

Abstract
Melatonin has been shown to be produced by nonpineal cells and possess anti-inflammatory actions in animal models. In the present study, we tested the hypothesis that melatonin suppresses the expression of proinflammatory genes such as cyclooxygenase-2 (COX2) and inducible nitric oxide synthase (INOS) by a common transcriptional mechanism. Melatonin but not tryptophan or serotonin inhibited lipopolysaccharide (LPS)–induced COX-2 and iNOS protein levels and promoter activities in RAW 264.7 cells in a time- and concentration-dependent manner. LPS or LPS plus interferon-γ (IFNγ) increased binding of all 5 isoforms of NF-κB to COX-2 and iNOS promoters. Melatonin selectively inhibited p52 binding without affecting p100 expression, p52 generation from p100, or p52 nuclear translocation. p52 acetylation was enhanced by LPS, which was abrogated by melatonin. Melatonin inhibited p300 histone acetyltransferase (HAT) activity and abrogated p300-augmented COX-2 and iNOS expression. HAT inhibitors suppressed LPS-induced p52 binding and acetylation to an extent similar to melatonin, and melatonin did not potentiate the effect of HAT inhibitors. These results suggest that melatonin inhibits COX-2 and iNOS transcriptional activation by inhibiting p300 HAT activity, thereby suppressing p52 acetylation, binding, and transactivation.
Introduction
Melatonin is produced primarily in the pineal gland and secreted into circulating blood. It has been shown that melatonin is also produced by cells including monocytes and macrophages and has been implicated in protection against inflammation (for a review, see Reiter1). Depletion of endogenous melatonin has been shown in rats to aggravate carrageenan-induced pleurisy, which is reversed by exogenous melatonin,2 while exogenous melatonin administration has been demonstrated to reduce acute inflammatory reactions in zymosan-activated plasma-induced paw inflammation3 and carrageenan-induced edema and pleurisy.4 Its anti-inflammatory actions are attributed to its ability to scavenge reactive oxygen species and suppress proinflammatory genes, including inducible nitric oxide synthase (INOS) and cyclooxygenase-2 (COX2).5,6 iNOS (also known as NOS-2) and COX-2 play an important role in diverse inflammatory conditions including vascular inflammation, atherosclerosis, and plaque stability.7-10 iNOS catalyzes the robust synthesis of nitric oxide (NO) from l-arginine.11 NO induces inflammatory reactions and tissue injury by diverse mechanisms including activation of soluble guanylyl cyclase, protein nitration, and nitrosylation.12-14 COX-2 occupies a key position in production of proinflammatory prostanoids such as prostaglandin E2 (PGE2).15 iNOS and COX-2 share similar cellular and molecular properties.16 Their expressions are highly inducible by cytokines and lipopolysaccharide (LPS).17-20 They are coinduced in inflammatory cells and their products work in concert to cause tissue inflammation and damage.7
Transcriptional regulation of COX2 and INOS in murine RAW 264.7 cells has been extensively investigated and the reported data indicate that they share common transcriptional properties.17-22 The 5′-flanking region of both genes harbors a canonical TATA motif and a multitude of regulatory elements responsive to LPS and cytokines, among which the κB and CCAAT/enhancer binding protein (C/EBP) elements are demonstrated to be essential for the promoter activation of both genes.17-20 LPS up-regulates binding of NF-κB and C/EBPβ to these 2 sites, respectively. There are 5 isoforms of NF-κB, 3 belonging to the Rel family (RelA, RelB, and C-Rel) and 2 to the NF-κB family (NF-κB1 or p50 and NF-κB2 or p52).23 It has been shown that LPS combined with interferon-γ (IFNγ) increases p50, RelA (p65), and C-Rel (p75) binding to the iNOS promoter.24,25 It is unclear whether LPS/IFNγ or LPS alone up-regulates the binding of the same isoforms to the COX-2 promoter. The effect of LPS on p52 or RelB binding to the INOS or COX2 promoter is also unclear. In this study, we postulate that melatonin inhibits iNOS and COX-2 transcriptional activation by LPS or LPS/IFNγ by blocking selectively the binding of identical transactivators to the respective promoters of INOS and COX2. The results show that melatonin selectively inhibited p52 NF-κB binding, thereby suppressing LPS-induced iNOS and COX-2 expression. The results further show that melatonin reduced p52 acetylation and inhibited p300 histone acetyltransferase (HAT) activity. Two melatonin structural analogs, tryptophan and serotonin, did not inhibit transcriptional activation of iNOS or COX-2 nor did they inhibit p300 HAT or p52 acetylation or binding.
Materials and methods
Cell culture
The murine macrophage cell line RAW 264.7 was obtained from American Type Culture Collection (ATCC, Manassas, VA) and cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1:100 dilution of an antibiotic-antimycotic solution (Invitrogen, Grand Island, NY). For all experiments, 80% to 90% confluent cells were cultured in serum-free medium for 24 hours, washed with PBS, and incubated in fresh medium in the presence or absence of 2 μg/mL LPS (E coli 026:B6; Sigma, St Louis, MO) and/or 400 U/mL IFNγ (Sigma) at 37°C for the indicated time period. The cells were washed with chilled PBS 3 times and then harvested. All the tissue-culture reagents were obtained from Life Technologies (Grand Island, NY).
Analysis of INOS and COX2 promoter activity
RAW 264.7 cells were transfected with a 1.63-kb murine promoter construct (–1486 to +145) constructed into a basic luciferase expression vector according to a procedure previously described.25 In brief, 4 μg promoter vector was mixed with 10 μL lipofectamine 2000 (Invitrogen, Carlsbad, CA), and the mixture was slowly added to cells in a 6-well plate and incubated for 24 hours. The cells were then washed, incubated in serum-free medium for 24 hours, and treated with LPS or LPS/IFNγ for 8 hours. Cells were lysed and luciferase activity was measured using an assay kit from Promega (Madison, WI) in a luminometer (TD 20/20). COX-2 promoter activity was analyzed by a similar procedure as previously described.26
Overexpression of p300 and E1A
A full-length p300 expression vector pCL.p300 was kindly provided by Dr Joan Boyes (Institute of Cancer Research, London, United Kingdom), and a full-length 12S E1A was provided by Dr P. Raychaudhuri (University of Illinois, Chicago). Transfection was done as previously described.25 In brief, 10 μg p300 was mixed with 25 μL lipofectamine 2000 reagent (Invitrogen), and the mixture was added to RAW 264.7 cells cultured in a 10-cm dish and incubated at 37°C for 48 hours.
