

Abstract
Most patients with acute promyelocytic leukemia (APL) express PML-RARα, the fusion product of t(15;17)(q22;q11.2). Transgenic mice expressing PML-RARα develop APL with long latency, low penetrance, and acquired cytogenetic abnormalities. Based on observations that 4% to 10% of APL patients harbor oncogenic ras mutations, we coexpressed oncogenic K-ras from its endogenous promoter with PML-RARα to generate a short-latency, highly penetrant mouse model of APL. The APL disease was characterized by splenomegaly, leukocytosis, extramedullary hematopoiesis (EMH) in spleen and liver with an increased proportion of immature myeloperoxidase-expressing myeloid forms; transplantability to secondary recipients; and lack of cytogenetic abnormalities. Bone marrow cells showed enhanced self-renewal in vitro. This model establishes a role for oncogenic ras in leukemia pathogenesis and thus validates the oncogenic RAS signaling pathway as a potential target for therapeutic inhibition in leukemia patients. This mouse model should be useful for investigating signaling pathways that promote self-renewal in APL and for testing the in vivo efficacy of RAS signaling pathway inhibitors in conjunction with other targeted therapies such as ATRA (all trans retinoic acid) and arsenic trioxide.
Introduction
Acute promyelocytic leukemia (APL) comprises 10% to 15% of all cases of adult acute myelogenous leukemia (AML). APL cases (> 90%) are associated with the t(15;17)(q22;q11.2), in which sequences of the promyelocytic leukemia (PML) gene are fused to those of retinoic acid receptor α (RARα) to generate the PML-RARα fusion protein. PML-RARα expression is associated with impaired myeloid differentiation, due to increased affinity for the nuclear repressor protein complex (NcoR); alteration of chromatin structure by histone deacetylase (HDAC); and inhibition of transcription.1 Treatment with all trans retinoic acid (ATRA) is a highly effective treatment strategy in APL and acts as a differentiating agent by promoting release of the NCoR/HDAC complex, thereby restoring normal transcription. Arsenic trioxide also has efficacy in treating relapsed or refractory APL.2,3
The PML-RARα fusion protein is necessary, but not sufficient, for the development of AML, as demonstrated in studies with PML-RARα transgenic mice4-6 and murine bone marrow transplant (BMT) models with retrovirally transduced PML-RARα.7 Transgenic mice expressing PML-RARα under the control of the cathepsin G promoter develop asymptomatic myeloid hyperplasia, with a subset of these mice progressing to APL with a long latency of 9 to 12 months and penetrance of 15% to 30%.4 Coexpression of the reciprocal RARα-PML and PML-RARα cDNAs from the cathepsin G promoter in double-transgenic mice increases disease penetrance to approximately 60% but does not shorten latency.8 A knock-in model in which PML-RARα cDNA is expressed from the endogenous cathepsin G promoter causes APL with a penetrance of more than 90% but still requires a long latency of 6 to 16 months.9 These data indicate that additional mutations are required for APL induction. Consistent with this notion, recurring, nonrandom cytogenetic abnormalities have been observed in PML-RARα transgenic mice that progress to APL.10-12
Oncogenic RAS mutations, present in 25% to 44% of patients with AML, are candidates for cooperating second mutations in leukemogenesis. Initial studies with small patient cohorts show that APL patients have coincident oncogenic RAS mutations.13-15 Recently, 2 larger studies identified oncogenic N-RAS and K-RAS mutations in 4% of 97 APL patients16 and in 10% of APL patients, respectively (8 of 146 [5.5%] N-RAS and 5 of 114 [4.4%] K-RAS).17 In these studies, there were no differences between APL patients with or without oncogenic RAS mutations with regard to age upon diagnosis, presenting white blood cell (WBC) count, prognosis, or frequency of the M3 variant subtype.16,17
Conditional expression of oncogenic K-ras from its endogenous promoter in the mouse hematopoietic system results in a myeloproliferative disease but is not sufficient to cause AML.18,19 We tested the ability of oncogenic K-ras to cooperate with PML-RARα in inducing APL in mice. We hypothesized that oncogenic ras would provide proliferative and/or cell survival signals and would cooperate with PML-RARα mutation associated with impaired hematopoietic differentiation to generate an APL-like phenotype with increased proliferation and impaired hematopoietic differentiation. We hypothesized that mice expressing oncogenic K-ras and PML-RARα would develop an APL-like disease with increased penetrance and decreased latency.
Materials and methods
Mouse strains
LSL-K-ras G12D mice20 were crossed to cathepsin G-PML-RARα mice4 to generate LSL-K-ras G12D+/-/cathepsin G-PML-RARα+/- mice (KP mice). KP mice (in mixed BALB/c, C57BL/6, and 129Sv/Jae backgrounds) were crossed to Mx1-Cre mice21 on a BALB/c background to generate triple-transgenic LSL-K-ras G12D+/-/cathepsin G-PML-RARα+/-/Mx1-Cre+/- mice (KPM mice) and control littermates in a mixture of BALB/c, C57BL/6, and 129Sv/Jae genetic backgrounds. To induce Cre expression, 4- to 7-week old mice received intraperitoneal injections of 250 μg of polyinosinic-polycytidylic acid (pI-pC; Sigma-Aldrich, St Louis, MO) every other day for a total of 3 doses. All mice were maintained in microisolator cages with daily monitoring for evidence of disease. All experiments were conducted with the ethical approval of the Harvard Medical Area Standing Committee on Animals.
Molecular and biochemical analysis
Mice were genotyped by polymerase chain reaction (PCR) amplification of genomic DNA from tail tissue to identify the LSL-K-ras G12D allele,20 cathepsin G PML-RARα transgene,4,22 and Mx1-Cre transgene,21 as previously described. Cre-mediated recombination of the LSL-K-ras G12D allele was verified by PCR amplification of DNA from mouse bone marrow, liver, and spleen, as well as from individual colonies from primary bone marrow methylcellulose cultures.20 Wild-type and K-ras G12D proteins were detected by immunoprecipitation and Western blotting of spleen cell lysates as previously described,23,24 using a Y13-259 agarose conjugate (Oncogene Research Products, Boston, MA) for ras immunoprecipitation, and polyclonal antibodies recognizing wild-type (G12) or oncogenic ras (D12; codon 12 glycine-to-aspartic acid mutation) for Western blotting (kind gift from Leisa Johnson).
