

Abstract
Both genetic and environmental influences contribute to the wide variation in plasma von Willebrand factor (VWF) levels observed in humans. Inbred mouse strains also have highly variable plasma VWF levels, providing a convenient model in which to study genetic modifiers of VWF. Previously, we identified a major modifier of VWF levels in the mouse (Mvwf1) as a regulatory mutation in murine Galgt2. We now report the identification of an additional murine VWF modifier (Mvwf2). Mvwf2 accounts for approximately 16% of the 8-fold plasma VWF variation (or ∼ 25% of the genetic variation) observed between the A/J and CASA/RkJ strains and maps to the murine Vwf gene itself. Twenty SNPs were identified within the coding regions of the A/J and CASA/RkJ Vwf alleles, and in vitro analysis of recombinant VWF demonstrated that a single SNP (+7970G>A) and the associated nonsynonymous amino acid change (R2657Q) confers a significant increase in VWF biosynthesis from the CASA/RkJ Vwf allele. This change appears to represent a unique gain of function that likely explains the mechanism of Mvwf2 in vivo. The identification of a natural Vwf gene variant among inbred mice affecting biosynthesis suggests that similar genetic variation may contribute to the wide range of VWF levels observed in humans.
Introduction
von Willebrand factor (VWF), a central component of the blood coagulation system, is secreted from the vascular endothelium to generate the circulating plasma pool of multimeric VWF. Plasma VWF serves to mediate platelet adhesion to the vascular endothelium after injury and also acts as a carrier protein for circulating factor VIII. Significant qualitative or quantitative deficiencies in VWF result in von Willebrand disease (VWD), the most common inherited bleeding disorder in humans.1 Estimates of VWD prevalence have ranged as high as 1%,2,3 although these numbers and the precise criteria for diagnosis remain controversial.4 Both the wide distribution of VWF levels observed in the general population (between 50% and 200% of the population mean) and the reduced penetrance and variable expressivity associated with type 1 VWD can contribute to diagnostic difficulties.1 Type 1 VWD is characterized by a mild bleeding phenotype that results from a mild to moderate reduction of plasma VWF (20%-50% of the population mean). More than 70% of VWD cases are considered type 1, whereas qualitative defects (type 2) and severe quantitative defects (type 3) account for the remaining cases. In contrast to the bleeding associated with low VWF levels, elevated factor VIII levels, caused at least in part by associated high VWF, have been recently identified as a major risk factor for thrombosis.5
Previous efforts to define the determinants of plasma VWF level variability in humans have identified both environmental and genetic factors. Age, stress, exercise, pregnancy, and hormonal changes are among some of the environmental factors known to alter VWF levels.6 Heritability estimates from twin studies suggest that the majority of VWF variation (66%-75%) is genetically determined,7,8 although the responsible genes are not well defined.6 The primary genetic modifier of plasma VWF levels in humans identified to date is ABO blood group, although it only accounts for approximately 30% of the genetically determined VWF level variation.7 Sequence differences within or around the VWF gene may also be important determinants of total plasma VWF. Causative mutations within VWF are well documented in type 2 and type 3 VWD,1 although the correlation between VWF mutations and type 1 VWD is less complete.9,10 The degree to which VWF polymorphisms contribute to variation within the general population is unclear, because published linkage and association studies comparing VWF genotype and plasma protein levels have been inconsistent.11,12
As observed in humans, plasma VWF levels within inbred mouse strains are also highly variable, and VWF levels between strains may differ by as much as 20-fold.13 We previously used this wide variation to identify a genetic determinant of plasma VWF level in the laboratory mouse. Analysis of the RIIIS/J strain, originally reported as a mouse model for type 1 VWD, identified the first murine genetic modifier of plasma VWF (termed Mvwf1 for Modifier of von Willebrand factor 1) as a regulatory mutation in the Galgt2 gene.14
We have now extended our analysis to identify additional genetic factors controlling plasma VWF levels in the mouse. Using an (A/J × CASA/RkJ) F2 population, we have mapped a second candidate modifier (Mvwf2) to murine chromosome 6. We also show that Mvwf2 is a CASA/RkJ-specific sequence variant within the murine VWF gene (Vwf), resulting in enhanced VWF biosynthesis and/or secretion.
Materials and methods
Mice
With the exception of DDK, all inbred mouse strains were obtained from The Jackson Laboratory (Bar Harbor, ME). DDK mice were the generous gift of C. Sapienza, Temple University. F1 animals in Figure 1 were generated by intercrossing the parental strains. For the (A/J × CASA/RkJ) F2 population, F1 animals were generated by breeding CASA/RkJ males to A/J females, and F2 animals were obtained by intercrossing F1 animals. Mice were housed at The University of Michigan, supervised by the Unit for Laboratory Animal Medicine. All study protocols were approved by the University Committee on Use and Care of Animals (UCUCA) at the University of Michigan.
Figure 1.

Plasma VWF levels in selected inbred strains and CASA/RkJ intercrossed animals. VWF levels were calculated as a percentage of the CASA/RkJ strain, which is arbitrarily defined as 100%. The RIIIS/J, SWR/J, and DDK strains all share the low Mvwf1 allele (Galgt2 mutation).14 Average VWF levels for each pure strain (□ and ▪) as well as F1 animals resulting from CASA/RkJ intercrosses ( ) are shown (± SD, standard deviation).
) are shown (± SD, standard deviation).
