

Abstract
Hepcidin, the principal iron regulatory hormone, regulates the absorption of iron from the diet and the mobilization of iron from stores. Previous studies indicated that hepcidin is suppressed during anemia, a response that would appropriately increase the absorption of iron and its release from stores. Indeed, in the mouse model, hepcidin-1 was suppressed after phlebotomy or erythropoietin administration but the suppression was reversed by inhibitors of erythropoiesis. The suppression of hepcidin necessary to match iron supply to erythropoietic demand thus requires increased erythropoiesis and is not directly mediated by anemia, tissue hypoxia, or erythropoietin.
Introduction
It has long been thought that intestinal iron absorption and the mobilization of iron from stores is controlled by the combined action of 2 regulators: the stores regulator and the erythroid regulator.1 The stores regulator controls intestinal iron absorption and is responsible for meeting the body's normal iron requirement and for accumulating and controlling iron stores. The erythroid regulator maintains the production of erythrocytes irrespective of the body's iron balance. In persistent anemia due to blood loss, this process increases iron absorption and depletes iron stores. In anemias with ineffective erythropoiesis, the erythroid regulator also increases iron absorption but, in the absence of iron losses, the accumulation of iron eventually results in iron overload. It has been demonstrated that the erythroid regulator can facilitate the absorption of iron up to 40 mg/d in severe anemia with oral iron supplementation compared with the stores regulator, which seems to only permit absorption of up to 2 mg iron/d.1
More recently, the molecular basis of systemic iron regulation began to be understood. Hepcidin, the principal iron regulatory hormone, blocks the intestinal absorption of iron and the release of iron from stores by inducing the internalization and degradation of the cellular iron exporter ferroportin.2 Ferroportin is the sole known cellular iron exporter and is found on the basolateral surface of duodenal enterocytes as well as on macrophages and hepatocytes.3 Hepcidin is regulated both by iron and anemia and has been proposed as the final mediator of both the stores and erythroid regulators.
The amount of iron in red blood cells is so large that increases in erythropoiesis require a considerable increase in the flow of iron from the diet or storage pools. For example, in patients with postoperative anemia, hemoglobin (Hgb) reportedly increases from 106 to 130 g/L (10.6-13.0 g/dL) during postoperative days 7 to 21.4 Given an average blood volume of 5 L, this increment obligates 420 mg iron, or 30 mg/d. Given that baseline iron absorption is only 1 mg/d, iron absorption and release from stores must increase dramatically to compensate for this massive increase in iron consumption by erythropoietic precursors. Similarly, iron consumption for erythropoiesis increases greatly after stimulation with erythropoietin (EPO) or in response to hypoxemia. EPO treatment at doses of 100 U/kg 3 times a week leads to an average increase in Hgb of 1.8 g/L/d (0.18 g/dL/d)5 again requiring more than 30 mg of additional iron per day. Athletes training for 3 weeks at an elevation of 2050 meters increased their Hgb concentrations by 12 g/L (1.2 g/dL), which would consume 10 mg of additional iron per day.
After phlebotomy, EPO administration, or hemolysis, hepcidin production is decreased,6-8 which in turn allows increased iron absorption and release of iron from stores. Therefore, the erythroid regulator exerts its activity, at least in part, by suppressing hepcidin. As shown in Figure 1, anemia induces a cascade of changes that individually or in combination suppress hepcidin expression. Anemia could directly regulate hepcidin, or when sufficiently severe, could lead to tissue hypoxia. Tissue hypoxia is an appealing mechanism for the regulation of hepcidin because hypoxia has been shown to regulate hepcidin at the cellular and organismic levels6 and a similar mechanism regulates the production of EPO.9 Increased erythropoiesis could directly influence hepcidin production through the concomitant generation of soluble mediators. For example, soluble transferrin receptor-1 increases during erythropoiesis10,11 and correlates inversely with urinary hepcidin excretion in patients with thalassemic syndromes.12 However, when the soluble transferrin receptor was transiently overexpressed in the mouse, there was no change in hepcidin expression.13 Nevertheless, other erythropoiesis-related signals could regulate hepcidin.
Figure 1.