Western blot analysis
Western blot analysis was performed as previously described.27 In brief, proteins in cell lysates were separated by electrophoresis in a 4% to 15% sodium dodecyl sulfate–polyacrylamide gradient minigel (SDS-PAGE; Bio-Rad, Hercules, CA) and electrophoretically transferred to a nitrocellulose membrane (Amersham Pharmacia Biotech, Piscataway, NJ). Western blots were probed with affinity-purified rabbit polyclonal IgG against iNOS, COX-2, p50, p52, RelA, RelB, C-Rel, C/EBPβ, c-Jun, and IRF-1 (Santa Cruz Biotechnology, Santa Cruz, CA) at 1 μg/mL each. The protein bands were detected by enhanced chemiluminescence (Amersham Pharmacia Biotech) and analyzed by densitometry.
Streptavidin-agarose pulldown assay
Binding of transactivators to iNOS and COX-2 promoter DNA was assayed by streptavidin-agarose pulldown as described previously.28 The procedure allows for quantitative determination of binding of multiple transactivators to a specific probe. In this study, we determined binding of multiple transactivators to a biotinylated double-strand DNA probe corresponding to COX-2 promoter sequence –30/–508 or iNOS promoter sequence +1/–1168, which contains functionally important enhancer elements. To evaluate specific binding to κB, we used a 22-nucleotide sequence containing κB binding site in COX-2 promoter (5′-GGGAGAGGGCATTCCCTGCGCC-3′). A κB mutant was included as a control (5′-GGGAGAGGCGATTCCCTGCGCC-3′). In all experiments, we included a 22-nucleotide sequence that does not contain any known enhancer element as a negative control (5′-AGAGTGGTCACTACCCCCTCTG-3′). All the biotin-labeled double-strand DNA probes were synthesized by Integrated DNA Technologies (Coralville, IA). The binding assay was performed by mixing 400 μg nuclear extract proteins, 4 μg biotinylated DNA probe, and 40 μL streptavidin-conjugated agarose beads. The mixture was incubated at room temperature for 1 hour with shaking and centrifuged to pull down the DNA-protein complex. DNA-bound transactivators were dissociated and analyzed by Western blotting using antibodies specific for the candidate transactivators.
Chromatin immunoprecipitation (ChIP)
The assay was done as previously described.27 Confluent cells (80%-90%) were serum-starved for 24 hours and treated with or without LPS/IFNγ at 37°C for the time indicated. Formaldehyde (1%) was added to the culture medium, and after incubation for 20 minutes at 37°C, the cells were washed twice in phosphate-buffered saline, scraped, and lysed in lysis buffer (1% SDS, 10 mM Tris-HCl, pH 8.0, with 1 mM phenylmethylsulfonyl fluoride, pepstatin A, and aprotinin) for 10 minutes at 4°C. The lysates were sonicated 5 times for 10 seconds each time, and the debris was removed by centrifugation. One third of the lysate was used as DNA input control. The remaining two thirds of the lysate were diluted 10-fold with a dilution buffer (0.01% SDS, 1% Triton X-100, 1 mM EDTA, 10 mM Tris-HCl, pH 8.0, and 150 mM NaCl) followed by incubation with antibodies against p52, or a non–immune rabbit IgG control (Santa Cruz Biotechnology) overnight at 4°C. Immunoprecipitated complexes were collected by using protein A/G plus agarose beads. The precipitates were extensively washed and incubated in an elution buffer (1% SDS and 0.1 M NaHCO3) at room temperature for 20 minutes. Cross-linking of protein-DNA complexes was reversed at 65°C for 5 hours, followed by treatment with 100 μg/mL proteinase K for 3 hours at 50°C. DNA was extracted 3 times with phenol/chloroform and precipitated with ethanol. The pellets were resuspended in TE buffer and subjected to polymerase chain reaction (PCR) amplification using specific COX-2 promoter primers (5′ primer: -709CTGT-TGAAAGCAACTTAGCT-690 and 3′ primer: -32AGACTGAAAAC-CAAGCCCAT-51), or using specific iNOS promoter primers (5′ primer: -498CTGCCCAAGCTGACTTACTAC-478 and 3′ primer: -1GACCCTGGCAGCAGCCATCAG-21). The resulting product of 678 bp for COX-2 or 498 bp for iNOS was separated by agarose gel electrophoresis.
Analysis of HAT activity
RAW 264.7 cells were transfected with a FLAG-p300 vector and the transfected cells were treated with melatonin, tryptophan, or vehicle for 30 minutes followed by LPS/IFNγ for 8 hours. Nuclear extracts prepared from the treated cells were precipitated with an anti-FLAG M2 affinity gel (Sigma). FLAG-p300 was eluted with FLAG peptides (Sigma). HAT of the isolated FLAG-p300 was assayed as previously described.29 In brief, FLAG-p300 was incubated for 30 minutes at 30°C in 100 μL HAT buffer (100 mM Tris HCl, pH 8, 20% glycerol, 2 mM DTT, 2 mM PMSF, 0.2 mM EDTA, 0.1 M NaCl, and 0.02 M butyric acid) containing 100 μg calf thymus histone (Sigma) and 35 pmol 3H-acetyl Coenzyme A (Amersham Pharmacia Biotech). The samples were spotted on P81 filters (Whatman, Florham Park, NJ), and [3H] acetyl histone was measured in a scintillation counter.
Determination of acetylated p52
p52 in nuclear extracts was immunoprecipitated with a specific antibody of p52 (Santa Cruz Biotechnology) and the immunoprecipitate was pulled down with protein A/G agarose beads. After extensive washing, p52 proteins were separated in a 4% to 20% SDS-PAGE system and acetylated p52 was detected with a monoclonal antibody against acetyl-lysine (1:1000 dilution; Cell Signaling Technology, Beverly, MA).
