

Abstract
A number of agents reducing interleukin-1β (IL-1β) activity are being developed as novel immunomodulatory and anti-inflammatory therapies. However, the elucidation of their molecular mechanism of action is required in the context of medical management of inflammatory diseases. Inhibitors of histone deacetylases (HDACs) are promising anticancer agents with pleiotropic activities. Of these, suberoylanilide hydroxamic acid has been reported to inhibit the production of several proinflammatory cytokines. In the present study, we investigated the effects of 2 HDAC inhibitors on IL-1β secretion: suberoylanilide hydroxamic acid and a newly developed hydroxamic acid-derived compound ITF2357. These HDAC inhibitors do not affect the synthesis or intracellular localization of IL-1β but both strongly reduce the levels of extracellular IL-1β by preventing the exocytosis of IL-1β-containing secretory lysosomes. At nanomolar concentrations, ITF2357 reduces the secretion of IL-1β following ATP activation of the P2X7 receptor. Whereas the inhibition of HDACs results in hyperacetylation of tubulin, acetylation of HSP90 was unaffected. The reduction in IL-1β secretion appears to be due to disruption of microtubules impairing lysosome exocytosis. Together, these observations indicate that a functional microtubule network is required for IL-1β secretion and suggest that disruption of tubulin is the mechanism by which inhibitors of HDACs reduce the secretion of IL-1β.
Introduction
Interleukin-1β (IL-1β) is a potent proinflammatory cytokine; subnanomolar concentrations following intravenous injection into humans are sufficient to cause several inflammatory responses.1 Thus it is not surprising that the production of active IL-1β is tightly controlled, on more than one level.2 A first level is transcriptional: IL-1β is not expressed in healthy individuals. A second is translational, as high levels of IL-1β mRNA can be present in cells in the absence of translation.3 The third and perhaps the most complex is the pathway by which the initial translational product, the inactive IL-1β precursor, is cleaved into an active form and released extracellularly. The cleavage of the inactive IL-1β precursor is accomplished by caspase-1 and both cleavage and secretion appear to be linked. However, the activation of caspase-1 itself from an inactive form is also tightly controlled by mechanisms that have only recently been partially elucidated.4,5 Similar to other clinically relevant cytokines, IL-1β lacks a secretory signal peptide and avoids the classical exocytotic route.6 Two major steps can be envisaged in the IL-1β secretion process in human monocytes, the primary sources of IL-1β. First, toll-like-receptor ligands such as lipopolysaccharide (LPS), peptidoglycans, or the intracellular nucleotide oligomerization domain (NOD) agonists induce gene expression and synthesis of the IL-1β precursor2; however, it accumulates in the cytosol and only a portion of the total synthesis enters a specialized subset of secretory lysosomes, where inactive procaspase-1 is also present.4,7 Once stimulated, monocytic cells release approximately 20% of the IL-1β slowly over 24 to 48 hours into the extracellular compartment, unless a stimulus-triggered secretion event takes place. A powerful stimulus of secretion is exogenous ATP, which acting in an autocrine manner on P2X7 receptors expressed on the surface of monocytic cells,8 facilitates IL-1β secretion. In vivo, ATP accumulates at the site of inflammation, released by dying cells or actively secreted by monocytes or other inflammatory cells, such as platelets, and thus accelerates the secretion of the mature cytokine.8
Engagement of P2X7 receptors triggers a series of events, leading to IL-1β processing and secretion, both of which have been dissected and partially clarified. Fundamental to the engagement of the purinergic P2X7 receptor is the efflux of K+ from the cell, which occurs simultaneously with ATP activation. K+ efflux appears crucial for the generation of active caspase-1 from its inactive precursor9-11 through activation of calcium-independent phospholipase A2 (iPLA2).12 The exit of K+ is then followed by Ca2+ entry13 and the sequential activation of phosphatidylcholine-specific phospholipase C (PC-PLC) and calcium-dependent phospholipase A2 (cPLA2), which is required for lysosome exocytosis and secretion of IL-1β.4 The small peptide LL37, released by activated neutrophils and epithelial cells, triggers P2X7 receptors, and thus also can facilitate secretion of IL-1β.14
An important cellular process that remains to be clarified is whether and how the cytoskeleton contributes to IL-1β-containing vesicle movement and/or exocytosis. The involvement of the cytoskeleton in vesicle and organelle transport is a longstanding concept. However, the molecular constituents vary considerably depending on the cell type or the cargo vesicles themselves, and the complete picture of the roles of the cytoskeleton in vesicle transport and exocytosis remains unclear. For instance, the combined action of actin-myosin and microtubule networks is required for intracellular lysosome movement.15 In contrast, only the microtubule network seems involved in secretory lysosomes and secretory granule exocytosis in different cells16; in mast cells, actin filaments have been shown to down-regulate degranulation.17
Despite the tight regulation of processing and release of IL-1β, several inflammatory disorders are known, each of which result from a defective secretion of IL-1β. Among these are the autoinflammatory syndromes such as Muckle-Wells syndrome, familial cold-induced autoinflammatory syndrome, familial mediterranean fever as well as systemic juvenile rheumatoid arthritis, which affect growing children and are particularly devastating.5 Importantly, blocking the IL-1 receptors in each of these diseases with treatment by the IL-1 receptor antagonist (IL-1Ra) returns these patients to normalcy,18-20 thus confirming the exquisite dependency of excessive secretion of IL-1β in the pathogenesis of these syndromes.
Although IL-1Ra therapy is safe and effective, it requires daily injections and its efficacy is directly related to occupancy of IL-1 type I receptors, which are present on nearly all cells.2 Furthermore, the relatively short in vivo half-life of the antagonist and urinary excretion necessitates the use of large quantities of the recombinant protein. For these reasons, blocking the secretion of IL-1β would offer a specific therapy for the inflammation due to IL-1β. Although developed to increase proapoptotic genes for the treatment of cancer,21 the inhibitor of histone deacetylases (HDACs), suberoylanilide hydroxamic acid (SAHA), reduces secretion of mature IL-1β from human peripheral blood mononuclear cells in vitro and the level of circulating IL-1β in mice that had been given injections of LPS, without affecting the steady-state levels of IL-1β mRNA or the intracellular levels of precursor IL-1β.22 However, SAHA does not target caspase-1, at least in vitro, and the mechanism underlying its ability to reduce the secretion of IL-1β remains to be clarified.22 Most HDACs are nuclear enzymes, involved in the epigenetic regulation of gene expression by modulating the acetylation state of core histones.23 Accordingly, the major and most studied substrates of HDACs are histones. Several transcription factors and nuclear import factors have also been shown to be regulated by processes of acetylation and deacetylation.24 In addition, 2 cytoplasmic targets of HDACs have been identified: α-tubulin25 and heat shock protein 90 (HSP90).26 Deacetylation of both proteins is mediated by HDAC6, which acts cytoplasmically.25,27 In addition, α-tubulin is a target of the NAD-dependent histone deacetylase SIRT2.28 Based on this information we hypothesized that the inhibition of HDAC activity could alter the microtubule organization leading to the inhibition of IL-1β secretion. Here we investigated the molecular components of the IL-1β secretory pathway, which may be affected by 2 HDAC inhibitors, SAHA and ITF2357, both hydroxamic acid-derived, orally active compounds with known anticancer activities29 but also anti-inflammatory properties.30
Materials and methods
Chemicals and antibodies
LPS, ATP, trichostatin A (TSA), HC-toxin, paclitaxel (Taxol), nocodazole, proteinase K, Ponceau S, and Tween-20 were purchased from Sigma-Aldrich (Milan, Italy). Arachidonyl trifluoromethylketone (AACOCF3) and bromoenol lactone were obtained from Alexis Biochemicals (Lausen, Switzerland). SAHA, ITF2357, and ITF-nil (ITF2375) were synthesized by Italfarmaco, Cinisello Balsamo, Italy.29,30 Tubacin31,32 was a kind gift from Drs R. Mazitschek and S. L. Schreiber (Harvard University, Cambridge, MA). The following antibodies were used: 3ZD anti-IL-1β monoclonal antibody (mAb) (IgG1, obtained from the National Cancer Institute Biological Resources Branch, Frederick, MD); rabbit anti-caspase-1 anti-serum R105 (kind gift from Dr D. K. Miller, Merck Research Laboratories, Rahway, NJ); anti-cathepsin D mAb (IgG2a, Calbiochem, Milan, Italy), anti-α-tubulin and anti-acetyl α-tubulin mAb (Sigma-Aldrich); rabbit anti-histone H4 and rabbit anti-acetylated histone H4 (Upstate, Lake Placid, NY), anti-HSP90 mAb (Stressgen, Victoria, BC, Canada) and anti-acetyl-lysine mAb (Cell Signaling Technology, Beverly, MA). Horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit IgG were from DAKO A/S, Glostrup, Denmark; fluorescein isothiocyanate (FITC)- or cyanine 3 (Cy3)-labeled secondary reagents were from Jackson ImmunoResearch, Soham, United Kingdom.