Histopathology
Tissue sections (4 μm) of mouse organs were processed for staining with hematoxylin and eosin solutions or immunohistochemical analysis for myeloperoxidase, as previously described.18,25 The samples were analyzed using an Olympus BX41 microscope with the objective lens of 40 ×/0.75 Olympus UPlanFL (Olympus, Melville, NY). The pictures were taken using Olympus QColor3 and analyzed with acquisition software QCapture v2.60 (QImaging, Burnaby, BC, Canada) and Adobe Photoshop 6.0 (Adobe, San Jose, CA).
Flow cytometric analysis
Spleen and bone marrow single-cell suspensions were prepared as previously described.18 Antibodies used included allophycocyanin (APC)-conjugated Gr-1, fluorescein isothiocyanate (FITC)-conjugated Mac-1, and phycoerythrin (PE)-conjugated CD117 (c-Kit). A 4-color FACSCalibur cytometer (Becton Dickinson, Mountain View, CA) was used to acquire a minimum of 10 000 events. Data were analyzed with CELLQuest software. Myeloid progenitors were analyzed as previously described26 to distinguish Lin-Sca1-c-Kit+CD34+FcγRII/IIIlo CMPs, Lin-Sca1-c-Kit+CD34+FcγRII/IIIhi GMPs, and Lin-Sca1-c-Kit+CD34-FcγRII/IIIlo MEPs. Data acquisition was performed on a highly modified double laser (488 nm/350 nm Enterprise II + 647-nm Spectrum) high-speed fluorescence-activated cell-sorter scanner (FACS) (Moflo-MLS, Cytomation, Fort Collins, CO), and data were analyzed with FlowJo software (Treestar, San Carlos, CA). For all analyses, dead cells were excluded by propidium iodide staining.
Colony-forming activity, serial replating, and ATRA differentiation assays
Primary bone marrow methylcellulose cultures were performed in 3 independent experiments using MethoCult GF M3434 media (StemCell Technologies, Vancouver, BC, Canada) containing 50 ng/mL murine stem cell factor (SCF), 10 ng/mL murine interleukin-3 (IL-3), 10 ng/mL human IL-6, and 3 U/mL human erythropoietin (EPO), or MethoCult GF M3231 from which SCF, IL-3, IL-6, and EPO were absent. Duplicate cultures containing 4 × 103 to 1 × 105 cells were harvested after 7 days. Individual colonies were harvested for Wright-Giemsa staining of cytospin preparations and for DNA samples. Serial replating assays were performed in 3 independent experiments with 104 cells replated in duplicate every 7 days in MethoCult GF M3434 for each round of replating. ATRA-induced differentiation assays with KPM+ cells (from rounds 4 and 5 of serially replated methylcellulose cultures) were performed in 2 independent experiments. Duplicate cultures containing 104 KPM+ cells in MethoCult GF M3434 media were grown in the presence or absence of 1 μM ATRA (Sigma-Aldrich) and harvested after 5 days for Wright-Giemsa staining of cytospin preparations. Data shown are representative of 2 independent experiments.
Spectral karyotyping
Single-cell spleen suspensions from APL-diseased KPM+, KM+, and nondiseased KP+ mice were cultured as described previously. Metaphase cell preparations, and spectral karyotyping analysis were performed as described previously.11 A minimum of 10 metaphase cells was analyzed for each mouse.
Secondary transplantation experiments
Spleen cells were harvested from APL-diseased KPM+ mice. After red blood cell lysis, cells were washed in phosphate-buffered saline (PBS), resuspended in Hanks balanced salt solution (Life Technologies, Bethesda, MD), and injected (106 or 107 spleen cells per mouse) into the lateral tail vein of sublethally irradiated (650 centi-Gray [cGy]) or lethally irradiated (2 × 450 cGy) wild-type littermates. Mice were housed in microisolator cages with autoclaved chow and acidified water.
Results
Physiologic expression of oncogenic K-ras from its endogenous promoter in the hematopoietic system results in a completely penetrant, rapid onset myeloproliferative disease but is not sufficient to induce AML.18,19 These studies used conditional knock-in Lox-stop-lox (LSL)-K-ras G12D mice harboring an oncogenic K-ras allele containing a glycine-to-aspartic acid mutation at codon 12, whose expression is regulated by Cre-mediated excision of an upstream floxed transcriptional stop cassette (Figure 1A),20 and Mx1-Cre mice that express Cre from an interferon α/β-inducible Mx1 promoter in hematopoietic tissues.21
Figure 1.

Generation of mice coexpressing oncogenic K-ras and a cathepsin G-PML-RARα transgene, survival analysis, and spleen WBC differentials. (A) Schematic of wild-type, LSL-K-ras G12D, and lox-K-ras G12D alleles, depicting K-ras exons 0, 1, and 2. Gene targeting to the endogenous K-ras locus generated the LSL-K-ras G12D allele, containing a floxed transcriptional termination codon upstream of an oncogenic mutation in codon 12 (glycine-to-aspartic acid) in exon 1. Cre recombinase-mediated excision of the stop cassette expresses the oncogenic lox-K-ras G12D allele. *G12D mutation. (B) Generation of LSL-K-ras G12D+/PML-RARα+/Mx1-Cre+ mice and controls. Progeny of crosses between LSL-K-ras G12D+/PML-RARα+ and Mx1-Cre+ mice were treated with pI-pC to generate KPM+, KM+, KP+, PM+, P+, and K+ mice. K indicates LSL-K-ras G12D; P, cathepsin G-PML-RARα; M, Mx1-Cre. + indicates pI-pC treatment. (C) Kaplan-Meier comparative survival analysis of littermate KPM+, KM+, and control KP+, PM+, and P+ mice. Analysis was performed using large numbers of mice and littermate controls to minimize strain effects. Cumulative survival is plotted against days after treatment with pI-pC for KPM+ (n = 13), KM+ (n = 20), KP+ (n = 11), PM+ (n = 12), and P+ (n = 20) mice over an observation period of more than 200 days. (D) Manual differential counts performed on 200 nucleated cells in myeloid rich sections of splenic red pulp demonstrate an increased percentage of morphologically immature myeloid cells (blasts + promyelocytes) in the spleens of KPM+ mice with APL, compared to KPM+ mice with myeloproliferative disease, KM+, nondiseased KP+, or K+ mice. Median values are represented by horizontal bars. Indicated P values are determined by Mann-Whitney test.