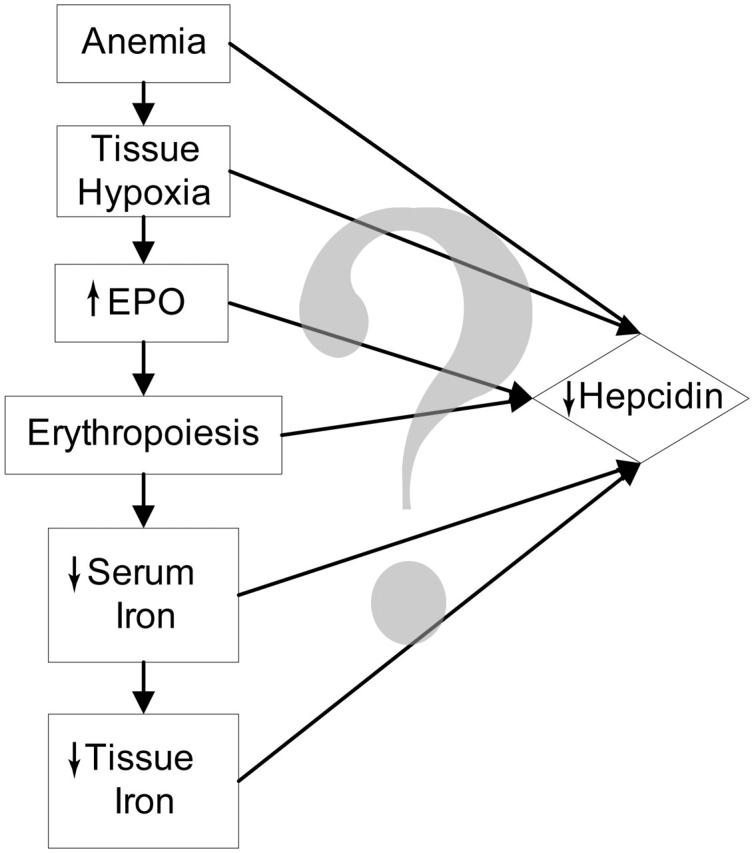
Possible regulators of hepcidin during increased erythropoiesis.
As erythropoiesis increases, substantial quantities of iron will be used to make Hgb for the new erythrocytes. This will cause a fall in serum iron level. Iron is known to regulate hepcidin,14-18 but it is not certain whether the iron signal is plasma iron or intracellular iron (iron stores) or both. Because serum iron is consumed for erythropoiesis, tissue iron content must also fall and this too could be a signal for the down-regulation of hepcidin.
To determine how anemia regulates hepcidin, we administered erythropoiesis inhibitors after phlebotomy to dissociate the effects of anemia, hypoxia, and EPO from the effects of increased erythropoiesis and iron use. Phlebotomized mice develop anemia, tissue hypoxia, increased EPO, increased erythropoiesis, and a fall in serum iron levels, but inhibitors of erythropoiesis should prevent or blunt the erythropoietic response and the consequent fall in serum iron.
Materials and methods
Animals
Equal numbers of male and female 8-week-old C57Bl/6 mice were obtained from Charles River Laboratories (Wilmington, MA) or bred in our facility. Mice were maintained on one of 2 normal iron diets: NIH 31 rodent diet (333 ppm iron, Harlan Teklad, Indianapolis, IN) or Prolab RMH breeder diet (440 ppm iron, PMI International, Brentwood, MO). Mice were switched to a diet containing less than 4 ppm iron (Harlan Teklad) for 10 days prior to all experiments to decrease the stimulation of hepcidin synthesis by the iron-rich “normal” diet. We had previously observed that this duration of iron deprivation decreases endogenous hepcidin without significant hypoferremia. All studies were approved by the Animal Research Committee at UCLA.
Inhibitors of erythropoiesis
Carboplatin or doxorubicin (Sigma-Aldrich, St Louis, MO) dissolved in 0.25 mL water was administered intraperitoneally 2 days before phlebotomy or 1 day prior to EPO administration at doses of 2.5 mg/mouse and 0.25 mg/mouse, respectively. Goat anti–mouse EPO antibody (α-EPO; R&D Systems, Minneapolis, MN) was given intraperitoneally at a dose of 80 μg/mouse on the day prior to the first phlebotomy and prior to the second phlebotomy. Control mice received 0.25 mL bacteriostatic 0.9% sodium chloride (saline, NS; Hospira, Lake Forest, IL).
Phlebotomy
Mice were anesthetized prior to phlebotomy with isoflurane (Abbott Laboratories, North Chicago, IL) and blood withdrawn from the retroorbital sinus with a heparinized capillary tube (Fisher, Hampton, NH). The goal of phlebotomy was to remove approximately 400 μL blood on each of 2 successive days. The quantity of blood withdrawn was estimated by weighing the mice before and after phlebotomy and dividing the change in weight by 1.06 (density of blood19).
