

Abstract
We previously identified JAZ as a novel zinc finger (ZF) protein by screening a murine interleukin-3 (IL-3)–dependent NFS/N1.H7 myeloid cell cDNA library. JAZ is a member of a new class of ZFPs that is evolutionarily conserved and preferentially binds to dsRNA, but its function was unknown. Now, we report that the stress of IL-3 growth factor withdrawal up-regulates JAZ expression in hematopoietic cells in association with p53 activation and induction of cell death. Biochemical analysis reveals that JAZ associates with p53 to stimulate its transcriptional activity in p53-expressing cells, but not in p53-null cells unless complemented with p53. JAZ functions to mediate G1 cell-cycle arrest followed by apoptosis in a p53-dependent mechanism that is associated with up-regulation of p21 and BAX, dephosphorylation of Rb, and repression of cyclin A. Of importance, siRNA “knockdown” of endogenous JAZ inhibits p53 transcriptional activity, decreases the G1/G0 population, and attenuates stress-induced cell death. While JAZ directly binds p53 in vitro in a mechanism requiring p53's C-terminal regulatory domain but independent of dsRNA, the dsRNA-binding ZF domains are required for JAZ's stimulatory role of p53 in vivo by dictating its nuclear localization. Thus, JAZ is a novel negative regulator of cell growth by positively regulating p53.
Introduction
The p53 tumor-suppressor gene is the most frequent target of genetic inactivation in human cancer.1 p53 is a homotetrameric transcription factor with several distinct domains for its function and regulation.2,3 Most of the tumor-associated mutations in p53 occur in the core DNA-binding domain and disrupt the DNA-binding/transactivational activity of p53.1 p53 transactivation-deficient mice develop spontaneous tumors, indicating that the transcriptional activity of p53 is essential for its potent tumor-suppressor function.4,5 p53 can transactivate a number of genes containing p53-response elements including p21 and BAX, which play key roles in p53-mediated growth arrest and apoptosis.1,6 In addition, p53 can function extranuclearly by directly inhibiting Bcl2/BclXL or activating BAX at the mitochondria to induce apoptosis.7-9
p53 acts as a central negative regulator of cell growth by integrating genotoxic stress signals.1 However, p53 is also reported to respond to nongenotoxic stresses, but the mechanism(s) is not well understood.10 In response to DNA damage, growth factor depletion, chromosomal aberrations, telomere erosion, oncogene activation, and hypoxia, p53 is activated to induce growth arrest, differentiation, or apoptosis.1,10 p53 has also been reported to be necessary for efficient hematopoietic growth factor withdrawal–induced apoptosis.11-16 Furthermore, regulation of p53 is central to normal cell growth and tumor suppression, but the mechanism by which p53 is regulated is complex and still not fully understood.17,18 However, it is clear that interaction with cellular proteins plays an important role in p53 regulation.17 For example, an increasing number of cellular regulators of p53 have been identified that include ARF, ASPP, HIPK2, HMG-1, L11, MDM2, PC4, Pin1, PML, Ref-1, TAFII31, YY1, and ZPB-89.19-31 These regulators mediate/enhance p53's growth inhibitory and proapoptotic function through apparently different mechanisms.
We initially discovered JAZ as a novel mammalian ZFP by screening a murine interleukin-3 (IL-3)–dependent NFS/N1.H7 myeloid cell cDNA library.32 Both murine and human JAZ encode a 294–amino acid polypeptide that contains 4 homologous C2H2-type ZF domains connected by an unusually long linker sequence (ie, 28-38 amino acids) compared with most known ZFPs with a 6– to 8–amino acid linker sequence.32,33 Moreover, unlike other classic C2H2-type ZFPs that usually bind DNA, JAZ preferentially binds dsRNA, at least in vitro.32 A 65– to 70–amino acid “consensus” dsRNA-binding motif (dsRBM) has been identified in a number of proteins that specifically recognize and bind dsRNA.34,35 For example, PKR plays a fundamental role in regulating protein synthesis and apoptosis and contains 2 dsRNA-binding motifs within its regulatory domain.36,37 In addition, the first cellular activator of PKR, RAX/PACT, consists of 3 such dsRBMs.38,39 However, while JAZ preferentially binds dsRNA, it does not contain such a consensus dsRBM but rather requires its ZF domains to bind dsRNA.32 Of interest, exportin-5, a nuclear export receptor for specific classes of dsRNAs as well as the RNA-binding protein ILF3, was recently reported to bind and “export” JAZ as a cargo protein.40 However, while JAZ is a nuclear protein at steady state,32,40 the significance of JAZ, if any, in an exporting function is unknown.
In addition to JAZ, PAG608/Wig-1, a mammalian p53-inducible ZFP that contains 3 C2H2-type ZF domains, and dsRBP-Zfa, a Xenopus ZFP with 7 such ZFs, have also been reported to preferentially bind dsRNA in vitro.41-45 Of interest, these 2 dsRNA-binding ZFPs also contain unusually long linker sequences (ie, 34-44 amino acids for dsRBP-Zfa and 54-77 amino acids for PAG608/Wig-1).41,42,44 While the function of dsRBP-Zfa remains unknown, JAZ and PAG608/Wig-1 can induce apoptosis when ectopically expressed in cells.32,41,46 In addition, PAG608/Wig-1 was shown to inhibit cell growth in a colony-formation assay, but the mechanism is not known.47 We now report that JAZ, whose expression is responsive to IL-3 growth factor withdrawal that induces apoptosis, negatively regulates cell growth by acting as a novel, positive regulator of p53 transcriptional activity.
Materials and methods
Cell culture, transfection, IL-3/serum deprivation, and cell viability
NFS/N1.H7 murine myeloid cells and M1 murine myeloid leukemic cells were grown as described.36,48 All other cell lines used were grown in DMEM supplemented with 10% fetal bovine serum. Transfection was performed using LipofectAMINE (Invitrogen, Frederick, MD).
At various points of time following withdrawal of IL-3 or serum from H7 or M1 cells, cell viability was measured by the trypan blue exclusion method.49
Flow cytometry analysis
Green fluorescent protein (GFP)–JAZ (10 μg) in pEGFPN1 (BD CLONTECH, Palo Alto, CA) as described previously32 was transfected into p53+/+ and p53–/– isogenic mouse embryonic fibroblasts (MEFs) and NIH3T3 cells that were grown in 10-cm–diameter culture plates. By 24, 48, or 72 hours, cells were harvested for flow cytometry analysis as described.50
Immunoprecipitation, immunoblotting, and subcellular fractionation
All antibodies used were obtained from Santa Cruz Biotechnology (Santa Cruz, CA), except for p21 (Ab-1; Oncogene, Cambridge, MA) and FLAG (Sigma, St Louis, MO). Coimmunoprecipitation (IP) and immunoblotting (IB) studies were carried out similarly as described.36 For subcellular fractionation, the nuclei of p53+/+ MEFs or CV-1 cells were isolated as described previously.32 The nuclei were lysed in the lysis buffer,32 and Triton X-100 was also added into cytoplasmic fractions to 1% to obtain cytoplasmic lysates.
A rabbit polyclonal antibody (JAZ111) was produced and affinity purified using a JAZ-specific synthetic peptide (ETIKGDVKRLDSDQKSSRSK; ALPHA DIAGNOSTIC, San Antonio, TX). The control Ab for JAZ111 was produced by antigen competition. The specificity of JAZ111 was verified by demonstrating that JAZ111 can specifically coimmunoprecipitate and immunoblot FLAG-JAZ that was transfected in p53–/– MEFs (Figure S1, available on the Blood website; see the Supplemental Figures link at the top of the online article).
In vitro GST “pull-down” assay
Glutathione S-transferase (GST)–JAZ was prepared as described.32 GST only or GST-JAZ glutathione-Sepharose beads (20 μL) were then incubated at 4°C for 1 hour with 100 ng purified recombinant p53 (rp53; ACTIVE MOTIF, Carlsbad, CA) in the binding buffer (20 mM Tris [pH 8.0], 10% glycerol, 0.5 mM EDTA, 0.2% NP-40, and 200 mM NaCl) followed by washing beads and IB using a p53 antibody (DO-1). In control experiments, the specificity and activity of RNase V1 were verified (Figure S2), and 1 U/mL RNase V1 (Ambion, Austin, TX) was used to treat GST-JAZ or rp53 similarly as described.40 Alternatively, 0.1 μg/mL poly(I) · poly(C) (Amersham Pharmacia Biotech, Piscataway, NJ) was added in the binding mixture.
