

Abstract
Kaposi sarcoma (KS) remains the most common AIDS-associated malignancy worldwide. In sub-Saharan Africa especially, this aggressive endothelial-cell tumor is a cause of widespread morbidity and mortality. Infection with Kaposi sarcoma-associated herpesvirus (KSHV) is now known to be an etiologic force behind KS and primary-effusion lymphoma (PEL). Over time, KSHV has pirated many human genes whose products regulate angiogenesis, inflammation, and the cell cycle. One of these, the KSHV vGPCR, is a lytic product that is a constitutively active homolog of the IL-8 receptor. Although it is considered a viral oncogene and causes KS-like lesions in mice, vGPCR expression results in cell-cycle arrest of KSHV-infected PEL cells. In the present study, we show that this arrest is mediated by p21 in a p53-independent manner; the resulting Cdk2 inhibition decreases the efficiency of chemical induction of KSHV lytic transcripts ORF 50 and 26. Importantly, Cdk2 activity is also essential for replication in other human herpesviruses. The ability of vGPCR to delay or abort KSHV replication may explain how despite being a lytic product, this potent signaling molecule has a vital role in tumor formation via its induction of various KS-associated cytokines.
Introduction
Kaposi sarcoma (KS), an endothelial-cell tumor, is the most common AIDS-related malignancy worldwide. It is a major cause of morbidity and mortality, particularly in several sub-Saharan African countries where it is the most common cancer overall. In 1994, Kaposi sarcoma-associated herpesvirus (KSHV) was discovered and soon established as an etiologic agent in all forms of KS.1-3 KSHV is a lymphotropic γ2-herpesvirus that infects not only the endothelial cells of KS but has also been detected in several hematopoietic cell types. It is detected in a subpopulation of the infiltrating mononuclear inflammatory cells seen in KS and also gives rise to primary effusion lymphoma (PEL). PEL is an AIDS-associated non-Hodgkin lymphoma that is characterized by lymphomatous effusions of serous cavities and only occasionally presents with a definable mass.4 Furthermore, KSHV infection has been linked to a more aggressive phenotype in another B-cell-proliferative disease called multicentric Castleman disease and its associated plasmablastic lymphoma.5,6
During coevolution with its human host, KSHV has acquired homologues of several human genes involved in angiogenesis, inflammation, and cell-cycle regulation.7-18 One of these is the product of ORF 74, a G protein-coupled receptor (vGPCR) that is a constitutively active homologue of human CXCR1, CXCR2, and of herpesvirus saimiri ECRF3. Such agonist-independent activity is also a characteristic of the ORF 74 product of murine γherpesvirus 68. Notably, there exist several constitutively active GPCR mutants associated with human disease.19-23 The KSHV vGPCR has mutations of its cytoplasmic tail and of transmembrane helices 2 and 3 that confer its constitutive activity.24,25 Even so, vGPCR signaling can be fine-tuned by various ligands, and this modulation may prove essential to its pathogenic role in KSHV-mediated disease.26-31 Multiple mouse models have now shown that vGPCR expression causes KS-like lesions, and in one study a mutant vGPCR lacking the ligand-binding domain failed to produce tumors.32-35 The mechanism of vGPCR-induced KS-like tumors is not fully understood. Although vGPCR directly transforms primary endothelial cells and fibroblasts in vitro,36,37 histology of vGPCR-derived tumors in mice shows that relatively few vGPCR-expressing endothelial or hematopoietic cells drive endothelial-cell outgrowth. This observation concurs with early KS tumors in which a relatively small subset of cells are KSHV-infected and even fewer express lytic genes such as vGPCR.38-41 For these reasons, many postulate that vGPCR has its primary tumorigenic effects via paracrine pathways rather than via direct transformation. Indeed, vGPCR expression results in the elaboration of various cytokines that are known to be essential KS pathogenesis.25,42-50
In addition to the histologic evidence, vGPCR expression patterns also make it difficult to argue for a directly transforming role for vGPCR in KSHV-mediated disease. vGPCR is a lytic KSHV gene and as such is expressed mainly in cells destined to die secondary to viral replication.51,52 However, in addition to its paracrine-mediated proliferative effects, vGPCR influences the transcription of viral genes, thereby giving it potential to affect the viral life cycle.43,53 Furthermore, recent evidence supports the possibility of vGPCR expression outside the context of KSHV lytic phase; this shows potential for vGPCR to have a more prolonged effect on host and viral gene transcription than may be assumed by a model in which vGPCR is expressed only after the latent-lytic switch has been activated.54 Given these data together with our own that show vGPCR causes cell-cycle arrest in PEL cells,43 we sought to more completely characterize this arrest and to study the effect of vGPCR on the induction of 2 lytic transcripts, ORF 50 and ORF 26. The ORF 50 product is necessary and sufficient for the latent-lytic switch in KSHV,55,56 and ORF 26 is a late lytic transcript encoding a capsid protein.57,58 Both these transcripts are detectable after treatment of KSHV-infected PEL cells with tetradecanoyl phorbol acetate (TPA) or butyrate. These chemicals are used to induce production of KSHV virions from various PEL lines and have been used extensively to study the latent-lytic switch.59-63 Using a KSHV-infected PEL line engineered to express vGPCR under a doxycycline-inducible promoter, we show that vGPCR signaling results in an inhibition of S-phase entry with increased transcription of p21cip that is p53 independent. Furthermore, kinase assays show that this arrest is mediated by decreased activity of cyclin-dependent kinase (Cdk) 2, but not Cdk4 or Cdk6. We also show that vGPCR overexpression results in a drastic decrease in TPA- or butyrate-induced ORF 50 and ORF 26 messages. Using a dominant-negative Cdk2 construct, we conclude that it is likely the inhibition of Cdk2 at least partially mediates vGPCR's inhibition of chemical induction of ORF 50 and 26 transcripts. These studies add significantly to the knowledge of how vGPCR, despite being characterized as a lytic transcript, can influence the KSHV lytic cycle and the pathogenesis of KSHV-mediated disease.
Materials and methods
Study protocol was approved by University College London (UCL) Safety Committee GM688.
Cell culture
The PEL cell lines used were maintained in RPMI 1640 plus 40 mg/L gentamicin (Invitrogen, Carlsbad, CA) with 10% FBS (Atlanta Biologicals, Norcross, GA) at 37°C, 5% CO2.
Cell-cycle analysis
BC3.14 cells in exponential growth were incubated for 48 hours with or without doxycycline and then fixed with 70% ethanol overnight. Fortyeight hours was chosen as the time point based on our previous work with BC3.14 cells showing that vGPCR signaling and phenotypic effects are not seen prior to 48 hours. They were then stained with propidium iodide at a final concentration of 50 μg/mL with RNaseA 100 U/mL. When nocodazole (Sigma, St Louis, MO) was used to inhibit mitosis, it was added for 17 hours to cells at a final concentration of 0.2 μg/mL. Ten thousand cells were then analyzed using CellQuest and ModFit on a FACScalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ). To quantify S phase, cells were incubated in the dark at 37°C for 4 hours in the presence of 100 μM BrdU (Sigma). They were then plated using a Cytospin 2 (Shandon, Thermo Electron Corporation, Waltham, MA) and fixed for 30 minutes with 70% ethanol. Cells were washed in PBS and incubated for 15 minutes with 2N HCL to denature genomic DNA. After several more PBS washes, 70 μL staining solution was added (50 μL PBS with 0.5% BSA and 0.5% Tween-20, 20 μL anti-BrdU-FITC [BD Pharmingen, Franklin Lakes, NJ]), and cells were incubated for 1 hour in a dark humid chamber. Cells were then washed, and mounting medium with DAPI was applied (Vector Laboratories, Burlingame, CA). At least 100 cells were counted, and S phase was expressed as the percentage of FITC-staining cells relative to total number of DAPI-staining cells. Images were obtained using an Axiovert 100 microscope, LD Acroplan 20×/0.04 numeric aperture objective, Axiocam camera, and Axiovision 3.0.6 software (all from Zeiss, Oberkochen, Germany).