Generation of mice coexpressing oncogenic K-ras and PML-RARα in the hematopoietic system
Conditional oncogenic K-ras (LSL-K-ras G12D) knock-in mice (K mice)20 were bred to cathepsin G-PML-RARα transgenic mice (P mice)4 to generate doubly transgenic mice (KP mice), which were then crossed with Mx1-Cre (M) mice.21 Four- to seven-week old progeny were treated with synthetic double-stranded RNA polyinosinic-polycytidylic acid (pI-pC) to generate experimental KPM+, KM+, KP+, PM+, P+, and K+ mice (Figure 1B).
Oncogenic K-ras and PML-RARα coexpression results in a short latency, high-penetrance, APL-like disease
As expected, control KP+ (2 of 11), PM+ (5 of 12), or P+ (8 of 20) mice developed APL-like disease with long latency (median, 170 days) and low combined penetrance (35%) (Figure 1C). By contrast, littermate KPM+ mice coexpressing oncogenic K-ras and PML-RARα developed APL-like disease with a penetrance of 69% (9 of 13 mice with APL; the remaining 4 of 13 mice had myeloproliferative disease) and a much shorter latency with a median survival of 39 days (P < .001, KPM+ vs KP+, PM+ and P+ controls by log-rank test; range, 22-63 days). All KM+ mice developed myeloproliferative disease, with median survival of 36 days (P < .001, KM+ vs KP+,PM+ and P+ controls by log-rank test; range, 22-67 days). KPM+ mice with APL had greater than or equal to 20% immature myeloid cells (blasts + promyelocytes) in the spleen, in contrast to KPM+ mice with myeloproliferative disease, KM+, nondiseased KP+, or K+ mice that had fewer than 20% immature myeloid cells (Figure 1D).
KPM+ mice showed splenomegaly and leukocytosis (Table 1). KPM+ spleens were significantly enlarged (range, 840-2308 mg), compared to KM+ spleens (270-1690 mg; P < .001 by Mann-Whitney test) and nondiseased KP+ spleens (240-300 mg; P < .001 by Mann-Whitney test). KPM+ mice with APL had significant leukocytosis, with a median WBC count of 181 × 103/μL, compared to KM+ mice with median WBC counts of 59 × 103/μL and nondiseased KP+ mice with median WBC counts of 4.4 × 103/μL (P = .002, KPM+ vs KM+; P = .001, KPM+ vs KP+ by Mann-Whitney tests). The leukocytosis in KPM+ mice was due to an increase in the granulocyte population, with an increase in immature forms compared to KM+ mice (Figure 2). KPM+ mice with APL and KM+ mice were anemic, with comparable median hematocrit levels of 30% (P = .001, KPM+ vs KP+ by Mann-Whitney test), but had normal platelet counts (Table 1). Overall, the KPM+ mice that developed APL had higher WBC counts than the KPM+ mice with myeloproliferative disease but were not otherwise distinguishable on the basis of spleen weights, hematocrit levels, or platelet counts.
Table 1.
Phenotype of KPM+ mice and controls
| Genotype | pl-pC | WBC × 103 per μL (median) | Hct % (median) | Platelets per μL (median) | Spleen weight mg (median) | Liver weight mg (median) | Phenotype |
|---|---|---|---|---|---|---|---|
| KPM | + | 61-512 (181) n = 9 | 14-45 (30) n = 9 | 81-1151 (501) n = 9 | 840-2308 (1324) n = 9 | 1220-2880 (2131) n = 9 | APL* |
| KPM | + | 30-603 (211) n = 4 | 21-34 (28) n = 4 | 452-811 (563) n = 4 | 1170-1810 (1395) n = 4 | 2000-3240 (2482) n = 4 | MPD |
| KM | + | 15-111 (59) n = 18 | 9-44 (30) n = 18 | 515-1456 (891) n = 18 | 270-1690 (606) n = 19 | 790-2110 (1390) n = 19 | MPD |
| KP | + | 2.5-6.2 (4.4) n = 5 | 42-48 (45) n = 5 | 306-746 (598) n = 5 | 240-300 (268) n = 6 | 1050-1660 (1318) n = 6 | Myeloid hyperplasia (APL†) |
Normal mouse spleen weight was 70 to 130 mg. Normal mouse WBC count was 3 to 9 × 103 per μL
High penetrance disease with short latency
Low penetrance disease with long latency
Figure 2.
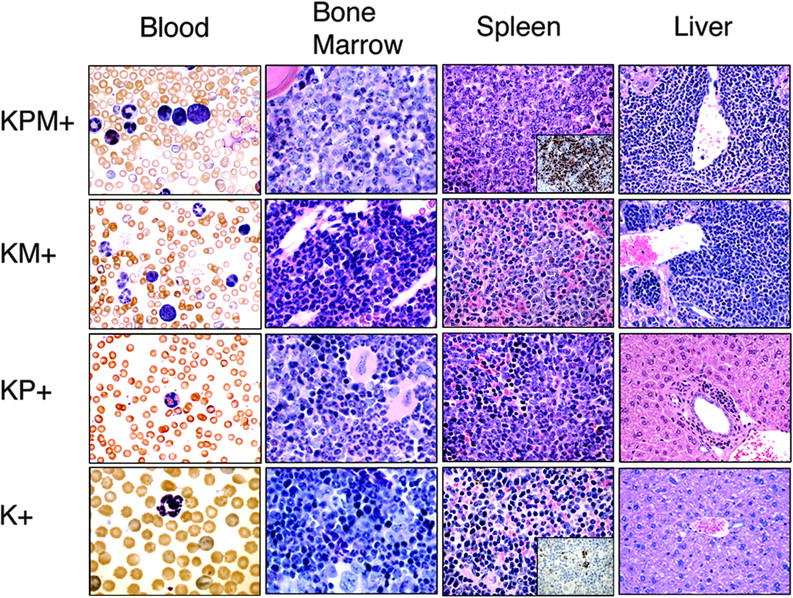
KPM+ mice coexpressing oncogenic K-ras and PML-RARα develop a lethal APL-like disease. Representative peripheral blood (1000× original magnification), bone marrow (1000× original magnification), spleen (400× original magnification), and liver (400× original magnification) histopathology from age-matched KPM+, KM+, nondiseased KP+, and K+ mice. Insets show representative myeloperoxidase immunohistochemistry of KPM+ and K+ spleen (400× original magnification). Note the predominance of immature mononuclear cells within the marrow and spleen of KPM+ mice compared to KM+, nondiseased KP+, and K+ mice.