Cell cultures
Human monocytes isolated from buffy coats from healthy donors, enriched by adherence in RPMI 1640 medium containing 10% fetal bovine serum (all from Sigma-Aldrich) were activated with 1 μg/mL LPS for 4 hours or 18 hours at 37°C in RPMI 1640 medium supplemented with 1% Nutridoma-HU (Roche Applied Science, Monza, Italy) as described.4,7 In each experiment, supernatants were first collected and then replaced with RPMI/1% Nutridoma in the presence or absence of 1 mM ATP or other drugs as indicated and incubated for the indicated times. After the addition of ATP, supernatants were collected and cells were lysed in 1% Triton X-100 lysis buffer.4,7
Microglial N9 cells were obtained from Dr C. Verderio (Consiglio Nazionale delle Ricerche-Institute of Neuroscience, Milan, Italy) and maintained as described.33 One million cells were activated with 100 ng/mL LPS for 4 hours followed by 1 mM ATP for 20 minutes as described.33
Subcellular fractionation by differential ultracentrifugation
Subcellular fractionation was performed as described.4,7 Briefly, monocytes were washed, resuspended in homogenizing buffer (250 mM sucrose, 5 mM EGTA, 20 mM HEPES-KOH, pH 7.2) at 5 × 107/mL and disrupted in a Dounce homogenizer. The postnuclear supernatants (PNSs) were diluted 10-fold and centrifuged at 50 000g for 5 minutes, resulting in a pellet enriched in endo-lysosomes, as confirmed by electron microscopy (not shown); the pellet was treated with 0.1 mg/mL proteinase K in the presence or absence of 1% Triton X-100 for 30 minutes on ice. The 50 000g-generated supernatants were subjected to 40 minutes of centrifugation at 100 000g to obtain soluble cytosol.
ELISA analyses
IL-1β, IL-8, and TNF-α content of supernatants from human monocyte cultures was determined by enzyme-linked immunosorbent assay (ELISA; R&D Systems, Minneapolis, MN).
The IL-1β content of supernatants of N9 cells was assayed by ELISA (Pierce Endogen, Woburn, MA). Triton X-100 (2%) was added to supernatants to solubilize microvesicles.33
Western blot analysis
Cell lysates, pellets from subcellular fractionation, trichloroacetic acid-concentrated supernatants or cytosolic fractions were boiled in reducing Laemmli sample buffer, resolved on 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electrotransferred as described.4,7 Filters were probed with the different Abs followed by the relevant secondary Abs as indicated and developed with ECL-plus (Amersham Pharmacia Biotech, Milan, Italy). Densitometric analyses were performed by analyzing at least 3 different exposures of the same blot.
Flow cytometry
Cells were processed and stained with anti-α-tubulin or anti-acetyl-tubulin followed by the relevant secondary reagents as described.34 Data acquisition was performed using a FACSort cytometer (Becton Dickinson, Milan, Italy). The percentage of positive cells and mean values of fluorescence intensity were evaluated using CellQuest software (Becton Dickinson).
Determination of phospholipase activity
PC-PLC and PLA2 activity was measured using the Amplex Red PC-PLC assay kit provided by Molecular Probes Europe (Leiden, The Netherlands) and the cPLA2 Assay Kit by Cayman Chemical (Ann Arbor, MI) according to the manufacturer's instructions. Briefly, 50 μg cell lysates were used for each sample. PLC activity (mU/μg) was determined by comparison with the positive controls.4 PLA2 activity (nmol/μg per minute) was calculated as indicated in the kit instructions.4
Calcium mobilization assay
Monocytes adherent on a glass coverslip, activated with 1 μg/mL LPS for 4 hours at 37°C in the presence or absence of HDAC inhibitors, as indicated, were loaded with the acetoxymethyl-ester of Fura-2 (Fura-2-AM, 1 M) for 1 hour, placed under an AXIOVERT 10 microscope (Zeiss, Milan, Italy) maintained at 37°C, and stimulated with 1 mM ATP. Fura-2-AM was excited at 340 nm and 380 nm, and emitted light was filtered at 510 nm. The fluorescence ratio at 340:380 was evaluated by a charged-coupled device camera (Atto Instruments, Rockville, MD). Results are presented as the mean of fluorescence of at least 50 cells in each experiment monitored for 15 minutes. [Ca2+]i increase was calculated as described.4
Measurement of K+ efflux
Two million monocytes, which had been incubated for 4 hours with 1 μg/mL LPS in the presence or absence of HDAC inhibitors were exposed to 1 mM ATP for 30 minutes. The reaction was stopped by removal of the medium followed by lysis of the cells in 10% nitric acid. Cell extracts were then spun at 100 000g for 1 hour and the K+ content was assayed in an atomic absorption spectrophotometer.9,12
Cytolytic assay
Natural killer (NK)-cell cytolytic activity against K562 was tested in a 51Cr-release assay as described.34 Briefly, K562 cells were loaded with 51Cr and cocultured for 4 hours with NK cells (pretreated for 4 hours with the different drugs) used as effector cells, at a different effector-to-target (E/T) ratio. Results are expressed as percentage of cytotoxicity as described.34
Mixed lymphocyte reaction
Allospecific T cells were obtained by coculturing purified T cells with allogenic irradiated peripheral blood mononuclear cells as described.35 After 1 week of coculture, 105 allospecific T cells pretreated for 4 hours with the inhibitors were cocultured with 104 allogeneic dendritic cells (DCs) in 96-well plates for 3 days. Cells were pulsed with 1 μCi (0.037 MBq) [3H]thymidine (Amersham Pharmacia Biotech, Piscataway, NJ) per well for the last 18 hours of culture, harvested, and counted in a β-counter (Packard, Milan, Italy). Tests were conducted in triplicate, and results are expressed as mean cpm plus or minus standard deviation (SD).