Bleeding
Blood samples were obtained by retroorbital bleeds into heparinized capillary tubes (Fisher, Hampton, NH) and/or terminal inferior vena cava (IVC) blood draw into 0.5M EDTA, pH 8.0 (2.5% by volume). Mice were anesthetized using methoxyflurane (Metophane; Schering-Plough, Kenilworth, NJ), isoflurane (Aerrane; Baxter, Deerfield, IL), or isoflurane followed by pentobarbital (Abbott Laboratories, Abbott Park, IL). For the pure strain survey (Figure 1), 2 to 3 retroorbital bleeds on each of 3 to 6 mice were obtained. For the parental strains and (A/J × CASA/RkJ) F1 mice (Figure 2), 1 to 3 bleeds (both retroorbital and IVC) were performed on each of 8 to 13 mice between 1.5 and 8 months of age. Blood samples from mice in the (A/J × CASA/RkJ) F2 population (Figure 2) included 3 retroorbital bleeds and 1 IVC bleed, all taken between weaning (3 weeks) and 6 months of age, with at least 1 week between bleeds. Three exceptions to this were 1 IVC bleed taken 2 days after the third bleed in 1 animal and 2 others in which terminal IVC bleeds were not assayed. In the F2 population, the mean age at the terminal IVC bleed was 2.5 months (range, 1.5-6 months). Platelet-poor plasma samples were prepared by centrifugation of whole blood (∼ 2300g for 10 minutes) and stored at –80°C until use.
Figure 2.

Distribution of plasma VWF levels in 200 (A/J × CASA/RkJ) F2 mice. VWF levels were calculated as a percentage of the CASA/RkJ parental strain. The latter is arbitrarily defined as 100%. F2 values are summarized by the bar graph. Averages for the parental and F1 populations (± SD) are shown: A/J (open diamond), CASA/RkJ (black diamond), and F1 (gray diamond). Samples selected for high and low pools are indicated by the dotted ovals.
Determination of VWF levels
Plasma VWF was measured by enzyme-linked immunosorbent assay (ELISA) as previously described.13 Pooled C57BL/6J plasma (5-14 mice per standard) was used to generate a standard curve, and each new pooled standard was sequentially normalized to previous standards for consistency. VWF levels for each individual mouse were determined by the mean ELISA value from all plasma samples obtained. VWF levels for the pure strains and F1 populations represent the mean ELISA value of mice within the subgroups, equally weighted for male and female contributions. All VWF levels are expressed as a percentage of the mean parental CASA/RkJ value (arbitrarily defined as 100%). Subsequent reanalysis and a correction in the formula used to calculate VWF values caused a subtle shift in the F2 VWF distribution between the time of DNA pooling (see “Pooled genome scans”) and final analysis. As a result, individual mice initially assigned to the high and low pools would have each changed by a single mouse. All pooled genotype data are representative of the original pools, prior to the VWF level correction. A similar shift also results in small differences between the VWF levels in the strain survey (Figure 1) and the A/J, CASA/RkJ, and F1 values shown in Figure 2.
Pooled genome scans
For the (A/J × CASA/RkJ) F2 population, DNA was prepared from liver tissue using phenol/chloroform extraction, and samples from the upper and lower 5% of the VWF distribution (10 mice for each pool) were combined in equal proportions to form the high and the low pools, respectively (50 ng/μL) (Figure 2). A pooled genome scan was performed as previously described,13,15 and a panel of 45 polymorphic microsatellite markers (Table S1, available on the Blood website; see the Supplemental Tables link at the top of the online article) was used to cover the genome at an average interval of 31 cM.16 Markers with suggestive linkage were confirmed by genotyping individuals within the pools, followed by the total F2 population (n = 200).
Vwf sequencing
Vwf cDNA fragments from each mouse strain (A/J, CASA/RkJ, and F1) were amplified by reverse transcription–polymerase chain reaction (RT-PCR) from total lung RNA prepared with TRIzol reagent (Invitrogen, Carlsbad, CA) and sequenced at The University of Michigan sequencing core. Following completion of the Vwf sequence, primers were developed (nos. 1-14; Table S2) to amplify the full-length cDNA in 4 fragments. Initial amplification was performed using the Superscript One-Step RT-PCR system (Invitrogen) (50 ng lung RNA + 0.5 μL RNAse inhibitor [Roche, Indianapolis, IN] in 50-μL reactions: 50°C for 30 minutes; 94°C for 2 minutes; 35 cycles of 94°C for 20 seconds, 55°C for 30 seconds, 72°C for 2 minutes; 72°C for 10 minutes). The extension cycles were repeated with Pfu Hotstart (Stratagene, La Jolla, CA) and nested primers as noted (Table S2).
For identification of 5′ and 3′ UTR sequences, 5′ and 3′ rapid amplifications of cDNA ends (RACEs) were performed using total lung RNA from A/J, CASA/RkJ, and F1 animals. The 5′ UTR was determined by using the RLM-RACE kit (Ambion, Austin, TX) and Vwf gene-specific primers nos. 15, 16 (Table S2). The 3′ UTR was determined by using the 3′/5′ RACE kit (Roche) and gene-specific primer no. 17 (Table S2) as per the manufacturer's instructions.
Portions of several introns (3, 12, 13, 21, 50, 51, as well as ∼ 500 base pair [bp] of downstream genomic sequence and ∼ 450 bp of upstream genomic sequence) were also sequenced using total genomic DNA (gDNA: A/J, CASA/RkJ, and F1 animals). A list of intronic primers is included (Table S3).