Deletion mutational analysis of JAZ-p53 binding
GST-p53 or GST-JAZ deletion mutants were constructed by polymerase chain reaction (PCR)–based subcloning using appropriate primers and wt p53 or JAZ cDNA as a template. GST-p53 beads were prepared and incubated at 4°C for 2 hours with 250 μg lysate of H1299 cells (that were transfected by FLAG-JAZ/pcDNA3) followed by IB using a FLAG antibody.
Luciferase assays
pp53-TA–(containing the wt p53 response elements) and pTA-control luciferase reporter vectors were obtained from BD CLONTECH. PG13 or MG15 (containing the wt or mutant p53 response elements) and p21-promoter (containing 2.3 kb endogenous p21 promoter) luciferase reporter constructs were kindly provided by Bert Vogelstein (Johns Hopkins University).51
To determine the effect of JAZ on p53 transcriptional activity, 10 μg JAZ-GFP/pEGFPN1 (pEGFPN1 as a control) was cotransfected with 1.0 μg pp53-TA or p21-promoter luciferase vector into COS-7, CV-1, or NIH3T3 cells, p53+/+ or p53–/– MEFs that were grown in 10-cm–diameter culture plates. After 24 or 48 hours, fluorescence-activated cell sorting (FACS) was used to sort out GFP-expressing cells followed by luciferase assays using the Luciferase Assay System (Promega, Madison, WI).
In addition, 1.0 μg FLAG-JAZ/pcDNA3 and 0.1 μg p53wt/pCMV or p53ΔC(30)/pcDNA3 were cotransfected with 0.1 μg PG13, MG15, or p21-promoter luciferase vector into SAOS-2 cells or p53–/– MEFs that were grown in 6-well plates. pAc-β-GAL vector (0.1 μg) containing a β-galactosidase gene under control of a β-actin promoter52 was also included as an internal control. After 24 hours, cells were lysed and assayed for luciferase activity and normalized by β-galactosidase activity.
siRNA interference
Using the Silencer siRNA Construction Kit (Ambion), JAZ-specific and control siRNAs were synthesized and selected. To test the effectiveness of the JAZ-siRNA in “knocking down” endogenous JAZ expression, 20 pmol JAZ-siRNA (or the control siRNA) was transfected into p53+/+ and p53–/– MEFs that were grown in 6-well plates using Oligofectamine (OLIGOENGINE, Seattle, WA). By 48 hours, cells were lysed followed by IB using JAZ111 and other antibodies against p53 (FL393), p21, and α-tubulin. To assess the effect of the JAZ-siRNA on p53 transcriptional activity, 20 pmol JAZ-siRNA was cotransfected with 0.1 μg pp53-TA-luc (plus 0.3 μg pEGFPN1 as a carrier vector) into p53+/+ and p53–/– MEFs using LipofectAMINE. By 48 hours, cells were harvested for luciferase assays.
A JAZ-siRNA– or control siRNA–expressing plasmid was also constructed using a pSUPER.retro.puro vector (OLIGOENGINE). Transient transfection of the JAZ-siRNA plasmid in p53+/+ and p53–/– MEFs could effectively knock down endogenous JAZ expression (data not shown). To assess the effect of the JAZ-siRNA on the cell cycle, cotransfection of 7.0 μg JAZ-siRNA/pSUPER with 3.0 μg pEGFPN1 was performed in the MEFs for 72 hours followed by flow cytometry analysis. Furthermore, the JAZ-siRNA or control siRNA expression plasmid was stably transfected in NFS/N1.H7 or M1 cells similarly as described,38,53 and the clones were deprived of IL-3/serum followed by the viability measurement.
Results
IL-3 growth factor or serum withdrawal up-regulates JAZ expression in association with p53 activation and induction of cell death
IL-3 is a multipotential hematopoietic growth factor that induces cell signaling responsible for growth and survival of hematopoietic cells.54,55 Since IL-3 withdrawal induces apoptosis in factor-dependent NFS/N1.H7 murine myeloid cells,38 we tested whether endogenous JAZ expression might be responsive to the stress of withdrawal. Both Northern and Western blot analyses reveal that IL-3 deprivation up-regulates JAZ expression that temporally precedes induction of cell death (Figure 1A). Since p53 has been reported to play a necessary role in efficient IL-3 withdrawal–mediated apoptosis in factor-dependent hematopoietic cells (eg, DA-1 and 32D cells),14-16 we assessed whether p53 expression was affected. Results reveal that IL-3 withdrawal can induce p53 expression and up-regulate its transcriptional activity as assessed by increased BAX expression (Figure 1A). Furthermore, while more rapid cell death in association with p53 activation can be induced by withdrawal of both IL-3 and serum from H7 cells (Figure 1B), M1 murine myeloid leukemic cells, which are IL-3 independent and p53 deficient,48 are highly insensitive to serum withdrawal–induced cell death (Figure 1C). Potentially of interest, either IL-3 or serum withdrawal appears to induce a biphasic pattern of JAZ and p53 up-regulation in H7 cells (Figure 1). Furthermore, JAZ expression appears as a doublet, which can be specifically blocked with the JAZ-specific peptide to which the JAZ111 antisera were raised (data not shown). While we speculate that the JAZ doublet may result from potential posttranslational modification(s), the nature of any such “modification(s)” and its significance for p53 activation remain to be tested. While IL-3 or serum deprivation induces JAZ up-regulation, the expression of JAZ is not altered following genotoxic treatment of cells with ionizing radiation, cisplatin, adriamycin, or 5-fluorouracil—all of which activate p53 responses (data not shown).10 Collectively, these data suggest that JAZ may specifically play a role in nongenotoxic stress (ie, IL-3 or serum withdrawal) that induces p53 activation and apoptosis.
Figure 1.
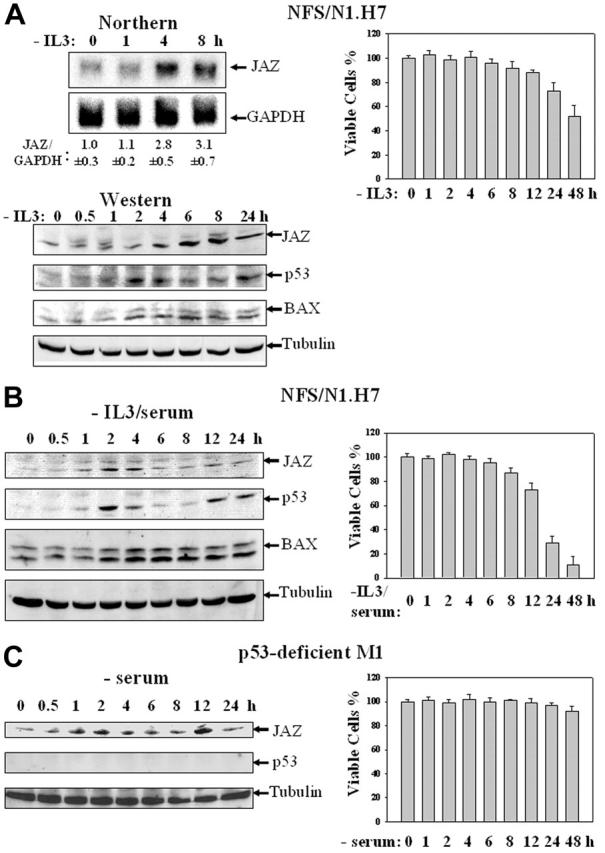
IL-3 growth factor or serum deprivation up-regulates the expression of endogenous JAZ in association with p53 activation and induction of cell death. (A) IL-3–dependent NFS/N1.H7 murine myeloid cells were deprived of IL-3 for various periods of time. (Left top panel) Northern analysis of JAZ's mRNA level was performed using 32P-labeled JAZ cDNA, and GAPDH expression was used for a loading control. Quantitation of JAZ mRNA level by densitometry is displayed relative to that of GAPDH, and the average ratio of JAZ/GAPDH is set to 1.0 at 0 hour upon IL-3 withdrawal (n = 3). (Left bottom panel) Western blot analysis of JAZ's protein level was performed using antibodies against JAZ (JAZ111), p53, and BAX. Tubulin expression was used for a load control. (Right panel) Cell viability was measured by trypan blue exclusion method. (B-C) NFS/N1.H7 or p53-deficient M1 murine myeloid leukemic cells were deprived of IL-3 and/or serum for various periods of time followed by Western blot analysis (left panels) using antibodies against JAZ, p53, BAX, and/or tubulin and the cell viability assay (right panels). Error bars represent standard deviations (n = 3).