Results
SAHA and ITF2357 inhibit the secretion of mature IL-1β but do not affect synthesis or intracellular distribution of the IL-1β precursor
Monocytes were stimulated for 4 hours with LPS in the presence or absence of increasing concentrations of SAHA or ITF2357, and then exposed for an additional 15 minutes to ATP. Secreted IL-1β was determined by Western blotting. Secretion of IL-1β during the 4 hours of LPS stimulation is low (Figure 1A, lane 6); in contrast, when LPS-stimulated monocytes are exposed to ATP, a marked increase in the secretion of IL-1β is observed (Figure 1A, lane 7). The 2 inhibitors do not affect the amount of precursor IL-1β synthesized by LPS-stimulation (compare Figure 1A, lanes 4 and 5 with Figure 1A, lane 2) but impressively reduced the portion of secreted IL-1β (Figure 1A, lanes 9 and 10). ATP-induced IL-1β secretion is paralleled by secretion of caspase-14,36 and of the lysosomal enzyme cathepsin D.4 Treatment with SAHA or ITF2357 prevents the ATP-induced secretion of the 3 proteins (Figure 1A-B), suggesting that the 2 compounds exert their inhibitory effects on IL-1β secretion at the level of lysosomal exocytosis. SAHA is active at 0.1 μM (70% of inhibition), but loses efficacy at lower concentrations (Figure 1B). In contrast, ITF2357 maintains its inhibitory effect at low concentrations; for example, at 20 nM ITF2357 inhibits IL-1β secretion by 60%.
Figure 1.
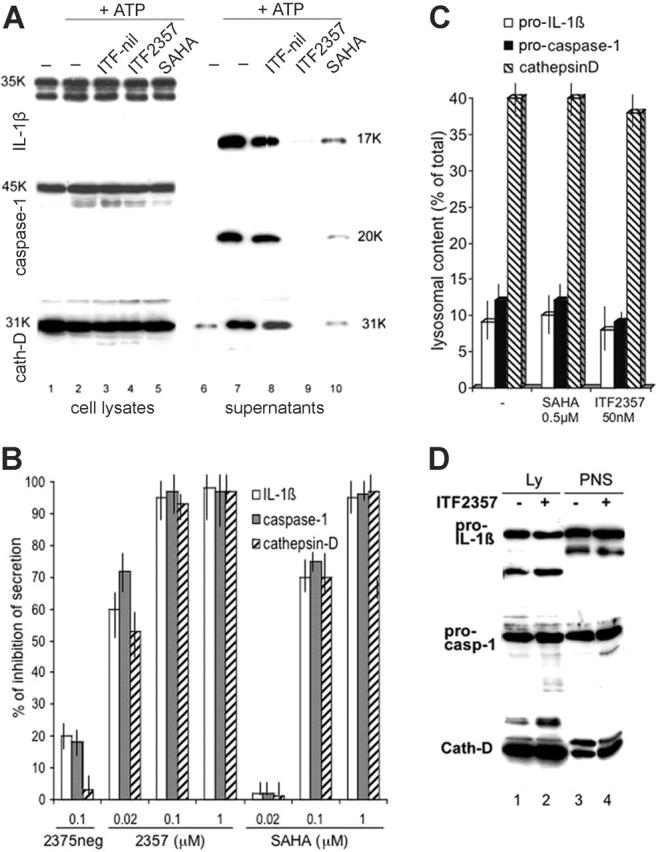
Both SAHA and ITF2357 inhibit IL-1β secretion but do not affect IL-1β intracellular accumulation and targeting. (A) Monocytes were activated 4 hours with LPS in the absence (lanes 1, 2, 6, and 7) or presence of 50 nM ITF2357 (lanes 4 and 9) or 50 nM of the negative compound ITF2375 (ITF-nil, lanes 3 and 8) or 0.5 μM SAHA (lanes 5 and 10), and then exposed to 1 mM ATP for 15 minutes (lanes 2-5 and 7-10). Supernatants and cell lysates were analyzed by Western blotting for the presence of IL-1β (top panel), caspase-1 (middle panel), and cathepsin D (cath-D, bottom panel). One representative experiment out of 10 performed is shown. (B) Densitometric analyses of Western blots of mature IL-1β, caspase-1, and cathepsin D secreted following exposure to different amounts of the 3 drugs as indicated. Results are expressed as percent of inhibition of secretion plus or minus standard error (SE) of a single experiment performed in triplicate. One representative experiment of at least 5 performed is shown. Data were validated by ELISA (not shown). In 13 different donors, IL-1β secreted in 15 minutes of ATP stimulation by 0.5 × 106 cells ranged between 5 ng and 15 ng (not shown). (C,D) Monocytes were cultured 4 hours with LPS, in the absence or presence of 50 nM ITF2357 or 0.5 μM SAHA, and a postnuclear supernatant (PNS) and an endolysosome enriched fraction were obtained as described in “Materials and methods.” (C) Densitometric analyses of Western blots showing the percent of pro-IL-1β, procaspase-1, and cathepsin-D in the lysosomal fractions of monocytes activated with LPS in the absence (-) or presence of SAHA or ITF2357. (D) Western blot of a representative experiment showing endolysosomal-enriched fractions (Ly, lanes 1 and 2) and aliquots (1/10) of postnuclear supernatants (PNS, lanes 3 and 4) from monocytes exposed for 4 hours to LPS (lanes 1 and 3) or to LPS and ITF2357 (lanes 2 and 4). Filters were hybridized with anti-IL-1β (top panel), anti-caspase-1 (middle panel), anti-cathepsin D (bottom panel). For each panel, one representative experiment of at least 3 performed is shown.
To further investigate the mechanism of action of the 2 compounds, we studied the effects of SAHA and ITF2357 on the intracellular localization of pro-IL-1β, procaspase-1, and cathepsin D. Monocytes stimulated with LPS in the presence or absence of the inhibitors were subcellularly fractionated and the presence of IL-1β in the lysosomal-enriched fraction was analyzed. Figure 1C shows that the relative amount of pro-IL-1β detected in the lysosomes of untreated cells (about 10% of the total cellular content) was equivalent to that found in the same fraction of cells exposed to SAHA or ITF. Similarly, the procaspase-1 content in the same lysosomal fraction was unaffected by treatment with the 2 compounds. As a control, the endolysosomal marker cathepsin D, which cofractionates with pro-IL-1β and procaspase-1 in comparable amounts (about 40% of the total content), is present in both untreated and treated cells. Figure 1D shows a representative experiment, with similar amounts of pro-IL-1β (upper panel), procaspase-1 (middle panel), and cathepsin D (lower panel) in lysosomal fractions (Ly) and postnuclear supernatants (PNSs) of untreated or ITF2357-treated monocytes.
These data indicate that the 2 compounds do not interfere with the intracellular targeting of pro-IL-1β and caspase-1.
Both SAHA and ITF prevent PLA2 but not PLC activation
We have previously shown that IL-1β secretion by monocytes is driven by a series of sequential events involving PC-PLC and cPLA2 activation.4 Therefore, we investigated whether SAHA or ITF2357 interfere with phospholipase functions. As shown in Figure 2, whereas PLC activation is unaffected by ITF2357 or SAHA, both compounds inhibit PLA2 activation by 50%.
Figure 2.

Both SAHA and 2357 prevent PLA2 activation. Monocytes stimulated for 4 hours with LPS in the absence or presence of 50 nM ITF2357 or 0.5 μM SAHA. The cells were then incubated for 2 minutes with 2 mM ATP and the presence of active PC-PLC or PLA2 in cell lysates was determined. Data are expressed as percent of PL activity of control (LPS-ATP stimulated) cells. PLC activity (mU/μg) and PLA2 activity (nmol/μg per minute) in a representative experiment were, respectively, 1.15 ± 0.002 and 0.44 ± 0.01 after LPS-ATP treatment and 1.21 ± 0.004 and 0.22 ± 0.018 after LPS-ATP treatment in the presence of ITF2357 or SAHA, respectively.