Histopathological analysis was performed in age-matched mice (Figure 2). K+ spleens contained normal cellular constituents of the red and white pulp. Nondiseased KP+ spleens showed mild mature myeloid hyperplasia as previously described.4 KM+ spleens showed increased extramedullary hematopoiesis (EMH) with expansion of red pulp by mature myeloid forms, consistent with a myeloproliferative disease.18,19 Of 13 KPM+ spleens, 9 showed expansion of red pulp by myeloid cells, with an increased proportion of immature forms compared to KM+ mice, most consistent with features of an acute leukemia (Figure 2). Immunohistochemical analysis of KPM+ spleens confirmed an increased proportion of immature, myeloperoxidase (MPO)-expressing myeloid forms, similar to findings in other murine models of APL-like disease (inset, Figure 2). KPM+ mice with APL had more intense MPO staining of mononuclear cells and a higher frequency of MPO+ cells than negative control K+ mice. There was no MPO staining of mature neutrophils. In the remaining 4 of 13 KPM+ cases examined, the splenic EMH was composed of predominantly mature granulocytic forms, similar to the myeloproliferative disease seen in KM+ mice (data not shown). In KPM+ mice with APL, the bone marrow also demonstrated an increased proportion of immature myeloid forms compared to KM+ bone marrow (Figure 2). KPM+ and KM+ livers showed periportal and perivascular infiltration by mature myeloid forms (Figure 2). In addition to the dominant APL phenotype, KPM+ mice also developed with lower penetrance the constellation of lymphomas, squamous papillomas of the ear, vulvo-vaginal/anal skin, or esophagus, and lung adenomas previously observed in KM+ mice (data not shown and Chan et al18).
Flow cytometric analysis of spleen cells from KPM+ mice with APL confirmed expansion of immature myeloid cells (Figure 3). Normal mouse spleens have 5% to 6% Gr-1+/Mac-1+ mature myeloid cells. KPM+, KM+, and nondiseased KP+ spleens each showed expansion of mature myeloid cells with 23%, 37%, and 24% Gr-1+/Mac-1+ cells, respectively. Only spleens from KPM+ mice with APL had an increased percentage of immature myeloid cells, as evidenced by 14% Gr-1-/c-Kit+ cells, compared with 3% Gr-1-/c-Kit+ cells in KM+ spleen. Flow cytometry of bone marrow cells from KPM+ mice with APL showed a more subtle expansion of immature Gr-1-/c-Kit+ myeloid cells, compared to KM+ and nondiseased KP+ bone marrow (Figure 3).
Figure 3.

Flow cytometric analysis of bone marrow and spleen cells from KPM+, KM+, and nondiseased KP+ mice. Bone marrow and spleen cells from APL-diseased KPM+ (n = 5), KM+ (n = 6), and nondiseased KP+ (n = 3) mice were stained with a combination of antibodies to Gr-1, Mac-1, and c-Kit. Dot plots are shown for viable cells gated on the basis of scatter properties and 7-aminoactinomycin D staining. Representative data are shown with the percentages of cells in quadrants of interest indicated.
Myeloid progenitor analysis
In the bone marrows of KPM+ mice with APL, there was an increase in the percentage of granulocyte-monocyte progenitors (GMPs) and a decrease in the percentages of common myeloid progenitors (CMPs) and megakaryocyte-erythrocyte progenitors (MEPs) (relative to the total bone marrow cell count), compared to control M+ bone marrow (Figure 4). Within the c-Kit+/Sca-1- population of myeloid progenitors, there was an increase in the relative proportion of GMPs in KPM+, KM+, and nondiseased KP+ bone marrow, compared with control M+ bone marrow.
Figure 4.

Analysis of myeloid progenitors in bone marrow and spleen of APL-diseased KPM+, KM+, nondiseased KP+, and control M+ (pI-pC treated Mx1-Cre) mice. Quadrants represent the gated populations of myeloid progenitors (IL-7Rα-Lin-Sca1-c-Kit+), CMPs (FcγRloCD34+), GMPs (FcγRhiCD34+), and MEPs (FcγRloCD34-), whose percentages relative to whole bone marrow and spleen are indicated.
In the M+ normal mouse control, MEPs comprise the major steady state myeloid progenitor population in the spleen. KPM+, KM+, and nondiseased KP+ spleens each showed increased percentages relative to the total splenocyte count and increased absolute numbers of CMP, GMP, and MEP populations (Figure 4 and data not shown) relative to control M+ spleen, reflecting increased EMH. Within the myeloid progenitor (c-Kit+/Sca-1-) population, there was a proportional increase in GMPs and a proportional decrease in MEPs. However, there were no significant differences in the percentages of myeloid progenitor populations between KPM+, KM+, and nondiseased KP+ spleens (Figure 4). In summary, expression of oncogenic K-ras or PML-RARα alone in nonleukemic KM+ and KP+ mice coincides with an increase in the GMP population, but leukemic transformation in KPM+ mice is not accompanied by significant changes in the distribution of myeloid progenitors.
Oncogenic K-ras expression
Cre-mediated excision of the stop cassette to generate the lox-K-ras G12D allele was verified by PCR analysis of genomic DNA from bone marrow, liver, and spleen (Figure 5A). DNA from KPM+ and KM+ tissues yielded the PCR product corresponding to the lox-K-ras G12D allele. Additional PCR analysis of genomic DNA samples confirmed the presence of the PML-RARα transgene in KPM+ and KP+ tissues (Figure 5A). Oncogenic K-ras protein expression in KPM+ and KM+ spleens was confirmed by Western blotting of anti-ras immunoprecipitates (Figure 5B).