EPO administration
Mice received 200 U human biosynthetic EPO-α (epoetin alfa, Ortho Biotech, Bridgewater, NJ) dissolved in 100 μL water by intraperitoneal injection on 3 successive days. Mice were killed 24 hours after the last injection.
Specimen collection
Mice were anesthetized with isoflurane and approximately 50 μL blood was collected by retro-orbital sinus puncture into a tube containing 1 μL of 1000 U/mL heparin (Baxter Healthcare, Deerfield, IL) for Hgb and reticulocyte determination. Mice were immediately killed with isoflurane and blood was collected into Microtainer serum separator tubes (Becton Dickinson, Franklin Lakes, NJ) by cardiac puncture. The abdomen was then opened and a sample of liver obtained for RNA isolation.
Blood analysis
Hgb was determined by a colorimetric assay using the cyanmethemoglobin technique according to the manufacturer's instructions (Pointe Scientific, Canton, MI). Reticulocyte counts were determined by staining blood with thiazole orange (Retic-COUNT, BD Biosciences, San Jose, CA) and performing fluorescence-activated cell sorting before and after staining according to the manufacturer's instructions. Blood was collected in serum separator tubes and allowed to clot for at least 30 minutes and then spun in a microcentrifuge at 13 000g for 2 minutes and the serum layer removed with a pipette. Serum iron content and total iron-binding capacity (TIBC) were then determined using a commercial colorimetric assay (Diagnostic Chemicals, Oxford, CT), which we modified to use 200 μL serum (50 μL for both total iron and TIBC, both run in duplicate). Serum EPO was determined by enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's instructions (R&D Systems).
Measurement of mRNA concentrations
A small piece (∼50 mg) of liver was homogenized in 0.5 mL TRIzol reagent (Invitrogen, Calrsbad, CA) and an additional 0.7 mL TRIzol added after homogenization. RNA was then isolated according to the manufacturer's instructions. Reverse transcription of mRNA was performed using the iScript cDNA synthesis kit as recommended by the manufacturer (Bio-Rad, Hercules, CA) to make cDNA. Quantitative real-time polymerase chain reaction (qRT-PCR) was performed in an iCycler thermal cycler (Bio-Rad) with an iQ5 qRT-PCR detection system attached (Bio-Rad) using iQ SYBR green supermix (Bio-Rad) as directed by the manufacturer. Primer sequences starting from the 5′ are: Hepc1 Fwd-CCTATCTCCATCAACAGATG, Rev-AACAGATACCACACTGGGAA; β-actin Fwd-ACCCACACTGTGCCCATCTA, Rev-CACGCTCGGTCAGGATCTTC; serum amyloid A-1 (SAA-1): Fwd-TGACCAGGAAGCCAACAG, Rev-GTAGGAAGACCAGACC; fibrinogen-γ (FGN-γ): Fwd-TGGCTGGGAAATGAGAAG, Rev-GAAGTAGGCATAGGTCAGG; and vascular endothelial growth factor (VEGF): Fwd-AGGCTGCTGTAACGATGAAG, Rev-TCTCTATGTGCTGGCTTTGG.
Tissue iron determination
Mice were exsanguinated prior to iron determination. Nonheme tissue iron concentration was determined using the method described by Rebouche and colleagues.20 A 200-mg or smaller piece of liver was homogenized in 4 mL iron-free water. An aliquot of this solution was mixed 1:1 with a solution containing 1 N HCl and 10% trichloroacetic acid. The resulting suspension was centrifuged and the supernatant analyzed for iron content using the chromogen reagent.20
Statistics
All statistics were calculated using SigmaStat (SyStat Software, Richmond, CA). Data are reported as mean ± 1 SD for parametric data and median (interquartile range) for nonparametric data. Parametric data were compared using the Student t test. Nonparametric data were compared using the Wilcoxon rank sum test. Correlation coefficients were determined using the Pearson correlation or Spearman correlation for parametric and nonparametric data, respectively.
All mRNA concentrations determined by qRT-PCR are reported as the relative expression of mRNA calculated using the method described by Pfaffl.21 During every qRT-PCR, a dilution series consisting of four 5-fold serial dilutions is used to determine the PCR efficiency of the target gene and a stable reference gene (housekeeping gene, β-actin). The relative concentration (expression) of mRNA is determined relative to the same sample in every run and normalized to the expression of the reference gene.