Kinetics of SAHA and ITF inhibitory effects
To investigate the effect of SAHA or ITF2357 on cells actively synthesizing pro-IL-1β, monocytes were stimulated with LPS and treated with the 2 compounds for the entire time of culture (4 hours) or exposed to the inhibitors for the last 2 hours or 1 hour. At the end of the incubation time cells were triggered for 10 minutes with ATP, and secretion of IL-1β, caspase-1, and cathepsin D was evaluated. As shown in Figure 3, 4 hours of treatment with SAHA or ITF2357 lead to a complete inhibitory effect on IL-1β secretion. Only about 25% of inhibition is obtained when monocytes cultured for 2 hours with LPS were exposed to the compounds for an additional 2 hours before triggering with ATP, and no inhibition is observed when treatment is restricted to the last hour before addition of ATP.
Figure 3.

Four hours of treatment with SAHA and ITF2357 are required to prevent IL-1β secretion. Monocytes were stimulated with LPS and treated without or with 50 nM ITF2357 or 0.5 μM SAHA for the entire time of culture (4h) or left untreated the first 2 hours and treated the last 2 hours (2h), or left untreated 3 hours and treated the last 1 hour (1h). At the end of the incubation time, cells were triggered for 10 minutes by ATP and secretion of IL-1β, caspase-1, and cathepsin D was evaluated by Western blot followed by densitometric analyses. Data are expressed as percent of inhibition of secretion of IL-1β, caspase-1, and cathepsin D. One representative experiment of 4 performed is shown.
HDAC inhibitors with different specificity block IL-1β secretion
SAHA possesses well-known HDAC inhibitory activities22,37; ITF2357 has recently been shown to cause H3 and H4 histone hyperacetylation in an hepatoma cell line and in PBMCs, respectively.29,30 We reasoned that the inhibition of lysosomal exocytosis observed with SAHA and ITF2357 could be due to their inhibitory effects on HDACs, and that the time elapsed between exposure to the inhibitors and block of IL-1β secretion (Figure 3) may be required for the hyperacetylation of a substrate, in turn responsible for blocking the exocytosis of the secretory lysosomes. We therefore investigated whether other HDAC inhibitors37 affect IL-1β secretion. Among the compounds tested, TSA, like SAHA, results in nonselective inhibition of all HDACs. In addition, the fungal metabolite, HC-toxin, is a trapoxin-related inhibitor, selective for class I HDACs, whereas tubacin specifically binds to the catalytic domain of HDAC6,31,32 which is responsible for the deacetylation of α-tubulin25,27,32 and HSP90.26 As shown in Figure 4A, exposures of monocytes to each of the different HDAC inhibitors resulted in a block of secretion of IL-1β without any effect on its intracellular accumulation. Similar inhibition was observed on caspase-1 and cathepsin D secretion (data not shown). HDAC inhibitors did not affect ATP-induced K+ efflux and Ca2+ entry, ruling out that their inhibitory effects on IL-1β secretion are due to interference with the proximal signaling responses to P2X7 activation (Figure 4B-C). Also, the different compounds decrease the amount of IL-1β secreted by monocytes during 18 hours of culture with LPS, without additional ATP stimulation, from 50% to 80% (Figure 4D).
Figure 4.

Different inhibitory effects of HDAC inhibitors on IL-1β, IL-8, and TNF-α secretion. (A) Monocytes were stimulated for 3 hours with LPS in the absence (lanes 1 and 2) or presence of TSA (1 μM, lane 3), HC-toxin (HC-T; 1 μM, lane 4), SAHA (0.5 μM, lane 5), or tubacin (Tub; 20 μM, lane 6) and then exposed to 1 mM ATP for 15 minutes (lanes 2-6). Supernatants and cell lysates were analyzed by Western blotting for the presence of IL-1β. (B) HDAC inhibitors do not affect ATP-induced K+ efflux. Monocytes as in panel A, untreated or exposed to 1 mM ATP for 20 minutes, were analyzed for K+ intracellular content. K+ depletion is expressed as percent of intracellular K+ in LPS-stimulated monocytes (control cells). (C) HDAC inhibitors do not affect ATP-induced [Ca2+]i rise. Monocytes as in panel A were loaded with Fura-2-AM and exposed to 1 mM ATP. Results are the mean of fluorescence of at least 50 cells in each experiment monitored for 15 minutes, and are expressed as increase in [Ca2+]i above non-ATP stimulated cells (Δ[Ca2+]i). (D) Monocytes were cultured for 18 hours with LPS and supernatants were assayed for the presence of IL-1β (□), IL-8 ( ), or TNF-α (▪) by ELISA. Results are expressed as percent of secretion by LPS-stimulated monocytes (control cells). One experiment of 3 performed is shown. In this experiment, 18 hours' secretion by 0.7 × 106 LPS-stimulated monocytes was: 9.5 ± 0.1 ng/mL for IL-1β; 305 ± 2 ng/mL for IL-8; 6.1 ± 0.3 ng/mL for TNF-α.
), or TNF-α (▪) by ELISA. Results are expressed as percent of secretion by LPS-stimulated monocytes (control cells). One experiment of 3 performed is shown. In this experiment, 18 hours' secretion by 0.7 × 106 LPS-stimulated monocytes was: 9.5 ± 0.1 ng/mL for IL-1β; 305 ± 2 ng/mL for IL-8; 6.1 ± 0.3 ng/mL for TNF-α.
To test the specificity of the effect of HDAC inhibitors on cytokine release, we compared the secretion of IL-1β with that of IL-8 and TNF-α. IL-8 is released by default via the ER-Golgi pathway.38 On the other hand, TNF-α is trafficked from the Golgi to the recycling endosomes, from which it is transported to the cell surface.39 As shown in Figure 4D, 18 hours of secretion of IL-8 was almost unaffected by HDACs, the highest inhibition being 25% and 30% upon treatment with TSA and HC-toxin, respectively. Secretion of TNF-α was decreased by the various HDAC inhibitors, although considerably less than that of IL-1β.
We then compared the effects of HDAC inhibitors on the acetylation state of nuclear (histones) and cytoplasmic (α-tubulin and HSP-90) targets of HDACs in activated monocytes. Treatment with each of the HDAC inhibitors except tubacin resulted in marked hyperacetylation of histone H4 (Figure 5A). In contrast, none of the compounds tested induced detectable acetylation of HSP90 (Figure 5B). Exposure to HDAC inhibitors resulted in different hyperacetylation of tubulin when evaluated both by cytofluorimetry (Figure 5C) and Western blotting (Figure 5D). However, HC-toxin did not result in a detectable change in the acetylation state of tubulin. Of note, ITF2357 induced H4 and tubulin acetylation, even if to a lesser extent than the other inhibitors, at a concentration greater than that required for inhibition of IL-1β secretion (100 nM versus 50 nM).
Figure 5.