Figure 5.
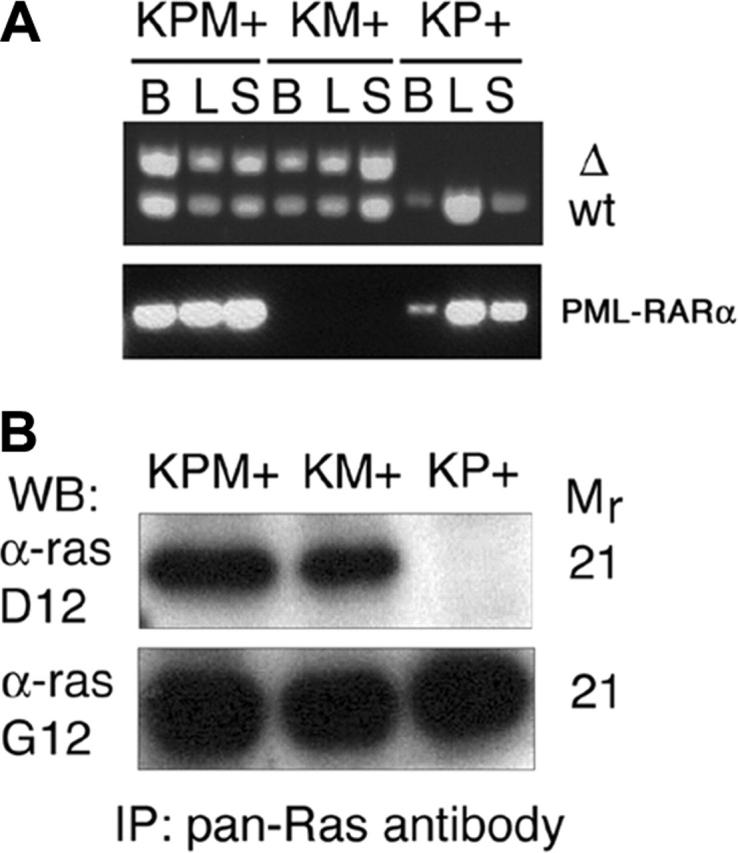
Cre-mediated activation and expression of the oncogenic K-ras allele. (A) PCR for 285-bp wild-type (WT) and 315-bp activated (Δ) K-ras alleles demonstrate presence of the activated allele in KPM+ and KM+ tissues. PCR demonstrates presence of PML-RARα transgene in KPM+ and KP+ tissues. B indicates bone marrow; L, liver; S, spleen. (B) Oncogenic K-ras expression in diseased KPM+ and KM+ spleens. Pan-ras antibody immunoprecipitates of spleen cell lysates were immunoblotted with antibodies specific for 21-kDa wild-type ras (α-ras G12) and oncogenic ras G12D (α-ras D12) proteins. WB indicates Western blot; IP, immunoprecipitation.
Colony-forming unit activity and ATRA differentiation
Primary bone marrow methylcellulose cultures were performed in which KPM+, KM+, or nondiseased KP+ bone marrow readily formed colonies in the presence of growth factors (IL-3, IL-6, SCF, and EPO; Figure 6A). In the absence of growth factors, nondiseased KP+ bone marrow did not form colonies. However, both KM+ and KPM+ bone marrow formed growth factor-independent colonies (Figure 6A), suggesting that expression of oncogenic K-ras provided a proliferation signal that bypassed the requirement for growth factors normally required for colony formation. KPM+, KM+, and nondiseased KP+ primary bone marrow methylcellulose cultures in the presence of growth factors generated each of the expected colony types. KM+ bone marrow cultures gave the expected distribution of burst-forming unit-erythroid (BFU-E), colony-forming unit-granulocyte/monocyte (CFU-GM), and colony-forming unit-granulocyte/erythrocyte/monocyte/megakaryocyte (CFU-GEMM) colonies, with a tendency toward generating colony-forming unit-monocyte (CFU-M) colonies (Figure 6D and Chan et al18). By contrast, KPM+ and nondiseased KP+ bone marrow methylcellulose cultures yielded a preponderance of colony-forming unit-granulocyte (CFU-G) colonies (Figure 6D).
Figure 6.

Bone marrow cells from KPM+ mice with APL demonstrate growth factor-independent colony forming activity and enhanced serial replating properties, and differentiate in response to ATRA in methylcellulose cultures. (A) Colony forming activity of KPM+, KM+, and nondiseased KP+ bone marrow cells in the presence (plus) or absence (minus) of growth factors (GF: IL-3, IL-6, SCF, EPO). Data shown are representative of 3 independent experiments. Values shown represent the mean of duplicate cultures from one representative experiment. (B) Serial replating activity of KPM+ bone marrow cells in the presence of growth factors. Data shown is representative of 3 independent experiments. Values shown are the mean of duplicate cultures from one representative experiment. (C) PCR for wild-type (WT) and oncogenic lox-K-ras G12D (Δ) K-ras alleles demonstrates presence of the oncogenic lox-K-ras G12D allele in individual methylcellulose colonies derived from KPM+ and KM+ bone marrow. M indicates molecular weight marker; c-, negative control DNA; c+, positive control DNA. (D) Quantitation of BFU-E, CFU-M, CFU-G, CFU-GM, and CFU-GEMM colonies from KPM+, KM+, and nondiseased KP+ bone marrow cultured in the presence of growth factors. Note predominance of CFU-G colonies derived from KPM+ and nondiseased KP+ bone marrow and CFU-M colonies from KM+ bone marrow. (E) Cytospins (Wright-Giemsa stain) of pooled methylcellulose colonies from KPM+ bone marrow cells cultures in the presence and absence of 1 μM ATRA. Original magnification, × 100.
In control experiments, PCR reactions on genomic DNA prepared from individual methylcellulose colonies (grown in the presence of growth factors) verified the presence of the activated lox-K-ras G12D allele, corresponding to excision of the stop cassette and activation of the oncogenic K-ras allele in KPM+ and KM+, but not KP+, colonies (Figure 6C). In the KPM+ and KM+ methylcellulose cultures, the Cre excision efficiency was 100% (n = 20 colonies each; Figure 6C and data not shown).
