosine (CpG) DNA (5′-TCCATGACGTTCCTGACGTT-3′) (Lofstrand Labs, Gaithersburg, MD), peptidoglycan from Staphylococcus aureus (Fluka, St Louis, MO), Poly I:C (Amersham, Freiburg, Germany), IFN-γ (Peprotech, Rocky Hill, NJ), fluorescein isothiocyanate (FITC)–conjugated albumin (Sigma), and WHI-P-154 (Calbiochem, San Diego, CA). Quantikine M enzyme-linked immunosorbent assay (ELISA) kits were from R&D Systems (Minneapolis, MN). The following monoclonal antibodies were purchased from Pharmingen (San Diego, CA): FITC-conjugated anti-CD11c, phycoerythrin (PE)–conjugated anti-CD4, PE-conjugated anti-Gr1, PE-conjugated anti–IFN-γ, peridinin-chlorophyII-protein complex (PerCP)–conjugated anti-CD8, PerCP-conjugated anti-B220, PE-conjugated antigoat, and PE-conjugated anti-CD25.
Cells and cell lines
DC-enriched low-density cell fractions from Liberase CI (Roche, Indianapolis, IN)–treated spleens were prepared using 30% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) as described.28 To generate BMDCs, cells were obtained from femurs and tibias of 6- to 12-week-old mice, and cultured at 1 × 106 cells/mL in RPMI 1640 medium with 10% fetal calf serum (Biosource, Camarillo, CA), 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM l-glutamine, 100 mM HEPES (N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid), 1 mM sodium pyruvate, and 5 μM 2-mercaptoethanol. The medium was supplemented with 40 ng/mL granulocyte-macrophage colony-stimulating factor (GM-CSF) and 20 ng/mL IL-4 (Peprotech). On days 3 and 5, fresh media equal to half of the initial culture volume containing 40 ng/mL GM-CSF was added to culture. On day 7, nonadherent cells were collected, and dead cells were eliminated by centrifugation over Ficoll (Amersham Biosciences, Uppsala, Sweden). CD11c+ DCs were isolated by positive selection with FITC-conjugated anti–mouse CD11c and magnetic separation using anti-FITC microbeads (Miltenyi Biotech, Auburn, CA) and an AUTOMACS (Miltenyi Biotech) according to the manufacturer's instructions. The purity of the cell population was determined to be more than 90% CD11c+ by flow cytometry.
Human monocyte-derived DCs (MoDCs) were prepared from elutriated monocytes (Department of Transfusion Medicine, NIH, Bethesda, MD) by culturing the cells with GM-CSF (50 ng/mL) and IL-4 (10 ng/mL; Peprotech) for 7 days. Nonadherent cells collected on day 7 were found to be CD14-. NIH3T3-SAMEN and NIH3T3-CD154 were generated by transduction of the murine NIH3T3 fibroblast line with an empty retroviral vector (SAMEN) or retroviral vector encoding full-length transmembrane murine CD40L and were kindly provided by Dr Patrick Hwu (University of Texas).
Western blotting, quantitative reverse transcriptase–polymerase chain reaction (RT-PCR), RNase protection assays, and ELISA
Human MoDCs and murine BMDCs were stimulated for 24 hours and lysed in buffer containing 50 mM Tris (tris(hydroxymethyl)aminomethane)–HCl (pH 7.5), 300 mM NaCl, 2 mM EDTA (ethylenediaminetetraacetic acid), 0.5% TritonX-100 (Sigma), 200 μM Na3VO4, 10 μg leupeptin, 10 μg aprotinin, and 2.5 μM p-nitrophenyl-p-guanidinobenzoate. Protein concentration was quantified with a bicinchonic acid (BCA) kit (Pierce, Rockford, IL). Lysates (100 μg) were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), transferred to nitrocellulose membranes, and immunoblotted with antibodies to Jak3. Total RNA was isolated from BMDCs with RNeasy minikit (Qiagen, Valencia, CA) and reverse-transcribed using a first-strand cDNA synthesis kit (Roche). Real-time PCR was performed using the ABI PRISM 7700 Sequence Detection System (Applied Biosystems, Foster City, CA). Amplification of 18s mRNA was done for each experimental sample as an endogenous control to account for differences in the amount and quality of total RNA added to each reaction. Relative levels of Bax and Bak were normalized to 18s levels. The data are presented as relative to the unstimulated control for each individual experiment, which was arbitrarily designated a value of 1.0.
RNase protection assays were performed with the RPAIII Ribonuclease Protection Assay Kit (Ambion, Austin, TX) with the mAPO-2 Multi-Probe Template Set (Pharmingen) according to the manufacturer's instructions. Protected RNA fragments were resolved on a 6% polyacrylamide-urea gel under denaturing conditions followed by densitometric quantification by Storm (Molecular Dynamics, Sunnyvale, CA). Internal standards (L32 and glyceraldehyde 3-phosphate [GAPDH]) were used to normalize for sample loading. For cytokine measurement, purified DCs (106/mL) were cultured in a 96-well plate with the appropriate stimulus. After 24 hours, supernatants were collected and Quantikine M ELISA kits (R&D Systems) were used to assess cytokine levels, according to the manufacturer's instructions. In selected experiments, a Cytometric bead array mouse inflammation kit (Pharmingen) was used.