Results
Time- and concentration-dependent inhibition of COX-2 and iNOS protein levels induced by LPS and IFNγ
LPS combined with IFNγ (LPS/IFNγ) induced COX-2 and iNOS-protein levels in a comparable, time-dependent manner (Figure 1A). COX-2 and iNOS proteins were increased at 2 hours; COX-2 reached plateau at 4 hours, while the iNOS protein level still increased at 8 hours. Melatonin at 1 mM abrogated the increase at all time points (Figure 1A). Melatonin inhibited LPS/IFNγ-induced COX-2 and iNOS proteins at 4 hours in a concentration-dependent manner (Figure 1B). COX-2 and iNOS proteins stimulated by LPS alone for 4 hours were also inhibited by melatonin in a concentration-dependent manner (Figure 1B). IFNγ alone induced a small increase in COX-2, which was concentration-dependently reduced by melatonin but had no effect on iNOS protein levels (Figure 1B). As melatonin, serotonin, and tryptophan share a common tryptophan backbone, we evaluated the effects of serotonin and tryptophan on LPS/IFNγ-, LPS-, or IFNγ-induced COX-2 or iNOS protein levels. Neither serotonin (Figure 1C) nor tryptophan (Figure 1D) exerted an effect on COX-2 protein levels, suggesting the requirement of melatonin side chains for inhibiting COX-2 and iNOS expression.
Figure 1.
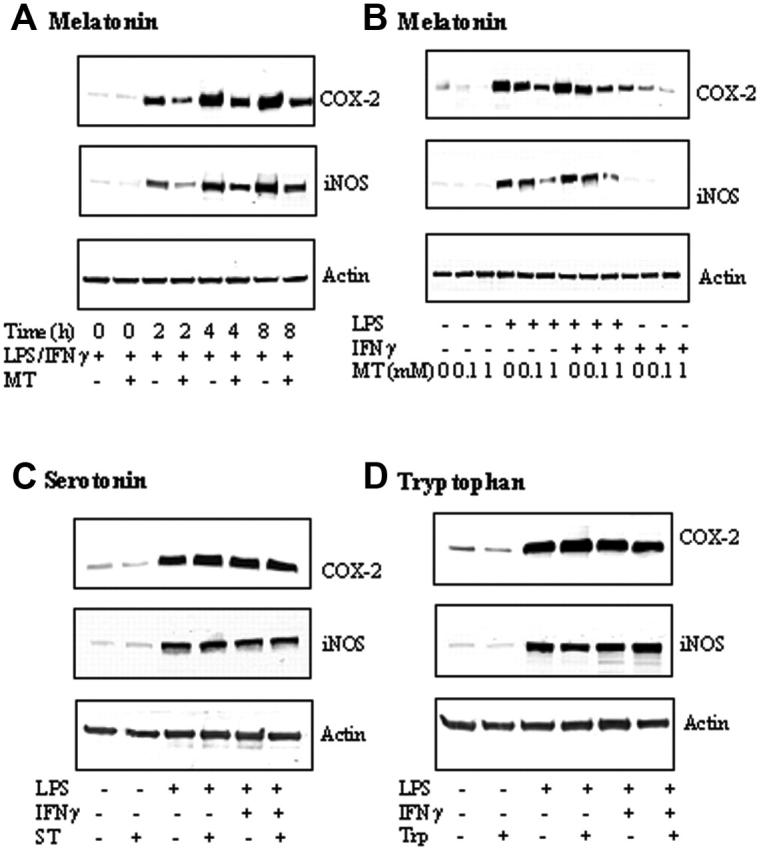
Melatonin (MT) but not tryptophan (Trp) or Serotonin (ST) inhibited COX-2 and iNOS protein levels. (A) Time-dependent inhibition by MT. RAW 264.7 cells were treated with MT (1 mM) for 30 minutes followed by LPS (2 μg/mL) combined with IFNγ (400 U/mL) (LPS/IFNγ) for up to 8 hours, and, at indicated time points, cells were lysed and COX-2 and iNOS in the cell lysates were analyzed by Western blotting. Actin was measured to serve as control. (B) Concentration-dependent inhibition of COX-2 and iNOS proteins by MT. Cells were treated with MT at 0.1 to 1 mM for 30 minutes followed by LPS, IFNγ, or LPS/IFNγ for 8 hours. Cells were homogenized and COX-2, iNOS, and actin were measured by Western blotting. (C-D) Neither serotonin nor tryptophan inhibited COX-2 or iNOS proteins. RAW 264.7 cells were treated with ST (1 mM) or Trp (1 mM) for 30 minutes followed by LPS, or LPS/IFNγ for 8 hours. COX-2, iNOS, and actin were analyzed by Western blotting. Each panel of figures is representative of data from 3 separate experiments.
Selective suppression of p52 NF-κB binding to COX-2 and iNOS promoter by melatonin
iNOS promoter activity in RAW 264.7 cells was determined by transfecting cells with a pGL3 luciferase expression vector containing a 1.63 kb (–1486 to +145) iNOS promoter fragment and measuring the expression of luciferase. Basal promoter activities detected in resting cells were increased by LPS/IFNγ or LPS treatment to a similar extent but were suppressed by melatonin in a concentration-dependent manner (Figure 2A). Neither serotonin nor tryptophan inhibited iNOS promoter activities (Figure 2B and 2C, respectively). COX-2 promoter activity was inhibited by melatonin but not serotonin or tryptophan in a manner similar to iNOS (data not shown).
Figure 2.

Melatonin but not serotonin or tryptophan inhibited iNOS promoter activity. RAW 264.7 cells transfected with luciferase expression vector containing iNOS promoter were pretreated with melatonin (A), serotonin (B), or tryptophan (C) (1 mM each) for 30 minutes followed by LPS, or LPS/IFNγ for 8 hours. iNOS promoter activity was measured as luciferase activity expressed as relative light unit (RLU)/μg proteins. Each bar denotes mean ± SEM of 3 experiments.