Results
During anemia, hepcidin mRNA expression could be regulated by anemia itself, tissue hypoxia, or EPO, as well as a change in erythropoiesis or iron use. We therefore used inhibitors of erythropoiesis to determine the contribution of erythropoiesis and its metabolic consequences to hepcidin regulation. Erythropoiesis was inhibited with either carboplatin or doxorubicin, agents known to be toxic to developing erythrocytes,22-24 or with the noncytotoxic EPO-blocking antibody (α-EPO).
Mice were pretreated with one of the inhibitors of erythropoiesis or saline (phlebotomy control) and phlebotomy was performed on 2 subsequent days. An average of 0.40 ± 0.11 mL blood was withdrawn at each phlebotomy. As seen in Figure 2A, there was no difference in the Hgb levels achieved after phlebotomy regardless of pretreatment with inhibitors of erythropoiesis. Carboplatin and doxorubicin markedly suppressed reticulocytosis (Figure 2B) despite the potent erythropoietic stimulus of phlebotomy. Although reticulocyte counts increased in phlebotomized mice pretreated with α-EPO, they were still markedly reduced compared with phlebotomized controls (Figure 2B).
Figure 2.

Hepcidin is regulated by erythropoietic activity, not anemia. Prior to phlebotomy (Phleb) mice were pretreated with NS (Phleb Alone), carboplatin (Cp + Phleb), doxorubicin (Dox + Phleb), or anti-EPO. Control mice (No Phleb) were pretreated with NS but did not undergo phlebotomy. (A) Hemoglobin concentration. (B) Reticulocyte fraction measured by flow cytometry. (C) Serum iron. (D) Hepcidin mRNA is the hepatic relative concentration normalized to the amount of β-actin relative to no phlebotomy samples. Means and SDs are shown, with superscripts indicating P < .001 compared to no phlebotomy by t test* or rank sum test&; #P < .001 compared to phlebotomy alone by rank sum test, %P < .01 by rank sum test compared to no phlebotomy.
Serum iron fell in response to phlebotomy (Figure 2C), and, as previously shown by Nicolas and colleagues,6 hepatic hepcidin mRNA decreased in response to phlebotomy (Figure 2D). Thus, any increased flux of iron into the plasma driven by a fall in hepcidin was outweighed by increased consumption of iron by the developing erythrocytes in phlebotomy-induced anemia. Serum iron rose in mice given inhibitors of erythropoiesis even with phlebotomy (Figure 2C). This is consistent with continued release of recycled iron from senescent erythrocytes in the face of decreased or absent incorporation of iron into new erythrocytes. The effect of erythropoietic activity on serum iron was further supported by the strong inverse correlation in all the mice of serum iron with reticulocytosis (Pearson R =–0.546, P < .001).
In the presence of inhibitors of erythropoiesis, hepcidin mRNA expression increased significantly, despite severe anemia (Figure 2D). Hepcidin mRNA concentration was proportional to serum iron (Spearman R = 0.530, P < .001) and inversely proportional to the reticulocyte fraction (Spearman R =–0.542, P < .001) but was not dependent on Hgb (Spearman R = 0.114, P = .44). Taken together, these data indicate that anemia does not directly regulate hepcidin expression but affects hepcidin indirectly, through an as yet uncharacterized signal from the expanding erythron and through increased iron consumption and the resulting fall in plasma iron. Mice treated with inhibitors of erythropoiesis but not phlebotomized had the highest hepcidin mRNA expression (data not shown), indicating that even basal erythropoiesis regulates hepcidin expression.
Unless sufficient iron is available in the diet, tissue stores of iron should change in parallel with serum iron during an increase in the rate of erythropoiesis as iron flows out of the storage pools to maintain iron homeostasis. Conversely, when erythropoiesis is blocked, excess iron from recycled erythrocytes should accumulate in the tissue stores. Although it has been shown that changes in total body iron load lead to changes in hepcidin,14-18 it is not known whether hepcidin is responding to changes in serum or tissue iron or both. Liver nonheme iron content did not decrease in phlebotomized mice compared to controls (0.47 ± 0.08 versus 0.49 ± 0.06 μmol/g wet tissue, respectively; P = .5 by t test). This may be because it was too early in the erythropoietic response to detect an appreciable fall in tissue iron. Tissue iron was significantly higher in phlebotomized mice that were pretreated with carboplatin (0.68 ± 0.07 μmol/g wet tissue) compared with nonphlebotomized mice (P < .001 by t test) or phlebotomized mice (P < .001 by t test).