Adoptive transfer of DCs
CD4+ T cells from OTII TCR transgenic mice (CD45.2+) were isolated from peripheral lymph nodes of donor mice by incubating with FITC-conjugated anti–I-Ab, anti-CD24, anti-CD16 (2.4G2), anti-CD8, anti-B220 and anti-NK1.1, followed by anti-FITC microbeads (Miltenyi Biotech). After magnetic separation using an LS column (Miltenyi Biotech), isolated T cells (3 × 106) were injected intravenously from tail vein into C57BL/6 congenic mouse (CD45.1+). Concomitantly, BMDCs from Jak3-/- and wild-type mice (1 × 10,6 CD45.2+) were pulsed with ovalbumin329-339 (OVA329-339) peptide (OVAp; Research Services Branch, NIAID, NIH) and injected into footpads of the mice that had received the T cells. Three and a half days later, axillary lymph nodes were collected and single-cell suspensions were made. The cells were restimulated with anti–mouse CD28 (3 μg/mL) and OVAp (1 μM) for 6 to 8 hours. Brefeldin A was added for the last 4 hours of culture, and the cells were fixed with 2% paraformaldehyde and permeabilized with PBS supplemented with 0.1% BSA plus 0.1% saponin, and intracellular staining was performed with FITC-conjugated anti-CD45.2, PE-conjugated anti–IFN-γ, and allophycocyanin (APC)–conjugated anti-CD4. Cells were gated on CD45.2+CD4+ cells to analyze IFN-γ production. Fluorescence was measured with a FACSCalibur (Becton Dickinson, Franklin Lakes, NJ) and was analyzed with FlowJo software (Treestar, Ashland, OR).
Bone marrow transplant
Six million total bone marrow cells were injected into the lateral tail vein of lethally irradiated Rag2 knock-out recipients, using a 1:1 mixture of wild-type (CD45.1+) and Jak3-/- (CD45.2+) bone marrow. After 6 to 8 weeks, spleens were harvested, stained for DC populations, and analyzed by flow cytometry. Cells generated from Jak3-/- and wild-type precursors were assessed by determining CD45.1 and CD45.2 expression on CD11c+ and CD11b+ populations using FlowJo software.
Results
Jak3 and receptors that use Jak3 are induced in DCs
To evaluate potential functions of Jak3 in DCs, we first examined Jak3 expression in these cells and assessed whether the levels correlated with differentiation. As shown in Figure 1A (top panel), Jak3 was expressed at low levels in immature MoDCs. However, upon stimulation with TLR ligands, including LPS, peptidoglycan (PGN), zymosan, and CpG oligonucleotides, Jak3 was markedly induced in a manner similar to that reported in macrophages.21 By contrast, in murine BMDCs, Jak3 was expressed in immature DCs and its expression level did not change upon maturation. The levels of Jak3 in BMDCs were confirmed by assessment of mRNA by real-time PCR and were consistent with the protein expression (Figure 1, lower panel). Concomitantly, receptors for IL-2 and IL-15, which use γc and Jak3, were induced in both MoDCs and BMDCs (Figure 1B-C), although we did not find alteration in expression of IL-7 receptor expression (Figure 1B). The levels of γc did not change with activation (data not shown).21,22 These results show that Jak3 and receptors that use this kinase are present in DCs, suggesting that they may play a role in DC development or function.
Figure 1.

Regulation of γc cytokine receptors and Jak3 in DCs. (A) Jak3 expression in activated DCs. Human MoDCs (top) and murine BMDCs (middle and bottom) were stimulated with LPS (1 μg/mL), PGN (5 μg/mL), or zymosan A (5 μg/mL) for 24 hours. Cell lysates (100 μg) were electrophoresed and blotted with anti-Jak3 antibody. Jak3 mRNA was assessed by real-time PCR in murine BMDCs stimulated with LPS (1 μg/mL) or CpG oligonucleotides (oligos) (1 μg/mL) for 6 hours (bottom). (B) Human MoDCs and murine BMDCs were stimulated with LPS (1 μg/mL) for 24 hours, stained with FITC-labeled antibodies, and then analyzed by flow cytometry. Broken line indicates unstimulated cells; solid line, activated cells. Cells stained with an isotype control antibody are depicted in the shaded histogram. (C) IL-15Rα expression by murine BMDCs was measured by real-time PCR in cells treated with LPS (1 μg/mL) for 6 hours or left unstimulated.