COX-2 and iNOS promoter activation by LPS or LPS/IFNγ was mediated by binding of several transactivators including NF-κB and C/EBPβ to their respective sequences on the promoter region.17-22 To determine whether melatonin suppresses iNOS and COX-2 expression through inhibition of the binding of common transactivators, we treated RAW 264.7 cells with melatonin (1 mM) for 30 minutes followed by LPS/IFNγ for 4 hours. Nuclear extracts prepared from these cells were incubated with a biotinylated COX-2 or iNOS promoter probe in a solution containing streptavidin-conjugated agarose beads. Nuclear transactivators that bind to the biotinylated probe were pulled down by streptavidin-agarose. Following centrifugation, proteins were dissociated and analyzed by Western blotting using antibodies against candidate transactivators of interests. In this study, we chose to study the following transactivators: NF-κB and C/EBPβ, which are required for both COX-2 and iNOS transcriptional activation as well as C-Jun and IRF-1. A biotinylated control probe that does not harbor any known regulatory element was included in each experiment. Only a trace amount of p52 (Figure 3A) and other NF-κB isoforms (Figure S1A, available on the Blood website; see the Supplemental Figure link at the top of the online article) was noted to bind iNOS promoter probe at the basal state, which was not altered by melatonin. Treatment with LPS alone or in combination with IFNγ resulted in increased binding of p52 (Figure 3A) as well as other isoforms of NF-κB but not C/EBPβ, C-Jun, or IRF-1 to iNOS promoter (Figure S1A). Melatonin reduced LPS or LPS/IFNγ-induced p52 binding by approximately 50% (Figure 3A) and had minimal or no effect on the binding of other NF-κB isoforms or other transactivators (Figure S1A). As LPS or LPS/IFNγ did not increase C-Jun binding, the effect of melatonin on reducing C-Jun binding was attributed to the reduction of basal binding (Figure S1A). There was no detectable binding to the control probe, consistent with specific iNOS binding. Melatonin also reduced p52 binding to the COX-2 promoter (Figure 3B) without an effect on the binding of other transactivators (Figure S1B). The effect of melatonin on p52 binding to κB was further evaluated by using a 22-bp probe containing κB sequence or mutant κB sequence. A 22-bp control probe without transactivator binding motifs was included as control. LPS/IFNγ increased p52 binding to the wild-type κB probe, which was reduced by approximately 50% by melatonin (Figure 3C). p52 binding was barely detectable with mutated κB probe and control probe (Figure 3C). These results are consistent with specific binding of p52 to κB site, which was suppressed by melatonin. We next performed ChIP assay to confirm the effect of melatonin in vivo. LPS/IFNγ-induced p52 binding to chromatin COX-2 promoter region and iNOS promoter region was suppressed by melatonin to a similar extent (Figure 3D). A control non–immune IgG did not precipitate COX-2 or iNOS chromatin promoter fragment. To determine whether reduced p52 binding by melatonin may be related to a decrease in p52 protein level in nuclear extracts, we analyzed p52 as well as other transactivators by Western blots. P52 protein level was elevated in LPS- or LPS/IFNγ-treated cells but was not altered by melatonin (Figure 3E). The protein levels of RelB, C-Rel, and, to a lesser extent, RelA were also elevated by LPS or LPS/IFNγ but were not influenced by melatonin (Figure S1C). The protein level of none of the other transactivators was altered by LPS or LPS/IFNγ in the presence or absence of melatonin (Figure S1C). Taken together, these results indicate that melatonin suppressed LPS-induced p52 binding to κB enhancer elements at iNOS and COX-2 promoters, without altering its protein levels.
Figure 3.

Inhibition of p52 DNA binding by MT. (A-C) RAW 264.7 cells were pretreated with MT (1 mM) for 30 minutes followed by LPS or LPS/IFNγ for 8 hours. Nuclear extracts from cells were incubated with (A) a biotinylated iNOS promoter probe corresponding to sequence + 1 to –1168, (B) a COX-2 promoter probe (–30/–508), or (C) a 22-bp κB-containing probe, a 22-bp κB-mutant probe, and a 22-bp control probe, which does not contain any known enhancer element (please see “Materials and methods” for their sequences). The DNA-protein complexes were pulled down with streptavidin-agarose beads, and p52 in the complex was analyzed by Western blots using a specific antibody. C denotes control probe; iNOS, iNOS promoter probe; COX-2, COX-2 promoter probe; mt-κB, mutant κB probe; and wt-κB, wild-type κB probe. (D) ChIP analysis of p52 binding to a 678-bp COX-2 or 498-bp iNOS promoter region. C-IgG refers to immunoprecipitation of chromatin with a non–immune IgG. Cell treatment protocol was identical to that described in panels A-C. (E) p52 protein levels in nuclear extracts in panels A-C were determined by Western blots. Each figure is representative of 2 to 3 experiments with similar results.
Melatonin time-dependently inhibited p52 binding but not p52 processing or nuclear translocation
To confirm that melatonin specifically suppresses p52 binding, we performed a kinetic experiment to determine changes in p100 and p52 levels in cytosolic versus nuclear fractions and binding of p52 to a κB-containing probe. Unstimulated RAW 264.7 cells contain a low level of p52 and p100 in cytosolic and/or nuclear fractions (Figure 4A and 4B, respectively). The cytosolic p52 and p100 levels were not changed at 1 or 2 hours but were markedly elevated at 4 hours after LPS/IFNγ treatment (Figure 4A). The p52 protein level was increased time dependently in the nuclear fraction by LPS/IFNγ, while very little p100 was detected in the nucleus (Figure 4B). p52 binding to COX-2 and iNOS promoter probes was increased by LPS/IFNγ in a time-dependent manner (Figure 4C). Melatonin did not have an effect on p100 or p52 proteins in cytosolic or nuclear fractions (Figure 4A and 4B, respectively). However, it inhibited p52 binding at 2 hours, which persisted for 4 hours (Figure 4C). These results are consistent with inhibition of LPS-induced p52 binding to COX-2 and iNOS promoters without alteration of p100 levels, p52 generation, or p52 nuclear translocation by melatonin.
Figure 4.

Kinetic analysis of p100 and p52 proteins. (A) Cytosol and (B) nuclear extracts of RAW 264.7 cells stimulated with LPS/IFNγ for 8 hours in the presence or absence of MT. (C) Kinetics of p52 binding to biotinylated COX-2 or iNOS promoter probe.