JAZ induces p53-dependent G1 cell cycle arrest followed by apoptosis
To further test a role for JAZ in p53-mediated cell death, GFP-tagged JAZ was transfected into isogenic p53+/+ or p53–/– MEFs. At 24, 48, and 72 hours after transfection, cells were harvested for flow cytometry analysis. Results reveal that after 48 or 72 hours, the JAZ-GFP–expressing cells undergo cell death as indicated by the increased presence of a sub-G1 population (Figure 2A, arrow). By contrast, GFP-only–expressing cells remain viable. Of interest, at 24 hours and prior to induction of cell death, G1 cell cycle arrest was already noted in p53+/+ cells expressing JAZ-GFP compared with those expressing GFP-only as indicated by a significant (∼ 25%) increase in the G1/G0 population (Figure 2A). No JAZ-mediated cell death or G1 arrest could be observed at any time point tested in p53–/– MEFs (Figure 2B). These results indicate that JAZ may mediate G1 cell-cycle arrest and apoptosis in a p53-dependent manner. In support of this, when JAZ-GFP but not GFP-only is expressed in NIH3T3 murine fibroblasts that express endogenous (wt) p53, potent G1 arrest occurs followed by cell death (Figure 2C, arrow). We previously demonstrated that cell death was due to apoptosis by the TUNEL assay.32 The apparent “2 peaks” of sub-G1 nonviable cells observed at 72 hours may result from subpopulations of apoptotic cells that contain different levels of DNA that has been fragmented and degraded as the molecular hallmark of apoptosis (Figure 2C).50
Figure 2.
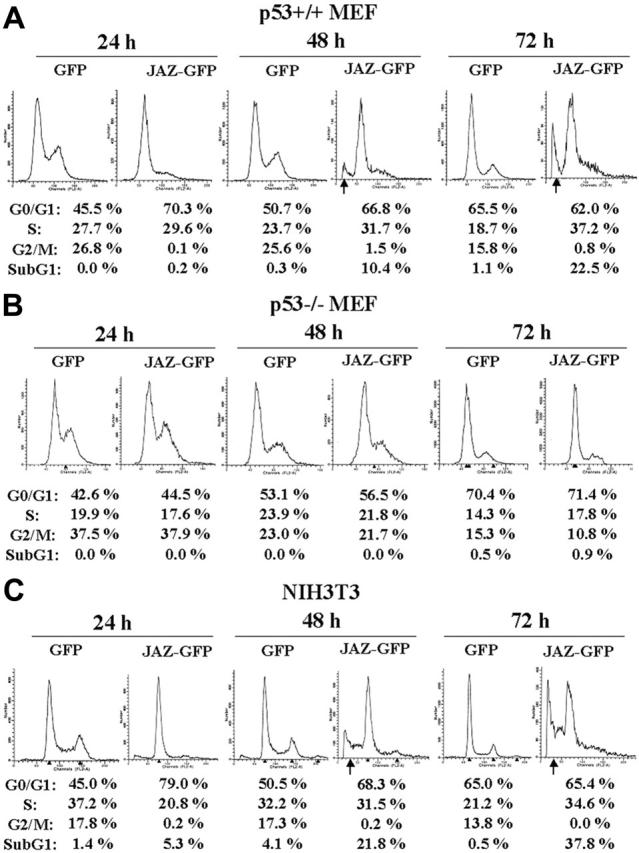
Expression of JAZ-GFP induces G1 cell-cycle arrest followed by apoptosis in a p53-dependent manner. JAZ-GFP/pEGFPN1 was transfected into p53+/+ (A) and p53–/– (B) MEFs and NIH3T3 cells (C). pEGFPN1 (GFP) alone was used as a control. At 24, 48, and 72 hours following transfection, cells were harvested, fixed by 1% formaldehyde at 4°C for 1 hour, permeabilized by 70% ethanol at 4°C overnight, and stained by propidium iodide followed by flow cytometry analysis as described.50 To normalize for transfection efficiency, only JAZ-GFP– or GFP-only–expressing cells were gated and then analyzed for a sub-G1 population and cell-cycle distribution. According to the program used here, the sub-G1 population represents the percentage of total gated (GFP or GFP positive) cells that are apoptotic, while the sum of the percentages of G1/G0, S, and G2/M is normalized to 100%. This is done in order to accurately compare the cell-cycle distribution of the viable cells expressing JAZ-GFP or GFP-only. The arrows indicate sub-G1 populations.
JAZ associates with p53 in vivo
To explore the mechanism, we investigated whether JAZ can interact with p53. Coimmunoprecipitation (IP) studies reveal that FLAG-JAZ can associate with p53 in either COS-7 or parental CV-1 monkey kidney cells and in the p53-null SAOS-2 human osteoblastoma cells when complemented with exogenous p53 (Figure 3A-B). We next tested whether endogenous JAZ and p53 can interact by producing a specific polyclonal JAZ antibody (JAZ111) that was used to coimmunoprecipitate endogenous JAZ from NFS/N1.H7 myeloid cells, NIH3T3 fibroblasts, or p53-deficient M1 leukemic cells. Results clearly indicate that endogenous JAZ and p53 can associate (Figure 3C). It should be noted that only a small percentage of endogenous (or exogenously expressed) JAZ is coimmunoprecipitated with p53. While the nature of the association and its “stability” following detergent lysis could account for this, a small fraction of p53 association has been observed for other p53 regulators YY1 and Pin1.26,30
Figure 3.

JAZ associates with p53 in various mammalian cell lines and directly binds p53 in vitro independent of dsRNA. (A-B) FLAG-JAZ in pcDNA3 (Invitrogen) was transfected into COS-7, CV-1, or p53-null SAOS-2 cells with or without p53wt/pCMV (BD CLONTECH). After 24 or 48 hours, cells were lysed in the lysis buffer (10 mM Hepes [pH 7.3], 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, and protease inhibitor cocktail σ). Coimmunoprecipitation (IP) followed by immunoblotting (IB) was performed using antibodies against FLAG and p53 (DO-1 or FL393). V indicates pcDNA3; F-J, FLAG-JAZ/pcDNA3; p53, p53wt/pCMV; and 20% or 50% input, 50 μg cell lysate. (C) The rabbit JAZ antibody (JAZ111 and the control antibody as described in “Materials and methods”) was used in IP and IB endogenous JAZ from 1- or 0.5-mg whole-cell lysate from NIH3T3, NFS/N1.H7, or p53-deficient M1 cells, and JAZ-associated p53 was immunoblotted using a p53 antibody (PAb 246). To facilitate detection of endogenous JAZ or p53 protein, H7 or M1 cells were deprived of IL-3/serum for 2 hours to increase JAZ or p53 expression (as shown in Figure 1). “10% input” indicates 100 or 50 μg cell lysate; +, JAZ111; and –, the control Ab. (D) Subcellular fractionation of p53+/+ MEFs and CV-1 cells was performed as described in “Materials and methods.” The JAZ111 antibody was used to coimmunoprecipitate endogenous JAZ from 1 mg lysate of the nuclear or cytoplasmic fraction, followed by IB using JAZ111 and antibodies against p53 and PCNA (a nuclear protein as a control). Nuc indicates nuclear fraction; Cyt, cytoplasmic fraction; and 10% Input, 100 μg nuclear or cytoplasmic lysate. (E) GST and GST-JAZ glutathione-Sepharose beads were incubated with 100 ng purified, recombinant p53 in the in vitro binding buffer followed by IB using DO-1 (top panel). The purified proteins were treated by RNase V1 as described in “Materials and methods.” The bottom panel represents Fast-green staining of the blot to show expression of GST or GST-JAZ. rp53 indicates recombinant p53; 20% Input, 20 ng rp53. Densitometric quantitation of bound rp53 versus GST-JAZ was performed following treatment with or without RNase V1. The average ratio of p53/JAZ from 3 independent experiments is shown (the ratio is set to 1.00 for the control/no treatment sample). (F) The GST-JAZ NF beads containing the mutant in which all 4 zinc fingers are point mutated by changing C2H2→C2AH32 were also used to pull down rp53.