Abnormalities in splenic DC subsets associated with Jak3 deficiency
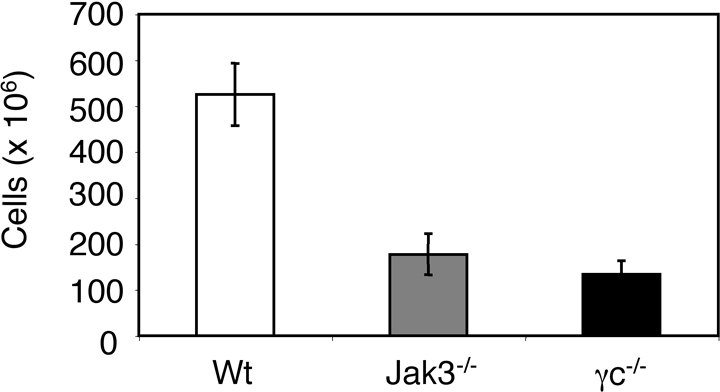
Murine splenic DCs express the surface molecule CD11c. As shown in Figure 2A, approximately 40% of the DC-enriched fraction from splenocytes in normal mice expresses this marker (upper left panel). In contrast, the proportion of CD11c+ DCs in Jak3-/- mice was significantly reduced (Figure 2A [lower left panel] and Figure 2B). In addition, the number of splenocytes was reduced in Jak3-/- mice, and thus the absolute number of CD11c+ cells was also reduced (Figure 2B; also see Supplemental Figure S1, available at the Blood website by clicking on the Supplemental Figures link at the top of the online article). Furthermore, γc-/- mice also had reduced numbers of splenocytes and also exhibited similar reduction in the proportions and absolute numbers of CD11c+ DCs (Supplemental Figure S1; data not shown). Within the spleen, at least 4 DC subsets are recognized: CD11chiCD4-CD8+, CD11chiCD4+CD8-, CD11chiCD4-CD8- DCs, and CD11cintB220+Gr1+.29-32 The CD4+CD8α- population (CD4+ DCs) was found to be the predominant DC subset (46%) in control mice (Figure 2A, upper middle panel), consistent with previous reports, whereas the CD4-CD8α+ population (CD8+ DCs) represented approximately 20% of cells.29,30 In contrast, the proportion and absolute number of CD8+ DCs were greatly reduced in spleens from Jak3-/- mice. The proportion and numbers of CD4+ DCs in Jak3-/- mice were also reduced (Figure 2A-B). However, plasmacytoid DCs (CD11cint, Gr-1+, B220+) were observed with the same frequency in spleens of Jak3-/- mice as wild-type mice (Figure 2A, right panel), and there was no significant difference in absolute number of cells (Figure 2B). Normally, in the absence of stimulation, CD11c+ DCs express low levels of the costimulatory molecules CD80, CD86, and CD40, and express moderate levels of MHC class II (Figure 2C, dashed lines); such DCs are referred to as immature DCs. However, splenic DCs from Jak3-/- mice constitutively expressed high levels of these activation markers (Figure 2C, solid lines). Thus, Jak3-/- mice had reduced numbers of DCs, which appeared to be activated.
Figure 2.

Abnormal proportions of splenic DC subsets in Jak3-/- mice. (A) DC-enriched low-density cells from wild-type (Wt) and Jak3-/- spleens were stained with anti-CD4, anti-CD8, anti-B220, anti-Gr1, and anti-CD11c. The proportion of CD11c+ cells is presented in the left column. Gating on CD11c+ cells, the proportions of different DC subsets are illustrated in the middle and right columns. (B) The absolute number (mean ± SD) of each DC subset in wild-type and Jak3-/- spleens from 8 separate experiments is shown. *P < .05 and **P < .01 compared with wild type (n = 8). (C) Activated phenotype of Jak3-/- splenic DCs. DC-enriched low-density cells from wild-type (broken line) and Jak3-/- (solid line) spleens were gated on CD11c+ and analyzed for MHC class II, CD40, CD80, and CD86 expression. Membrane expression of these proteins was assessed by flow cytometry and is compared with staining with an isotype control antibody (shaded histograms). Data are representative of 5 separate experiments with similar results.
Normal in vitro development and maturation of Jak3-/- DCs
Given the abnormalities in splenic DC populations in Jak3-/- mice, we next sought to determine if this was a cell-intrinsic defect. Lymphocytes from Jak3-/- mice have an activated phenotype.33 These mice also exhibit T-cell–dependent expansion of myeloid cells.27 Thus, it was possible that the DC alterations were not due to a cell-intrinsic defect but could be the result of the immune dysregulation seen in Jak3-/- mice. To determine if Jak3-/- DCs had the capacity to develop and function normally, we isolated bone marrow precursors and differentiated DCs in vitro. As shown in Figure 3A, both wild-type and Jak3-/- BM cells showed similar capacity to generate CD11c+ cells. These cells also showed normal expression of DC maturation markers, confirming that immature DCs were generated (Figure 3B). DCs can also be generated in vitro using bone marrow cultured with Flt3 ligand (Flt3L).34 Both wild-type and Jak3-/- BM cells showed similar capacity to generate immature CD11c+ cells when this stimulus was used (Supplemental Figure S2A). Additionally, these cells had similar expression of maturation markers, before and after stimulation with TLR ligands (data not shown). We confirmed these findings by transplanting irradiated mice with wild-type and Jak3-/- bone marrow and found that equivalent numbers and proportions of DCs were generated (Supplemental Figure S2B-C). Thus, we conclude from these data that Jak3 is not required for DC development. The abnormal populations and maturation of DCs in spleens of Jak3-/- mice are not the result of an intrinsic defect; rather they are likely secondary effects due to the immune abnormalities evident in these mice.
Figure 3.