To determine whether the increase in tissue iron in response to carboplatin could be completely accounted for by the decreased erythropoiesis, we compared mice treated with carboplatin with saline-treated mice in the absence of the confounding influences associated with phlebotomy. The average Hgb concentration was 139 ± 23 g/L (13.9 ± 2.3 g/dL; there was no significant difference between carboplatin- and saline-treated mice). Given an average blood volume of 2.3 mL,25 this would mean that there is 20 000 nmol iron contained in red blood cells. Assuming that carboplatin completely inhibited erythropoiesis but had no effect on the recycling of iron and that the life span of the mouse red blood cell is 54 days,26 approximately 1500 nmol iron would be released from red blood cells over the 4 days after carboplatin treatment. To determine the average amount of iron in the livers of our mice, we measured nonheme iron content of the livers (470 ± 80 nmol/g wet tissue) and multiplied the result by the average weight of the liver in a 25-g mouse (1.2 g27), yielding 564 nmol iron. After carboplatin treatment, liver nonheme iron rose to 814 ± 119 nmol/g wet tissue, a change of 413 nmol iron. Therefore, the rise in liver iron seen in response to carboplatin can be completely explained by decreased iron use for Hgb synthesis. It is notable, however, that a large part of iron released from senescent erythrocytes is not accounted for by increased liver iron content. This suggests that in the presence of high hepcidin, iron may be stored in macrophages in the spleen and elsewhere.
Hepcidin is strongly up-regulated by inflammation6,28-31 and during liver regeneration.32 Therefore we explored the possibility that inhibitors of erythropoiesis and their cytotoxic or acute immune effects present an inflammatory stimulus that increases hepcidin expression, unrelated to the severity of anemia. Hepatic production of SAA-1 and FGN-γ is a sensitive indicator of inflammation.33-35 SAA-1 expression increased in response to phlebotomy, but there was no further increase in response to the inhibitors of erythropoiesis (Figure 3A). FGN-γ expression did not change in response to phlebotomy or the inhibitors of erythropoiesis. Therefore, the increase in hepcidin expression in response to the inhibitors of erythropoiesis is not caused by inflammatory stimulation.
Figure 3.

Inhibitors of erythropoiesis do not act by inducing a hepatic acute-phase response or interfering with hypoxic sensing or EPO production. Prior to phlebotomy (Phleb), mice were pretreated with NS (Phleb Alone), carboplatin (Cp + Phleb), doxorubicin (Dox + Phleb), or anti-EPO. Control mice (No Phleb) were pretreated with NS but did not undergo phlebotomy. Messenger RNAs for SAA-1 (A, ▪), FGN-γ (A,  ), and VEGF (B) were measured by real-time qRT-PCR relative to actin mRNA concentrations, and expressed as a ratio to no phlebotomy samples. Serum EPO concentration (C) was measured by enzyme-linked immunosorbent assay. Means and SDs are shown with superscripts indicating *P = .001, #P < .05, &P < .001 by rank sum test compared to no phlebotomy, and %P = .003 by rank sum test compared to phlebotomy alone.
), and VEGF (B) were measured by real-time qRT-PCR relative to actin mRNA concentrations, and expressed as a ratio to no phlebotomy samples. Serum EPO concentration (C) was measured by enzyme-linked immunosorbent assay. Means and SDs are shown with superscripts indicating *P = .001, #P < .05, &P < .001 by rank sum test compared to no phlebotomy, and %P = .003 by rank sum test compared to phlebotomy alone.
Tissue hypoxia has been shown to suppress hepcidin expression directly.6 Because anemia could cause tissue hypoxemia and thereby suppress hepcidin, we sought to determine whether the degree of anemia we induced by phlebotomy led to liver hypoxia. VEGF mRNA expression is up-regulated during tissue hypoxia.36 Therefore, we used hepatic expression of VEGF mRNA as a marker of hepatic hypoxia. VEGF expression was increased in the phlebotomized mice (Figure 3B) indicating that tissue hypoxia could be one signal by which anemia suppresses hepcidin expression. However, VEGF expression was maintained or slightly increased (with doxorubicin) when mice were pretreated with the inhibitors of erythropoiesis. Therefore, the increase in hepcidin expression in response to the inhibitors of erythropoiesis could not be due to interference of the cytotoxic agents with the sensing of tissue hypoxia.