Vwf mRNA expression analysis
Initial analysis of allele-specific expression was determined by the quantification of sequencing chromatogram peaks using PolyPhred (University of Washington, Seattle, WA). Exon 11 was amplified using F1 gDNA and mRNA (primer nos. 18-21; Table S2), and peak area ratios (CASA/RkJ:A/J) were determined for SNPs +1188, +1236, and +1264.
Relative levels of mRNA expression from the A/J and CASA/RkJ alleles were also determined by comparing the ratio of F1 gDNA with F1 mRNA in a primer extension assay using fluorescently labeled extension primers as previously described.17,18 Briefly, fragments of exons 7 and 48 were amplified from gDNA and mRNA using the One-Step RT-PCR kit (Roche) and primers no. 22 to 23 and no. 26 to 27 (Table S2). Additional primers were designed to amplify across introns for detection of RNA contamination in the gDNA (primer nos. 24, 28). A T/C SNP (+774) in exon 7 and a G/A SNP (+7970) in exon 49 were subjected to primer extension using the Cy-5–labeled primers no. 25 and no. 29 (Table S2), respectively, (MWG Biotech, High Point, NC) in the presence of a nucleotide mix containing ddATP.
Generation of Vwf cDNA expression constructs and mutants
Vwf fragments 1 through 4 were amplified from lung RNA using RT-PCR (see “Vwf sequencing”) and assembled to form strain-specific full-length expression constructs (Figure 6A). The first fragment was flanked by NotI and EcoRI sites, the second by EcoRI and NheI sites, the third by NheI and SphI sites, and the fourth by SphI and XbaI sites. Three different vectors (pCR4Blunt-Topo [Invitrogen], pBluescript II SK vector [Stratagene], and pcDNA3.1+ [Invitrogen]) were used for cloning and assembly, the latter 2 with a modified multiple cloning site. An adaptor, designated “A” (primers nos. 30, 31; Table S2), was synthesized and introduced into pBluescript cut with XbaI and DraII, and pcDNA3.1 cut with NheI and DraII to form “pBlueA” and “pcDNA3.1A,” respectively. The pcDNA3.1A vector was further modified with the “+” adaptor (primers nos. 32, 33; Table S2) to introduce a Myc-tag at the C-terminus of VWF via XbaI and DraII digestion. This “pcDNA3.1A+” construct was used for all transfection analyses.
Figure 6.

In vitro transfection of full-length Vwf cDNA constructs and mutagenized variants. (A) Reference full-length Vwf cDNA is shown with the Myc-tag, relevant restriction enzyme sites, and SNPs noted (lines below construct). Larger representations of the NotI/EcoRI fragment and the region flanking R2657Q are shown below in CASA/RkJ and A/J VWF cDNA full-length constructs (no. 1 and no. 2, respectively) and mutagenized variants (nos. 3-10). In the individual constructs, black boxes represent CASA/RkJ sequence and gray boxes represent A/J sequence. The various chimeras are depicted as mixed gray and black, with corresponding amino acid designations below. VWF levels in conditioned media from COS-1 transfections are shown at right, represented as a percentage of CASA/RkJ transfection levels ± SD. (B) VWF levels in conditioned media following transfection of COS-1 cells with full-length A/J (pA) and CASA/RkJ (pC) cDNA. A strain-specific increase (1.43-fold) is noted. Error bars represent ± 95% confidence interval. *P < .001. (C) Expression of Vwf plasmids are summarized by pooling the results for all transfections by the sequence differences at each amino acid position. For example, in the T20A columns, the gray bar summarizes VWF expression in the 5 constructs which have the A/J-specific amino acid at position 20 (T), and the black bar summarizes VWF levels in the 5 constructs which have the CASA/RkJ amino acid (A) at the equivalent location. A similar pattern is repeated for each amino acid. An amino acid–specific increase (1.32-fold) is noted. Error bars represent ± 95% confidence interval. †P < .001.
Site-directed mutagenesis was used both to introduce the A/J 3′ SNP into the full-length constructs (Gene Editor mutagenesis kit; [Promega, Madison, WI]; and primer no. 34; Table S2) and to alter the nucleotide sequence conferring the +122 amino acid change (QuickChangeII Site-Directed Mutagenesis Kit [Stratagene]; and primers nos. 35-38; Table S2). Other plasmid modifications were introduced by mixing restriction-digested fragments between the strain-specific plasmids. EcoRI/NotI digestions were used to create multiple combinations of the 5′ and 3′ variable regions, and a BamHI digestion enabled recombination within the 5′ variable region. Plasmid sequences were verified prior to transfections.
Vwf transfections and protein analysis
One Shot TOP10 Escherichia coli (Invitrogen) containing the transfection constructs were grown overnight in LB medium at 37°C and purified using the Hi-Speed Maxiprep kit (Qiagen, Valencia, CA). DNA was quantified using either a spectrophotometer (DU530; Beckman Coulter, Fullerton, CA) or the PicoGreen dsDNA quantification kit (Invitrogen) and a fluorometer (Wallac Victor2; Perkin-Elmer, Wellesley, MA). COS-1 and HEK-293T cells were grown in Dulbecco Modified Eagle Medium, + 5% fetal bovine serum, and 1 × penicillin-streptomycin-glutamine (Gibco, Carlsbad, CA). Cells were maintained at 37°C in 5% CO2.