Transfections and luciferase assays
Transfections were performed on exponentially growing cells by electroporation (Bio-Rad Gene Pulser II; Bio-Rad, Hercules, CA) at settings of 270 mV and 975 mF in 0.8-cm cuvettes (Invitrogen), using 8 × 106 cells/cuvette resuspended in 0.8 mL RPMI 1640. Cuvettes were prechilled on ice, and after transfection cells were quickly plated in full culture medium. For luciferase assays, 10 μg p53-TA-luc or pTA-luc (Clontech BD, San Jose, CA) were transfected, and cells were then divided evenly and plated with or without doxycycline (Calbiochem, EMD Biosciences, Darmstadt, Germany). After 48 hours, lysates were prepared using 1 × Cell Culture Lysis Reagent as per the manufacturer's directions (Promega, Madison, WI). Assays were performed using 10 μL lysate and 50 μL beetle luciferin in a Microtiters Plate Luminometer (Dynex Tech, Chantilly, VA), using a 10-second read time. Protein concentration was used for normalization and was determined by the Bradford method with Bio-Rad DC Protein Assay Reagent after diluting samples and standards 1:1 in PBS. Because cells were divided after transfection, transfection efficiency was inherently controlled for, and protein equalization was deemed sufficient.
Western blotting and antibodies
BC3.14 cells were plated in full growth medium at 4 × 105/mL in a 24-well plate and subjected to 2 μg/mL doxycycline for various time periods. Cells were diluted to keep below 1.0 × 106/mL to minimize effects of crowding. Cells were lysed with standard RIPA buffer with 1 mg/mL each of aprotinin, leupeptin, and pepstatin, 0.5 mM PMSF, and 1 mM each of NaVO4 and NaF (Sigma). Protein was quantitated by the Bradford method and loaded onto sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using 10% to 12% gels. Semidry transfer to PVDF (Millipore, Billerica, MA) was performed using transfer buffer, 48 mm Tris, 39 mm glycine, 0.037% SDS, and 20% methanol. Blots were probed with primary antibody (Ab) overnight at 4°C. HRP-conjugated secondary Ab was added after washing and detected by an enhanced chemiluminescence system (Amersham, Little Chalfont, United Kingdom). Antibodies used included anti-p21 (F-5), anti-p27 (C-19), anti-p53 (FL393), anti-mdm2 (smp14), anti-cyclinA (H-432), and anti-cyclinD2 (C-17) (Santa Cruz Biotechnology, Santa Cruz, CA).
Kinase assays
Cells were lysed in IP buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM EDTA, 2.5 mM EGTA, 10% glycerol, 1 mM DTT, 0.1% Tween-20, 10 mM β-glycerophosphate, 1 mM NaF, 0.1 mM sodium orthovanadate, 2 μg/mL aprotinin, 5 μg/mL leupeptin, 0.1 mM PMSF); 800 μg protein was precleared and then incubated overnight at 4°C with 2 μL of the appropriate anti-Cdk antibody and 20 μL protein G plus agarose beads (Upstate Biotechnology, Lake Placid, NY). Beads were then washed 4 times in IP buffer and twice in 50 mM HEPES with 1 mM DTT. Kinase reactions were carried out in 30 μL kinase buffer (50 mM HEPES, 10 mM MgCl2, 1 mM DTT, 10 mM β-glycerophosphate) along with 10 μg substrate, GST-RbC-pocket (aa 773-928). Cold ATP to final concentration of 50 μM with 10 μCi (0.37 MBq) γ-32P ATP (6000 Ci/mmol [222.0 × 1012 Bq/mmol]) was added, and the mixture was incubated at 30°C for 30 minutes. The reaction was stopped by addition of Laemmli buffer and brief boiling. The reaction product was loaded and run on a 10% SDS-PAGE gel, dried, and exposed overnight. Blots were also incubated with anti-Cdk2 antibody to check for equal loading. Antibodies used included anti-Cdk2 (M2), anti-Cdk4 (C22), anti-Cdk6 (C21) (Santa Cruz Biotechnology).
Quantitative RT-PCR (qRT-PCR)
Total RNA from cells was treated with DNase I (RNase-Free; Ambion, Oxon, United Kingdom) before cDNA synthesis according to the manufacturer's instructions. cDNA was generated from 1 μg total RNA by using the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen). Real-time qRT-PCR was performed on an ABI PRISM 7700 sequence detector (Applied Biosystems, Foster City, CA) by using the SYBR Green PCR Master Mix (Applied Biosystems) in duplicate, with triplicate nontemplate controls in a 25-μL PCR reaction. One microliter of cDNA was used in a 25-μL PCR mixture containing 1 × SYBR Green PCR Master Mix and 0.3 μM GAPDH primers (5′-GGA GTC AAC GGA TTT GGT CGT A and 3′-GGC AAC AAT ATC CAC TTT ACC AGA GT), or 0.5 μM ORF 26 primers (5′-GAT TCC ACC ATT GTG CTC GAA T and 3′-CCC AGT TGC TGA GGC ACG), or 0.5 μM ORF 50 primers (5′-CAC AAA AAT GGC GCA AGA TGA and 3′-TGG TAG AGT TGG GCC TTC AGT T), or 0.7 μM p27 primers (5′-CGG TGG ACC ACG AAG AGT TAA and 3′-GGC TCG CCT CTT CCA TGT C), or 0.3 μM p21 primers (5′-ACA CCT TCC AGC TCC TGT AAC ATA CT and 3′-GAA ACG GGA ACC AGG ACA CAT), or 0.3 μM vGPCR primers (5′-GTG CCT TAC ACG TGG AAC GTT and 3′-GGT GAC CAA TCC ATT TCC AAG A). The comparative Ct method was used as described previously.64
Construction, production, and infection of lentivirus-expressing Cdk2-DN
The 900-bp Cdk2-DN sequence was obtained by BamH1 digestion of pCMV-cdk2-dn. The fragment was then ligated either in forward or reverse direction into the BamH1 site of pHR-CSGW (kind gift of Adrian Thrasher, Institute of Child Health, London, United Kingdom) which had been modified to replace the eGFP sequence with a multiple cloning site. To obtain pseudotyped lentivirus (recombinant HIV-1 with vesicular stomatitis virus G [VSV-G] envelope protein), which expresses the Cdk2-DN protein, we used the gene delivery and production system developed by Naldini et al.65
Results
KSHV vGPCR signaling results in cell-cycle arrest of PEL cells with inhibition of transition to S phase
Although KSHV vGPCR has been shown to enhance cell proliferation in fibroblasts and endothelial cells,36,37 we have shown that it inhibits PEL cell growth when overexpressed in the context of KSHV infection.43 This was an unexpected finding, but it reinforced that cell-signaling molecules often have very cell-specific effects. To better describe this growth inhibition, we examined the cell-cycle profile of vGPCR-expressing PEL cells. As in our previous work, we used BC3.14 cells which are derived from the PEL line BC3.43 BC3.14 cells express vGPCR when exposed to doxycycline. Cells were inoculated at identical concentrations and then incubated as normal for 48 hours with or without doxycycline to express the vGPCR. The microtubule inhibitor nocodazole was added for 17 hours, and cell-cycle position was determined by propidium iodide staining and flow cytometric analysis. Figure 1A shows that at baseline, BC3.14 cells progress rapidly through S phase and accumulate in G2/M in the presence of nocodazole. However, when vGPCR is expressed, the cells arrest prior to S phase and do not accumulate in G2/M. To accurately assess the S-phase compartment over time, BC3.14 cells with or without vGPCR overexpression were incubated with bromodeoxyuridine (BrdU), plated, then stained with anti-BrdU antibody, and counted. Figure 1B shows a vGPCR-induced exit from S phase from 50% to less than 20%. Figure 1C graphically represents multiple such experiments using 2 different doses of doxycycline over 5 days. As suspected, a very low dose of doxycycline had no effect. However, full-dose doxycycline resulted in a marked inhibition of S phase by 72 hours. Groα is a known vGPCR agonist, and we noted that downstream signaling effects of vGPCR are easier to detect in the presence of Groα.66 However, in this case, the phenotypic effect of cell-cycle arrest was not appreciably enhanced by Groα. This simply attests to the strong constitutive activity of vGPCR and is not unexpected.