HDAC inhibitors induce acetylation of different intracellular targets. (A) Monocytes stimulated for 3 hours with LPS in the absence or presence of ITF2357 (20 nM or 100 nM), TSA (1 μM), HC-toxin (HC-T; 1 μM), SAHA (0.5 μM), or tubacin (20 μM) were fractionated to obtain nuclei, which were subjected to SDS-PAGE and Western blotting. Filters were hybridized with anti-histone H4 (bottom panel) or anti-acetyl-H4 (top panel). (B) Monocytes stimulated for 3 hours with LPS in the absence or presence of SAHA (0.5 μM), ITF2357 (0.1 μM), HC-toxin (HC-T; 1 μM), or tubacin (tuba; 20 μM) were lysed and cell lysates were analyzed by Western blotting with anti-HSP90 (top panel) or anti-acetyl-lysine (bottom panel). The arrow in the top panel indicates HSP90; the arrow in the bottom panel indicates a 50-kDa band, probably corresponding to acetylated tubulin-α. (C) Monocytes treated as in panel A were fixed, permeabilized, and stained with anti-tubulin-α (not shown) or anti-acetyl-tubulin-α and analyzed by cytofluorimetry. Monocytes treated with the different inhibitors stained equally with anti-tubulin-α (not shown) but differently with anti-acetyl-tubulin-α. Data are expressed as mean fluorescence intensity (mean FI) of cells stained with anti-acetyl-tubulin-α. (D) Monocytes treated as in panel B were lysed and analyzed by Western blotting with anti-tubulin-α (top panel) or anti-acetyl-tubulin-α (bottom panel). Arrows indicate tubulin-α and acetyl-tubulin-α, respectively. For each panel, one representative experiment of at least 4 performed is shown.
Cytoskeletal inhibitors interfere with lysosome exocytosis
To further explore the role of cytoskeleton on lysosomal exocytosis, we studied the effects of different cytoskeleton-perturbing agents on the secretion of IL-1β. As shown in Figure 6, treatment with the actin depolymerizing fungal metabolite cytochalasin D17 increased IL-1β secretion in the absence of ATP triggering (Figure 6A, lane 3), confirming our previous data.40 We next examined the effects of 2 different substances, nocodazole and taxol, which act as microtubule disrupting and stabilizing agents, respectively, the net effect being the impairment of microtubular function.41 The results show that distinct from cytochalasin D, both drugs only slightly influenced basal secretion (Figure 6A, lanes 2, 4), but substantially impaired ATP-induced secretion (Figure 6A, lanes 6, 8). However, at high concentrations of taxol (20 nM and 40 mM), a paradoxical effect was often observed, with increased lysosomal exocytosis and secretion (Figure 6A, lanes 9, 10). As shown, taxol-induced hyperacetylation of tubulin was consistently dose dependent (Figure 6B). Together, these results indicate that microtubules participate in lysosomal exocytosis and that the levels of tubulin acetylation must be tightly balanced to allow a correct vesicle transport.
Figure 6.
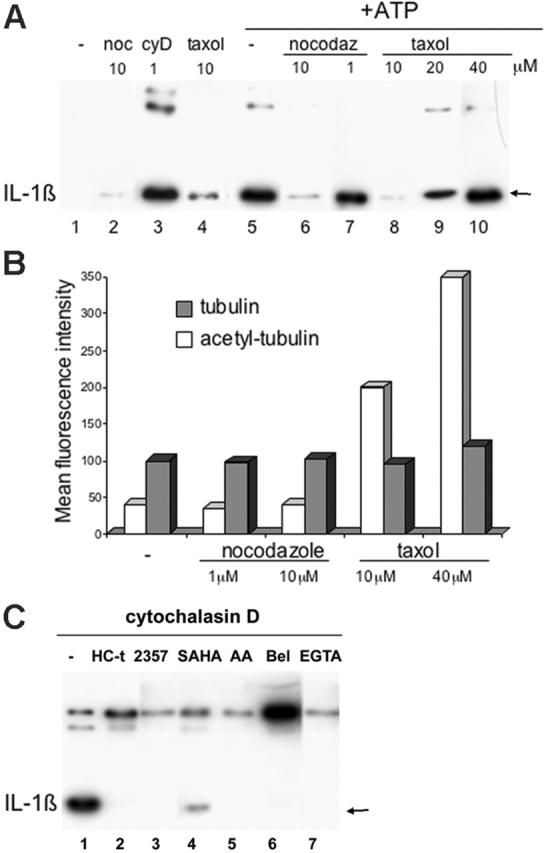
Microtubules are involved in IL-1β secretion. (A) Monocytes were stimulated for 4 hours with LPS in the absence (lanes 1 and 5) or presence of 1 μM cytochalasin D (cyD; lane 3), 1 and 10 μM nocodazole (noc; lanes 2, 6, and 7) or 10, 20, and 40 μM taxol (lanes 4, 8-10). At the end of the 4-hour incubation, supernatants were analyzed as such (lanes 1-4) or 1 mM ATP was added for 15 minutes (+ATP; lanes 5-10) before Western blot analysis for the presence of IL-1β. (B) Monocytes treated as in panel A were fixed, permeabilized, and stained with anti-tubulin-α ( )or anti-acetyl-tubulin-α (□) and analyzed by cytofluorimetry. Data are expressed as mean fluorescence intensity. For each panel, one representative experiment of at least 5 performed is shown. (C) Monocytes were activated 4 hours with LPS and 1 μM cytochalasin D in the absence (-; lane 1) or presence of HC-toxin (1 μM, lane 2), ITF2357 (50 nM, lane 3), SAHA (0.5 μM, lane 4), AACOCF3 (AA; 40 μM, lane 5), BEL (100 μM, lane 6), EGTA (5 mM, lane 7). At the end of the 4-hour incubation, supernatants were analyzed by Western blot for the presence of IL-1β.
)or anti-acetyl-tubulin-α (□) and analyzed by cytofluorimetry. Data are expressed as mean fluorescence intensity. For each panel, one representative experiment of at least 5 performed is shown. (C) Monocytes were activated 4 hours with LPS and 1 μM cytochalasin D in the absence (-; lane 1) or presence of HC-toxin (1 μM, lane 2), ITF2357 (50 nM, lane 3), SAHA (0.5 μM, lane 4), AACOCF3 (AA; 40 μM, lane 5), BEL (100 μM, lane 6), EGTA (5 mM, lane 7). At the end of the 4-hour incubation, supernatants were analyzed by Western blot for the presence of IL-1β.
In an attempt to understand the molecular basis of the stimulatory effect of cytochalasin D on IL-1β processing and secretion, we exposed LPS-treated monocytes to cytochalasin D together with compounds that block processing and/or secretion of IL-1β at different levels. Figure 6C shows that Ca2+ depletion, which prevents ATP-triggered IL-1β secretion,7,13 also inhibits the secretion induced by cytochalasin D (Figure 6C, lane 7 versus lane 1). Likewise, AACOCF3, which blocks cPLA2, essential for IL-1β externalization,4 strongly inhibits secretion (Figure 6C, lane 5). Interestingly, cytochalasin D-induced secretion is also prevented by pretreatment of monocytes with HDAC inhibitors (Figure 6C, lanes 2-4). However, treatment with BEL, an inhibitor of iPLA2 that prevents processing of the precursor form of IL-1β,4,12 results in increased secretion of unprocessed pro-IL-1β (Figure 6C, lane 6).
Effects of deacetylase inhibitors on T-lymphocyte and NK-cell activity
Secretory lysosomes and tubulin rearrangement are involved in a number of immunologic-related responses, including NK cell-mediated cytotoxicity and antigen presentation.16,42 To assess whether HDAC inhibitors have any effects on these responses, we tested the different compounds in 2 in vitro assays, NK killing and a secondary mixed lymphocyte reaction (MLR). As shown in Figure 7A, neither SAHA nor ITF2357 have significant effects on NK-cell lysis of the tumor cell line K562. In contrast, a small reduction in the killing was observed after exposure to TSA, HC-toxin, or tubacin. Of note, a clear correlation with induction of tubulin acetylation in NK cells is absent (Figure 7B). It remains unclear why ITF2357-treated NK cells, unlike monocytes, do not display detectable acetylation of tubulin even at higher concentrations (100 nM). When the different compounds were tested on alloreactive T cells toward allogenic DCs, ITF2357 and HC-toxin did not reduce or enhance proliferation; low levels of inhibition were observed with SAHA, TSA, and tubacin (Figure 7C).