Normal development and function of Jak3-/- BMDCs. (A) Normal in vitro generation of CD11c+ BMDCs from Jak3-/- mice. Bone marrow cells from wild-type (left) and Jak3-/- (right) mice were cultured for 7 days with GM-CSF and IL-4. The resultant cells were stained with anti-CD11c antibody (solid line). Shaded histograms represent samples stained with an isotype control. (B) Jak3-/- BMDCs have normal expression of costimulatory and MHC molecules. Purified Jak3-/- (solid line) and wild-type (broken line) BMDCs (> 90% CD11c+) were stained with the indicated antibodies after 7 days of culture in GM-CSF and IL-4. (C) Normal micropinocytosis by Jak3-/- BMDCs. BMDCs from wild-type and Jak3-/- mice were pulsed with 5 μg/mL FITC-conjugated albumin for 60 minutes. Broken line indicates background uptake of cells incubated at 0°C; solid line, albumin uptake at 37°C. The shaded histograms represent samples in which FITC-conjugated albumin was omitted. (D) Normal induction of CD86 in Jak3-/- BMDCs. BMDCs from wild-type and Jak3-/- mice were stimulated with CD40L-bearing cells (NIH3T3CD40L) and LPS (1 μg/mL) for 24 hours (solid line) and analyzed for CD86 expression compared with unstimulated cells (broken line). Shaded histograms are samples stained with an isotype control antibody. (E) Nonspecific inhibition of CD86 expression by WHI-P-154. Wild-type and Jak3-/- BMDCs were pretreated with dimethyl sulfoxide (DMSO; broken line) or WHI-P-154 (10 μg/mL; solid line) for 30 minutes, stimulated with CD40L and LPS for 24 hours, and stained with anti-CD86. Note that the putative Jak3 inhibitor effectively blocked CD86 expression in Jak3-/- cells, indicative of nonspecific effects.
While the in vitro differentiated Jak3-/- DCs were phenotypically normal, we did not know if their function was normal. Since immature DCs are highly active phagocytic cells, we first analyzed this aspect of DC function. When wild-type and Jak3-/- DCs were pulsed with FITC-labeled albumin, both showed similar capability of micropinocytosis (Figure 3C).
Using a nonspecific inhibitor, WHI-P-154, it has been proposed that Jak3 is important for DC maturation and cytokine production.35 To examine the role for Jak3 in this process, we analyzed Jak3-/- DCs treated with the inhibitor. As shown in Figure 3D, stimulation of BMDCs with LPS and CD40L up-regulated the expression of the costimulatory molecule CD86 (left panel), and the inhibitor completely blocked this induction (Figure 3E, left panel). However, CD86 was induced in Jak3-/- BMDCs at levels comparable with wild-type cells (Figure 3D, right panel). Moreover, the putative inhibitor effectively blocked expression of CD86 even on cells that lack Jak3 (Figure 3E, right panel). Thus, we conclude that Jak3 is not required for DC maturation, and the reported effects of the inhibitor are due to its lack of specificity.36
Prolonged survival of Jak3-/- DCs
Because γc cytokines regulate the survival of lymphoid cells, we next examined whether γc or Jak3 is involved in DC survival. Since the numbers of DCs generated in vitro and in bone marrow transplant studies were normal, we did not expect major defects in cell viability; however, we wanted to directly assess DC survival. To promote DC apoptosis, we cultured the cells in the absence of cytokines. As shown in Figure 4A-B, 90% of DCs became annexin V and propidium iodide (PI) positive 24 hours after cytokine withdrawal. As expected, the number of viable cells continued to decline, such that there were essentially no live cells by 48 hours. This is in marked contrast to Jak3-/- DCs, which were remarkably resistant to cytokine withdrawal; at 48 hours, 30% to 60% of DCs were still viable as determined by annexin V/PI staining. Because of their association, we expected that Jak3 and γc deficiency would be identical. As is evident in Figure 4A-B, this appears to be the case; γc-/- DCs were also resistant to cytokine withdrawal–induced apoptosis. To help define which γc cytokines might regulate DC survival, we analyzed DCs from IL-15-/- mice. As shown in Figure 4A-B, IL-15-/- BMDCs also showed enhanced survival compared with wild-type BMDCs.
Figure 4.

Enhanced survival of Jak3-/- and γc-/- BMDCs. (A-B) Purified BMDCs from wild-type, Jak3-/-, γc-/-, and IL-15-/- mice were cultured in medium lacking cytokines and stained with APC-conjugated annexin V and propidium iodide at the indicated time points. Cells that were negative for staining of both annexin V or PI were designated as live cells. These data are representative of more than 5 separate experiments. (C) Bak and Bax mRNA expression. Purified BMDCs were cultured in medium lacking cytokines for 12 hours and RNA was isolated. Bak and Bax levels were determined by RNase protection assay. L32 probes were used to normalize sample load in each well. The data are representative of 3 separate experiments.
To begin to explore the mechanism responsible for the prolonged survival of Jak3-/- and γc-/- BMDCs, we next analyzed the expression of proapoptotic proteins. We observed decreased expression of Bax and Bak in Jak3-/- BMDCs, while no significant differences were observed in Bcl-2 and Bcl-xL expression. Thus, Jak3 seems to negatively regulate apoptosis by up-regulating Bak and Bax expression in DCs.