Transfection experiments were conducted using COS-1 cells in 96-well plates or HEK-293T cells in 10-cm plates. For 96-well transfections, confluent plates (10 cm) were split 1:7 and transfected 12 hours later using Fugene6 (Roche) as per the manufacturer's instructions. Vwf cDNA constructs were transfected in replicates (5-6 wells/construct), using 50 ng plasmid DNA in each well. Media samples were collected at 48 hours after transfection, diluted 1:16 in 1% BSA/PBS, and assayed by VWF ELISA.
Western blots were performed on conditioned media from transfection experiments. Samples were separated under denaturing conditions on a 4% to 12% Tris-Glycine gel (Invitrogen) and assayed as previously described,19 using either a rabbit anti–human VWF polyclonal antibody (Dako, Glostrup, Denmark) or a c-Myc rabbit polyclonal IgG antibody (Santa Cruz Biotechnology, Santa Cruz, CA) as the primary antibody. Immunogenicity of the 2 antibodies was compared using quantitative densitometry (ImageQuant software; Molecular Dynamics, Sunnyvale, CA).
Statistical analyses
Suggestive linkage in the pooled genome scan was determined by visual assessment of parental allele distribution in the high and low pools following gel electrophoresis. Suggestive markers were analyzed for significance by performing a chi-square statistic on the distribution of individual genotypes in the pools when compared with the Mendelian distribution of an unlinked marker (25% A/J homozygotes, 25% CASA/RkJ homozygotes, 50% heterozygotes). Significance at the population level was determined by analysis of variance, considering VWF levels in each of the 3 genotype classes (A/J homozygotes, heterozygotes and CASA/RkJ homozygotes). All measures of significance in the genome scan were adjusted for multiple observations using a Bonferroni correction. Potential differences in Vwf expression and transfection experiments were assayed for significance by 2-tailed t tests (2 sample, assuming equal variances).
For measures of in vivo VWF variance, the natural log (ln) of the VWF values was considered to correct for a slight skewing of the data. Analysis of variance was performed to assess the ELISA assay variance (variation in replicate measurements on the same plasma sample), environmental variance (variation among replicate measurements taken on the same animal), genetic variance (variation among animals), and total F2 population variance. Heritability was estimated by comparing genetic variance with total F2 population variance after correcting for assay variance. Assay variance was determined to be approximately 6% of total variance.
Results
(A/J × CASA/RkJ) F2 VWF levels are independent of Mvwf1 and highly heritable
To eliminate the RIIIS/J-dependent Mvwf1 effect when searching for additional novel VWF modifiers, we first surveyed several inbred strains for both plasma VWF level and Mvwf1 phenotype. The A/J strain was identified as a strain with low VWF levels that were independent of the Mvwf1 allele (Figure 1).14 When A/J mice were crossed with CASA/RkJ mice (high VWF), VWF levels in F1 animals were unlike either parent strain, additionally suggesting that VWF level regulation in these strains is not under the control of one dominant modifier analogous to Mvwf1.
(A/J × CASA/RkJ) F1 mice were intercrossed to generate 200 F2 progeny for further analysis. As depicted in Figure 2, a wide distribution of VWF levels was observed in the F2 population, with individual values ranging from 11% to 83% of CASA/RkJ levels. Reassessment of A/J, CASA/RkJ, and (A/J × CASA/RkJ) F1 VWF levels found A/J mice to average approximately 12% of CASA/RkJ and F1 animals approximately 36% of CASA/RkJ. Heritability of VWF levels in the (A/J × CASA/RkJ) F2 population was determined to be approximately 65%.
Although age has been positively associated with plasma VWF level increases in humans,6 the F2 population examined did not demonstrate elevated VWF levels over time, and no significant difference in population averages was noted among bleeds (population averages: bleed no. 1 = 41.6%; no. 2 = 37.7%; no. 3 = 38.4%; no. 4 = 39.0; P > .08).
Mvwf2 maps to the Vwf locus itself
To identify genes important for the regulation of plasma VWF levels within the (A/J × CASA/RkJ) F2 population, we conducted a genome scan comparing pooled DNA samples from opposite ends of the F2 VWF distribution curve (Figure 2). Of the 45 polymorphic markers used to identify linkage, 80% of the genotyping reactions resulted in well-defined bands that were subsequently assessed by visual gel analysis. Enrichment of the A/J (low VWF) alleles in the low pool and CASA/RkJ (high VWF) alleles in the high pool suggests linkage between the corresponding polymorphic markers and the VWF phenotype. Within the pooled DNA samples, unequal allelic distribution was noted in 5 genotyping reactions, suggesting possible linkage (Figure 3). None of the markers mapped to the previously described murine modifier, Mvwf1 (chromosome 11). Further genotyping of the individual mice comprising the high and low pools was consistent with statistically significant linkage at only D6Mit12 (P < .002) (Figure 3). Linkage at the 4 other loci (D3Mit219, D4Mit204, D9Mit67, and D18Mit14) was not significant after correcting for multiple observations (P > .9, P > .06, P > .4, and P > .9, respectively). When the entire F2 population was examined, highly significant linkage at D6Mit12 was confirmed (P < .001), with approximately 25% of the genetic variance within the (A/J × CASA/RkJ) F2 population attributable to D6Mit12 genotype (Figure 4). We termed this locus Mvwf2. D6Mit12 is located at 59.6 cM, only 1.2 cM from the Vwf gene, suggesting a mutation within the Vwf gene as the molecular mechanism underlying Mvwf2.