Figure 7.

HDAC inhibitors interfere barely with T-lymphocyte or NK cell activity. (A) NK cells were pretreated for 4 hours with ITF2357 (50 nM), SAHA (0.5 μM), TSA (1 μM), HC-toxin (1 μM), or tubacin (10 μM) and coincubated with 51Cr-labeled K562 cells for 4 hours at a different effector-target (E/T) ratio, in the presence of the same HDAC inhibitors. Results are expressed as percentage of cytotoxicity as described. (B) NK cells treated for 4 hours with ITF2357 (50 nM), SAHA (0.5 μM), TSA (1 μM), HC-toxin (1 μM), or tubacin (10 μM) were fixed, permeabilized, and stained with anti-tubulin-α (not shown) or anti-acetyl-tubulin-α and analyzed by cytofluorimetry. The different cultures stained equally with anti-tubulin-α (not shown). Data are expressed as mean fluorescence intensity (mean FI) of cells stained for acetyl-tubulin-α. (C) Allospecific T cells from 7-day MLR (105) were pretreated for 4 hours with ITF2357 (50 nM), SAHA (0.5 μM), TSA (1 μM), HC-toxin (1 μM), or tubacin (10 μM) and coincubated with 104 allogeneic DCs for 18 hours in the presence of the same HDAC inhibitors and of 1 μCi (0.037 MBq) of [3H]thymidine per well. Cells were harvested and counted in a beta counter. Tests were conducted in triplicate, and results are expressed as mean cpm ± SD. For each panel, one representative experiment of at least 3 performed is shown.
Discussion
The development of novel therapeutic strategies for inhibition of IL-1β activity is central for the treatment of many systemic inflammatory diseases caused by this cytokine. Understanding the mechanisms that control IL-1β processing and release is therefore of considerable importance not only as a matter of basic mechanisms but also for the development of innovative anti-inflammatory therapies. We have previously identified a subset of secretory lysosomes as vehicles for the extracellular delivery of IL-1β, and outlined the key role of the sequential activation of phospholipases C and A2 in regulating exocytosis.4,7
Here we extend the understanding of the IL-1β secretion pathway by defining the involvement of the cytoskeleton in the process of exocytosis. We report that microtubule targeting agents, including taxol and nocodazole, impair IL-1β secretion, indicating that a functional microtubule network is required for the release of this cytokine. Microtubules play important roles in the immuneinflammatory response. Tubulin rearrangement in dendritic cells is critical not only for the organization of the immune synapse at the interface with T cells42 but also for the polarized release of cytokine-containing secretory lysosomes at the synaptic cleft.34,35 This is in agreement with the function of microtubules as tracks along which organelles and vesicles are transported through the cell, and supports previous observations that the microtubular cytoskeleton is essential for exocytosis of secretory lysosomes in hemopoietic cells.16
A major finding of this study is that HDAC inhibitors are highly effective in reducing the secretion of IL-1β. Remarkably, all the HDAC inhibitors assessed in this study, apart from HC-toxin, prevent IL-1β secretion and induce tubulin acetylation. Tubulin undergoes acetylation by the cytoplasmic HDAC625,27 or by the recently described type III HDAC SIRT2.28 Since the HDAC inhibitors we explored are directed to class I or II enzymes, with no activity on class III, as it is for ITF2357 (not shown), their target is likely to be HDAC6 rather than HDAC SIRT2.
A role for tubulin acetylation has been proposed in cell motility27 but the true significance of this posttranslational modification as well as its role in other microtubule functions is still debated.43 It is conceivable that tubulin acetylation affects the activity of microtubule-associated proteins or motors, for this would result in an impaired vesicle transport. Interestingly, the microtubule stabilizing agent taxol, which induces a concentration-dependent acetylation of tubulin, is less effective in blocking secretion of IL-1β at concentrations resulting in greater acetylation. This is reminiscent of the paradoxical effect reported for SAHA on IL-1β secretion: at high concentrations (which results in stronger tubulin acetylation, not shown) the inhibitory effects on IL-1β secretion decreased or even reversed.22 These observations suggest that the degree of tubulin acetylation must be under a tight balance to allow correct progression of exocytosis. For example, an excessive acetylation may result in a loss of contact with regulatory proteins associated to microtubules. Alternatively, high doses of taxol or HDAC inhibitors may deregulate other posttranslational modifications of tubulin,43 resulting in enhanced rather than reduced exocytosis.
The lack of detection of intracellular mature IL-1β and caspase-1 upon treatment with HDAC inhibitors suggests that these drugs also attenuate the activity of either caspase-1 or other inflammasome regulatory proteins.5 As iPLA2, which is implicated in caspase-1 activation,7,11,12 is also involved in various cytoskeleton-mediated processes of membrane trafficking,44 it is tempting to speculate that tubulin hyperacetylation inhibits iPLA2 activity.
The discrepancy between the effects of HC-toxin on lysosomal exocytosis, which is blocked, and tubulin acetylation, which apparently is unaffected (Figure 5 and Haggarty et al32), suggests that either a cellular target different from tubulin is responsible for the inhibitory effects of this compound on IL-1β secretion, or that low levels of tubulin acetylation, undetectable in our assays (cytofluorimetry and Western blot), are sufficient to prevent lysosomal exocytosis. Consistent with the latter possibility, high concentrations of ITF2357 induced tubulin acetylation, but low concentrations effectively blocked IL-1β secretion without detectable modifications on the acetylation state of tubulin. In any case, the kinetics of inhibition of IL-1β secretion by HDAC inhibitors (Figure 3 and not shown) reveals that 4 hours of exposure to the drugs are required to achieve the inhibitory effect, suggesting that one or more hyperacetylated substrates participate in reducing lysosomal exocytosis. The possibility that HDAC inhibitors, acting on nuclear substrates, promote the synthesis of unknown proteins able to contrast lysosome exocytosis is unlikely, as blocking protein synthesis in LPS-activated monocytes does not prevent the secretion of previously synthesized IL-1β.6 Our data do not, however, rule out the possibility that other HDAC6-sensitive molecules may also (or alternatively) participate in the secretion inhibitory process.
Treatment of monocytes with HDAC inhibitors does not affect the proximal signaling responses to P2X7 activation (eg, K+ efflux and Ca2+ influx),8 but inhibits the activity of cPLA2 (Figure 2), which is required for IL-1β-containing lysosome exocytosis.4 Functional association between microtubular cytoskeleton and phospholipases has been demonstrated in several models.45-48 In particular, cPLA2 is implicated in EGF- or FGF-induced cytoskeletal reorganization and has been found to interact with microtubules.47,48 Based on these observations, it is possible that in monocytes the activation of cPLA2, as discussed for iPLA2, requires a microtubule rearrangement: induction of tubulin acetylation even if weak, such as in the case of ITF2357, could prevent this activation.