Jak3-/- BMDCs overproduce IL-12 and IL-10
Upon encounter with pathogens, DCs are activated to produce a number of immunoregulatory cytokines that have critical functions in directing immune responses. We therefore assessed the ability of Jak3-/- DCs to produce various cytokines. To this end, wild-type, Jak3-/-, and γc-/- BMDCs were treated with either LPS or CpG oligonucleotides alone or in combination with IFN-γ, and the production of IL-12, IL-10, tumor necrosis factor α (TNF-α), and IL-6 was assessed (Figure 5). Upon stimulation with LPS or CpG oligos, wild-type DCs showed induction of IL-12p70 production, which was enhanced when IFN-γ was added. Of interest, Jak3-/- and γc-/- BMDCs showed spontaneous production of IL-12p70, which was induced to significantly greater levels following stimulation compared with wild-type DCs (Figure 5A). Stimulation of wild-type BMDCs with LPS or CpG oligos also induced IL-10 production, but the addition of IFN-γ suppressed the production. Conversely, Jak3-/- and γc-/- BMDCs showed spontaneous production of IL-10, which was enhanced 2- to 4-fold upon stimulation, and was 3- to 5-fold higher compared with wild-type BMDCs. IFN-γ attenuated this induction, but both Jak3-/- and γc-/- cells still showed 3- to 8-fold higher production of IL-10 compared with wild-type BMDCs (Figure 5B). In contrast, the production of TNF-α and IL-6 was similar in wild-type and Jak3-/- and γc-/- cells (Figure 5C-D). We also found similar results with cytometric bead array (data not shown). Thus, we conclude that Jak3-dependent signals are essential for constraining the production of selected cytokines in DCs.
Figure 5.

Enhanced IL-12 and IL-10 production by Jak3-/- and γc-/- BMDCs. BMDCs were cultured in medium or stimulated with LPS (1 μg/mL) or CpG oligos (1 μg/mL) with IFN-γ (10 ng/mL). After 24 hours, the production of the following cytokines was determined by ELISA: IL-12p70 (A), IL-10 (B), TNF-α (C), and IL-6 (D). *P < .05 and **P < .01 compared with wild-type BMDCs. ND indicates not detectable; n = 5.
Jak3-/- BMDCs promote enhanced Th1 differentiation in vivo
To understand the in vivo correlate of the dysregulated cytokine production, we next examined the ability of Jak3-/- BMDCs to regulate CD4+ T-cell differentiation in an adoptive transfer model. OVAp-specific OTII transgenic CD4+ T cells were administered intravenously to C57BL/6 congenic mouse. Simultaneously, peptide-pulsed BMDCs were injected subcutaneously and the frequency of IFN-γ–producing T cells in draining lymph nodes was enumerated 3 days later. In the absence of injected BMDCs, IFN-γ production was virtually absent (Figure 6). When wild-type BMDCs were injected in the absence of cognate antigen, less than 1% of T cells produced IFN-γ. In contrast, the number of IFN-γ–producing cells was enhanced to 3% when OVAp-pulsed wild-type BMDCs were used as a stimulus. However, when Jak3-/- BMDCs were used, the proportion of IFN-γ–producing cells increased to more than 12% (P < .001). Thus, we conclude that loss of Jak3-mediated signals can promote DC IL-12 production, which can cause exaggerated Th1 differentiation.
Figure 6.

Exaggerated Th1 development by Jak3-/- BMDCs. CD4+ T cells (3 × 106) from OTII transgenic mice (CD45.2+) were injected intravenously into C57BL/6 congenic mice (CD45.1+). Concomitantly, wild-type and Jak3-/- BMDCs cultured with or without OVA peptide (OVAp, 1 μM) for 2 hours were injected into footpads. Three days later, draining lymph nodes were obtained. Then cells were isolated and restimulated in vitro with anti–mouse CD28 and OVAp (1 μM). The frequency of IFN-γ–producing cells was assessed by gating on CD45.2+CD4+ cells staining for intracellular IFN-γ. Each circle indicates an individual mouse, and the mean is depicted by a horizontal line. **P < .001 compared with wild-type BMDCs pulsed with OVAp; n = 5.
Discussion
To explore the role of Jak3 in DC development and function, herein we analyzed DCs from Jak3-/- and γc-/- mice. Our studies showed that γc-associated cytokine receptors and Jak3 expression were induced by TLR ligands. However, neither γc nor Jak3 was essential for DC development and maturation. CD8+ DCs were reduced in spleens from Jak3-/- mice; however, Jak3-/- BMDCs developed normally in vitro, suggesting that this was not the result of an intrinsic defect. Unexpectedly, Jak3 was found to be important for DC function by negatively regulating apoptosis and cytokine production. The overproduction of IL-12 by Jak3-/- BMDCs resulted in enhancement of T-cell IFN-γ production. In all cases, γc deficiency was entirely congruent with Jak3 deficiency.