Next, we determined whether endogenous JAZ and p53 can associate within the nucleus where both proteins are localized. Nuclear and cytoplasmic fractions of p53+/+ MEFs and CV-1 cells were isolated for IP studies. Results reveal that both JAZ and p53, as expected, are expressed virtually exclusively in the nucleus and that nuclear JAZ can specifically associate with and coimmunoprecipitate with p53 but not PCNA, a nuclear protein used as a control (Figure 3D). These results indicate that nuclear JAZ-p53 association is specific and strongly support a potentially direct interaction between JAZ and p53 in vivo. Furthermore, JAZ-p53 interaction is neither cell-type nor species limited.
JAZ directly binds p53 in vitro in a mechanism requiring p53's C-terminus but independent of dsRNA binding
A GST pull-down assay was performed to test whether purified GST-JAZ and recombinant p53 (rp53) can directly bind in vitro, and results demonstrate a direct JAZ-p53 interaction (Figure 3E, lane 4). Since JAZ is a dsRNA-binding ZFP,32 we next tested whether binding may depend on dsRNA. RNase V1, which specifically cleaves dsRNAs associated with GST-JAZ in vitro,40 was used to treat both GST-JAZ and rp53 prior to attempting the pull downs. Results clearly show that RNase V1 treatment fails to affect JAZ-p53 binding (Figure 3E, lane 5). Furthermore, addition of poly(I · C) (ie, a form of free dsRNA) to the binding mixture also has no effect on JAZ-p53 interaction (data not shown). Finally, the GST-JAZ NF mutant, which contains point mutations in all 4 ZFs that render JAZ unable to bind dsRNA,32 fully retains p53-binding capacity compared with wt GST-JAZ (Figure 3F). It should be noted that the NF point mutant appears to migrate more slowly than wt JAZ. This may result from the mutational loss of Zn2+ binding by the NF mutant (ie, loss of positive charges) as occurs in Bcl2 phosphomimic point mutants with increased negative charge(s).9 Collectively, these data support the notion that JAZ and p53 do directly interact in a mechanism that is independent of JAZ's dsRNA-binding capacity, at least in vitro.
To determine which domain(s) of p53 is required for interaction with JAZ, GST-p53 constructs containing various p53 deletion mutations were prepared (Figure 4A). GST-p53 beads were used to pull down FLAG-JAZ after transient transfection in p53-null H1299 cells. Results reveal that while no binding was detected between FLAG-JAZ and the GST-p53 N(1-100) mutant, FLAG-JAZ interacts strongly with both the GST-p53 C(309-393), which contains the C-terminal region of p53, and the GST-p53 M+C(101-393) mutant (Figure 4B, lanes 4, 6, and 8). In addition, neither GST-p53 M(101-308), which contains its core DNA-binding domain, nor GST-p53 N+M(1-308) can bind, or can only weakly bind, FLAG-JAZ (Figure 4B, lanes 5 and 7). Collectively, these data indicate that the C-terminal region of p53, which contains both its oligomerization domain (OD) and C-terminal regulatory domain (C-RD), is necessary for JAZ interaction. To test this further, deletion mutants lacking either the OD(325-355) or the C-RD(364-393) were used (Figure 4A). Results reveal that while GST-p53ΔOD(1-308&365-393) binds strongly to FLAG-JAZ, the GST-p53ΔC(30)(1-363) mutant that lacks the C-RD demonstrates markedly reduced binding (Figure 4C), indicating that p53's C-terminal regulatory domain but not its oligomerization sequence is necessary for efficient JAZ binding.
Figure 4.

Deletion mutational analysis of JAZ-p53 binding. (A) A schematic representation3 of p53 deletion mutants N(1-100), M(101-308), C(309-393), N+M(1-308), M+C(101-393), p53ΔOD(1-308&365-393), and p53ΔC(30)(1-363). TAD indicates N-terminal transactivation domain; DBD, sequence-specific DNA-binding domain; OD, oligomerization domain; and RD, C-terminal regulatory domain. While ++++ and +++ represent strong or relatively strong JAZ binding, respectively, + and – stand for weak and no binding, respectively. (B-C) GST or wt and mutant GST-p53 glutathione-Sepharose beads were incubated with the cell lysate obtained from p53-null H1299 cells that transiently express FLAG-JAZ as described in “Materials and methods.” The GST or GST-p53 beads were then immunoblotted using a FLAG antibody. “20% Input” indicates 50 μg cell lysate. Bottom panels represent Fast-green staining of GST-p53 proteins (arrows). (D) A schematic representation of JAZ deletion mutants J(1-118), J(123-171), J(167-221), J(214-294), J(1-171), J(1-221), J(167-294), and J(112-294). (E) GST-JAZ (containing deletion mutants) beads were used in vitro to pull down rp53 as described in Figure 3E. Bottom panel represents Fast-green staining of GST-JAZ proteins (arrows).
In similar studies, we also tested several GST-JAZ deletion mutants for their ability to pull down rp53 (Figure 4D-E). While GST full-length JAZ strongly interacts with rp53, various GST-JAZ deletion mutants exhibit only weak binding (Figure 4E), suggesting that full-length JAZ may be required for maximum/stable interaction with p53, at least in vitro.
JAZ stimulates p53 transcriptional activity in vivo
Since JAZ and p53 can interact in the nucleus where p53 functions as a transcription factor, we tested whether JAZ can modulate p53 transcriptional activity. While p53 transcriptional activity is stimulated approximately 10-fold when JAZ-GFP is expressed in CV-1 and NIH3T3 cells, no such stimulation is observed in COS-7 cells where p53 is expressed but is functionally inactivated by the SV40 large T antigen56 (Figure 5A). While JAZ binds p53's C-terminus (Figure 4A), the large T antigen interacts with p53's core DNA-binding domain to inhibit its transcriptional activity.56
Figure 5.

Effects of JAZ on p53 transcriptional activity. (A) JAZ-GFP/pEGFPN1 or pEGFPN1 (GFP-only) was cotransfected with a p53-luciferase reporter vector (p53-TA-luc) into COS-7, CV-1, and NIH3T3 cells. After 24 or 48 hours, JAZ-GFP– or GFP-positive COS-7, CV-1, and NIH3T3 cells were sorted by fluorescence-activated cell sorting (FACS). The same numbers of such sorted GFP-only and JAZ-GFP–positive cells were lysed and then assayed for luciferase activity. G indicates GFP; J-G, JAZ-GFP. Luciferase activity is displayed relative to that of the GFP control. (B) FLAG-JAZ/pcDNA3 (empty vector pcDNA3 as a control) was cotransfected with PG13 or MG15 luciferase reporter vector into p53-null SAOS-2 cells, in the presence or absence of p53wt/pCMV. pAc-β-GAL internal control vector was also included to normalize transfection efficiencies. After 24 hours, cells were lysed and assayed for luciferase activity; V indicates pcDNA3; F-J, FLAG-JAZ/pcDNA3; and p53, p53wt/pCMV; PG13 and MG15 luciferase vectors contain the wild-type and the mutant p53 DNA-binding site, respectively.51 Luciferase activity is displayed relative to that of the pcDNA3 vector. (C) JAZ-GFP/pEGFPN1 was cotransfected with a p21-promoter luciferase vector (p21-P-luc) into p53+/+ and p53–/– MEFs followed by FACS and luciferase assays. (D) FLAG-JAZ/pcDNA3 was cotransfected with p53wt/pCMV or p53ΔC(30)/pcDNA3 plus PG13 and pAc-β-GAL vectors into p53–/– MEFs following luciferase assays and normalization. (E) Mutational analysis of JAZ stimulation of p53 transcriptional activity in CV-1 cells. To determine the role of JAZ's ZF domains, JAZ-GFP ZF mutants (in pEGFPN1) containing a single or multiple C2H2→C2AH32 mutations were also used. Wt JAZ-GFP and its ZF mutants were cotransfected with the PG13 vector into CV-1 cells. After 24 hours, cells were sorted by FACS for luciferase activity. The ZF mutations H91A, H152A, H203A, and H257A are in JAZ's first, second, third, and fourth ZF motifs, respectively. NF represents the mutant in which all 4 zinc fingers are point mutated. JAZ-GFP deletion mutants were also used. J(1-171) is a JAZ-GFP deletion mutant containing 1 to 171 amino acids of JAZ, while J(167-294) deletion mutant contains 167 to 294 amino acids. Error bars represent standard deviations (n = 3).