Melatonin abrogated p52 binding augmented by p300 overexpression
It was reported that forced overexpression of p300 augmented COX-2 and iNOS expression induced by LPS/IFNγ and other agonists.27,30 We determined here whether melatonin altered p300-induced COX-2 and/or iNOS protein levels. Melatonin abrogated LPS and LPS/IFNγ-induced COX-2 and iNOS protein levels in cells transfected with p300 vector compared with those transfected with control vectors (Figure 5A), without changing the p300 protein levels (Figure 5B). p300 overexpression augmented LPS/IFNγ-induced p52 binding both to COX-2 and iNOS promoter probes and to a κB probe, all of which were abrogated by melatonin (Figure 5C). Cotransfection of cells with adenoviral E1A and p300 or treatment of p300-transfected cells with roscovitine, an indirect inhibitor or p300 HAT, reduced p52 binding to an extent close to that of melatonin treatment (Figure 5C). Furthermore, addition of melatonin to E1A-transfected cells or roscovitine-treated cells did not suppress p52 binding further (Figure 5C). These binding results are confirmed by ChIP assay. Melatonin, E1A, and roscovitine inhibited p52 binding to chromatin COX-2 or iNOS promoter region to a similar extent and addition of melatonin to E1A or roscovitine did not suppress the chromatin-binding further (Figure 5D). Taken together, these results suggest that melatonin inhibits p300 HAT, thereby reducing p52 binding and suppressing COX-2 and iNOS expressions.
Figure 5.

Melatonin abrogated p300-augmented COX-2 or iNOS proteins and p52 DNA binding stimulated by LPS/IFNγ. (A-B) RAW 264.7 cells transfected with p300 or control vectors were treated with MT or Trp for 30 minutes followed by LPS or LPS/IFNγ for 8 hours. COX-2, iNOS, and p300 protein levels were analyzed by Western blotting. (C) Cells were cotransfected with p300 and E1A as described in “Materials and methods,” or transfected with p300 followed by treatment with roscovitine (RST, 25 μM) for 30 minutes with or without MT. Binding of nuclear extract p52 to COX-2 or iNOS promoter probe or control probe was analyzed by streptavidin-agarose pulldown assay. (D) Cells were treated by an identical protocol as described in panel C. Chromatin was immunoprecipitated by a p52 antibody (anti-p52) or non–immune IgG (C-IgG). The targeted promoter regions of COX-2 and iNOS were amplified by PCR.
Melatonin inhibited p300 HAT activity
To determine whether melatonin inhibited p300 HAT activity, we transfected RAW 264.7 cells with a FLAG-p300 vector and treated the transfected cells with LPS/IFNγ in the presence or absence of melatonin or tryptophan. Nuclear extracts were immunoprecipitated with a FLAG antibody, and FLAG-p300 in the precipitate was eluted using FLAG peptides. HAT activity of the purified p300 was determined. Melatonin concentration-dependently inhibited p300 HAT activity, whereas tryptophan had no effect (Figure 6A). In separate experiments, we transfected cells with FLAG-p300 and treated them with LPS/IFNγ, and FLAG-p300 proteins were isolated. The purified p300 proteins were treated with melatonin or tryptophan. p300 HAT was assayed. Melatonin inhibited HAT activity by approximately 70%, while tryptophan did not (Figure 6B). These results are consistent with direct inhibition of p300 HAT by melatonin.
Figure 6.

Melatonin but not tryptophan inhibited p300 HAT activity. (A) RAW 264.7 cells transfected with FLAG-p300 were treated with LPS/IFNγ in the presence or absence of melatonin or tryptophan. Nuclear extracts were immunoprecipitated with an anti-FLAG M2 Gel, and FLAG-p300 was eluted with a FLAG peptide. HAT activity of p300 was measured. (B) FLAG-p300–transfected cells were treated with LPS/IFNγ for 8 hours. FLAG-p300 was isolated and treated with MT or Trp for 30 minutes and HAT activity was measured. Each bar denotes mean ± SEM of 3 experiments.
Melatonin suppressed p52 acetylation
As NF-κB has been shown to be acetylated by p300 HAT,30 we determined whether melatonin might influence LPS/IFNγ-induced p52 acetylation. Nuclear extracts from RAW 264.7 cells treated with LPS/IFNγ in the presence or absence of melatonin were immunoprecipitated with a p52 antibody or a control non–immune IgG. Acetylated p52 (Ac-p52) and total p52 proteins were analyzed by Western blots. As a control, acetylated CREB-2 was analyzed. Melatonin inhibited basal and LPS/IFNγ-induced Ac-p52 without influencing p52 levels (Figure 7A). Control IgG did not precipitate detectable Ac-p52 or p52. CREB-2 proteins were detected, but Ac-CREB-2 was undetectable (Figure 7B). To determine whether p300 HAT is involved in p52 acetylation, we transfected cells with p300 vector and treated the transfected cells with LPS/IFNγ in the presence or absence of melatonin, roscovitine, or E1A. Ac-p52 induced by p300 was inhibited by melatonin to a similar extent as E1A transfection or roscovitine treatment (Figure 7C). Furthermore, the addition of melatonin to E1A or roscovitine did not potentiate the action of either HAT inhibitor.
Figure 7.

Melatonin inhibited p52 acetylation. (A-B) Nuclear extracts prepared from RAW 264.7 cells treated with LPS/IFNγ in the presence or absence of MT were immunoprecipitated with a p52 antibody (anti-p52) or non–immune IgG (IgG). Acetylated p52 (Ac-p52) was analyzed by Western blots using an acetyl-lysine antibody. To serve as a control, CREB-2 was immunoprecipitated with a CREB-2 antibody, and Ac-CREB-2 was analyzed by Western blots using acetyl-lysine antibody. (C) Cells were treated by E1A or roscovitine (RST) according to procedures described in Figure 5. Ac-p52 in immunoprecipitates was analyzed as described above.