Loss of CD8+ splenic DCs
Although there was marked reduction in CD8+ DCs in spleens of Jak3-/- mice, in vitro culture of BMDCs suggested that the decreased number of CD4+ DCs and loss of CD8+ DCs was not an intrinsic defect (Figures 2A-B, 3A). In agreement with this conclusion, Berg and colleagues have analyzed chimeric mice reconstituted with a mixture of wild-type and Jak3-/- bone marrow and found that Jak3-/- DCs developed normally (Leslie Berg et al, manuscript submitted). This is in agreement with our results (Supplemental Figure S2A-B). An alternative explanation, therefore, might be that the loss of CD8+ DCs is the result of the T-cell–dependent immunodysregulation noted in Jak3-/- mice.27,33 Since T cells are known to kill DCs,37-39 we speculate that this might be one mechanism by which Jak3-/- DCs are removed. Splenic DCs in Jak3-/- mice also have higher expression of maturation markers compared with wild-type splenic DCs. One explanation for this phenotype may be that abnormally expanded and activated Jak3-/- T cells both activate and kill DCs. CD8+ splenic DCs are known to have a shorter lifespan compared with other subsets of DCs in spleen and this may also contribute to the preferential loss of splenic CD8+ DCs in Jak3-/- mice.40
Jak3 and γc as negative regulators of DCs
For lymphoid cells, a critical, nonredundant function of Jak3 appears to be its role in providing survival signals. Jak3-/- mice display high levels of spontaneous apoptosis, and Jak3 is thought to be required for the proper regulation of the Bcl-2 family proteins.41,42 Surprisingly, we found that in DCs, loss of Jak3 was associated with down-regulation of the proapoptotic proteins Bax and Bak, and prolonged survival of Jak3-/- BMDCs in conditions of cytokine withdrawal. IL-15 deficiency also enhanced survival, suggesting that this cytokine promoted DC apoptosis (Figure 4A-B). While these effects may seem paradoxical, it should be kept in mind that promotion of apoptosis by γc cytokines is not without precedent. Even in T cells, γc cytokines can promote apoptosis under specific circumstances.43 It is intriguing to consider what the mechanism of this proapoptotic effect might be. The major cytokines that promote DC development and keep the cells alive in vitro are GM-CSF and IL-4. In this regard, it should be noted that IL-4 signaling can occur independently of γc and Jak3.44 Precisely how activation of Jak3 might promote apoptosis in the setting of cytokine withdrawal is not clear. Suppressor of cytokine signaling (SOCS) proteins are cytokine-inducible negative regulators of cytokine signaling. One could imagine a scenario in which γc cytokines could enhance the expression of such negative regulators of cytokine signaling; however, we did not see alterations in the level of SOCS-1 and SOCS-3 in Jak3-/- or γc-/- BMDCs (data not shown). In addition to direct effects on cell proliferation and survival, cytokines also regulate cell metabolism.45 It is intriguing to consider that Jak3 activation in the context of cytokine starvation might also have metabolic effects that augment apoptosis.
Another surprising observation in our study is the enhanced IL-12 and IL-10 production in the Jak3-/- and γc-/- BMDCs. Transcription factors that contribute to IL-12 p40 regulation include: nuclear factor κ-B (NF-κB), c-Rel, interferon regulatory factor 8 (IRF-8), and Ets family proteins. NF-κB and IRF family members also promote cell survival. It is intriguing to speculate that if Jak3 deficiency enhanced expression of such a transcription factor, this could promote IL-12 production and inhibit apoptosis. However, it is also not clear how Jak3 might negatively regulate such transcription factors. The mechanisms underlying the regulation of IL-12 are better understood than those of IL-10. Indeed, it has been suggested that a major aspect of IL-10 regulation is posttranscriptional control.46,47 It is therefore difficult to envision a single mechanism that links up-regulation of IL-12 and IL-10 production and enhanced survival.
Clinical implications
Existing inhibitor studies argue that inhibition of Jak3 blocks DC function. However, we conclude that this is not likely to be the case and relates more to the lack of specificity of the inhibitor. This was most dramatically established by the finding that the putative Jak3 inhibitor very effectively inhibits activation of Jak3-deficient cells. This underscores that this putatively selective Jak3 inhibitor has effects unrelated to Jak3 and indicates that studies using this inhibitor need to be interpreted with great caution. A new potent and highly selective Jak3 inhibitor has been developed.36 The expectation is that patients treated with such an inhibitor should have normal DC development and antigen presentation. However, DC survival and cytokine production might be enhanced. This could enhance immune responses, although overproduction of IL-10 would be immunosuppressive. Nonetheless, it is not clear which dysregulated cytokine (IL-12 vs IL-10) would predominate in the clinical setting of Jak3 inhibition. Indeed, it will be important to determine whether dysregulation of either cytokine is clinically meaningful. This may also be pertinent to Jak3-SCID and X-SCID patients who have received hematopoietic stem cell transplants and are known to possess recipient origin monocytes. One wonders whether these cells would also exhibit enhanced survival and cytokine production and whether there might be circumstances where this could be clinically relevant.
In summary, our findings demonstrate that Jak3 is not essential for DC development and that Jak3 deficiency is entirely in line with γc deficiency. These findings are consistent with the lack of requirement for γc and Jak3 in myeloid-cell development. However, these molecules are critical for maintaining normal DC function by negatively regulating DC survival and cytokine production. This is an unexpected finding that points to new functions of γc cytokines in regulating DC functions. Elucidating the mechanism(s) underlying this negative regulation will be important and should lead to new insights in the control of DC function.