Figure 3.

Location of polymorphic DNA markers used for the (A/J × CASA/RkJ) F2 genome scan. Black circles represent unlinked markers, open circles represent failed or indeterminate markers, and gray circles designate markers with suggestive linkage by visual assessment of pooled samples. After examining individual genotypes within the pools, only D6Mit12 achieved statistically significant linkage (asterisk). This linkage was subsequently confirmed after extended genotype analysis of the entire F2 population (n = 200).
Figure 4.

Distribution of plasma VWF levels as a function of F2 genotype at the significantly linked marker D6Mit12. VWF levels in mice homozygous for the A/J allele at D6Mit12 are represented by the open bars with the open triangle representing the mean value for this population (error bars represent the 95% confidence interval). Similar distributions are shown in gray for mice heterozygous at D6Mit12 and in black for mice homozygous for the CASA/RkJ allele.
Twenty Vwf cDNA single nucleotide variants (SNPs) distinguish the CASA/RkJ and A/J alleles
To identify potential causative mutations in murine Vwf, we sequenced the full-length cDNA in the A/J and CASA/RkJ strains (GenBank accession nos. DQ355287 [A/J]; DQ355288 [CASA/RkJ]). 5′ RACE identified a similar major transcriptional start as predicted by Guan et al,20 with the addition of a single upstream adenine. The presence of one or more additional weak transcription start site(s) cannot be excluded.
As shown in Table 1, of the 8813 total base pairs comprising the Vwf cDNA, 20 nucleotide differences (SNPs) were identified between these strains, 5 of which resulted in nonsynonymous amino acid substitutions. There is a significant clustering of SNPs at the 5′ end of the transcript with only one amino acid difference (R2657Q) present within the mature protein. For comparison, we also analyzed these VWF amino acid sequences in both the published human sequence (GenBank accession no. NP_000543) and several other inbred mouse strains as shown in Table 2.
Table 1.
Nucleotide and amino acid sequence variants between A/J and CASA/RkJ VWF
| Nucleotide | VWF domain | Exon | A/J | CASA/RkJ | AA Δ |
|---|---|---|---|---|---|
| -205 | 5′ UTR | 1 | T | C | — |
| -157 | 5′ UTR | 1 | C | T | — |
| -130 | 5′ UTR | 1 | T | C | — |
| -91 | 5′ UTR | 1 | A | C | — |
| +36 | Signal peptide | 2 | T | C | — |
| +58 | Signal peptide | 3 | A | G | T20A |
| +158 | Propeptide, D1 | 3 | T | C | — |
| +339 | Propeptide, D1 | 5 | C | T | — |
| +365 | Propeptide, D1 | 5 | T | G | L122R |
| +366 | Propeptide, D1 | 5 | C | T | — |
| +525 | Propeptide, D1 | 5 | G | C | — |
| +758 | Propeptide, D1 | 7 | T | C | 1253T |
| +774 | Propeptide, D1 | 7 | T | C | — |
| +1110 | Propeptide, D1 | 10 | T | C | — |
| +1188 | Propeptide, D2 | 11 | C | T | — |
| +1236 | Propeptide, D2 | 11 | T | C | — |
| +1264 | Propeptide, D2 | 11 | A | T | T422S |
| +1395 | Propeptide, D2 | 12 | C | G | — |
| +1404 | Propeptide, D2 | 12 | T | C | — |
| +7970 | Mature VWF, C2 | 48 | G | A | R2657Q |
Nucleotides are numbered from the A of the ATG start codon and amino acids from the initiation methionine.36 Amino acid changes (AA Δ) are designated as A/J residue, amino acid number, CASA/RkJ residue.
— indicates that the observed nucleotide change did not alter the amino acid sequence.
Table 2.
VWF amino acid sequence comparison between the published human sequence and selected inbred mouse strains
| T20A | L122R | I253T | T422S | R2657Q | |
|---|---|---|---|---|---|
| Mouse | |||||
| A/J | T | L | I | T | R |
| CASA/RkJ | A | R | T | S | Q |
| C57BL/6J | T | R | I | T | R |
| RIIIS/J | ND | R | T | T | R |
| 129/Sv | ND | R | T | T | R |
| Spretus | ND | R | I | T | R |
| Human | T | T | T | S | Q |
ND indicates sequence was not determined.
To examine whether the apparent increased sequence identity downstream of +1404 was limited to the exons, selected intronic sequences were also analyzed. Consistent with the cDNA pattern of sequence variation, all A/J and CASA/RkJ noncoding genomic sequences examined 5′ of exon 13 contain several SNPs (77 sequence variants in ∼ 4.0 kB [kilobase] examined; GenBank accession nos, DQ834886 [A/J]; DQ834887 [CASA/RkJ]). Except for a few closely flanking SNPs noted in intron 48, intronic sequences examined 3′ of exon 13 are completely identical (∼ 6.9 kB examined; GenBank accession nos., DQ834886 [A/J]; DQ834887 [CASA/RkJ]) (Table 3). The latter identity also extends at least 500 bp beyond the terminal exon. Taken together, these data suggest a small region of sequence divergence flanking the single coding SNP at +7970 (R2657Q) exists within an implied larger haplotype block of more than 86 kB.21
Table 3.