To confirm whether JAZ stimulation of p53 is specific, FLAG-JAZ was cotransfected with or without wt p53 into p53-null SAOS-2 cells. Results show that in the absence of p53 complementation, no stimulatory effect on p53 transcriptional activity is observed, while coexpression of FLAG-JAZ and p53 significantly stimulates p53's transactivation of the PG13 p53-binding element but not the MG15 mutant element to which p53 is unable to bind (Figure 5B). Next, to test for functional relevance of p53 transactivation, a luciferase reporter containing the p21 promoter51 was used. Results clearly demonstrate that JAZ-GFP stimulates transcription of the p21 promoter exclusively in p53+/+ but not p53–/– MEFs (Figure 5C). Furthermore, Western blot analysis of lysates of cells expressing JAZ-GFP or GFP-only demonstrates no effect on p53 protein levels (Figure 6A), indicating that JAZ may directly stimulate p53 transcriptional activation in a mechanism that does not enhance p53 stability.
Figure 6.

Ectopic expression of JAZ up-regulates expression of p21 and BAX, induces Rb dephosphorylation, and represses cyclin A expression. Cotransfection of JAZ-GFP or FLAG-JAZ in p53+/+ and p53–/– MEFs (A,B,D) and NIH3T3 cells (C) was performed as described in Figure 5. JAZ-GFP– or GFP-positive cells were sorted by FACS. Western blot analysis was performed using antibodies against GFP; FLAG; cyclins D, E, and A; p53; p21; p27; BAX; Rb; and/or tubulin (as a loading control). (A) The densitometric quantitation of protein expression was performed. The average ratio of p21 or BAX compared with tubulin is shown. The ratio is set to 1.00 for the GFP-only–expressing p53+/+ cells. Three independent experiments with similar results were performed, and a representative figure is displayed. G indicates GFP; J-G, JAZ-GFP; V, pcDNA3; F-J, FLAG-JAZ/pcDNA3; p53, p53wt/pCMV; and p53ΔC, p53ΔC(30)/pcDNA3.
These data support the notion that JAZ specifically stimulates p53 transcriptional activity and that JAZ's stimulatory effect is neither cell-type specific nor species limited.
p53's C-terminal regulatory domain and JAZ's ZF domains are necessary for JAZ stimulation of p53 transcriptional activity
p53's C-terminal regulatory domain (C-RD) is a negative regulatory domain of p53 transcriptional activity.3 Removal of the C-RD (or binding antibodies or peptides targeted to this domain) activates the transactivation potential of p53.3,57,58 Since the C-RD is necessary for efficient JAZ-p53 binding (Figure 4C), we tested whether the C-RD may play a role in the functional outcome of JAZ-p53 binding. FLAG-JAZ and either wt p53 or p53ΔC(30) (lacking the C-RD) were cotransfected into p53–/– MEFs. Results confirm that FLAG-JAZ stimulates wt p53 transcriptional activity and, as expected, the truncated p53ΔC(30) mutant by itself displays increased transcriptional activity compared with wt p53 (Figure 5D). While it has been reported that 2 other p53 regulators, HMG-1 and PC4, can further increase p53ΔC(30)'s transcriptional activity,22,25 FLAG-JAZ does not display such an enhancing effect (Figure 5D). Therefore, we can propose that p53's C-RD is required for JAZ binding and stimulation of p53.
Since JAZ's ZF domains are not only required for dsRNA binding but also are essential for nuclear localization,32 we next tested whether these ZF domains may also play a role in JAZ stimulation of p53 transcription activity in vivo. Results reveal that while mutation of a single ZF fails to affect JAZ stimulation of p53, mutation of more than 2 ZF loci, which leads to loss of JAZ's ability to localize to the nucleus,32 profoundly reduces/abolishes its stimulatory effect (Figure 5E). Therefore, while JAZ's ZF domains are not required for p53 binding in vitro (Figure 3F), they are necessary for JAZ's stimulation of p53 transcriptional activity in vivo likely because they are necessary for JAZ's nuclear localization.
In addition, the JAZ-GFP deletion mutants J(1-171) and J(167-294), which contain either the N- or C-terminal half sequence of wt JAZ and bind p53 only weakly (Figure 4E), also fail to stimulate p53 transcriptional activity (Figure 5E). These data indicate that full-length JAZ is necessary for functional stimulation of p53.
JAZ induces up-regulation of p21 and BAX as well as dephosphorylation of Rb and repression of cyclin A
In association with stimulation of p53 transcriptional activity and induction of G1 cell cycle arrest and apoptosis, Western blot analysis demonstrates that JAZ-GFP up-regulates p21 and BAX in p53+/+ but not p53–/– MEFs (Figure 6A). Moreover, FLAG-JAZ expression in p53–/– MEFs enhances up-regulation of p21 mediated by exogenous wt p53 but not when the p53ΔC(30) mutant that lacks ability to efficiently bind JAZ is coexpressed (Figure 6B). Thus, JAZ functionally stimulates p53 transcriptional activity in a mechanism requiring p53's C-RD.
To verify the specificity of JAZ in mediating p53-dependent G1 arrest (Figure 2), we examined whether JAZ may affect expression of other G1 regulatory proteins. For example, cyclins D, E, and A are 3 important regulators that sequentially control cell-cycle progression in G1 phase.59 Western blot analysis demonstrates that JAZ-GFP expression is associated with down-regulation of cyclin A but not cyclins D or E in NIH3T3 cells (Figure 6C). Since cyclin A is essential for initiation of DNA replication,59 it is possible that JAZ-mediated G1 cell-cycle arrest may occur at the G1/S boundary. Moreover, since Rb is the major regulator of G1 cell-cycle progression,60 the effect of JAZ on Rb's phosphorylation status was also assessed. In JAZ-GFP–positive cells, more Rb protein was found to migrate as a faster band (ie, hypophosphorylated form61; Figure 6C), suggesting an induction of Rb dephosphorylation. In addition to p21, we also assessed expression of p27, another inhibitor of CDK2, a major Rb kinase.59,60 While JAZ-GFP expression increases the expression of the p53 target gene and inhibitor of CDK2, p21, no change was observed in p27 expression that is not transcriptionally regulated by p5359 (Figure 6C). The effect of JAZ-GFP on cyclin A or Rb phosphorylation was also observed exclusively in p53+/+ but not isogenic p53–/– MEFs (Figure 6D). Therefore, from these data, we propose that JAZ mediates cell-cycle arrest at the G1/S boundary in a p53-dependent manner.
siRNA knockdown of endogenous JAZ down-regulates p53 transcriptional activity, decreases G1/G0 population, or attenuates IL-3/serum withdrawal–induced cell death
To test the role of endogenous JAZ in regulating p53 transcriptional activity, we used siRNA interference to knock down expression of endogenous JAZ. Western blot analysis demonstrates that endogenous JAZ is effectively knocked down in both p53+/+ and p53–/– MEFs (by ∼ 70%, Figure 7A). Cotransfection of a p53-luciferase reporter reveals that JAZ knockdown cells expressing p53 display approximately 65% reduced p53 transcriptional activity and, as expected, there is no effect in p53–/– MEFs (Figure 7A). In addition, at 72 hours, expression of JAZ-siRNA decreases the G1/G0 population in p53+/+ but not p53–/– MEFs (Figure 7B), supporting a necessary role for JAZ in positively regulating p53 transcriptional activity and negatively regulating G1 cell-cycle progression.