ATRA-differentiation assays were performed in vitro using pooled cells derived from rounds 4 and 5 of serially replated KPM+ bone marrow methylcellulose cultures (Figure 6E). In the absence of ATRA, most KPM+ cells maintained the immature blastic/promyelocytic morphology, as also observed in other mouse models of APL.4,5,8 Mature myeloid cells arising from spontaneous differentiation were rarely observed. In the presence of ATRA, KPM+ cells readily differentiated into mature myeloid cells, without evidence of proliferation (Figure 6E).
Serial replating activity
KPM+ cells showed a striking difference in phenotype in serial replating assays. When cells from the primary bone marrow methylcellulose cultures were serially replated, KM+ and nondiseased KP+ cells had normal serial replating activity; that is, they did not form any colonies beyond round 3 of replating (Figure 6B). By contrast, KPM+ bone marrow cells readily replated, forming more than 400 colonies on rounds 6 through 12 and more of replating (Figure 6B and data not shown). In additional experiments, bone marrow from PM+ and P+ mice that developed APL with a long latency of 182 and 191 days, respectively, exhibited growth factor-independent colony forming activity and enhanced serial replating activity (data not shown). These results suggest that immortalization and/or enhanced self-renewal properties are conferred by the cooperative effects between oncogenic K-ras and PML-RARα but not by either oncogene alone.
Transplantation assays
In consonance with the findings in the serial replating assays, the APL-like disease from KPM+ mice was transplantable into secondary recipient mice using the criteria of injecting 106 spleen cells into the tail veins of sublethally irradiated recipient mice. Of 15 sublethally irradiated, wild-type littermate secondary recipient mice that received transplants of 106 KPM+ spleen cells, 10 died with an APL-like disease, with median survival of 84 days (range, 42-114), average spleen weight of 950 mg, and average WBC count of 152 × 103/μL (data not shown). Of nine lethally irradiated, wild-type littermate secondary recipient mice that received transplants of 107 KPM+ spleen cells, all died with the APL-like disease with median survival of 75 days (range, 69-91), average spleen weight of 1300 mg, and average WBC count of 139 × 103/μL. In contrast, we have previously reported that the KM+ myeloproliferative disease is not transplantable into secondary recipients18 and also that spleen cells from nondiseased cathepsin G-PML-RARα mice do transplant the APL-like disease but only with a long latency (> 180 days) and low penetrance.22
Spectral karyotyping
Analysis of spleen cells from KPM+ mice with APL (n = 4) revealed a normal karyotype in all cases. This contrasts with previous findings of recurring chromosomal abnormalities in the long-latency, low-penetrance models of APL expressing PML-RARα alone10,11 and the short-latency, high-penetrance retroviral transduction APL models of PML-RARα + activated FLT3 or PML-RARα + BCL2.11,12,27,28 These data suggest that expression of oncogenic K-ras alone from its endogenous promoter may subserve requirements for acquisition of additional mutations for leukemia development in this model. As expected from previous studies, there was no evidence of clonal karyotypic abnormalities in KM+ mice (n = 1) or nondiseased KP+ mice (n = 3).10,11,19
Discussion
Conditional expression of oncogenic K-ras from its endogenous promoter cooperates with PML-RARα in mice to induce a rapid-onset and highly penetrant, lethal APL-like disease. There are several interesting and novel features of this disease phenotype. Oncogenic K-ras expression is capable of conferring factor-independent colony outgrowth but is incapable of conferring self-renewal potential when expressed alone. However, coexpression of oncogenic K-ras and PML-RARα engenders anAPL phenotype characterized by enhanced self-renewal potential of hematopoietic progenitors, including serial replating activity in the absence of stroma, and serial transplantability into secondary recipient mice, whereas neither allele alone has this capacity. These findings indicate that cooperation between alleles is requisite for generation of leukemia cells with self-renewing potential and provide tools for assessing the translational and posttranscriptional regulatory mechanisms that activate these pathways. These findings also provide strategies for prospective identification and purification of the elusive and enigmatic “leukemia stem cell” activity in APL.29
In all other published murine models of APL, a spectrum of karyotypic abnormalities are identified with variable frequencies that include del(2), trisomy 15, trisomy 8, trisomy 10, and -X/-Y.10-12,27,28 The variability between reports may be related to differences in the models (eg, MRP8 vs cathepsin G promoter) or in the method of karyotypic analysis. Specifically, in the cathepsin G-PML-RARα model of APL, the most frequently observed chromosomal abnormalities are monosomy 11,-X/-Y, trisomy 6, trisomy 15, and del(2). The frequency of these chromosomal abnormalities is significantly increased in APL cells from mice coexpressing both PML-RARα and the reciprocal fusion protein RARα-PML, notably -X/-Y, trisomy 15, and del(2).10 By contrast, no karyotypic abnormalities could be identified in the APL induced by expression of oncogenic K-ras from its endogenous promoter and PML-RARα. There are several potential explanations, including the possibility that physiologic expression of oncogenic K-ras may subserve requirements for second mutations. Alternatively, there may be subcytogenetic abnormalities that contribute to phenotype. However, the characteristic 2p deletions in which loss of function of PU.1 has been implicated in disease progression in mice30 were not observed, suggesting that oncogenic K-ras expression may phenocopy loss of PU.1 in this model of disease.