Figure 1.
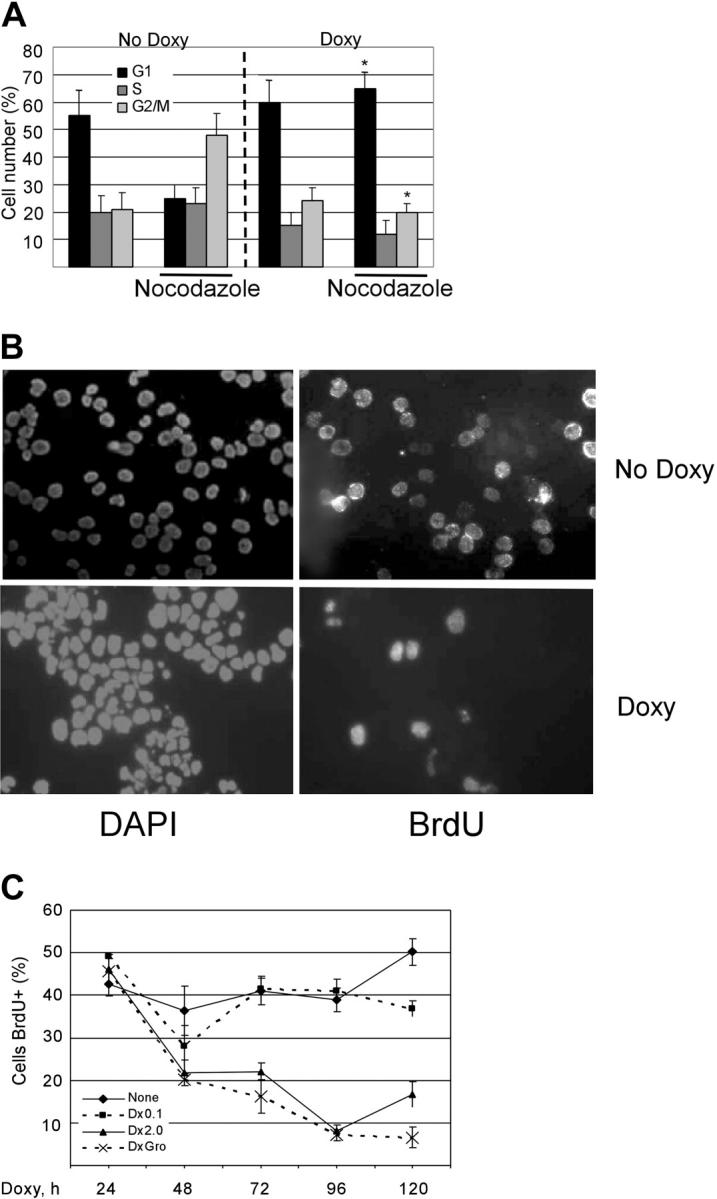
KSHV vGPCR expression results in cell-cycle arrest with decrease in proportion of cells in S phase. (A) BC3.14 cells were exposed to 2 μg/mL doxycycline where indicated for 48 hours to induce vGPCR expression. Cells were then stained with propidium iodide and analyzed by FCM. The percentage of cells in each cell phase is shown. Average of 2 independent experiments is shown. *P ≤ .05 compared with corresponding point in “No Doxy” panel. (B) BC3.14 cells were exposed to doxycycline where indicated for 48 hours and then fixed to glass slide by cytocentrifugation. Cells were incubated with BrdU for 6 hours followed by FITC-conjugated anti-BrdU. (C) BC3.14 cells were exposed to doses of doxycycline as shown for up to 120 hours. BrdU staining was performed at each time point. Cells were manually counted, and the average of 3 experiments is shown. Error bars indicate SD.
vGPCR causes a p53-independent transcriptional up-regulation of p21cip in PEL cells
Cell-cycle progression is regulated through a precise temporal activation of Cdk's. Two classes of Cdk inhibitors (CDIs) are crucial to this ordered Cdk activation and include the CIP/KIP and INK4 families.67 Using Western blotting and quantitative reverse transcriptase-PCR (qRT-PCR), we examined the effect of vGPCR signaling on p21cip and p27kip, CDIs known to inhibit the S-phase cyclins, E and A. Figure 2A shows that doxycycline-induced expression of vGPCR results in increased protein levels of both p21 and p27. Supporting the specificity of this vGPCR-mediated effect is that Groα results in slight enhancement of protein accumulation when added to vGPCR-expressing cells but has no independent effect. It is possible that accumulation of p21 and p27 merely reflect an accumulation of cells in G0/G1. We therefore used qRT-PCR to look for transcriptional up-regulation of these CDIs. Figure 2B shows a clear increase in p21 message that would argue for a causal effect on cell-cycle arrest rather than a secondary accumulation. Changes in p27 transcript are not significant in the absence of Groα stimulation. Because cell-cycle arrest does not require the addition of a vGPCR agonist (see Figure 1), we conclude that vGPCR signaling mediates an increase in p21, but that increases in p27 are a secondary effect of cell-cycle arrest and not likely to be an important part of the vGPCR signaling pathway. Furthermore, using qRT-PCR we assayed for p15, p16, p18, and p19 but found no detectable effect of vGPCR on the INK family of CDIs (data not shown). Of note, BC3, the parent line of BC3.14, has been shown to express no p16, and our experiments confirmed this for BC3.14.68
Figure 2.

KSHV vGPCR causes increase in p21 and p27. (A) BC3.14 cells were exposed to 2 μg/mL doxycycline for length of time shown in hours. Western blot shows vGPCR-dependent increases in p21 and p27 over 96 hours. Shown is 1 of 3 independent experiments. (B) Quantitative RT-PCR shows vGPCR-dependent transcriptional regulation of p21 and to a lesser extent, p27. Groα (100 nm), a vGPCR agonist, is included to assure specificity of observed effects. Average of 2 independent experiments each done in duplicate is shown. Error bars indicate SD. *Significant increase over baseline, P ≤ .05.