Figure 7.

Effects of siRNA knockdown of endogenous JAZ expression. Several 21-nt JAZ-siRNAs that targeted different regions of the JAZ sequence and a control siRNA that is based on a JAZ-irrelevant 21-nt sequence in pCDNA3's ampicillin ORF were synthesized. The JAZ-siRNA that targets the central region of JAZ mRNA (5′-AAGAGGCTAGACTCAGATCAG-3′) was selected, since it could effectively reduce expression of JAZ-GFP (but not GFP-only) in NIH3T3 cells (by ∼ 70%, data not shown). (A) siRNA-mediated knockdown of endogenous JAZ expression inhibits p53 transcriptional activity in p53+/+ but not p53–/– MEFs. The JAZ-siRNA (or the control siRNA) was transiently transfected into p53+/+ and p53–/– MEFs. After 48 hours, cells were lysed followed by Western blot analysis using antibodies against JAZ (JAZ111), p53, p21, and tubulin. It should be noted that in order to accurately evaluate the effectiveness of JAZ-siRNA (compared with the control siRNA), a longer exposure of the film (∼ 3 hours) following the enhanced chemiluminescence (ECL) reaction was performed to facilitate detection of the markedly “decreased” endogenous JAZ level in JAZ-siRNA–transfected cells. Endogenous JAZ expression was knocked down by approximately 70%, as determined by densitometry. The JAZ-siRNA was also cotransfected into the MEFs with p53-TA (containing the wt p53 response elements)–luciferase reporter vector and a carrier vector pEGFPN1 (GFP). After 48 hours, cells were lysed and assayed for luciferase activity. p53 transcriptional activity (luciferase reporter) is displayed relative to the control siRNA–expressing p53+/+ MEFs. Con indicates control siRNA. In addition, the control experiment indicates that the JAZ-siRNA had no effect on the activity of the pTA-luciferase control vector (data not shown). (B) The JAZ-siRNA or the control siRNA expression plasmid was transiently cotransfected into p53+/+ and p53–/– MEFs along with a carrier vector pEGFPN1. After 72 hours, cells were harvested for flow cytometry analysis as described in “Materials and methods.” The GFP-positive cells were gated and analyzed for their cell-cycle distribution. (C) NFS/N1.H7 or M1 hematopoietic cells (as described in Figure 1) were stably transfected with the JAZ-siRNA (or the control siRNA) expression plasmid. After IL-3 and/or serum withdrawal for various periods of time, cells were harvested for the viability measurement by trypan blue exclusion method. siR-C indicates the control siRNA; siR-J, the JAZ-siRNA. Bottom panel shows Western blot analysis of siRNA knockdown of endogenous JAZ in H7 or M1 cells. IL-3/serum was present (+) or removed (–) for 2 hours. Error bars represent standard deviations (n = 3).
The JAZ-siRNA expression plasmid also stably knocks down expression of endogenous JAZ in p53-expressing NFS/N1.H7 myeloid cells and results in a marked delay in cell death (∼ 40% at 48 hours) following IL-3/serum withdrawal (Figure 7C). However, in spite of the stable knockdown of JAZ in p53-deficient M1 myeloid leukemic cells, cells fail to display cell death even by 48 hours following serum withdrawal (Figure 7C).
Discussion
IL-3 withdrawal–induced apoptosis plays a fundamental role in developmental and steady-state hematopoiesis.11,54,55 We have discovered that JAZ, whose expression is up-regulated in hematopoietic cells upon IL-3 growth factor withdrawal, mediates p53-dependent G1 cell-cycle arrest and apoptosis by directly stimulating p53 transcriptional activity. Of interest, JAZ and 2 other dsRNA-binding ZFPs, dsRBP-ZFa and PAG608/Wig-1, appear to belong to a novel class of C2H2-type ZFPs that feature an unusually long linker sequence and preferentially bind to dsRNA.32,41-45
JAZ stimulates p53 transcriptional activity and mediates p53-dependent G1 cell-cycle arrest by up-regulating p21 (Figures 2, 5, and 6). Alternatively, knockdown of endogenous JAZ down-regulates p53 transcriptional activity and promotes G1 cell-cycle progression (Figure 7). Of interest, as reported for the ARF tumor suppressor,62,63 the pathway for how JAZ may mediate p53-dependent G1 growth arrest may also involve dephosphorylation of Rb and repression of cyclin A, which are downstream of p2159 (Figure 6).
JAZ appears to mediate p53-dependent apoptosis in a mechanism that may result, at least in part, from up-regulation of BAX (Figures 2 and 6). Thus, while other p53 proapoptotic target genes could also be involved (but were not tested), it is well appreciated that BAX (or BAK) is required for dysregulating mitochondrial integrity and activating the intrinsic apoptotic pathway.64 Of interest, IL-3/serum withdrawal from p53-expressing NFS/N1.H7 myeloid cells induces endogenous JAZ expression, p53 activation, BAX up-regulation, and cell death (Figure 1). By contrast, no such effect of serum withdrawal occurs in the p53-deficient M1 myeloid leukemic cells even though JAZ is up-regulated. Moreover, siRNA knockdown of endogenous JAZ attenuates/decelerates factor-deprived cell death in H7 but not M1 cells (Figure 7). These data strongly suggest a regulatory role for JAZ in IL-3/serum withdrawal–induced apoptosis in a p53-dependent mechanism. Therefore, in addition to its well-described role in DNA damage–induced responses, p53 can also play a regulatory role in nongenotoxic stress that leads to apoptosis.1,10 The mechanism(s) may involve JAZ activation of p53 transcriptional activity. Of interest, recent reports indicate that other cellular regulators of p53 may also play similar roles in non–DNA damage–induced p53 stress responses.17 For example, ARF, a positive regulator of p53,65,66 is activated by hyperproliferative and oncogenic signals,19 while ZBP-89 and L11 can mediate p53 activation upon growth inhibitory signals including serum depletion.23,31,67
Of importance, JAZ was found to associate with p53 in vivo and specifically in the nucleus (Figure 3A-D). Mechanistically, JAZ and p53 directly interact in a dsRNA-independent manner, at least in vitro (Figure 3E-F). Also, of interest, JAZ has recently been reported to bind to ILF3 (whose function remains unclear), independent of dsRNA, but interacts with exportin-5 in a dsRNA- and Ran-GTP–dependent manner.40 Therefore, while the mechanism(s) by which dsRNA may be involved in these interactions is not clear, dsRNA may play different roles in JAZ's interaction with different cellular proteins. Moreover, full-length JAZ was reported to be necessary for efficient binding to ILF3.40 Our studies also find that full-length JAZ is required for both efficient binding to p53 in vitro and functional stimulation of p53 activity in vivo (Figures 4E and 5E).
We previously reported that JAZ's dsRNA-binding ZF domains are required for its nuclear localization.32 While mutational analysis reveals that the ZF domains are necessary for JAZ's stimulatory function of p53 transcriptional activity in vivo (Figure 5E), they are apparently not required for JAZ-p53 binding in vitro (Figure 3F). Therefore, the most plausible explanation is that the presence of the intact ZF domains serves to dictate JAZ's nuclear localization, which would be necessary for p53 association and activation in vivo but not for their direct interaction in vitro. It has also been reported that the C-terminal dsRNA-binding domain of RAX/PACT is not necessary for the direct interaction with its target kinase PKR, but plays an essential role in enzymatically activating PKR after binding.68 Thus, it is formally possible that in addition to their necessary role in nuclear localization, the JAZ's dsRNA-binding ZF domains may also play a direct role in activating/enhancing p53 transcriptional activity. This important mechanistic possibility remains to be tested.
Deletion analysis reveals that p53's C-terminal regulatory domain (C-RD) is necessary for JAZ binding and enhancement of p53 transcriptional activity (Figures 4C and 5E). Mechanistically, the C-RD of p53 has been reported to repress p53's DNA binding and transcriptional activity by maintaining p53 in a latent state.3 Of interest, the latent population of p53 in the MEFs has recently been reported to play an essential role in executing the stress-induced apoptosis program.69-71 Since JAZ does not appear to affect p53 expression at least in the cell lines tested here (Figure 6), we propose that JAZ may directly activate latent p53 by binding and inhibiting the negative regulatory function of the C-RD, resulting in p53 transcriptional activation with growth arrest and apoptosis. Further studies are now required to elucidate the mechanism.