There are several differences between this model of cooperativity in APL and others that have been reported.22,27,28 These differences may be due in part to use of alleles that serve as pathway surrogates rather than known APL disease alleles (such as overexpression of BCL-2) or to the use of retroviral transduction of APL disease alleles like FLT3-ITD, in which retroviral integration site effects and aberrant dysregulated expression of FLT3-ITD may ensue. This model of APL is the first in which a known cooperating allele is expressed from its endogenous promoter. It is likely that analysis of transformation and assessment of various targeted therapeutic strategies, as discussed briefly below, will benefit from use of accurate models of disease based on physiological expression of disease alleles. Further improvements in the model could include the use of FLT3-ITD conditional knock-in alleles, similar to the oncogenic K-ras expression strategy used here and expression of PML-RARα in a cathepsin G knock-in model that has been described.9
Through use of model systems in which oncogenic K-ras is expressed from its endogenous promoter, detailed biochemical characterization of the activation state of ras downstream effectors in KPM+ cells may yield insights into additional candidate proteins to target for drug development. Ras activates multiple downstream effectors, including the Raf/MEK/ERK, RalGEF, and PI3-kinase/AKT pathways. Selective mutation of the ras effector domain (residues 32-40) can disrupt binding and activation of individual effector pathways31,32 with differential effects on cellular transformation.33,34 Analysis of conditional oncogenic K-ras effector domain mutant knock-in alleles, such as T35S (Raf pathway activation), E37G (RalGEF activation), and Y40C (PI3 kinase activation), in cooperation with PML-RARα, may be a fruitful approach to evaluate the importance of these ras effector pathways in promoting APL. More expansively, a comparative analysis of global patterns of gene and protein expression in KPM+ (APL) versus KM+ (myeloproliferative disease) cells will promote our understanding of molecular mechanisms of acute myeloid leukemogenesis.
This mouse model of APL validates oncogenic RAS and the RAS signaling pathway as important targets for therapeutic inhibition in patients with AML. Previous mouse models also have underscored critical contributions of oncogenic ras in the pathogenesis of adenocarcinomas of the lung,20,24,35,36 pancreas,37,38 and ovary.39 While awaiting the development of pharmacological agents that specifically target ras proteins, currently available inhibitors of ras downstream signaling (MEK inhibitors, PI3 kinase/AKT pathway inhibitors) and ras posttranslational modifications (eg, farnesyltransferase, geranylgeranyltransferase, or isoprenylcysteine carboxylmethyltransferase inhibitors) may be tested for their therapeutic efficacy in these mouse models of cancer.40,41 Currently, MEK inhibitors and farnesyltransferase inhibitors are being tested in phase 1 and phase 2 clinical trials in myeloid leukemias.40,42-44 Treating the APL-like disease in KPM+ mice with ras signaling pathway inhibitors alone and in combination with ATRA or arsenic trioxide may reveal promising novel therapeutic strategies for patients with APL.
Acknowledgments
We gratefully acknowledge administrative assistance from Alexis Bywater and helpful discussions with members of the Gilliland lab. We thank Tyler Jacks and David Tuveson for providing the LSL-K-ras G12D mice, Leisa Johnson for polyclonal antibodies specific to wild-type and oncogenic ras, and Brian Huntly for statistical analysis.
Prepublished online as Blood First Edition Paper, May 4, 2006; DOI 10.1182/blood-2006-04-015040.
Supported in part by National Institutes of Health grants DK51564 and CA66996 (D.G.G.), K08 CA109117 (I.T.C.), and U01 CA84221 (M.M.L.B.); the Irving W. Janock Fellowship (I.T.C.), and the Leukemia and Lymphoma Society (D.G.G., I.T.C.). D.G.G. is an Investigator of the Howard Hughes Medical Institute.
I.T.C. and D.G.G. designed the research; I.T.C., I.R.W., S.C., S.M., H.S., and M.M.L.B. performed the research; T.J.L. contributed vital reagents; I.T.C., J.L.K., I.R.W., H.S., K.A., M.M.L.B., and D.G.G. analyzed the data; and I.T.C. and D.G.G. wrote the paper.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Grignani F, De Matteis S, Nervi C, et al. Fusion proteins of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukaemia. Nature. 1998;391: 815-818. [DOI] [PubMed] [Google Scholar]
- 2.Soignet SL, Maslak P, Wang ZG, et al. Complete remission after treatment of acute promyelocytic leukemia with arsenic trioxide. N Engl J Med. 1998;339: 1341-1348. [DOI] [PubMed] [Google Scholar]
- 3.Chen GQ, Shi XG, Tang W, et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL), I: As2O3 exerts dose-dependent dual effects on APL cells. Blood. 1997; 89: 3345-3353. [PubMed] [Google Scholar]
- 4.Grisolano JL, Wesselschmidt RL, Pelicci PG, Ley TJ. Altered myeloid development and acute leukemia in transgenic mice expressing PML-RAR alpha under control of cathepsin G regulatory sequences. Blood. 1997;89: 376-387. [PubMed] [Google Scholar]
- 5.He LZ, Tribioli C, Rivi R, et al. Acute leukemia with promyelocytic features in PML/RARalpha transgenic mice. Proc Natl Acad Sci U S A. 1997; 94: 5302-5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown D, Kogan S, Lagasse E, et al. A PML-RARalpha transgene initiates murine acute promyelocytic leukemia. Proc Natl Acad Sci U S A. 1997;94: 2551-2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Minucci S, Monestiroli S, Giavara S, et al. PML-RAR induces promyelocytic leukemias with high efficiency following retroviral gene transfer into purified murine hematopoietic progenitors. Blood. 2002;100: 2989-2995. [DOI] [PubMed] [Google Scholar]
- 8.Pollock JL, Westervelt P, Kurichety AK, Pelicci PG, Grisolano JL, Ley TJ. A bcr-3 isoform of RARalpha-PML potentiates the development of PML-RARalpha-driven acute promyelocytic leukemia. Proc Natl Acad Sci U S A. 1999;96: 15103-15108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Westervelt P, Lane AA, Pollock JL, et al. High-penetrance mouse model of acute promyelocytic leukemia with very low levels of PML-RARα expression. Blood. 2003;102: 1857-1865. [DOI] [PubMed] [Google Scholar]