Increases in p21 can be either dependent or independent of the tumor suppressor p53.69-72 We therefore assessed the vGPCR-mediated effect on p53 in our PEL cell line. Figure 3A shows that, despite prolonged incubation with doxycycline, the resultant vGPCR expression did not result in accumulation of p53 or mdm2 as would be expected with p53 activity.73,74 Furthermore, luciferase reporter assays using a p53-responsive promoter show some decrease in p53 activity. The cause of this decrease is unclear but certainly argues against a p53-mediated increase in p21.
Figure 3.

KSHV vGPCR-induced cell-cycle arrest is not mediated by p53. (A) BC3.14 cells were exposed to doxycycline for the specified length of time to induce vGPCR, after which protein lysates (30 μg) were loaded onto 12% SDS-PAGE gels and probed for p53 and mdm2. Shown is a representative of 3 independent Western blot experiments. (B) BC3.14 cells were transfected with p53-TA-luc, a commercially available p53 reporter construct. Transfected cells were then divided and incubated with doxycycline (2 μg/mL for 48 hours), Groα (100 nM), or without additives as shown. Protein lysates were harvested, and equal amounts were assayed. Cells were divided after transfection, so no further control for transfection efficiency was performed. As expected, vGPCR expression has no effect on pTA-luc the control plasmid (inset). Shown is the average of 3 independent experiments. Error bars indicate SD.
vGPCR signaling inhibits Cdk2 but not Cdk4 or Cdk6 activity
Having identified p21 as the CDI responsible for vGPCR-mediated cell-cycle arrest, we next investigated the level of various cyclins and, more importantly, the activity of the relevant Cdk's. Cdk's are expressed at more or less constant levels throughout the cycle but are regulated through the synthesis and degradation of the cyclins.75 The D-type cyclins are short lived and assemble with Cdk4 or Cdk6 in G1 in the presence of ongoing mitogenic stimuli.76 Cyclin E peaks at the transition from G1 to S phase followed by cyclin A during S phase. Both these cyclins activate Cdk2.77 Although multiple approaches failed to accurately quantify cyclin E levels in these cells, Western blotting (Figure 4A) shows that ongoing vGPCR expression results in decreased levels of cyclin A and cyclin D2. This decrease in levels of the G1- and S-phase cyclins are consistent with the data in Figure 1, showing a G0/G1 arrest. However, a more definitive functional assay of the cyclin-dependent kinases was performed.78 Using the C-terminus of retinoblastoma (Rb) protein as a substrate, we assayed the kinase activity of Cdk2, Cdk4, and Cdk6 (Figure 4B). Although vGPCR had no effect on Cdk4 or Cdk6, it produced a marked reduction in Cdk2 activity. This is in keeping with the known inhibitory effects of p21 on Cdk2. The effect of p21 on Cdk4/6 is more complex with some models showing inhibition but others arguing that the cdk/cyclinD interaction with p21 is also important to sequester p21 away from the Cdk2/cyclin E complex.79
Figure 4.

KSHV vGPCR causes decreased levels of cyclin A, D2 complexes, and decreased cdk2 kinase activity. (A) To express vGPCR, BC3.14 cells were exposed to 2 μg/mL doxycycline for the time shown in hours. Protein lysates were run on 10% SDS-PAGE gels and probed with appropriate antibodies. (B) BC3.14 cells were incubated for 48 hours with doxycycline and then lysed in kinase buffer. Antibodies were then used to immunoprecipitate each kinase as indicated. After resuspension in kinase buffer, the substrate GST-pRb (C term) was added along with [γ-32P] ATP. Phosphorylated product was detected by autoradiography after separation on a 12% SDS-PAGE gel (top). Ten percent of the protein input for the IP was used to Western blot for each kinase (bottom). A representative of 3 independent experiments is shown.
KSHV vGPCR inhibits chemically mediated lytic gene induction via inactivation of Cdk2
Herpesviruses need to manipulate the host cell cycle to their own replicative ends. It is known that both CMV and EBV require the S-phase Cdk's (eg, Cdk2) for successful replication. Furthermore, very recent data suggests that cells in S phase produce KSHV virions more efficiently when chemically induced.80-82 Having shown that vGPCR overexpression inhibits Cdk2, we used qRT-PCR to quantitate the production of KSHV ORF 50 and ORF 26 in chemically induced BC3.14 cells in the presence or absence of vGPCR expression. The product of ORF 50 is vital to KSHV replication, and ORF 26 is a late lytic gene that encodes a capsid protein. Both butyrate and TPA induce PEL lines to produce new virions, although levels of class II and III transcripts vary depending on the stimulus.61 Using standard doses of butyrate and TPA we found that induction of both ORF 50 and ORF 26 transcripts was markedly reduced in BC3.14 cells that were made to express vGPCR prior to induction (Figure 5A). The effect was most dramatic with TPA and high-dose butyrate. Because vGPCR is itself a lytic gene and chemically inducible, we compared the levels of vGPCR transcript achieved with doxycycline induction versus chemical induction. We felt it important to do so because vast overexpression of exogenous signaling proteins can force otherwise unnatural associations and downstream effects. We found that at 48 to 96 hours of doxycycline induction, levels of vGPCR transcript were comparable to those induced by standard TPA and butyrate doses at 48 hours; doses known to bring about successful virion production (Figure 6). Although we have never seen a phenotypic effect of doxycycline on control cell lines (ie, BC3), we also performed control experiments on BC3 cells to ensure that doxycycline was not having an unexpected effect on the chemical induction of ORF 50 or ORF 26. Indeed, doxycycline had no independent inhibitory effect on ORF 50 or ORF 26 when assayed by qRT-PCR; curiously however, there was a trend of higher butyrate-induced levels in the presence of doxycycline (Table 1). The reason for this is unclear but does not detract from the data presented in Figure 5.
Figure 5.

KSHV vGPCR expression inhibits lytic gene induction by TPA and butyrate. BC3.14 cells were treated for 48 hours with 2 μg/mL doxycycline where indicated to express vGPCR, after which cells were also exposed to butyrate (1 mM or 2 mM) (A) or TPA (20 ng/mL) (B) to induce KSHV lytic gene transcription. q RT-PCR was performed for ORF 50 and ORF 26 message. Results are normalized to untreated cells (lane 1). Shown in both panels are 2 independent experiments each done in duplicate. Error bars indicate SD. *Significant increase over baseline, P ≤ .05; **Significant decrease from maximum induction, P ≤ .05.
Figure 6.

Doxycycline induction of KSHV vGPCR in BC3.14 cells results in increases in vGPCR message similar to levels obtained with lysis-inducing chemicals. BC3.14 cells were treated for 48 hours with TPA (20 ng/mL), butyrate (1 mM and 2 mM), or 2 μg/mL doxycycline (48 and 96 hours). Quantitative RT-PCR was then done using primers for KSHV vGPCR. Results were normalized to untreated cells in lane 1. Error bars indicate SD.
Table 1.
Quantitative RT-PCR results for ORF 50 and ORF 26 in BC3 cells
|
Fold increase in mRNA
|
||
|---|---|---|
| ORF 50 | ORF 26 | |
| Butyrate | 42.8 | 400.2 |
| TPA | 12.6 | 100.1 |
| Doxycycline | 1.1 | 0.8 |
| Butyrate + doxycycline | 85.4 | 812.3 |
| TPA + doxycycline | 17.4 | 125.1 |
BC3 cells were treated for 48 hours with butyrate (2 mM), TPA (20 ng/mL), doxycycline (2 μg/mL), or combinations as shown. Results are an average of 2 independent experiments and indicate that doxycycline has no independent negative effect on induction of ORF 50 or ORF 26.