Sequence variation within flanking and noncoding regions of murine Vwf
|
Intron
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| 5′ gDNA | 3 | 12 | 13 | 21 | 48 | 50 | 51 | 3′ gDNA | |
| SNPs | 6 | 36 | 31 | 0 | 0 | 3 | 0 | 0 | 0 |
| Insertion/deletion events | 2 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Total length sequenced, bp | 455 | 2313 | 1239 | 1892 | 2342 | 44 | 1641 | 438 | 500 |
CASA/RkJ and A/J Vwf mRNAs are equally expressed in vivo
Initial visual assessment of sequencing chromatograms comparing gDNA and lung mRNA in an F1 animal did not suggest significant expression differences between the A/J and CASA/RkJ alleles (Figure 5A). PolyPhred analysis estimating the allelic expression by calculating the area under several SNP curves in the original sequencing chromatograms was consistent with equal allelic expression (data not shown). Quantitative primer extension analysis on gDNA and mRNA samples confirmed equivalent expression for each Vwf allele in an F1 animal (Figure 5B). Deviations from the gDNA allelic extension ratio averaged 10% or less. These data demonstrate that the Mvwf2 effect is not due to allelic differences in Vwf mRNA expression and implies a posttranscriptional mechanism, such as differences in VWF protein biosynthesis, secretion, or plasma clearance.
Figure 5.
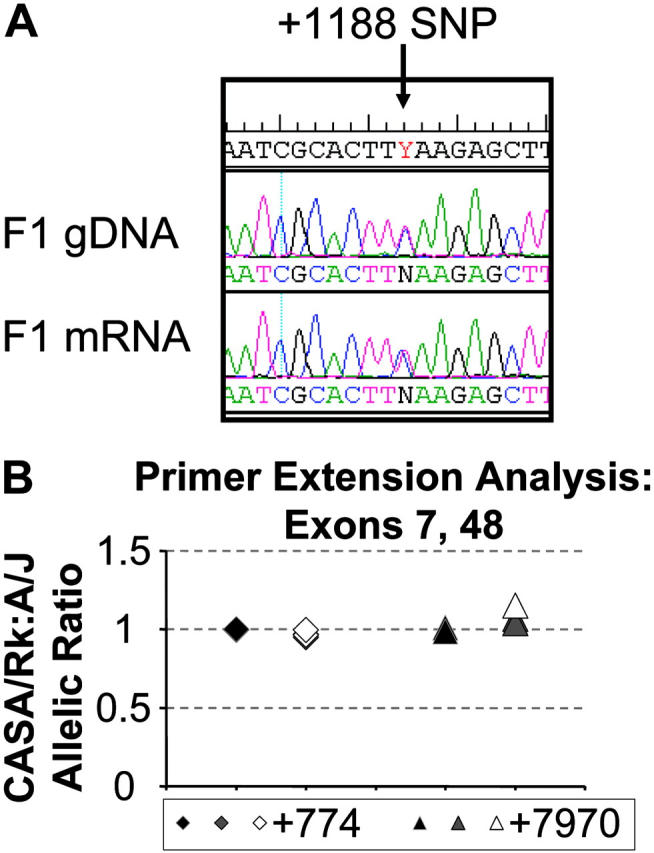
Analysis of VWF allelic expression. (A) Chromatogram comparison between F1 gDNA sequence (top) and F1 mRNA sequence (bottom) flanking the +1188 SNP. CASA/RkJ sequence encodes a T at this position, A/J a C. The F1 gDNA and mRNA show similar sized superimposed peaks of C and T at this position, consistent with similar mRNA expression from both alleles. (B) Primer extension analysis on gDNA (black symbols), mRNA (gray symbols), and mRNA amplified across an intron (open symbols) at SNP +774 (diamonds) and SNP +7970 (triangles). Symbols represent individual measurements, and all but one sample were assayed in duplicate.
R2657Q substitution in CASA/RkJ VWF results in increased biosynthesis of recombinant VWF in COS-1 cells
To analyze allele-specific effects on VWF biosynthesis, we transfected COS-1 cells with strain-specific full-length recombinant A/J and CASA/RkJ Vwf cDNAs (no. 1 and no. 2; Figure 6A). As shown in Figure 6B, conditioned media from cells transfected with CASA/RkJ Vwf contained VWF levels 1.43-fold higher than those transfected with A/J Vwf (P < .001), confirming our identification of Mvwf2 as a Vwf gene variant. This strain-specific difference in VWF production was also observed in HEK-293T cells transfected with the same constructs. In this cell line a 1.6-fold increase of VWF in conditioned media was observed in transfections using CASA/RkJ Vwf compared with A/J Vwf. These results demonstrate that this strain-specific difference in VWF protein expression is not limited to COS-1 cells and likely reflects a more general change in VWF biosynthesis efficiency not restricted to a specific cell type.
To determine which SNP or combination of SNPs among the 20 identified in Table 1 is responsible for the Mvwf2 effect, a panel of chimeric constructs was generated and analyzed (Figure 6A). The overall transfection results are summarized with regard to amino acid content in Figure 6C. Constructs containing the CASA/RkJ SNP at +7970 (Q2657) averaged a 1.32-fold increase of VWF levels in the conditioned media when compared with constructs containing the A/J SNP (R2657) at the same position (P < .001). No statistically significant difference was observed for any other single amino acid substitution or combination thereof.