Discussion
Results from this study show that LPS/IFNγ increased the nuclear levels of p52, RelA, RelB, and C-Rel and enhanced the binding to these NF-κB isoforms to COX-2 and iNOS promoters, consistent with the involvement of multiple NF-κB isoforms in regulating COX-2 and iNOS transcriptional activation. Melatonin selectively inhibited p52 binding to those 2 promoters. Although LPS has been shown to induce NF-κB binding to diverse genes, previous studies have reported regulation of RelA, C-Rel, and p50 by LPS/IFNγ in RAW 264.7 cells.24 To our knowledge, this is the first report of the involvement of p52 NF-κB2 isoform in LPS-induced COX-2 and iNOS transcriptional activation in a nonlymphoid cell such as RAW 264.7 macrophages. p52 has been reported to be selectively involved in lymphoid development and function.31,32 Its generation and activation in lymphocytes are reported to be under tight regulation. p52 is generated from its precursor protein p100 by a cotranslational mechanism requiring de novo synthesis of p100.33 LPS was reported to increase p100 expression and p52 generation in lymphocytes only after lymphocytes had been treated with LPS for 3 hours, suggesting a slower process than the generation of p50.33 Our results are consistent with this previous report. We have found that LPS increased cytosolic p100 and p52 only after the macrophages had been treated with LPS for 4 hours. This slow kinetics of p52 generation is attributed to a complex process via which p52 is generated from p100. p100 contains the NF-κB2 domain as well as an IκB-like domain that provides intramolecular control of the NF-κB2 activity.34 Upon cell activation by only a selected group of agonists such as LPS and lymphotoxin, p100 is phosphorylated in an NIK (NF-κB inducing kinase)–dependent and IKKα (IκB kinase β)–dependent manner,35-38 and the phosphorylated p100 is polyubiquitinylated and degraded via the proteasome to remove the IκB domain.39,40 p52 generated from this process forms heterodimers with NF-κB isoforms, preferably with RelB.23 Our results show that RelB binding was also increased by LPS/IFNγ, which may enhance p52/RelB heterodimer binding. Melatonin selectively blocked p52 binding without perturbing RelB or other isoform binding, and reduced p52 binding was correlated with reduction in COX-2 or iNOS transcriptional activation induced by LPS/IFNγ. Thus, p52 plays an important role in LPS/IFNγ-induced COX-2 and iNOS expressions. As p52 is generated from p100 by a tightly regulated and coordinated program, we suspected that melatonin might inhibit p52 binding by interfering with p100 processing. However, our results show that this is not the case. Kinetic analysis shows that melatonin did not influence p100 protein expression, p52 generation, or p52 nuclear translocation but had a selective effect on p52 binding to COX-2 or iNOS promoter. A previous study has provided suggestive evidence for inhibition of NF-κB binding by melatonin,41 but the isoform target was not reported. Our results clearly show that melatonin targets the binding process of p52.
Little is known about the regulation of p52 binding to its DNA motif. Our results suggest that p52 binding to κB enhancer element is regulated by acetylation by p300 HAT. Overexpression of p300 resulted in increased acetylated p52 accompanied by enhanced p52 binding to the κB site by in vitro and in vivo binding assays. p300-induced augmentation of p52 acetylation and binding activity was abrogated by E1A and roscovitine, which are known to inhibit p300 HAT. Furthermore, melatonin, which directly inhibits p300 HAT, also abrogated p300-induced augmentation of p52 acetylation and binding. It was previously shown that p300 HAT is also involved in regulating acetylation of p50 and p50 binding to iNOS promoter induced by LPS/IFNγ.27 It is unclear why melatonin did not inhibit p50 binding in the present study. Further studies are needed to resolve this paradox.
An important finding of this study is that melatonin inhibits p300 HAT activity. p300 has been shown to play an essential role in transcriptional activation of INOS and COX2 induced by diverse proinflammatory mediators including LPS.24-28 p300 interacts with promoter-bound transactivators such as NF-κB and basal transcription factors such as TFIIB in the transcription machinery.42 Besides serving as a bridge molecule to transmit the message to the transcription machinery, p300 contains a HAT domain, which acetylates histone to open the chromatin structure making the cis-activating elements on the promoter accessible to the trans-activating nuclear factors.42 Furthermore, p300 HAT has been shown to acetylate transactivators thereby enhancing their binding activity.43 Results from recent reports have shown that deletion of p300 HAT greatly reduces the p300-mediated iNOS and COX-2 transcriptional activities.27,44 It is therefore of importance to note that melatonin inhibits p300 HAT activity. The results suggest that melatonin exerts its inhibition of iNOS and COX-2 by targeting p300 HAT, thereby suppressing p52 acetylation and binding activity. It is unclear how melatonin inhibits p300 HAT activity. Melatonin may represent a valuable molecule for studying the mechanism of p300 HAT catalytic action.
Findings from this study show that melatonin at 1 mM inhibited p300 HAT activity, p52 acetylation, and COX-2 and iNOS expressions by approximately 50%, while at 0.1 mM it had lesser and minimal effect. Previous reports have shown that melatonin inhibits iNOS expression and lipid peroxidation at 1 mM or higher.41,45 Numerous cell types besides pineal gland cells are capable of producing melatonin. For example, bone marrow cells have been reported to produce a high level of melatonin.46,47 However, the quantity of melatonin produced by those cells has not been clearly defined. Hence, the physiologic concentrations of melatonin produced by cells remain unknown. Pharmacologically, infusion of melatonin into rats intravenously or intraperitoneally has been shown to control inflammation and tissue injury at doses up to 60 mg/kg.48,49 However, the in vivo cellular concentrations have not been determined.6 Nevertheless, as high doses of melatonin administered to rats did not produce overt side effects,50 melatonin may have therapeutic potential for treating inflammatory disorders and ischemia-reperfusion injury by suppressing the expression of p300 HAT-mediated genes including COX-2 and iNOS.
Supplementary Material
Acknowledgments
We thank Susan Mitterling for editorial assistance.
Prepublished online as Blood First Edition Paper, April 11, 2006; DOI 10.1182/blood-2005-09-3691.