As discussed, Cdk2 activity is necessary for both EBV and CMV replication. We therefore assessed specifically whether the vGPCR-induced inactivation of Cdk2 could be at least partially responsible for the decrease in chemically induced transcription of ORF 50 and 26. Using lentiviral transduction, we infected BC3.14 cells with either a Cdk2 dominant-negative (DN) construct or a reverse Cdk2-DN sequence as a control.83 Cells were incubated with butyrate; ORF 50 and ORF 26 transcripts were quantified by qRT-PCR. Figure 7 shows that the Cdk2 dominant negative caused an approximately 50% reduction in both transcripts. Of note these data allow us to conclude that it is the vGPCR-induced inhibition of Cdk2 that results in inhibition of ORF 50 and ORF 26 transcripts, strongly suggesting an impairment of successful lytic replication.
Figure 7.

Cdk2 activity is necessary for full chemical induction of KSHV lytic genes. BC3 cells were infected with lentivirus expressing the Cdk2 dominant-negative coding sequence in either forward or reverse (control) orientation; 72 hours later, cells were treated with 1 mM butyrate for 48 hours as indicated. Quantitative RT-PCR was performed for ORF 50 (top), and ORF 26 (bottom). Figure represents 2 independent experiments, each done in duplicate. Error bars indicate SD. *Significant increase over baseline, P ≤ .05; **Significant decrease from maximum induction, P ≤ .05.
Discussion
KSHV vGPCR signaling and function have been assayed in several cell types, but few studies have been done in hematopoietic cells. KSHV-infected hematopoietic cells are a vital source of new virion production in all KSHV-mediated disease. Furthermore, ongoing virion production is essential to maintain KSHV-derived tumors. For that reason we studied vGPCR signaling, transcription factor activation, and phenotypic effect in PEL cells (KSHV-infected lymphoma cells of B-cell lineage). Our initial studies showed that overexpression of vGPCR results in decreased proliferation.43 This finding was surprising because vGPCR had already been classified as a viral oncogene that transforms fibroblasts and endothelial cells. We also noted that when overexpressed in uninduced, latently infected cells, vGPCR modestly increases the transcription of ORF 50 and ORF 57. Because cell-cycle manipulation and lytic gene product expression are intimately connected in herpesvirus replication, we sought to better understand the mechanism of vGPCR-induced cell-cycle arrest and its effect on lytic gene transcription in KSHV.
Using standard techniques we show that vGPCR signaling results in a dramatic arrest in G0/G1 with subsequent exit from S phase. It has been shown that ERK1/2 and p38 can act in concert to cause a p53-independent, p21-mediated G1 arrest.84 Furthermore, we have previously shown that in PEL cells, vGPCR induces both ERK1/2 and p38 activity.43 We therefore hypothesized that p21 was the CDI primarily responsible for vGPCR-induced arrest. Our data here support such a signaling pathway in PEL cells. Consistent with activation of p21, we show that the S-phase kinase Cdk2 is markedly inhibited by vGPCR. Although cell-cycle arrest generally coincides with herpesviral latent-lytic switch, Cdk2 activity remains essential for replication.85 Using lytic gene transcription as a surrogate for successful replication, our data strongly support a vGPCR-driven inhibition of viral replication via down-regulation of Cdk2. It remains possible, however, that there are additional vGPCR-mediated events that also contribute to cell-cycle arrest or lytic-phase inhibition. For example, it has been shown many times that vGPCR induces NFκB activity; NFκB has in turn been shown to inhibit KSHV-lytic activation.86
Several studies support the idea that vGPCR may be expressed outside the context of the KSHV lytic cycle. HIV Tat increases the expression of vGPCR, and recent work from the Ganem laboratory (Yen-Moore et al87 and Liang and Ganem88) showed that vGPCR may be regulated by RBP-J, a transcription factor and target of the Notch pathway.87,88 Furthermore, abortive lytic-cycle progression in which a subset of lytic genes products is expressed has been shown in other herpesviruses. If such deregulated expression of vGPCR does occur, our data have implications for a longstanding conundrum in the vGPCR literature: as a lytic gene, it is difficult to reconcile either a directly transforming role, or indeed a paracrine role for vGPCR despite the many KS-related cytokines elaborated as a result of its signaling. Presumably a lytically activated KSHV-infected cell that is expressing vGPCR is destined to die and therefore not able to sustain a prolonged paracrine effect on surrounding cells. If, however, a sustained up-regulation of vGPCR were to occur prior to a latent-lytic switch, vGPCR may delay or even prevent full lytic transcription and cell death. Consequently, the proliferative and angiogenic potential of vGPCR would have time to be biologically significant in the tumor microenvironment. Furthermore, vGPCR-driven recruitment of new infectible cells would ensure viral propagation, producing as a byproduct the abnormal cellular proliferation characteristic of KSHV-mediated tumors (Figure 8). So although it may be that a few short-lived lytically activated cells can have an effect on the tumor microenvironment, the potential of a vGPCR-induced prolongation of an aberrant lytic phase exists and requires further investigation. The potential effects of a dysregulated (ie, nonlytically expressed) vGPCR has been described in a recent review by Sodhi et al.89
Figure 8.

Schematic representation of vGPCR function. Extracellular and intra-cellular factors both up-regulate vGPCR signaling, which results in cell-cycle arrest with inhibition of viral replication and cell death. This allows more prolonged effect of vGPCR on surrounding cells than would otherwise be possible if the lytic replication phase were to occur unhindered.
Aside from possible dysregulated expression outside the lytic cycle, the “normal” lytic expression of vGPCR may also play a significant role in KSHV replication. KSHV has evolved mechanisms to carefully regulate vGPCR. Its expression is restricted in that it is transcribed within the 3′ end of a bicistronic message; furthermore, KSHV encodes vMIP II, an inverse agonist of vGPCR.90 Precise control of vGPCR signaling suggests that the timing and level of vGPCR signaling is crucial to KSHV propagation, and it is likely that vGPCR plays more than one role in the course of KSHV-mediated disease. This may explain why we have previously found that in the setting of latent infection, vGPCR overexpression results in modest increased ORF 50 activity,43 but in the setting of lytic induction, vGPCR downwardly modulates the transcription of ORF 50. An interesting finding by Dezube et al91 is that during early de novo KSHV infection of primary endothelial cells, vGPCR transcription fluctuates in a cyclic pattern with a 48- to 72-hour time course consistent with viral replication. vGPCR-induced changes in viral transcription patterns or in host-cell function (including cell cycle) may be required during initial infection to establish successful latency or perhaps to kick start the initial rounds of lytic replication. Whether the cyclic pattern of vGPCR transcription is specific to endothelial cells remains to be studied.
We have established the ability of vGPCR to mediate cell-cycle arrest of infected B cells and provide compelling evidence that this occurs via Cdk2 inhibition. Furthermore, we provide initial evidence that this results in abnormal lytic phase induction. Independent inhibition of Cdk2 suggests that, as with other herpesviruses, this kinase is required for normal KSHV lytic gene induction. Although this helps explain how a lytic gene product could have important paracrine effects on surrounding cells, there remain questions about the exact role of vGPCR in the establishment of KSHV infection, maintenance of latency, and the latent to lytic switch. Our novel findings presented here are currently being extended to determine how vGPCR expression affects the quantity and infectivity of new virions. Furthermore, vGPCR-mediated effects on other KSHV lytic genes are being investigated and will shed light on the remaining questions about this pirated chemokine receptor and its potential as a target of rationally designed anti-KSHV treatment.