CASA/RkJ and A/J represent distinct murine subspecies and extensive sequence variation (∼ 1/200 bp) is expected between these 2 inbred lines.22 Surprisingly, a large region of apparent sequence conservation (> 86 kB) flanks the causative nucleotide change (+7970G>A), dividing the Vwf locus into 2 distinct regions of high-sequence variation (5′ end) and low-sequence variation (3′ end) (Table 3; Figure 6A). Similar shared haplotype blocks between CASA/RkJ and other inbred lines have been previously observed and are thought to have arisen from cross-contamination during the process of inbreeding.22,23 The observation that the CASA/RkJ sequence is unique among all other inbred strains examined (Table 2) suggests that the +7970G>A SNP may represent a recent gain-of-function mutation event in CASA/RkJ or a closely related ancestral strain. Indeed, the CASA/RkJ allelic sequence at +7970 is consistent with a C to T transition at a CpG dinucleotide, a known hot spot for mutation in the mammalian genome.24
As shown in Table 2, similar to CASA/RkJ, human VWF shares the CASA/RkJ amino acid sequence at the orthologous position (Q2657). To exclude the possibility that apparently elevated levels of VWF in CASA/RkJ may have resulted from greater immunoreactivity with the polyclonal rabbit anti–human VWF antibody used in the ELISA assays, conditioned media samples were also assayed by Western blot analysis with an anti-Myc antibody directed against the Myc-tags engineered in both the CASA/RkJ and A/J recombinant constructs. Quantitative analysis revealed equivalent immunoreactivity with both the anti–human VWF and anti-Myc antibodies and was consistent with the VWF levels detected by ELISA for each construct.
Discussion
The laboratory mouse provides a powerful tool for dissecting the complex genetic factors contributing to a number of important phenotypes relevant to many human diseases,25 including VWF and VWD. Previous use of this approach identified the first modifier of murine VWF levels as Mvwf1, corresponding to a regulatory mutation in the Galgt2 gene resulting in accelerated VWF clearance from plasma.14 We have now extended this strategy to identify Mvwf2, an additional locus contributing to the regulation of plasma VWF level in the mouse. Mvwf2 results from a single amino acid substitution within the mature VWF monomer (R2657Q), leading to a subtle difference in the efficiency of VWF biosynthesis and/or secretion (Figure 6C). Similar linkage to VWF has been reported in humans, with as much as 20% of plasma VWF level variability potentially linked to promoter and/or intronic polymorphisms within the human VWF gene.11,26,27 Although more than 150 common SNPs and VWD-associated amino acid substitutions in human VWF have been identified,28,29 none determined thus far are located at the orthologous position of the Mvwf2 murine mutation reported here (+7970).
Comparative sequence analysis of the Mvwf2 substitution (R2657Q) in A/J, CASA/RkJ, and several other inbred strains (Table 2) suggests that R2657 is the ancestral residue and that the Q2657 allele arose in CASA/RkJ or a related founder strain, conferring a gain-of-function enhancement in VWF biosynthesis and elevated plasma VWF. This novel gain-of-function effect of the CASA/RkJ Q2657 mutation is in obvious contrast to the more typical loss of function associated with VWF gene mutations identified in patients with type 1 and type 3 VWD. This observation raises the possibility that the Vwf sequence may not be optimized for maximally efficient protein folding and/or secretion, and that similar gain-of-function mutations may contribute to the variation of plasma VWF levels in human populations.
Although the Mvwf2 R2657Q variant is the major genetic factor identified in our cross between A/J and CASA/RkJ, it only accounts for approximately 25% of the genetic variation in plasma VWF levels between these 2 strains. Thus, a considerable amount of variation remains to be explained. We did not identify additional loci with similar or greater effects, suggesting that the regulation of plasma VWF levels in the mouse may be a complex genetic trait resulting from the combination of many smaller effects. We cannot exclude the possibility, however, that one or more loci of comparable effects may have been missed because of our pooling strategy or gaps in our primary genome scan (Figure 4). Our previous characterization of Mvwf1,14 together with recent studies in humans,30,31 suggests that accelerated VWF clearance from plasma could be an important mechanism for modifying plasma VWF levels. Although we did not directly examine the rate of VWF clearance in the A/J and CASA/RkJ strains, one or more additional unidentified genetic loci contributing to the differences in VWF levels between these 2 strains may operate at the level of plasma clearance.
Taken together with the high prevalence of VWD in humans1 and the identification of VWD in a number of other mammalian species,32 our identification of at least 2 natural genetic variants in inbred mice (Mvwf1 and Mvwf2) conferring significant changes in plasma VWF suggests that altered VWF levels may be advantageous under certain circumstances and subject to positive selection. Elevated VWF levels may ameliorate VWD-associated bleeding in some forms of human type 1 VWD, whereas the low VWF levels noted in pigs with VWD appear to be protective against experimental infective endocarditis.33 Variants in plasminogen and fibrinogen have also been shown to markedly alter host susceptibility to bacterial infection,34,35 suggesting a critical role for hemostatic function in host defense against microbial pathogens. Genetic variants maintained in the population by infectious disease selection could contribute to the wide variation observed in plasma VWF levels in mice and humans. These same variants would also be expected to function as genetic modifiers of bleeding severity in VWD and potentially other hemorrhagic and thrombotic disorders.
Supplementary Material
Acknowledgments
We thank D. Siemieniak for assistance with the PolyPhred analysis; J. Bernat for assistance with 5′ RACE analysis; C. Chow for assistance with the primer extension assay; and J. Johnsen, D. Motto, and B. Zhang for careful reading of the manuscript.
Prepublished online as Blood First Edition Paper, July 27, 2006; DOI 10.1182/blood-2006-04-014688.