Supplementary Material
Prepublished online as Blood First Edition Paper, July 14, 2005; DOI 10.1182/blood-2005-02-0769.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Nicola NA, Hilton DJ. General classes and functions of four-helix bundle cytokines. Adv Protein Chem. 1998;52: 1-65. [DOI] [PubMed] [Google Scholar]
- 2.Hofmann SR, Ettinger R, Zhou YJ, et al. Cytokines and their role in lymphoid development, differentiation and homeostasis. Curr Opin Allergy Clin Immunol. 2002;2: 495-506. [DOI] [PubMed] [Google Scholar]
- 3.Fry TJ, Mackall CL. Interleukin-7: from bench to clinic. Blood. 2002;99: 3892-3904. [DOI] [PubMed] [Google Scholar]
- 4.Moser M, Murphy KM. Dendritic cell regulation of TH1-TH2 development. Nat Immunol. 2000;1: 199-205. [DOI] [PubMed] [Google Scholar]
- 5.Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. 2002;2: 933-944. [DOI] [PubMed] [Google Scholar]
- 6.Szabo SJ, Sullivan BM, Peng SL, Glimcher LH. Molecular mechanisms regulating Th1 immune responses. Annu Rev Immunol. 2003;21: 713-758. [DOI] [PubMed] [Google Scholar]
- 7.Noguchi M, Yi H, Rosenblatt HM, et al. Interleukin-2 receptor gamma chain mutation results in X-linked severe combined immunodeficiency in humans. Cell. 1993;73: 147-157. [DOI] [PubMed] [Google Scholar]
- 8.Russell SM, Johnston JA, Noguchi M, et al. Interaction of IL-2R beta and gamma c chains with Jak1 and Jak3: implications for XSCID and XCID. Science. 1994;266: 1042-1045. [DOI] [PubMed] [Google Scholar]
- 9.Puel A, Leonard WJ. Mutations in the gene for the IL-7 receptor result in T(-)B(+)NK(+) severe combined immunodeficiency disease. Curr Opin Immunol. 2000;12: 468-473. [DOI] [PubMed] [Google Scholar]
- 10.Leonard WJ. Cytokines and immunodeficiency diseases. Nat Rev Immunol. 2001;1: 200-208. [DOI] [PubMed] [Google Scholar]
- 11.O'Shea JJ, Gadina M, Schreiber RD. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell. 2002;109(suppl): S121-S131. [DOI] [PubMed] [Google Scholar]
- 12.Schindler C, Darnell JE Jr. Transcriptional responses to polypeptide ligands: the JAK-STAT pathway. Annu Rev Biochem. 1995;64: 621-651. [DOI] [PubMed] [Google Scholar]
- 13.Nosaka T, van Deursen JM, Tripp RA, et al. Defective lymphoid development in mice lacking Jak3. Science. 1995;270: 800-802. [DOI] [PubMed] [Google Scholar]
- 14.Macchi P, Villa A, Giliani S, et al. Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID). Nature. 1995;377: 65-68. [DOI] [PubMed] [Google Scholar]
- 15.Thomis DC, Gurniak CB, Tivol E, Sharpe AH, Berg LJ. Defects in B lymphocyte maturation and T lymphocyte activation in mice lacking Jak3. Science. 1995;270: 794-797. [DOI] [PubMed] [Google Scholar]
- 16.Notarangelo LD. Immunodeficiencies caused by genetic defects in protein kinases. Curr Opin Immunol. 1996;8: 448-453. [DOI] [PubMed] [Google Scholar]
- 17.Park SY, Saijo K, Takahashi T, et al. Developmental defects of lymphoid cells in Jak3 kinase-deficient mice. Immunity. 1995;3: 771-782. [DOI] [PubMed] [Google Scholar]
- 18.Russell SM, Tayebi N, Nakajima H, et al. Mutation of Jak3 in a patient with SCID: essential role of Jak3 in lymphoid development. Science. 1995;270: 797-800. [DOI] [PubMed] [Google Scholar]
- 19.Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404: 193-197. [DOI] [PubMed] [Google Scholar]
- 20.Traver D, Akashi K, Manz M, et al. Development of CD8alpha-positive dendritic cells from a common myeloid progenitor. Science. 2000;290: 2152-2154. [DOI] [PubMed] [Google Scholar]
- 21.Musso T, Johnston JA, Linnekin D, et al. Regulation of JAK3 expression in human monocytes: phosphorylation in response to interleukins 2, 4, and 7. J Exp Med. 1995;181: 1425-1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mohamadzadeh M, Ariizumi K, Sugamura K, Bergstresser PR, Takashima A. Expression of the common cytokine receptor gamma chain by murine dendritic cells including epidermal Langerhans cells. Eur J Immunol. 1996;26: 156-160. [DOI] [PubMed] [Google Scholar]
- 23.Standiford TJ, Strieter RM, Allen RM, Burdick MD, Kunkel SL. IL-7 up-regulates the expression of IL-8 from resting and stimulated human blood monocytes. J Immunol. 1992;149: 2035-2039. [PubMed] [Google Scholar]
- 24.Ziegler SF, Tough TW, Franklin TL, Armitage RJ, Alderson MR. Induction of macrophage inflammatory protein-1 beta gene expression in human monocytes by lipopolysaccharide and IL-7. J Immunol. 1991;147: 2234-2239. [PubMed] [Google Scholar]