In summary, findings here indicate that JAZ directly interacts with p53's C-terminus to stimulate its transcriptional activity and mediate G1 cell-cycle arrest and apoptosis. The results specifically indicate a regulatory role for JAZ in nongenotoxic stress response (ie, IL-3 growth factor or serum withdrawal) that mediates hematopoietic cell death. Therefore, we propose that the nuclear factor JAZ may be a novel regulator of p53 in the hematopoietic cell response to stress leading to apoptosis. Since only about 15% of hematologic malignancies express mutant (ie, transcriptionally inactive) p53,72 it may be possible to therapeutically target wild-type p53 through a mechanism involving JAZ.
Authorship
Contribution: M.Y. designed research, performed research, analyzed data, and wrote the paper; S.W. performed research and analyzed data; X.S. collected data; W.S.M. designed research and revised paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Supplementary Material
Acknowledgments
This work was supported by Public Health Service grant CA44649 from the National Cancer Institute (W.S.M.) and grants from the American Cancer Society (Florida Division) and Stop! Children's Cancer (M.Y.).
We would like to thank Amanda Shanks for providing technical assistance with the luciferase assay and Western blot analysis.
Prepublished online as Blood First Edition Paper, August 24, 2006; DOI 10.1182/blood-2006-06-029645.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
References
- 1.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408: 307-310. [DOI] [PubMed] [Google Scholar]
- 2.Michael D, Oren M. The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol. 2003; 13: 49-58. [DOI] [PubMed] [Google Scholar]
- 3.Ahn J, Prives C. The C-terminus of p53: the more you learn the less you know. Nat Struct Biol. 2001;8: 730-732. [DOI] [PubMed] [Google Scholar]
- 4.Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene. 2003;22: 9030-9040. [DOI] [PubMed] [Google Scholar]
- 5.Jimenez GS, Nister M, Stommel JM, Beeche M, et al. A transactivation-deficient mouse model provides insights into Trp53 regulation and function. Nat Genet. 2000;26: 37-43. [DOI] [PubMed] [Google Scholar]
- 6.Slee EA, O'Connor DJ, Lu X. To die or not to die: how does p53 decide? Oncogene. 2004;23: 2809-2818. [DOI] [PubMed] [Google Scholar]
- 7.Mihara M, Erster S, Zaika A, et al. p53 has a direct apoptotic role at the mitochondria. Mol Cell. 2003;11: 577-590. [DOI] [PubMed] [Google Scholar]
- 8.Chipuk JE, Kuwana T, Bouchier-Hayes L, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303: 1010-1014. [DOI] [PubMed] [Google Scholar]
- 9.Deng X, Cao F, Flagg T, Anderson J, May WS. Bcl2's flexible loop domain regulates p53 binding and survival. Mol Cell Biol. 2006;26: 4421-4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer. 2002;2: 594-604. [DOI] [PubMed] [Google Scholar]
- 11.Ritchie A, Broxmeyer HE. Suppression of p53-mediated growth factor withdrawal-induced apoptosis in the myeloid compartment by hematopoietic cytokines: an overview of hematopoiesis and apoptosis with a presentation of thrombopoietin and the M07E cell line as a model system. Crit Rev Oncol Hematol. 1999;31: 169-191. [DOI] [PubMed] [Google Scholar]
- 12.Yonish-Rouach E, Resnitzky D, Lotem J. et al. Wild-type p53 induces apoptosis of myeloid leukemic cells that is inhibited by interleukin-6. Nature. 1991;35: 345-347. [DOI] [PubMed] [Google Scholar]
- 13.Canman CE, Gilmer TM, Coutts SB, et al. Growth factor modulation of p53-mediated growth arrest versus apoptosis. Genes Dev. 1995;9: 600-611. [DOI] [PubMed] [Google Scholar]
- 14.Blandino G, Scardigli R, Rizzo MG, et al. Wild-type p53 modulates apoptosis of normal, IL-3 deprived, hematopoietic cells. Oncogene. 1995;10: 731-737. [PubMed] [Google Scholar]
- 15.Prisco M, Hongo A, Rizzo MG, et al. The insulin-like growth factor I receptor as a physiologically relevant target of p53 in apoptosis caused by interleukin-3 withdrawal. Mol Cell Biol. 1997;17: 1084-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gottlieb E, Oren M. p53 facilitates pRb cleavage in IL-3-deprived cells: novel pro-apoptotic activity of p53. EMBO J. 1998;17: 3587-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vousden KH. Activation of the p53 tumor suppressor protein. Biochim Biophys Acta. 2002;1602: 47-59. [DOI] [PubMed] [Google Scholar]
- 18.Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005; 24: 2899-2908. [DOI] [PubMed] [Google Scholar]
- 19.Sherr CJ. The INK4a/ARF network in tumor suppression. Nat Rev Mol Cell Biol. 2001;2: 731-737. [DOI] [PubMed] [Google Scholar]
- 20.Samuels-Lev Y, O'Connor DJ, Bergamaschi D, et al. ASPP proteins specifically stimulate the apoptotic function of p53. Mol Cell. 2001;8: 781-794. [DOI] [PubMed] [Google Scholar]
- 21.Hofmann TG, Moller A, Sirma H, et al. Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat Cell Biol. 2002;4: 1-10. [DOI] [PubMed] [Google Scholar]
- 22.Jayaraman L, Moorthy NC, Mutthy KG, Manley JL, Bustin M, Prives C. High mobility group protein-1 (HMG-1) is a unique activator of p53. Genes Dev. 1998;12: 462-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lohrum MA, Ludwig RL, Kubbutat MH, Hanlon M, Vousden KH. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell. 2003;3: 577-587. [DOI] [PubMed] [Google Scholar]
- 24.Dond GL, Hu W, Levine AJ. MDM2 is a central node in the p53 pathway: 12 years and counting. Curr Cancer Drug Targets. 2005;5: 3-8. [DOI] [PubMed] [Google Scholar]
- 25.Banerjee S, Kumar BRP, Kundu TK. General transcriptional coactivator PC4 activates p53 function. Mol Cell Biol. 2004;24: 2052-2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng H, You H, Zhou XZ, et al. The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature. 2002;419: 849-853. [DOI] [PubMed] [Google Scholar]
- 27.Fogal V, Gostissa M, Sandy P, et al. Regulation of p53 activity in nuclear bodies by a specific PML isoform. EMBO J. 2000;19: 6185-6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gaiddon C, Moorthy NC, Prives C. Ref-1 regulates the transactivation and pro-apoptotic functions of p53 in vivo. EMBO J. 1999;18: 5609-5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu H, Levine AJ. Human TAFII31 protein is a transcriptional coactivator of the p53 protein. Proc Natl Acad Sci U S A. 1995;92: 5154-5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sui G, Affar el B, Shi Y, et al. Yin Yang 1 is a negative regulator of p53. Cell. 2004;117: 859-872. [DOI] [PubMed] [Google Scholar]
- 31.Bai L, Merchant JL. ZBP-89 promotes growth arrest through stabilization of p53. Mol Cell Biol. 2001;21: 4670-4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang M, May WS, Ito T. JAZ requires the double-stranded RNA-binding zinc finger motifs for nuclear localization. J Biol Chem. 1999;274: 27399-27406. [DOI] [PubMed] [Google Scholar]
- 33.Klug A, Rhodes D. `Zinc fingers': a novel protein motif for nucleic acid recognition. Trends Biochem Sci. 1987;12: 464-469. [Google Scholar]
- 34.St Johnston D, Brown NH, Gall JG, Jantsch M. A conserved double-stranded RNA-binding domain. Proc Natl Acad Sci U S A. 1992;89: 10979-10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Draper DE. Protein-RNA recognition. Annu Rev Biochem. 1995;64: 593-620. [DOI] [PubMed] [Google Scholar]