In contrast to microtubule poisons, cytochalasin D induces IL-1β secretion, confirming our previous results.40 This enhanced secretion is likely to be mediated by Ca2+ entry and cPLA2 activation, since monocytes exposed to cytochalasin D in the presence of Ca2+ chelators or PLA2 inhibitors do not exhibit increased secretion. These findings are in agreement with previous reports indicating that microfilament disruption leads to increased activity of cPLA217 and that cytochalasin D elicits Ca2+ mobilization and activates cPLA2 in polymorphonuclear leukocytes.49
Studies on macrophage or microglia cell lines have suggested that low efficiency ATP-triggered IL-1β secretion is mediated by microvesicles shed from the plasma membrane.33,50 When HDAC inhibitors were added to the N9 microglia cell line, the release of IL-1β was unaffected (data not shown), suggesting that tubulin and cPLA2 are not implicated in this pathway.
The HDAC inhibitors explored in this paper display a relevant specificity for IL-1β release, induced either by LPS only or by LPS plus exogenous ATP. Indeed, the same HDAC inhibitors only slightly affect the release of the chemokine IL-8, a proinflammatory protein secreted through the classical exocytotic pathway.38 In contrast, the amount of TNF-α39 secreted by HDAC-treated monocytes is decreased, although to a lesser extent than IL-1β. In the case of IL-1β, HDAC inhibitors prevent the secretion but not the synthesis of the precursor (Figures 1A, 4A). In the case of TNF-α, inhibition occurs at the level of gene expression (not shown), as reported for SAHA22 and ITF2357.30
HDAC inhibitors have recently emerged as novel agents with potential therapeutic value for the treatment of cancer, and are currently in development or early-phase clinical investigation.51 Our present data suggest that these drugs can also be suitable for the treatment of inflammation. In particular, ITF2357, given its very high activity and specificity, is a good candidate to be developed as an anti-inflammatory drug that could have wide utility in numerous IL-1β-mediated inflammatory conditions.
Acknowledgments
We thank Drs S. L. Schreiber and R. Mazitschek (Cambridge, MA) as well as the Howard Hughes Medical Institute (Harvard University, Cambridge, MA) and the Initiative for Chemical Genetics, National Cancer Institute, for kindly providing us with tubacin; the National Cancer Institute Biological Resources Branch (Frederick, MD) for providing 3ZD anti-IL-1β mAb; Dr D. K. Miller (Merck Research Laboratories, Rahway, NJ) for the kind gift of the rabbit anti-caspase-1 antiserum R105; Dr A. Poggi and Mr G. Rossi (Genoa, Italy) for Ca2+ and K+ measurement, respectively, and the Blood Centers of Ospedale Galliera and S. Martino (Genoa, Italy) for buffy coats.
Prepublished online as Blood First Edition Paper, May 9, 2006; DOI 10.1182/blood-2006-03-014126.
Supported in part by grants from Associazione Italiana per la Ricerca sul Cancro, Ministero della Salute, Comitato Interministeriale di Programmazione Economica (CIPE) (02/07/2004, Centro di Biotecnologie Avanzate [CBA] project) and National Institutes of Health grants AI-15 614 (C.A.D.) and HL-68 743 (C.A.D.).
G.F. and P.M. are employed by Italfarmaco (Milan, Italy), whose potential product ITF2357 was studied in the present work. Currently, Italfarmaco has no commercial products related to the compounds discussed in the present article.
A.R., S.C., S.T., and C.S. designed research; S.C., S.T., and C.S. performed research; P.M. and G.F. contributed ITF2357 and ITF2375 and critically reviewed the paper; C.A.D. contributed to the design of the research, and to the critical review of the experiments and the manuscript; A.R. supervised the experiments and wrote the paper.
S.C. and S.T. contributed equally to this study.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87: 2095-2147. [PubMed] [Google Scholar]
- 2.Dinarello CA. Interleukin-1, interleukin-1 receptors and interleukin-1 receptor antagonist. Int Rev Immunol. 1998;16: 457-499. [DOI] [PubMed] [Google Scholar]
- 3.Schindler R, Gelfand JA, Dinarello CA. Recombinant C5a stimulates transcription rather than translation of interleukin-1 (IL-1) and tumor necrosis factor: translational signal provided by lipopolysaccharide or IL-1 itself. Blood. 1990;76: 1631-1638. [PubMed] [Google Scholar]
- 4.Andrei C, Margiocco P, Poggi A, Lotti LV, Torrisi MR, Rubartelli A. Phospholipases C and A2 control lysosome-mediated IL-1 beta secretion: implications for inflammatory processes. Proc Natl Acad Sci U S A. 2004;101: 9745-9750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117: 561-574. [DOI] [PubMed] [Google Scholar]
- 6.Rubartelli A, Cozzolino F, Talio M, Sitia R. A novel secretory pathway for interleukin-1beta, a protein lacking a signal sequence. EMBO J. 1990;9: 1503-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andrei C, Dazzi C, Lotti L, Torrisi MR, Chimini G, Rubartelli A. The secretory route of the leaderless protein interleukin 1beta involves exocytosis of endolysosome-related vesicles. Mol Biol Cell. 1999;10: 1463-1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Di Virgilio F, Chiozzi P, Ferrari D, et al. Nucleotide receptors: an emerging family of regulatory molecules in blood cells. Blood. 2001;97: 587-600. [DOI] [PubMed] [Google Scholar]
- 9.Kahlenberg JM, Dubyak GR. Mechanisms of caspase-1 activation by P2X7 receptor-mediated K+ release. Am J Physiol Cell Physiol. 2004;286: 1100-1108. [DOI] [PubMed] [Google Scholar]
- 10.Cheneval D, Ramage P, Kastelic T, et al. Increased mature interleukin-1beta (IL-1 beta) secretion from THP-1 cells induced by nigericin is a result of activation of p45 IL-1beta-converting enzyme processing. J Biol Chem. 1998;273: 17846-17851. [DOI] [PubMed] [Google Scholar]
- 11.Walev I, Reske K, Palmer M, Valeva A, Bhakdi S. Potassium-inhibited processing of IL-1 beta in human monocytes. EMBO J. 1995;14: 1607-1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walev I, Klein J, Husmann M, et al. Potassium regulates IL-1 beta processing via calcium-independent phospholipase A2. J Immunol. 2000; 164: 5120-5124. [DOI] [PubMed] [Google Scholar]
- 13.Gudipaty L, Munetz J, Verhoef PA, Dubyak GR. Essential role for Ca2+ in regulation of IL-1beta secretion by P2X7 nucleotide receptor in monocytes, macrophages, and HEK-293 cells. Am J Physiol Cell Physiol. 2003;285: 286-299. [DOI] [PubMed] [Google Scholar]
- 14.Elssner A, Duncan M, Gavrilin M, Wewers MD. A novel P2X7 receptor activator, the human cathelicidin-derived peptide LL37, induces IL-1 beta processing and release. Immunol. 2004;172: 4987-4994. [DOI] [PubMed] [Google Scholar]
- 15.Cordonnier MN, Dauzonne D, Louvard D, Coudrier E. Actin filaments and myosin I alpha cooperate with microtubules for the movement of lysosomes. Mol Biol Cell. 2001;12: 4013-4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bossi G, Griffiths GM. CTL secretory lysosomes: biogenesis and secretion of a harmful organelle. Semin Immunol. 2005;17: 87-94. [DOI] [PubMed] [Google Scholar]