- 25.Bosco MC, Espinoza-Delgado I, Schwabe M, et al. Regulation by interleukin-2 (IL-2) and interferon gamma of IL-2 receptor gamma chain gene expression in human monocytes. Blood. 1994;83: 2995-3002. [PubMed] [Google Scholar]
- 26.Villa A, Sironi M, Macchi P, et al. Monocyte function in a severe combined immunodeficient patient with a donor splice site mutation in the Jak3 gene. Blood. 1996;88: 817-823. [PubMed] [Google Scholar]
- 27.Grossman WJ, Verbsky JW, Yang L, et al. Dysregulated myelopoiesis in mice lacking Jak3. Blood. 1999;94: 932-939. [PubMed] [Google Scholar]
- 28.Aliberti J, Schulz O, Pennington DJ, et al. Essential role for ICSBP in the in vivo development of murine CD8alpha + dendritic cells. Blood. 2003;101: 305-310. [DOI] [PubMed] [Google Scholar]
- 29.Vremec D, Pooley J, Hochrein H, Wu L, Shortman K. CD4 and CD8 expression by dendritic cell subtypes in mouse thymus and spleen. J Immunol. 2000;164: 2978-2986. [DOI] [PubMed] [Google Scholar]
- 30.Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nat Rev Immunol. 2002;2: 151-161. [DOI] [PubMed] [Google Scholar]
- 31.Kelsall BL, Biron CA, Sharma O, Kaye PM. Dendritic cells at the host-pathogen interface. Nat Immunol. 2002;3: 699-702. [DOI] [PubMed] [Google Scholar]
- 32.Asselin-Paturel C, Boonstra A, Dalod M, et al. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat Immunol. 2001;2: 1144-1150. [DOI] [PubMed] [Google Scholar]
- 33.Bradley LM, Croft M, Swain SL. T-cell memory: new perspectives. Immunol Today. 1993;14: 197-199. [DOI] [PubMed] [Google Scholar]
- 34.Laouar Y, Welte T, Fu XY, Flavell RA. STAT3 is required for Flt3L-dependent dendritic cell differentiation. Immunity. 2003;19: 903-912. [DOI] [PubMed] [Google Scholar]
- 35.Saemann MD, Diakos C, Kelemen P, et al. Prevention of CD40-triggered dendritic cell maturation and induction of T-cell hyporeactivity by targeting of Janus kinase 3. Am J Transplant. 2003;3: 1341-1349. [DOI] [PubMed] [Google Scholar]
- 36.Changelian PS, Flanagan ME, Ball DJ, et al. Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science. 2003;302: 875-878. [DOI] [PubMed] [Google Scholar]
- 37.Hermans IF, Ritchie DS, Yang J, Roberts JM, Ronchese F. CD8+ T cell-dependent elimination of dendritic cells in vivo limits the induction of antitumor immunity. J Immunol. 2000;164: 3095-3101. [DOI] [PubMed] [Google Scholar]
- 38.Ronchese F, Hermans IF. Killing of dendritic cells: a life cut short or a purposeful death? J Exp Med. 2001;194: F23-F26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loyer V, Fontaine P, Pion S, Hetu F, Roy DC, Perreault C. The in vivo fate of APCs displaying minor H antigen and/or MHC differences is regulated by CTLs specific for immunodominant class I-associated epitopes. J Immunol. 1999;163: 6462-6467. [PubMed] [Google Scholar]
- 40.Kamath AT, Pooley J, O'Keeffe MA, et al. The development, maturation, and turnover rate of mouse spleen dendritic cell populations. J Immunol. 2000;165: 6762-6770. [DOI] [PubMed] [Google Scholar]
- 41.Thomis DC, Lee W, Berg LJ. T cells from Jak3-deficient mice have intact TCR signaling, but increased apoptosis. J Immunol. 1997;159: 4708-4719. [PubMed] [Google Scholar]
- 42.Wen R, Wang D, McKay C, et al. Jak3 selectively regulates Bax and Bcl-2 expression to promote T-cell development. Mol Cell Biol. 2001;21: 678-689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng L, Trageser CL, Willerford DM, Lenardo MJ. T cell growth cytokines cause the superinduction of molecules mediating antigen-induced T lymphocyte death. J Immunol. 1998;160: 763-769. [PubMed] [Google Scholar]
- 44.Oakes SA, Candotti F, Johnston JA, et al. Signaling via IL-2 and IL-4 in JAK3-deficient severe combined immunodeficiency lymphocytes: JAK3-dependent and independent pathways. Immunity. 1996;5: 605-615. [DOI] [PubMed] [Google Scholar]
- 45.Plas DR, Thompson CB. Cell metabolism in the regulation of programmed cell death. Trends Endocrinol Metab. 2002;13: 75-78. [DOI] [PubMed] [Google Scholar]
- 46.Powell MJ, Thompson SA, Tone Y, Waldmann H, Tone M. Posttranscriptional regulation of IL-10 gene expression through sequences in the 3'-untranslated region. J Immunol. 2000;165: 292-296. [DOI] [PubMed] [Google Scholar]
- 47.Tone M, Powell MJ, Tone Y, Thompson SA, Waldmann H. IL-10 gene expression is controlled by the transcription factors Sp1 and Sp3. J Immunol. 2000;165: 286-291. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}