Nicolas and colleagues reported hepcidin suppression in mice treated with EPO.7 If EPO is an important direct or indirect regulator of hepcidin expression, blocking the effects of EPO would increase hepcidin expression. Indeed, when mice were treated with EPO antibody, hepcidin expression increased even in the face of severe anemia (Figure 1D). To determine whether cytotoxic agents caused a decrease in blood EPO concentrations leading directly to increased hepcidin, we measured serum EPO levels in mice treated with carboplatin (Figure 3C). Carboplatin did not reduce EPO levels, indicating that hepcidin is not regulated directly by EPO but rather by a signal downstream from EPO.
To explore the effects of EPO on hepcidin regulation further, mice were treated with high-dose EPO for 3 days (Figure 4). High-dose EPO elicited vigorous reticulocytosis that was inhibited by pretreatment with carboplatin (Figure 4A). The duration of EPO treatment was insufficient to cause an increase in the Hgb or hematocrit but mice pretreated with carboplatin had slightly lower values than untreated mice (Hgb = 124 ± 16 [1.24 ± 1.6], 111 ± 11 [11.1 ± 1.1], and 107 ± 9 [10.7 ± 0.9] for untreated, EPO treated, and carboplatin followed by EPO, respectively; P = .039 by 1-way ANOVA). Serum iron fell in response to EPO but this fall was completely blocked when mice were pretreated with carboplatin (Figure 4B). This is similar to what was seen in the phlebotomized mice; increased erythropoiesis causes a fall in serum iron, whereas inhibition of erythropoiesis leads to a rise in serum iron. As previously described,7 hepcidin expression decreased in response to exogenous EPO (Figure 4C). However, this effect was blocked by pretreatment with carboplatin.
Figure 4.

EPO suppresses hepcidin indirectly by inducing erythropoiesis. Reticulocyte fractions (A), serum iron (B), and hepcidin mRNA (C) were determined after each indicated treatment and compared by rank sum test to “no EPO” (*P < .005, #P = .01, %P < .05) or compared by rank sum test to “EPO” (&P ≤ .002). Hepcidin mRNA was normalized to actin mRNA and ratios to “no EPO” are shown. Error bars indicate one standard deviation.
Discussion
These experiments show that that the dominant regulators of hepcidin during increased erythropoiesis include a signal arising from the erythropoietic activity in the bone marrow or the effects of increased iron use on plasma or tissue iron. Hepcidin is known to be regulated by changes in iron status.14,29 With increasing erythropoiesis, there was a concomitant fall in serum and tissue iron. Conversely, when erythropoiesis was inhibited, serum and tissue iron rose dramatically. Therefore, the decrease in serum or tissue iron could contribute to the regulation of hepcidin during increased erythropoiesis.
Patients with β-thalassemia provide important insights into the relative effects of erythropoiesis and iron status on hepcidin levels. Patients with moderate or severe β-thalassemia, regardless of whether they are transfused, suffer from iron overload37,38 characterized by increased serum iron, increased tissue iron, and high ferritin levels. Even with chelation therapy, patients with β-thalassemia intermedia or major commonly have greatly increased ferritin levels.38 These patients also have dramatically increased erythropoiesis, including extramedullary sites.37 If iron were a dominant regulator of hepcidin, patients with β-thalassemia would be expected to have high hepcidin levels. To the contrary, patients with β-thalassemia intermedia have almost uniformly low urinary hepcidin but that hepcidin is higher in patients with thalassemia major who are given transfusions.38 Transfusions partially relieve anemia and decrease erythropoietic stimulation but also further increase the iron load. These and other clinical observations in iron-loading anemias would argue that erythropoiesis is able to suppress hepcidin production even in the face of severe iron overload. Phenylhydrazine-induced hemolytic anemia in mice also causes increased erythropoiesis with increases in tissue and serum iron.6,39 When Nicolas and colleagues treated mice with phenylhydrazine, they also observed a decrease in hepcidin expression,6 supporting the notion of a dominant erythropoietic regulator of hepcidin.
During the preparation of this manuscript, Vokurka and colleagues published data that are complementary to ours.8 When they irradiated mice to inhibit erythropoiesis, they saw a dramatic increase in hepcidin expression. However, they did not report on the effectiveness of inhibition of erythropoiesis or the extent to which radiation-induced cytokine release may have contributed to hepcidin stimulation. The increase in hepcidin expression during radiation-induced erythropoietic block persisted despite the administration of phenylhydrazine to induce hemolytic anemia or EPO to directly stimulate erythropoiesis.