- 17.Frigeri L, Apgar JR. The role of actin microfilaments in the downregulation of the degranulation response in RBL-2H3 mast cells. J Immunol. 1999;162: 2243-2250. [PubMed] [Google Scholar]
- 18.Verbsky JW, White AJ. Effective use of the recombinant interleukin 1 receptor antagonist anakinra in therapy resistant systemic onset juvenile rheumatoid arthritis. J Rheumatol. 2004;31: 2071-2075. [PubMed] [Google Scholar]
- 19.Hoffman HM, Rosengren S, Boyle DL, et al. Prevention of cold-associated acute inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist. Lancet. 2004;364: 1779-1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med. 2005;201: 1479-1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marks PA, Richon VM, Breslow R, Rifkind RA. Histone deacetylase inhibitors as new cancer drugs. Curr Opin Oncol. 2001;13: 477-483. [DOI] [PubMed] [Google Scholar]
- 22.Leoni F, Zaliani A, Bertolini G, et al. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc Natl Acad Sci U S A. 2002;99: 2995-3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370: 737-749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang XJ, Gregoire S. Class II histone deacetylases: from sequence to function, regulation, and clinical implication. Mol Cell Biol. 2005;25: 2873-2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y, Li N, Caron C, et al. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003;22: 1168-1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aoyagi S, Archer TK. Modulating molecular chaperone Hsp90 functions through reversible acetylation. Trends Cell Biol. 2005;15: 565-567. [DOI] [PubMed] [Google Scholar]
- 27.Hubbert C, Guardiola A, Shao R, et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417: 455-458. [DOI] [PubMed] [Google Scholar]
- 28.North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell. 2003;11: 437-444. [DOI] [PubMed] [Google Scholar]
- 29.Armeanu S, Pathil A, Venturelli S, et al. Apoptosis on hepatoma cells but not on primary hepatocytes by histone deacetylase inhibitors valproate and ITF2357. J Hepatol. 2005;42: 210-217. [DOI] [PubMed] [Google Scholar]
- 30.Leoni F, Fossati G, Lee J-K, et al. The histone deacetylase inhibitor ITF2357 reduces production of pro-inflammatory cytokines in vitro and systemic inflammation in vivo. Mol Med. 2005; 11: 1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haggarty SJ, Koeller KM, Wong JC, Butcher RA, Schreiber SL. Multidimensional chemical genetic analysis of diversity-oriented synthesis-derived deacetylase inhibitors using cell-based assays. Chem Biol. 2003;10: 383-396. [DOI] [PubMed] [Google Scholar]
- 32.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci U S A. 2003;100: 4389-4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bianco F, Pravettoni E, Colombo A, et al. Astrocyte-derived ATP induces vesicle shedding and IL-1β release from microglia. J Immunol. 2005; 174: 7268-7277. [DOI] [PubMed] [Google Scholar]
- 34.Semino C, Angelini G, Poggi A, Rubartelli A. NK/iDC interaction results in IL-18 secretion by DCs at the synaptic cleft followed by NK cell activation and release of the DC maturation factor HMGB1. Blood. 2005;106: 609-616. [DOI] [PubMed] [Google Scholar]
- 35.Gardella S, Andrei C, Costigliolo S, Olcese L, Zocchi MR, Rubartelli A. Secretion of bioactive interleukin-1beta by dendritic cells is modulated by interaction with antigen specific T cells. Blood. 2000;95: 3809-3815. [PubMed] [Google Scholar]
- 36.Laliberte RE, Eggler J, Gabel CA. ATP treatment of human monocytes promotes caspase-1 maturation and externalization. J Biol Chem. 1999; 274: 36944-36951. [DOI] [PubMed] [Google Scholar]
- 37.Dokmanovic M, Marks PA. Prospects: histone deacetylase inhibitors. J Cell Biochem. 2005;96: 293-304. [DOI] [PubMed] [Google Scholar]
- 38.Baggiolini M, Walz A, Kunkel SL. Neutrophil-activating peptide-1/interleukin 8, a novel cytokine that activates neutrophils. J Clin Invest. 1989;84: 1045-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murray RZ, Kay JG, Sangermani DG, Stow JL. A role for the phagosome in cytokine secretion. Science. 2005;310: 1492-1495. [DOI] [PubMed] [Google Scholar]
- 40.Rubartelli A, Bajetto A, Allavena G, Cozzolino F, Sitia R. Post-translational regulation of interleukin 1 beta secretion. Cytokine. 1993;5: 117-124. [DOI] [PubMed] [Google Scholar]
- 41.Nishida K, Yamasaki S, Ito Y, et al. Fc{epsilon}RI-mediated mast cell degranulation requires calcium-independent microtubule-dependent translocation of granules to the plasma membrane. J Cell Biol. 2005;170: 115-126. Erratum in: J Cell Biol. 2005;171:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Serrador JM, Cabrero JR, Sancho D, Mittelbrunn M, Urzainqui A, Sanchez-Madrid F. HDAC6 deacetylase activity links the tubulin cytoskeleton with immune synapse organization. Immunity. 2004;20: 417-428. [DOI] [PubMed] [Google Scholar]
- 43.Westermann S, Weber K. Post-translational modifications regulate microtubule function. Nat Rev Mol Cell Biol. 2003;4: 938-947. [DOI] [PubMed] [Google Scholar]
- 44.Drecktrah D, Brown WJ. Phospholipase A(2)antagonists inhibit nocodazole-induced Golgi ministack formation: evidence of an ER intermediate and constitutive cycling. Mol Biol Cell. 1999;10: 4021-4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang JS, Kim SK, Kwon TK, et al. Pleckstrin homology domains of phospholipase C-gamma1 directly interact with beta-tubulin for activation of phospholipase C-gamma1 and reciprocal modulation of beta-tubulin function in microtubule assembly. J Biol Chem. 2005;280: 6897-6905. [DOI] [PubMed] [Google Scholar]
- 46.Zheng XL, Gui Y, Du G, Frohman MA, Peng DQ. Calphostin-C induction of vascular smooth muscle cell apoptosis proceeds through phospholipase D and microtubule inhibition. J Biol Chem. 2004;279: 7112-7118. [DOI] [PubMed] [Google Scholar]
- 47.Cross MJ, Hodgkin MN, Roberts S, Landgren E, Wakelam MJ, Claesson-Welsh L. Tyrosine 766 in the fibroblast growth factor receptor-1 is required for FGF-stimulation of phospholipase C, phospholipase D, phospholipase A (2), phosphoinositide 3-kinase and cytoskeletal reorganisation in porcine aortic endothelial cells. J Cell Sci. 2000; 113: 643-651. [DOI] [PubMed] [Google Scholar]
- 48.Nakatani Y, Sunaga S, Murakami M, Kudo I. Cytosolic phospholipase A2 alpha interacts with microtubules. Adv Exp Med Biol. 2002;507: 21-24. [PubMed] [Google Scholar]
- 49.Fischer L, Poeckel D, Buerkert E, Steinhilber D, Werz O. Inhibitors of actin polymerisation stimulate arachidonic acid release and 5-lipoxygenase activation by upregulation of Ca2+ mobilisation in polymorphonuclear leukocytes involving Src family kinases. Biochim Biophys Acta. 2005;1736: 109-119. [DOI] [PubMed] [Google Scholar]
- 50.MacKenzie A, Wilson HL, Kiss-toth E, Dower SK, North RA, Surprenant A. Rapid secretion of interleukin-1β by microvesicle shedding. Immunity. 2001;8: 825-835. [DOI] [PubMed] [Google Scholar]
- 51.Drummond DC, Noble CO, Kirpotin DB, Guo Z, Scott GK, Benz CC. Clinical development of histone deacetylase inhibitors as anticancer agents. Annu Rev Pharmacol Toxicol. 2005;45: 495-528. [DOI] [PubMed] [Google Scholar]