Acknowledgments
We thank the Elton John AIDS Foundation for the donation of the ABI PRISM 7700 sequence detector to the WIBR. We also thank Heike Laman for technical advice on the cyclin-dependent kinase assays and Dimitra Bourboulia for technical assistance on qRT-PCR.
Prepublished online as Blood First Edition Paper, September 8, 2005; DOI 10.1182/blood-2005-06-2350.
Supported by the National Institutes of Health (NIH) (grant K08-AI53 971) (MC) and (grant CA068 939) (EC), by the Central Research and Development Committee (CRDC) of Middlesex University, London (grant F119) (MC), and by CRUK Program Grant (CB).
M.C. designed and performed the research and wrote the paper; E.C. and C.B. contributed vital reagents and funding.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Chang Y, Cesarman E, Pessin MS, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science. 1994;266: 1865-1869. [DOI] [PubMed] [Google Scholar]
- 2.Whitby D, Howard MR, Tenant-Flowers M, et al. Detection of Kaposi sarcoma associated herpesvirus in peripheral blood of HIV-infected individuals and progression to Kaposi's sarcoma. Lancet. 1995;346: 799-802. [DOI] [PubMed] [Google Scholar]
- 3.Boshoff C, Schulz TF, Kennedy MM, et al. Kaposi's sarcoma-associated herpesvirus infects endothelial and spindle cells. Nat Med. 1995;1: 1274-1278. [DOI] [PubMed] [Google Scholar]
- 4.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas [see comments]. N Engl J Med. 1995;332: 1186-1191. [DOI] [PubMed] [Google Scholar]
- 5.Dupin N, Diss TL, Kellam P, et al. HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8-positive plasmablastic lymphoma. Blood. 2000;95: 1406-1412. [PubMed] [Google Scholar]
- 6.Soulier J, Grollet L, Oksenhendler E, et al. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood. 1995;86: 1276-1280. [PubMed] [Google Scholar]
- 7.Chang Y, Moore PS, Talbot SJ, et al. Cyclin encoded by KS herpesvirus [letter]. Nature. 1996; 382: 410. [DOI] [PubMed] [Google Scholar]
- 8.Cesarman E, Nador RG, Bai F, et al. Kaposi's sarcoma-associated herpesvirus contains G protein-coupled receptor and cyclin D homologs which are expressed in Kaposi's sarcoma and malignant lymphoma. J Virol. 1996;70: 8218-8223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Russo J, Bohenzky R, Chien M, et al. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc Natl Acad Sci U S A. 1996; 93: 14862-14867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng EH, Nicholas J, Bellows DS, et al. A Bcl-2 homolog encoded by Kaposi sarcoma-associated virus, human herpesvirus 8, inhibits apoptosis but does not heterodimerize with Bax or Bak. Proc Natl Acad Sci U S A. 1997;94: 690-694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo HG, Browning P, Nicholas J, et al. Characterization of a chemokine receptor-related gene in human herpesvirus 8 and its expression in Kaposi's sarcoma. Virology. 1997;228: 371-378. [DOI] [PubMed] [Google Scholar]
- 12.Neipel F, Albrecht JC, Fleckenstein B. Cell-homologous genes in the Kaposi's sarcoma-associated rhadinovirus human herpesvirus 8: determinants of its pathogenicity? J Virol. 1997; 71: 4187-4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nicholas J, Ruvolo V, Zong J, et al. A single 13-kilobase divergent locus in the Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genome contains nine open reading frames that are homologous to or related to cellular proteins. J Virol. 1997;71: 1963-1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nicholas J, Ruvolo VR, Burns WH, et al. Kaposi's sarcoma-associated human herpesvirus-8 encodes homologues of macrophage inflammatory protein-1 and interleukin-6. Nat Med. 1997;3: 287-292. [DOI] [PubMed] [Google Scholar]
- 15.Sarid R, Sato T, Bohenzky RA, Russo JJ, Chang Y. Kaposi's sarcoma-associated herpesvirus encodes a functional bcl-2 homologue. Nat Med. 1997;3: 293-298. [DOI] [PubMed] [Google Scholar]
- 16.Swanton C, Mann DJ, Fleckenstein B, Neipel F, Peters G, Jones N. Herpes viral cyclin/Cdk6 complexes evade inhibition by CDK inhibitor proteins. Nature. 1997;390: 184-187. [DOI] [PubMed] [Google Scholar]
- 17.Wong WW. ICE family proteases in inflammation and apoptosis. Agents Actions Suppl. 1998;49: 5-13. [DOI] [PubMed] [Google Scholar]
- 18.Nicholas J, Zong JC, Alcendor DJ, et al. Novel organizational features, captured cellular genes, and strain variability within the genome of KSHV/HHV8. J Natl Cancer Inst Monogr. 1998;(23): 79-88 [DOI] [PubMed]
- 19.Ahuja SK, Murphy PM. Molecular piracy of mammalian interleukin-8 receptor type B by herpesvirus saimiri. J Biol Chem. 1993;268: 20691-20694. [PubMed] [Google Scholar]
- 20.Coughlin SR. Expanding horizons for receptors coupled to G proteins: diversity and disease. Curr Opin Cell Biol. 1994;6: 191-197. [DOI] [PubMed] [Google Scholar]
- 21.Spiegel AM. Defects in G protein-coupled signal transduction in human disease. Annu Rev Physiol. 1996;58: 143-170. [DOI] [PubMed] [Google Scholar]
- 22.Arvanitakis L, Geras-Raaka E, Gershengorn MC. Constitutively signaling G-protein-coupled receptors and human disease. Trends Endocrinol Metab. 1998;9: 27-31. [DOI] [PubMed] [Google Scholar]
- 23.Wakeling MN, Roy DJ, Nash AA, Stewart JP. Characterization of the murine gammaherpesvirus 68 ORF74 product: a novel oncogenic G protein-coupled receptor. J Gen Virol. 2001;82: 1187-1197. [DOI] [PubMed] [Google Scholar]
- 24.Ho HH, Ganeshalingam N, Rosenhouse-Dantsker A, Osman R, Gershengorn MC. Charged residues at the intracellular boundary of transmembrane helices 2 and 3 independently affect constitutive activity of Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor. J Biol Chem. 2001;276: 1376-1382. [DOI] [PubMed] [Google Scholar]
- 25.Schwarz M, Murphy PM. Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor constitutively activates NF-kappa B and induces proinflammatory cytokine and chemokine production via a C-terminal signaling determinant. J Immunol. 2001;167: 505-513. [DOI] [PubMed] [Google Scholar]
- 26.Rosenkilde MM, Schwartz TW. Potency of ligands correlates with affinity measured against agonist and inverse agonists but not against neutral ligand in constitutively active chemokine receptor. Mol Pharmacol. 2000;57: 602-609. [DOI] [PubMed] [Google Scholar]
- 27.Rosenkilde MM, Kledal TN, Brauner-Osborne H, Schwartz TW. Agonists and inverse agonists for the herpesvirus 8-encoded constitutively active seven-transmembraneoncogene product, ORF-74. J Biol Chem. 1999;274: 956-961. [DOI] [PubMed] [Google Scholar]
- 28.Geras-Raaka E, Arvanitakis L, Bais C, Cesarman E, Mesri EA, Gershengorn MC. Inhibition of constitutive signaling of Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor by protein kinases in mammalian cells in culture. J Exp Med. 1998;187: 801-806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geras-Raaka E, Varma A, Clark-Lewis I, Gershengorn M. Kaposi's sarcoma-associated herpesvirus (KSHV) chemokine vMIP-II and human SDF-1alpha inhibit signaling by KSHV G protein-coupled receptor. Biochem Biophys Res Commun. 1998;253: 725-727. [DOI] [PubMed] [Google Scholar]