Supported by grants from the National Institute of Neurological Disease and Stroke (P50 NS-23327) and the National Heart, Lung and Blood Institute (R01-HL 50675) of the National Institutes of Health.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Reiter RJ. Pineal melatonin: cell biology of its synthesis and of its physiological interactions. Endocr Rev. 1991;12: 151-180. [DOI] [PubMed] [Google Scholar]
- 2.Cuzzocrea S, Tan DX, Costantino G, Mazzon E, Caputi AP, Reiter RJ. The protective role of endogenous melatonin in carrageenan-induced pleurisy in the rat. FASEB J. 1999;13: 1930-1938. [DOI] [PubMed] [Google Scholar]
- 3.Costantino G, Cuzzocrea S, Mazzon E, Caputi AP. Protective effects of melatonin in zymosan-activated plasma-induced paw inflammation. Eur J Pharmacol. 1998;363: 57-63. [DOI] [PubMed] [Google Scholar]
- 4.Cuzzocrea S, Zingarelli B, Gilad E, Hake P, Salzman AL, Szabo C. Protective effect of melatonin in carrageenan-induced models of local inflammation: relationship to its inhibitory effect on nitric oxide production and its peroxynitrite scavenging activity. J Pineal Res. 1997;23: 106-116. [DOI] [PubMed] [Google Scholar]
- 5.Cuzzocrea S, Costantino G, Mazzon E, Caputi AP. Regulation of prostaglandin production in carrageenan-induced pleurisy by melatonin. J Pineal Res. 1999;27: 9-14. [DOI] [PubMed] [Google Scholar]
- 6.Crespo E, Macias M, Pozo D, et al. Melatonin inhibits expression of the inducible NO synthase II in liver and lung and prevents endotoxemia in lipopolysaccharide-induced multiple organ dysfunction syndrome in rats. FASEB J. 1999;13: 1537-1546. [PubMed] [Google Scholar]
- 7.Vane JR, Mitchell JA, Appleton I, et al. Inducible isoforms of cyclooxygenase and nitric-oxide synthase in inflammation. Proc Natl Acad Sci U S A. 1994;91: 2046-2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schonbeck U, Sukhova GK, Graber P, Coulter S, Libby P. Augmented expression of cyclooxygenase-2 in human atherosclerotic lesions. Am J Pathol. 1999;155: 1281-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burleigh ME, Babaev VR, Oates JA, et al. Cyclooxygenase-2 promotes early atherosclerotic lesion formation in LDL receptor-deficient mice. Circulation. 2002;105: 1816-1823. [DOI] [PubMed] [Google Scholar]
- 10.Buttery LD, Springall DR, Chester AH, et al. Inducible nitric oxide synthase is present within human atherosclerotic lesions and promotes the formation and activity of peroxynitrite. Lab Invest. 1996;75: 77-85. [PubMed] [Google Scholar]
- 11.Nathan C, Xie QW. Nitric oxide synthases: roles, tolls, and controls. Cell. 1994;78: 915-918. [DOI] [PubMed] [Google Scholar]
- 12.Stamler JS, Singel DJ, Loscalzo J. Biochemistry of nitric oxide and its redox-activated forms. Science. 1992;258: 1898-1902. [DOI] [PubMed] [Google Scholar]
- 13.Ischiropoulos H. Biological tyrosine nitration: a pathophysiological function of nitric oxide and reactive oxygen species. Arch Biochem Biophys. 1998;356: 1-11. [DOI] [PubMed] [Google Scholar]
- 14.Beckmann JS, Ye YZ, Anderson PG, et al. Extensive nitration of protein tyrosines in human atherosclerosis detected by immunohistochemistry. Biol Chem Hoppe Seyler. 1994;375: 81-88. [DOI] [PubMed] [Google Scholar]
- 15.Cipollone F, Prontera C, Pini B, et al. Overexpression of functionally coupled cyclooxygenase-2 and prostaglandin E synthase in symptomatic atherosclerotic plaques as a basis of prostaglandin E(2)-dependent plaque instability. Circulation. 2001;104: 921-927. [DOI] [PubMed] [Google Scholar]
- 16.Wu KK. Inducible cyclooxygenase and nitric oxide synthase. Adv Pharmacol. 1995;33: 179-207. [DOI] [PubMed] [Google Scholar]
- 17.Xie QW, Whisnant R, Nathan C. Promoter of the mouse gene encoding calcium-independent nitric oxide synthase confers inducibility by interferon gamma and bacterial lipopolysaccharide. J Exp Med. 1993;177: 1779-1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lowenstein CJ, Alley EW, Raval P, et al. Macrophage nitric oxide synthase gene: two upstream regions mediate induction by interferon gamma and lipopolysaccharide. Proc Natl Acad Sci U S A. 1993;90: 9730-9734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wadleigh DJ, Reddy ST, Kopp E, Ghosh S, Herschman HR. Transcriptional activation of the cyclooxygenase-2 gene in endotoxin-treated RAW 264.7 macrophages. J Biol Chem. 2000;275: 6259-6266. [DOI] [PubMed] [Google Scholar]
- 20.Herschman HR. Prostaglandin synthase 2. Biochim Biophys Acta. 1996;1299: 125-140. [DOI] [PubMed] [Google Scholar]
- 21.Dlaska M, Weiss G. Central role of transcription factor NF-IL6 for cytokine and iron-mediated regulation of murine inducible nitric oxide synthase expression. J Immunol. 1999;162: 6171-6177. [PubMed] [Google Scholar]
- 22.Goldring CE, Reveneau S, Algarte M, Jeannin JF. In vivo footprinting of the mouse inducible nitric oxide synthase gene: inducible protein occupation of numerous sites including Oct and NF-IL6. Nucleic Acids Res. 1996;24: 1682-1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dixit V, Mak TW. NF-kappaB signaling: many roads lead to madrid. Cell. 2002;111: 615-619. [DOI] [PubMed] [Google Scholar]
- 24.Xie QW, Kashiwabara Y, Nathan C. Role of transcription factor NF-kappa B/Rel in induction of nitric oxide synthase. J Biol Chem. 1994;269: 4705-4708. [PubMed] [Google Scholar]