Thus, anemia exerts its effects on hepcidin and iron metabolism predominantly through an as yet uncharacterized substance released during erythropoiesis. Secondary changes in plasma and tissue iron would also be expected to contribute to hepcidin regulation. The specific mediators and pathways by which these influences regulate hepcidin synthesis and release remain to be elucidated.
Authorship
M.P. and S.R. designed and performed the experiments, analyzed the data, and wrote the paper; M.A.L. and V.G. performed experiments; and T.G. helped analyze the data and write the paper.
The authors declare no competing financial interests.
Acknowledgments
This work was supported by National Institutes of Health grants K08 DK 07284-01 (S.R.) and RO1 DK 065029 (T.G.) and by the Will Rogers Fund (T.G.).
Prepublished online as Blood First Edition Paper, August 1, 2006; DOI 10.1182/blood-2006-06-028787.
An Inside Blood analysis of this article appears at the front of this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
References
- 1.Finch C. Regulators of iron balance in humans. Blood. 1994;84: 1697-1702. [PubMed] [Google Scholar]
- 2.Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306: 2090-2093. [DOI] [PubMed] [Google Scholar]
- 3.Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem. 2000;275: 19906-19912. [DOI] [PubMed] [Google Scholar]
- 4.Wallis JP, Wells AW, Whitehead S, Brewster N. Recovery from post-operative anaemia. Transfus Med. 2005;15: 413-418. [DOI] [PubMed] [Google Scholar]
- 5.Mosby's Drug Consult. Stat!Ref Online Electronic Medical Library (15). http://www.mosbydrugs.com. Accessed June 10, 2005.
- 6.Nicolas G, Chauvet C, Viatte L, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest. 2002;110: 1037-1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nicolas G, Viatte L, Bennoun M, et al. Hepcidin, a new iron regulatory peptide. Blood Cells Mol Dis. 2002;29: 327-335. [DOI] [PubMed] [Google Scholar]
- 8.Vokurka M, Krijt J, Sulc K, Necas E. Hepcidin mRNA levels in mouse liver respond to inhibition of erythropoiesis. Physiol Res. 2006; [Epub ahead of print.] [DOI] [PubMed]
- 9.Eckardt KU, Kurtz A. Regulation of erythropoietin production. Eur J Clin Invest. 2005;35(suppl 3): 13-19. [DOI] [PubMed] [Google Scholar]
- 10.Beguin Y. Soluble transferrin receptor for the evaluation of erythropoiesis and iron status. Clin Chim Acta. 2003;329: 9-22. [DOI] [PubMed] [Google Scholar]
- 11.Kohgo Y, Niitsu Y, Kondo H, et al. Serum transferrin receptor as a new index of erythropoiesis. Blood. 1987;70: 1955-1958. [PubMed] [Google Scholar]
- 12.Nemeth E, Origa R, Ganz T, Galanello R. Urinary hepcidin in thalassemic syndromes [abstract]. Blood. 2005;106. Abstract 3589.
- 13.Flanagan JM, Peng H, Lee PL, Beutler E. Analysis of the effect of soluble transferrin receptor 1 (sTfR1) on iron metabolism in mice [abstract]. 47th American Society of Hematology Annual Meeting and Exposition. 2005.
- 14.Pigeon C, Ilyin G, Courselaud B, et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem. 2001;276: 7811-7819. [DOI] [PubMed] [Google Scholar]
- 15.Anderson GJ, Frazer DM, Wilkins SJ, et al. Relationship between intestinal iron-transporter expression, hepatic hepcidin levels and the control of iron absorption. Biochem Soc Trans. 2002;30: 724-726. [DOI] [PubMed] [Google Scholar]
- 16.Frazer DM, Wilkins SJ, Becker EM, et al. Hepcidin expression inversely correlates with the expression of duodenal iron transporters and iron absorption in rats. Gastroenterology. 2002;123: 835-844. [DOI] [PubMed] [Google Scholar]
- 17.Ilyin G, Courselaud B, Troadec MB, et al. Comparative analysis of mouse hepcidin 1 and 2 genes: evidence for different patterns of expression and co-inducibility during iron overload. FEBS Lett. 2003;542: 22-26. [DOI] [PubMed] [Google Scholar]
- 18.Mazur A, Feillet-Coudray C, Romier B, et al. Dietary iron regulates hepatic hepcidin 1 and 2 mRNAs in mice. Metabolism. 2003;52: 1229-1231. [DOI] [PubMed] [Google Scholar]
- 19.Cutnell JD, Johnson KW. Physics. 4th ed. New York, NY John Wiley; 1997.