- 30.Geras-Raaka E, Varma A, Ho H, Clark-Lewis I, Gershengorn MC. Human interferon-gamma-inducible protein 10 (IP-10) inhibits constitutive signaling of Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor. J Exp Med. 1998;188: 405-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gershengorn MC, Geras-Raaka E, Varma A, Clark-Lewis I. Chemokines activate Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor in mammalian cells in culture. J Clin Invest. 1998;102: 1469-1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holst PJ, Rosenkilde MM, Manfra D, et al. Tumorigenesis induced by the HHV8-encoded chemokine receptor requires ligand modulation of high constitutive activity. J Clin Invest. 2001;108: 1789-1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang T, Chen S, Leach M, et al. Transgenic expression of the chemokine receptor encoded by human herpesvirus 8 induces an angioproliferative disease resembling Kaposi's sarcoma [see comments]. J Exp Med. 2000;191: 445-454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Montaner S, Sodhi A, Molinolo A, et al. Endothelial infection with KSHV genes in vivo reveals that vGPCR initiates Kaposi's sarcomagenesis and can promote the tumorigenic potential of viral latent genes. Cancer Cell. 2003;3: 23-36. [DOI] [PubMed] [Google Scholar]
- 35.Guo HG, Sadowska M, Reid W, Tschachler E, Hayward G, Reitz M. Kaposi's sarcoma-like tumors in a human herpesvirus 8 ORF74 transgenic mouse. J Virol. 2003;77: 2631-2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bais C, Van Geelen A, Eroles P, et al. Kaposi's sarcoma associated herpesvirus G protein-coupled receptor immortalizes human endothelial cells by activation of the VEGF receptor-2/KDR. Cancer Cell. 2003;3: 131-143. [DOI] [PubMed] [Google Scholar]
- 37.Arvanitakis L, Geras-Raaka E, Varma A, Gershengorn MC, Cesarman E. Human herpesvirus KSHV encodes a constitutively active G-protein-coupled receptor linked to cell proliferation. Nature. 1997;385: 347-350. [DOI] [PubMed] [Google Scholar]
- 38.Dupin N, Fisher C, Kellam P, et al. Distribution of human herpesvirus-8 latently infected cells in Kaposi's sarcoma, multicentric Castleman's disease, and primary effusion lymphoma. Proc Natl Acad Sci U S A. 1999;96: 4546-4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Linderoth J, Rambech E, Dictor M. Dominant human herpesvirus type 8 RNA transcripts in classical and AIDS-related Kaposi's sarcoma. J Pathol. 1999;187: 582-587. [DOI] [PubMed] [Google Scholar]
- 40.Staskus KA, Zhong W, Gebhard K, et al. Kaposi's sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J Virol. 1997;71: 715-719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teruya-Feldstein J, Zauber P, Setsuda JE, et al. Expression of human herpesvirus-8 oncogene and cytokine homologues in an HIV-seronegative patient with multicentric Castleman's disease and primary effusion lymphoma. Lab Invest. 1998;78: 1637-1642. [PubMed] [Google Scholar]
- 42.Montaner S, Sodhi A, Pece S, Mesri EA, Gutkind JS. The Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor promotes endothelial cell survival through the activation of Akt/protein kinase B. Cancer Res. 2001;61: 2641-2648. [PubMed] [Google Scholar]
- 43.Cannon ML, Philpott NJ, Cesarman E. The Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor has broad signaling effects in primary effusion lymphoma cells. J Virol. 2003;77: 57-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Masood R, Cai J, Zheng T, Smith DL, Naidu Y, Gill PS. Vascular endothelial growth factor/vascular permeability factor is an autocrine growth factor for AIDS-Kaposi sarcoma. Proc Natl Acad Sci U S A. 1997;94: 979-984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Samaniego F, Markham PD, Gendelman R, et al. Vascular endothelial growth factor and basic fibroblast growth factor present in Kaposi's sarcoma (KS) are induced by inflammatory cytokines and synergize to promote vascular permeability and KS lesion development. Am J Pathol. 1998; 152: 1433-1443. [PMC free article] [PubMed] [Google Scholar]
- 46.Aoki Y, Jaffe ES, Chang Y, et al. Angiogenesis and hematopoiesis induced by Kaposi's sarcoma-associated herpesvirus-encoded interleukin-6. Blood. 1999;93: 4034-4043. [PubMed] [Google Scholar]
- 47.Aoki Y, Jones KD, Tosato G. Kaposi's sarcoma-associated herpesvirus-encoded interleukin-6. J Hematother Stem Cell Res. 2000;9: 137-145. [DOI] [PubMed] [Google Scholar]
- 48.Jones KD, Aoki Y, Chang Y, Moore PS, Yarchoan R, Tosato G. Involvement of interleukin-10 (IL-10) and viral IL-6 in the spontaneous growth of Kaposi's sarcoma herpesvirus-associated infected primary effusion lymphoma cells. Blood. 1999;94: 2871-2879. [PubMed] [Google Scholar]
- 49.Sodhi A, Montaner S, Patel V, et al. The Kaposi's sarcoma-associated herpes virus G protein-coupled receptor up-regulates vascular endothelial growth factor expression and secretion through mitogen-activated protein kinase and p38 pathways acting on hypoxia-inducible factor 1alpha. Cancer Res. 2000;60: 4873-4880. [PubMed] [Google Scholar]
- 50.Shepard LW, Yang M, Xie P, et al. Constitutive activation of NF-kappa B and secretion of interleukin-8 induced by the G protein-coupled receptor of Kaposi's sarcoma-associated herpesvirus involve G alpha(13) and RhoA. J Biol Chem. 2001;276: 45979-45987 [DOI] [PubMed] [Google Scholar]
- 51.Nador RG, Milligan LL, Flore O, et al. Expression of Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor monocistronic and bicistronic transcripts in primary effusion lymphomas. Virology. 2001;287: 62-70. [DOI] [PubMed] [Google Scholar]
- 52.Sun R, Lin SF, Staskus K, et al. Kinetics of Kaposi's sarcoma-associated herpesvirus gene expression. J Virol. 1999;73: 2232-2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chiou CJ, Poole LJ, Kim PS, et al. Patterns of gene expression and a transactivation function exhibited by the vGCR (ORF74) chemokine receptor protein of Kaposi's sarcoma-associated herpesvirus. J Virol. 2002;76: 3421-3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liang Y, Chang J, Lynch SJ, Lukac DM, Ganem D. The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jkappa (CSL), the target of the Notch signaling pathway. Genes Dev. 2002;16: 1977-1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F, Miller G. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc Natl Acad Sci U S A. 1998;95: 10866-10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lukac DM, Renne R, Kirshner JR, Ganem D. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology. 1998;252: 304-312. [DOI] [PubMed] [Google Scholar]