- 25.Cieslik K, Zhu Y, Wu KK. Salicylate suppresses macrophage nitric-oxide synthase-2 and cyclooxygenase-2 expression by inhibiting CCAAT/enhancer-binding protein-beta binding via a common signaling pathway. J Biol Chem. 2002;277: 49304-49310. [DOI] [PubMed] [Google Scholar]
- 26.Schroer K, Zhu Y, Saunders MA, et al. Obligatory role of cyclic adenosine monophosphate response element in cyclooxygenase-2 promoter induction and feedback regulation by inflammatory mediators. Circulation. 2002;105: 2760-2765. [DOI] [PubMed] [Google Scholar]
- 27.Deng W-G, Wu KK. Regulation of nitric oxide synthase expression by p300 and p50 NF-κB acetylation. J Immunol. 2003;171: 6581-6588. [DOI] [PubMed] [Google Scholar]
- 28.Deng WG, Zhu Y, Montero A, Wu KK. Quantitative analysis of binding of transcription factor complex to biotinylated DNA probe by a streptavidin-agarose pulldown assay. Anal Biochem. 2003;323: 12-18. [DOI] [PubMed] [Google Scholar]
- 29.Marzio G, Tyagi M, Gutierrez MI, Giacca M. HIV-1 tat transactivator recruits p300 and CREB-binding protein histone acetyltransferases to the viral promoter. Proc Natl Acad Sci U S A. 1998;95: 13519-13524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deng WG, Zhu Y, Wu KK. Up-regulation of p300 binding and p50 acetylation in tumor necrosis factor-alpha-induced cyclooxygenase-2 promoter activation. J Biol Chem. 2003;278: 4770-4777. [DOI] [PubMed] [Google Scholar]
- 31.Caamano JH, Rizzo CA, Durham SK, et al. Nuclear factor (NF)-kappa B2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. J Exp Med. 1998;187: 185-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Franzoso G, Carlson L, Poljak L, et al. Mice deficient in nuclear factor (NF)-kappa B/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. J Exp Med. 1998;187: 147-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mordmuller B, Krappmann D, Esen M, Wegener E, Scheidereit C. Lymphotoxin and lipopolysaccharide induce NF-kappaB-p52 generation by a co-translational mechanism. EMBO Rep. 2003;4: 82-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rice NR, MacKichan ML, Israel A. The precursor of NF-kappa B p50 has I kappa B-like functions. Cell. 1992;71: 243-253. [DOI] [PubMed] [Google Scholar]
- 35.Xiao G, Harhaj EW, Sun SC. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol Cell. 2001;7: 401-409. [DOI] [PubMed] [Google Scholar]
- 36.Xiao G, Cvijic ME, Fong A, et al. Retroviral onco-protein Tax induces processing of NF-kappaB2/p100 in T cells: evidence for the involvement of IKKalpha. EMBO J. 2001;20: 6805-6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Senftleben U, Cao Y, Xiao G, et al. Activation by IKKal-pha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293: 1495-1499. [DOI] [PubMed] [Google Scholar]
- 38.Claudio E, Brown K, Park S, Wang H, Siebenlist U. BAFF-induced NEMO-independent processing of NF-kappa B2 in maturing B cells. Nat Immunol. 2002;3: 958-965. [DOI] [PubMed] [Google Scholar]
- 39.Fong A, Sun SC. Genetic evidence for the essential role of beta-transducin repeat-containing protein in the inducible processing of NF-kappa B2/p100. J Biol Chem. 2002;277: 22111-22114. [DOI] [PubMed] [Google Scholar]
- 40.Fong A, Zhang M, Neely J, Sun SC. S9, a 19 S proteasome subunit interacting with ubiquitinated NF-kappaB2/p100. J Biol Chem. 2002;277: 40697-40702. [DOI] [PubMed] [Google Scholar]
- 41.Gilad E, Wong HR, Zingarelli B, et al. Melatonin inhibits expression of the inducible isoform of nitric oxide synthase in murine macrophages: role of inhibition of NFκB activation. FASEB J. 1998;12: 685-693. [DOI] [PubMed] [Google Scholar]
- 42.Shikama N, Lyon J, La Thangue NB. The p300/CBP family: integrating signals with transcription factors and chromatin. Trends Cell Biol. 1997;7: 230-236. [DOI] [PubMed] [Google Scholar]
- 43.Boyes J, Byfield P, Nakatani Y, Ogryzko V. Regulation of activity of the transcription factor GATA-1 by acetylation. Nature. 1998;396: 594-598. [DOI] [PubMed] [Google Scholar]
- 44.Deng WG, Zhu Y, Wu KK. Role of p300 and PCAF in regulating cyclooxygenase-2 promoter activation by inflammatory mediators. Blood. 2004;103: 2135-2142. [DOI] [PubMed] [Google Scholar]
- 45.Gavazza M, Catala A. Melatonin preserves arachidonic and docosapentaenoic acids during ascorbate-Fe2+ peroxidation of rat testis microsomes and mitochondria. Int J Biochem Cell Biol. 2003;35: 359-366. [DOI] [PubMed] [Google Scholar]
- 46.Conti A, Conconi S, Hertens E, Skwarlo-Sonta K, Markowska M, Maestroni JM. Evidence for melatonin synthesis in mouse and human bone marrow cells. J Pineal Res. 2000;28: 193-202. [DOI] [PubMed] [Google Scholar]
- 47.Tan DX, Manchester LC, Reiter RJ, et al. Identification of highly elevated levels of melatonin in bone marrow: its origin and significance. Biochim Biophys Acta. 1999;1472: 206-214. [DOI] [PubMed] [Google Scholar]
- 48.Kunduzova OR, Escourrou G, Seguelas MH, et al. Prevention of apoptotic and necrotic cell death, caspase-3 activation, and renal dysfunction by melatonin after ischemia/reperfusion. FASEB J. 2003;17: 872-874. [DOI] [PubMed] [Google Scholar]
- 49.Cheung RT. The utility of melatonin in reducing cerebral damage resulting from ischemia and reperfusion. J Pineal Res. 2003;34: 153-160. [DOI] [PubMed] [Google Scholar]
- 50.Molina-Carballo A, Munoz-Hoyos A, Reiter RJ, et al. Utility of high doses of melatonin as adjunctive anticonvulsant therapy in a child with severe myoclonic epilepsy: two years' experience. J Pineal Res. 1997;23: 97-105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.