- 10.Zimonjic DB, Pollock JL, Westervelt P, Popescu NC, Ley TJ. Acquired, nonrandom chromosomal abnormalities associated with the development of acute promyelocytic leukemia in transgenic mice. Proc Natl Acad Sci U S A. 2000;97: 13306-13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le Beau MM, Bitts S, Davis EM, Kogan SC. Recurring chromosomal abnormalities in leukemia in PML-RARA transgenic mice parallel human acute promyelocytic leukemia. Blood. 2002;99: 2985-2991. [DOI] [PubMed] [Google Scholar]
- 12.Le Beau MM, Davis EM, Patel B, Phan VT, Sohal J, Kogan SC. Recurring chromosomal abnormalities in leukemia in PML-RARA transgenic mice identify cooperating events and genetic pathways to acute promyelocytic leukemia. Blood. 2003; 102: 1072-1074. [DOI] [PubMed] [Google Scholar]
- 13.Longo L, Trecca D, Biondi A, et al. Frequency of RAS and p53 mutations in acute promyelocytic leukemias. Leuk Lymphoma. 1993;11: 405-410. [DOI] [PubMed] [Google Scholar]
- 14.Kubo K, Naoe T, Kiyoi H, et al. Clonal analysis of multiple point mutations in the N-ras gene in patients with acute myeloid leukemia. Jpn J Cancer Res. 1993;84: 379-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chin YM, Bosco JJ, Koh CL. Analysis of ras gene mutations in acute myeloid leukemia by the polymerase chain reaction and oligonucleotide probes. Singapore Med J. 1992;33: 48-50. [PubMed] [Google Scholar]
- 16.Callens C, Chevret S, Cayuela JM, et al. Prognostic implication of FLT3 and Ras gene mutations in patients with acute promyelocytic leukemia (APL): a retrospective study from the European APL Group. Leukemia. 2005;19: 1153-1160. [DOI] [PubMed] [Google Scholar]
- 17.Bowen DT, Frew ME, Hills R, et al. RAS mutation in acute myeloid leukemia is associated with distinct cytogenetic subgroups but does not influence outcome in patients younger than 60 years. Blood. 2005;106: 2113-2119. [DOI] [PubMed] [Google Scholar]
- 18.Chan IT, Kutok JL, Williams IR, et al. Conditional expression of oncogenic K-ras from its endogenous promoter induces a myeloproliferative disease. J Clin Invest. 2004;113: 528-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Braun BS, Tuveson DA, Kong N, et al. Somatic activation of oncogenic Kras in hematopoietic cells initiates a rapidly fatal myeloproliferative disorder. Proc Natl Acad Sci U S A. 2004;101: 597-602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jackson EL, Willis N, Mercer K, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15: 3243-3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269: 1427-1429. [DOI] [PubMed] [Google Scholar]
- 22.Kelly LM, Kutok JL, Williams IR, et al. PML/RARalpha and FLT3-ITD induce an APL-like disease in a mouse model. Proc Natl Acad Sci U S A. 2002;99: 8283-8288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson L, Greenbaum D, Cichowski K, et al. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 1997;11: 2468-2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson L, Mercer K, Greenbaum D, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001; 410: 1111-1116. [DOI] [PubMed] [Google Scholar]
- 25.Rosenbauer F, Wagner K, Kutok JL, et al. Acute myeloid leukemia induced by graded reduction of a lineage-specific transcription factor, PU.1. Nat Genet. 2004;36: 624-630. [DOI] [PubMed] [Google Scholar]
- 26.Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404: 193-197. [DOI] [PubMed] [Google Scholar]
- 27.Sohal J, Phan VT, Chan PV, et al. A model of APL with FLT3 mutation is responsive to retinoic acid and a receptor tyrosine kinase inhibitor, SU11657. Blood. 2003;101: 3188-3197. [DOI] [PubMed] [Google Scholar]
- 28.Kogan SC, Brown DE, Shultz DB, et al. BCL-2 cooperates with promyelocytic leukemia retinoic acid receptor alpha chimeric protein (PMLRARalpha) to block neutrophil differentiation and initiate acute leukemia. J Exp Med. 2001;193: 531-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3: 730-737. [DOI] [PubMed] [Google Scholar]
- 30.Walter MJ, Park JS, Ries RE, et al. Reduced PU.1 expression causes myeloid progenitor expansion and increased leukemia penetrance in mice expressing PML-RARalpha. Proc Natl Acad Sci U S A. 2005;102: 12513-12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.White MA, Nicolette C, Minden A, et al. Multiple Ras functions can contribute to mammalian cell transformation. Cell. 1995;80: 533-541. [DOI] [PubMed] [Google Scholar]
- 32.Joneson T, White MA, Wigler MH, Bar-Sagi D. Stimulation of membrane ruffling and MAP kinase activation by distinct effectors of RAS. Science. 1996;271: 810-812. [DOI] [PubMed] [Google Scholar]
- 33.Hamad NM, Elconin JH, Karnoub AE, et al. Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev. 2002;16: 2045-2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rangarajan A, Hong SJ, Gifford A, Weinberg RA. Species- and cell type-specific requirements for cellular transformation. Cancer Cell. 2004;6: 171-183. [DOI] [PubMed] [Google Scholar]
- 35.Fisher GH, Wellen SL, Klimstra D, et al. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001;15: 3249-3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meuwissen R, Linn SC, van der Valk M, Mooi WJ, Berns A. Mouse model for lung tumorigenesis through Cre/lox controlled sporadic activation of the K-Ras oncogene. Oncogene. 2001;20: 6551-6558. [DOI] [PubMed] [Google Scholar]
- 37.Aguirre AJ, Bardeesy N, Sinha M, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17: 3112-3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4: 437-450. [DOI] [PubMed] [Google Scholar]
- 39.Dinulescu DM, Ince TA, Quade BJ, Shafer SA, Crowley D, Jacks T. Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat Med. 2005; 11: 63-70. [DOI] [PubMed] [Google Scholar]
- 40.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3: 11-22. [DOI] [PubMed] [Google Scholar]
- 41.Bergo MO, Gavino BJ, Hong C, et al. Inactivation of Icmt inhibits transformation by oncogenic K-Ras and B-Raf. J Clin Invest. 2004;113: 539-550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gotlib J. Farnesyltransferase inhibitor therapy in acute myelogenous leukemia. Curr Hematol Rep. 2005;4: 77-84. [PubMed] [Google Scholar]
- 43.Morgan MA, Ganser A, Reuter CW. Therapeutic efficacy of prenylation inhibitors in the treatment of myeloid leukemia. Leukemia. 2003;17: 1482-1498. [DOI] [PubMed] [Google Scholar]
- 44.English JM, Cobb MH. Pharmacological inhibitors of MAPK pathways. Trends Pharmacol Sci. 2002; 23: 40-45. [DOI] [PubMed] [Google Scholar]