- 57.O'Neill E, Douglas JL, Chien ML, Garcia JV. Open reading frame 26 of human herpesvirus 8 encodes a tetradecanoyl phorbol acetate- and butyrate-inducible 32-kilodalton protein expressed in a body cavity-based lymphoma cell line. J Virol. 1997;71: 4791-4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nealon K, Newcomb WW, Pray TR, Craik CS, Brown JC, Kedes DH. Lytic replication of Kaposi's sarcoma-associated herpesvirus results in the formation of multiple capsid species: isolation and molecular characterization of A, B, and C capsids from a gammaherpesvirus. J Virol. 2001;75: 2866-2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Renne R, Zhong W, Herndier B, et al. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat Med. 1996;2: 342-346. [DOI] [PubMed] [Google Scholar]
- 60.Miller G, Heston L, Grogan E, et al. Selective switch between latency and lytic replication of Kaposi's sarcoma herpesvirus and Epstein-Barr virus in dually infected body cavity lymphoma cells. J Virol. 1997;71: 314-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu Y, Black JB, Goldsmith CS, Browning PJ, Bhalla K, Offermann MK. Induction of human herpesvirus-8 DNA replication and transcription by butyrate and TPA in BCBL-1 cells. J Gen Virol. 1999;80: 83-90. [DOI] [PubMed] [Google Scholar]
- 62.Arvanitakis L, Mesri EA, Nador RG, et al. Establishment and characterization of a primary effusion (body cavity-based) lymphoma cell line (BC-3) harboring Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) in the absence of Epstein-Barr virus. Blood. 1996;88: 2648-2654. [PubMed] [Google Scholar]
- 63.Gaidano G, Cechova K, Chang Y, Moore PS, Knowles DM, Dalla-Favera R. Establishment of AIDS-related lymphoma cell lines from lymphomatous effusions. Leukemia. 1996;10: 1237-1240. [PubMed] [Google Scholar]
- 64.Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res. 1996;6: 986-994. [DOI] [PubMed] [Google Scholar]
- 65.Naldini L, Blomer U, Gallay P, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272: 263-267. [DOI] [PubMed] [Google Scholar]
- 66.Cannon ML, Cesarman E. The KSHV G protein-coupled receptor signals via multiple pathways to induce transcription factor activation in primary effusion lymphoma cells. Oncogene. 2004;23: 514-523. [DOI] [PubMed] [Google Scholar]
- 67.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13: 1501-1512. [DOI] [PubMed] [Google Scholar]
- 68.Platt G, Carbone A, Mittnacht S. p16INK4a loss and sensitivity in KSHV associated primary effusion lymphoma. Oncogene. 2002;21: 1823-1831. [DOI] [PubMed] [Google Scholar]
- 69.Akashi M, Osawa Y, Koeffler HP, Hachiya M. p21WAF1 expression by an activator of protein kinase C is regulated mainly at the post-transcriptional level in cells lacking p53: important role of RNA stabilization. Biochem J. 1999;337(Pt 3): 607-616. [PMC free article] [PubMed] [Google Scholar]
- 70.Zeng YX, el-Deiry WS. Regulation of p21WAF1/CIP1 expression by p53-independent pathways. Oncogene. 1996;12: 1557-1564. [PubMed] [Google Scholar]
- 71.Gartel AL, Tyner AL. Transcriptional regulation of the p21((WAF1/CIP1)) gene. Exp Cell Res. 1999; 246: 280-289. [DOI] [PubMed] [Google Scholar]
- 72.Kivinen L, Tsubari M, Haapajarvi T, Datto MB, Wang XF, Laiho M. Ras induces p21Cip1/Waf1 cyclin kinase inhibitor transcriptionally through Sp1-binding sites. Oncogene. 1999;18: 6252-6261. [DOI] [PubMed] [Google Scholar]
- 73.Barak Y, Juven T, Haffner R, Oren M. mdm2 expression is induced by wild type p53 activity. EMBO J. 1993;12: 461-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7: 1126-1132. [DOI] [PubMed] [Google Scholar]
- 75.Morgan DO. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol. 1997;13: 261-291. [DOI] [PubMed] [Google Scholar]
- 76.Hitomi M, Stacey DW. Cyclin D1 production in cycling cells depends on ras in a cell-cycle-specific manner. Curr Biol. 1999;9: 1075-1084. [DOI] [PubMed] [Google Scholar]
- 77.Lees EM, Harlow E. Sequences within the conserved cyclin box of human cyclin A are sufficient for binding to and activation of cdc2 kinase. Mol Cell Biol. 1993;13: 1194-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Laman H, Coverley D, Krude T, Laskey R, Jones N. Viral cyclin-cyclin-dependent kinase 6 complexes initiate nuclear DNA replication. Mol Cell Biol. 2001;21: 624-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sherr CJ. The Pezcoller lecture: cancer cell cycles revisited. Cancer Res. 2000;60: 3689-3695. [PubMed] [Google Scholar]
- 80.Kudoh A, Fujita M, Kiyono T, et al. Reactivation of lytic replication from B cells latently infected with Epstein-Barr virus occurs with high S-phase cyclin-dependent kinase activity while inhibiting cellular DNA replication. J Virol. 2003;77: 851-861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kudoh A, Daikoku T, Sugaya Y, et al. Inhibition of S-phase cyclin-dependent kinase activity blocks expression of Epstein-Barr virus immediate-early and early genes, preventing viral lytic replication. J Virol. 2004;78: 104-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bresnahan WA, Boldogh I, Chi P, Thompson EA, Albrecht T. Inhibition of cellular Cdk2 activity blocks human cytomegalovirus replication. Virology. 1997;231: 239-247. [DOI] [PubMed] [Google Scholar]
- 83.van den Heuvel S, Harlow E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science. 1993;262: 2050-2054. [DOI] [PubMed] [Google Scholar]
- 84.Todd DE, Densham RM, Molton SA, et al. ERK1/2 and p38 cooperate to induce a p21CIP1-dependent G1 cell cycle arrest. Oncogene. 2004; 23: 3284-3295. [DOI] [PubMed] [Google Scholar]
- 85.Flemington EK. Herpesvirus lytic replication and the cell cycle: arresting new developments. J Virol. 2001;75: 4475-4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Brown HJ, Song MJ, Deng H, Wu TT, Cheng G, Sun R. NF-kappaB inhibits gammaherpesvirus lytic replication. J Virol. 2003;77: 8532-8540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yen-Moore A, Hudnall SD, Rady PL, et al. Differential expression of the HHV-8 vGCR cellular homolog gene in AIDS-associated and classic Kaposi's sarcoma: potential role of HIV-1 Tat. Virology. 2000;267: 247-251. [DOI] [PubMed] [Google Scholar]
- 88.Liang Y, Ganem D. RBP-J (CSL) is essential for activation of the K14/vGPCR promoter of Kaposi's sarcoma-associated herpesvirus by the lytic switch protein RTA. J Virol. 2004;78: 6818-6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sodhi A, Montaner S, Gutkind JS. Does dysregulated expression of a deregulated viral GPCR trigger Kaposi's sarcomagenesis? FASEB J. 2004;18: 422-427. [DOI] [PubMed] [Google Scholar]
- 90.Talbot SJ, Weiss RA, Kellam P, Boshoff C. Transcriptional analysis of human herpesvirus-8 open reading frames 71, 72, 73, K14, and 74 in a primary effusion lymphoma cell line. Virology. 1999; 257: 84-94. [DOI] [PubMed] [Google Scholar]
- 91.Dezube BJ, Zambela M, Sage DR, Wang JF, Fingeroth JD. Characterization of Kaposi sarcoma-associated herpesvirus/human herpesvirus-8 infection of human vascular endothelial cells: early events. Blood. 2002;100: 888-896. [DOI] [PubMed] [Google Scholar]