- 36.Ito T, Jagus R, May WS. Interleukin 3 stimulates protein synthesis by regulating double-stranded RNA-dependent protein kinase. Proc Natl Acad Sci U S A. 1994;91: 7455-7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nicholson AW. Structure, reactivity, and biology of double-stranded RNA. Prog Nucleic Acid Res Mol Biol. 1996;52: 1-65. [DOI] [PubMed] [Google Scholar]
- 38.Ito T, Yang M, May WS. RAX, a cellular activator for double-stranded RNA-dependent protein kinase during stress signaling. J Biol Chem. 1999; 274: 15427-15432. [DOI] [PubMed] [Google Scholar]
- 39.Patel RC, Sen GC. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 1998;17: 4379-4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen T, Brownawell AM, Macara IG. Nucleocytoplasmic shuttling of JAZ, a new cargo protein for exportin-5. Mol Cell Biol. 2004;24: 6608-6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Israeli D, Tessler E, Haupt Y, et al. A novel p53-inducible gene, PAG608, encodes a nuclear zinc finger protein whose overexpression promotes apoptosis. EMBO J. 1997;16: 4384-4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Varneh-Ziaie S, Okan I, Wang Y, et al. Wig-1, a new p53-induced gene encoding a zinc finger protein. Oncogene. 1997;15: 2699-2704. [DOI] [PubMed] [Google Scholar]
- 43.Mendez-Vidal C, Wilhelm MT, Hellborg F, Qian W, Wiman KG. The p53-induced mouse zinc finger protein wig-1 binds double-stranded RNA with high affinity. Nucleic Acids Res. 2002;30: 1991-1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Finerty PJ, Bass BL. A xenopus zinc finger protein that specifically binds dsRNA and RNA-DNA hybrids. J Mol Biol. 1997;271: 195-208. [DOI] [PubMed] [Google Scholar]
- 45.Möller HM, Martinez-Yanout MA, Dyson HJ, Wright PE. Solution structure of the N-terminal zinc fingers of the Xenopus laevis double-stranded RNA-binding protein Zfa. J Mol Biol. 2005;351: 718-730. [DOI] [PubMed] [Google Scholar]
- 46.Higashi Y, Asanuma M, Miyazaki I, et al. The p53-activated gene, PAG608, requires a zinc finger domain for nuclear localization and oxidative stress-induced apoptosis. J Biol Chem. 2002; 277: 42224-42232. [DOI] [PubMed] [Google Scholar]
- 47.Hellborg F, Wang Q, Mendez-Vidal C, et al. Human wig-1, a p53 target gene that encodes a growth inhibitory zinc finger protein. Oncogene. 2001;20: 5466-5474. [DOI] [PubMed] [Google Scholar]
- 48.Yuan XM, Li W, Dalen H, et al. Lysosomal destabilization in p53-induced apoptosis. Proc Natl Acad Sci U S A. 2002;99: 6286-6291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cao X, Deng X, May WS. Cleavage of Bax to p18 Bax accelerates stress-induced apoptosis, and a cathepsin-like protease may rapidly degrade p18 Bax. Blood. 2003;102: 2605-2614. [DOI] [PubMed] [Google Scholar]
- 50.Schmid I, Sakamoto KM. Analysis of DNA content and green fluorescent protein expression. In: Robinson JP, Darzynkiewicz Z, Dean P, Orfao A, Rabinovitch P, Stewart C, Tanke H, Wheeless L, eds. Current Protocols in Cytometry. New York, NY: John Wiley & Sons; 2001: 7.16.1-7.16.10. [DOI] [PubMed]
- 51.el-Deiry WS, Tokino T, Velculescu VE, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75: 817-825. [DOI] [PubMed] [Google Scholar]
- 52.Pierani A, Pouponnot C, Calothy G. Transcriptional downregulation of the retina-specific QR1 gene pp60v-src and identification of a novel vsrc-responsible unit. Mol Cell Biol. 2004;13: 3401-3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bennett RL, Blalock WL, Abtahi DM, Pan Y, Moyer SA, May WS. RAX, the PKR activator, sensitizes cells to inflammatory cytokines, serum withdrawal, chemotherapy and viral infection. Blood. 2006;108: 821-829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.May WS. Control of apoptosis by cytokines. Adv Pharmacol. 1997;41: 219-246. [DOI] [PubMed] [Google Scholar]
- 55.Johnson DE. Regulation of survival pathways by IL-3 and induction of apoptosis following IL-3 withdrawal. Front Biosci. 1998;3: d313-d324. [DOI] [PubMed] [Google Scholar]
- 56.Ali SH, DeCaprio JA. Cellular transformation by SV40 large T antigen: interaction with host proteins. Semin Cancer Biol. 2001;11: 15-22. [DOI] [PubMed] [Google Scholar]
- 57.Hupp TR, Meek DW, Midgley CA, Lane DP. Regulation of the specific DNA binding function of p53. Cell. 1992;71: 875-886. [DOI] [PubMed] [Google Scholar]
- 58.Selivanova G, Iotsova V, Okan I, et al. Restoration of the growth suppression function of mutant p53 by a synthetic peptide derived from the p53 C-terminal domain. Nat Med. 1997;3: 632-638. [DOI] [PubMed] [Google Scholar]
- 59.Sherr CJ. The Pezcoller lecture: cancer cell cycles revisited. Cancer Res. 2000;60: 3689-3695. [PubMed] [Google Scholar]
- 60.Nevins JR. The Rb/E2F pathway and cancer. Hum Mol Genet. 2001;10: 699-703. [DOI] [PubMed] [Google Scholar]
- 61.DeCaprio JA, Ludlow JW, Lynch D, et al. The product of the retinoblastoma susceptibility gene has properties of a cell cycle regulatory element. Cell. 1989;58: 1085-1095. [DOI] [PubMed] [Google Scholar]
- 62.Kurokawa K, Tanaka T, Kato J. p19ARF prevents G1 cyclin-dependent kinase activation by interacting with MDM2 and activating p53 in mouse fibroblasts. Oncogene. 1999;18: 2718-2727. [DOI] [PubMed] [Google Scholar]
- 63.Modeston M, Puig-Antich V, Korgaonkar C, Eapen A, Quelle DE. The alternative reading frame tumor suppressor inhibits growth through p21-dependent and p21-independent pathways. Cancer Res. 2001;61: 3145-3150. [PubMed] [Google Scholar]
- 64.Wei MC, Zong WX, Cheng EH, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292: 727-730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53. Cell. 1998;92: 725-734. [DOI] [PubMed] [Google Scholar]
- 66.Pomerantz J, Schreiber-Agus N, Liégeois NJ, et al. The Ink4a tumor suppressor gene product, p19ARF, interacts with MDM2 and neutralizes MDM2's inhibition of p53. Cell. 1998;92: 713-723. [DOI] [PubMed] [Google Scholar]
- 67.Bhat KP, Itahana K, Jin A, Zhang Y. Essential role of ribosomal protein L11 in mediating growth inhibition-induced p53 activation. EMBO J. 2004;23: 2402-2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Peters GA, Hartmann R, Qin J, Sen GC. Modular structure of PACT: distinct domains for binding and activating PKR. Mol Cell Biol. 2001;21: 1908-1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Woo RA, Jack MT, Xu Y, Burma S, Chen DJ, Lee PW. DNA-damage-induced apoptosis requires the DNA-dependent protein kinase, and is mediated by the latent population of p53. EMBO J. 2002;21: 3000-3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jack MT, Woo RA, Hirao A, Cheung A, Mak TW, Lee PW. Chk2 is dispensable for p53-mediated G1 arrest but is required for a latent p53-mediated apoptotic response. Proc Natl Acad Sci U S A. 2002;99: 9825-9829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jack MT, Woo RA, Motoyama N, Takai H, Lee PW. DNA-dependent protein kinase and checkpoint kinase 2 synergistically activate a latent population of p53 upon DNA damage. J Biol Chem. 2004;279: 15269-15273. [DOI] [PubMed] [Google Scholar]
- 72.Stühmer T, Chatterjee M, Hildebrandt M, et al. Nongentoxic activation of the p53 pathway as a therapeutic strategy for multiple myeloma. Blood. 2006;106: 3610-3617. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}