- 20.Rebouche CJ, Wilcox CL, Widness JA. Micro-analysis of non-heme iron in animal tissues. J Biochem Biophys Methods. 2004;58: 239-251. [DOI] [PubMed] [Google Scholar]
- 21.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29: e2002-2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Golab J, Olszewska D, Mroz P, et al. Erythropoietin restores the antitumor effectiveness of photo-dynamic therapy in mice with chemotherapy-induced anemia. Clin Cancer Res. 2002;8: 1265-1270. [PubMed] [Google Scholar]
- 23.Hofland KF, Thougaard AV, Sehested M, Jensen PB. Dexrazoxane protects against myelosuppression from the DNA cleavage-enhancing drugs etoposide and daunorubicin but not doxorubicin. Clin Cancer Res. 2005;11: 3915-3924. [DOI] [PubMed] [Google Scholar]
- 24.Kalyanaraman B, Joseph J, Kalivendi S, et al. Doxorubicin-induced apoptosis: implications in cardiotoxicity. Mol Cell Biochem. 2002;235: 119-124. [PubMed] [Google Scholar]
- 25.Barbee RW, Perry BD, Re RN, Murgo JP. Microsphere and dilution techniques for the determination of blood flows and volumes in conscious mice. Am J Physiol. 1992;263: R728-R733. [DOI] [PubMed] [Google Scholar]
- 26.Neumann CA, Krause DS, Carman CV, et al. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature. 2003;424: 561-565. [DOI] [PubMed] [Google Scholar]
- 27.Inderbitzin D, Sidler D, Studer P, Gloor B. Mouse liver anatomy from a microsurgical point of view [abstract]. The Society for Surgery of the Alimentary Tract, May 14-19, 2005. Chicago, IL.
- 28.Nemeth E, Valore EV, Territo M, et al. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood. 2003;101: 2461-2463. [DOI] [PubMed] [Google Scholar]
- 29.Nemeth E, Rivera S, Gabayan V, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113: 1271-1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roy CN, Custodio AO, de Graaf J, et al. An Hfe-dependent pathway mediates hyposideremia in response to lipopolysaccharide-induced inflammation in mice. Nat Genet. 2004;36: 481-485. [DOI] [PubMed] [Google Scholar]
- 31.Shike H, Lauth X, Westerman ME, et al. Bass hepcidin is a novel antimicrobial peptide induced by bacterial challenge. Eur J Biochem. 2002;269: 2232-2237. [DOI] [PubMed] [Google Scholar]
- 32.Kelley-Loughnane N, Sabla GE, Ley-Ebert C, Aronow BJ, Bezerra JA. Independent and overlapping transcriptional activation during liver development and regeneration in mice. Hepatology. 2002;35: 525-534. [DOI] [PubMed] [Google Scholar]
- 33.Gervois P, Kleemann R, Pilon A, et al. Global suppression of IL-6-induced acute phase response gene expression after chronic in vivo treatment with the peroxisome proliferator-activated receptor-alpha activator fenofibrate. J Biol Chem. 2004; 279: 16154-16160. [DOI] [PubMed] [Google Scholar]
- 34.Ramadori G, Christ B. Cytokines and the hepatic acute-phase response. Semin Liver Dis. 1999;19: 141-155. [DOI] [PubMed] [Google Scholar]
- 35.Moshage H. Cytokines and the hepatic acute phase response. J Pathol. 1997;181: 257-266. [DOI] [PubMed] [Google Scholar]
- 36.Tsuchihashi S, Ke B, Kaldas F, et al. Vascular endothelial growth factor antagonist modulates leukocyte trafficking and protects mouse livers against ischemia/reperfusion injury. Am J Pathol. 2006;168: 695-705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rund D, Rachmilewitz E. Beta-thalassemia. N Engl J Med. 2005;353: 1135-1146. [DOI] [PubMed] [Google Scholar]
- 38.Papanikolaou G, Tzilianos M, Christakis JI, et al. Hepcidin in iron overload disorders. Blood. 2005; 105: 4103-4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rachmilewitz EA, Weizer-Stern O, Adamsky K, et al. Role of iron in inducing oxidative stress in thalassemia: can it be prevented by inhibition of absorption and by antioxidants? Ann N Y Acad Sci. 2005;1054: 118-123. [DOI] [PubMed] [Google Scholar]