Supported by the National Institutes of Health (grant R37-HL39693) (D.G.) and a Graduate Fellowship Award from the National Science Foundation (H.L.L.). D.G. is an investigator of the Howard Hughes Medical Institute.
H.L.L. and D.G. participated in the research design; H.L.L., G.G.L., and J.A.S. performed the research; S.M.C. and J.C.L. aided in data analysis; H.L.L. and D.G. wrote the paper.
An Inside Blood analysis of this article appears at the front of this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Nichols WC, Ginsburg D. von Willebrand disease. Medicine. 1997;76: 1-20. [DOI] [PubMed] [Google Scholar]
- 2.Rodeghiero F, Castaman G, Dini E. Epidemiological investigation of the prevalence of von Willebrand's disease. Blood. 1987;69: 454-459. [PubMed] [Google Scholar]
- 3.Werner EJ, Broxson EH, Tucker EL, et al. Prevalence of von Willebrand disease in children: a multiethnic study. J Pediatr. 1993;123: 893-898. [DOI] [PubMed] [Google Scholar]
- 4.Sadler JE. Von Willebrand disease type 1: a diagnosis in search of a disease. Blood. 2003;101: 2089-2093. [DOI] [PubMed] [Google Scholar]
- 5.Ginsburg D. Identifying novel genetic determinants of hemostatic balance. J Thromb Haemost. 2005;3: 1561-1568. [DOI] [PubMed] [Google Scholar]
- 6.Levy GG, Ginsburg D. Getting at the variable expressivity of von Willebrand disease. Thromb Haemost. 2001;86: 144-148. [PubMed] [Google Scholar]
- 7.Orstavik KH, Magnus P, Reisner H, et al. Factor VIII and factor IX in a twin population. Evidence for a major effect of ABO locus on factor VIII level. Am J Hum Genet. 1985;37: 89-101. [PMC free article] [PubMed] [Google Scholar]
- 8.de Lange M, Snieder H, Ariens RA, Spector TD, Grant PJ. The genetics of haemostasis: a twin study. Lancet. 2001;357: 101-105. [DOI] [PubMed] [Google Scholar]
- 9.James PD, Paterson AD, Notley C, et al. Genetic linkage and association analysis in type 1 von Willebrand disease: results from the Canadian Type 1 VWD Study. J Thromb Haemost. 2006;4: 783-792. [DOI] [PubMed] [Google Scholar]
- 10.Eikenboom J, Van Marion V, Putter H, et al. Linkage analysis in families diagnosed with type 1 von Willebrand disease in the European study, molecular and clinical markers for the diagnosis and management of type 1 VWD. J Thromb Haemost. 2006;4: 774-782. [DOI] [PubMed] [Google Scholar]
- 11.De Visser MC, Sandkuijl LA, Lensen RP, et al. Linkage analysis of factor VIII and von Willebrand factor loci as quantitative trait loci. J Thromb Haemost. 2003;1: 1771-1776. [DOI] [PubMed] [Google Scholar]
- 12.Souto JC, Almasy L, Soria JM, et al. Genomewide linkage analysis of von Willebrand factor plasma levels: results from the GAIT project. Thromb Haemost. 2003;89: 468-474. [PubMed] [Google Scholar]
- 13.Mohlke KL, Nichols WC, Westrick RJ, et al. A novel modifier gene for plasma von Willebrand factor level maps to distal mouse chromosome 11. Proc Natl Acad Sci U S A. 1996;93: 15352-15357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mohlke KL, Purkayastha AA, Westrick RJ, et al. Mvwf, a dominant modifier of murine von Willebrand factor, results from altered lineage-specific expression of a glycosyltransferase. Cell. 1999;96: 111-120. [DOI] [PubMed] [Google Scholar]
- 15.Taylor BA, Navin A, Phillips SJ. PCR-amplification of simple sequence repeat variants from pooled DNA samples for rapidly mapping new mutations of the mouse. Genomics. 1994;21: 626-632. [DOI] [PubMed] [Google Scholar]
- 16.Dietrich WF, Miller J, Steen R, et al. A comprehensive genetic map of the mouse genome. Nature. 1996;380: 149-152. [DOI] [PubMed] [Google Scholar]
- 17.Buchner DA, Trudeau M, Meisler MH. SCNM1, a putative RNA splicing factor that modifies disease severity in mice. Science. 2003;301: 967-969. [DOI] [PubMed] [Google Scholar]
- 18.Kearney JA, Buchner DA, De Haan G, et al. Molecular and pathological effects of a modifier gene on deficiency of the sodium channel Scn8a (Na(v)1.6). Hum Mol Genet. 2002;11: 2765-2775. [DOI] [PubMed] [Google Scholar]
- 19.Motto DG, Chauhan AK, Zhu G, et al. Shigatoxin triggers thrombotic thrombocytopenic purpura in genetically susceptible ADAMTS13-deficient mice. J Clin Invest. 2005;115: 2752-2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guan J, Guillot PV, Aird WC. Characterization of the mouse von Willebrand factor promoter. Blood. 1999;94: 3405-3412. [PubMed] [Google Scholar]
- 21.National Center for Biotechnology Information. Mouse Genome Resources. http://www.ncbi.nlm.nih.gov/genome/guide/mouse/. Accessed January 12, 2006.
- 22.Lindblad-Toh K, Winchester E, Daly MJ, et al. Large-scale discovery and genotyping of single-nucleotide polymorphisms in the mouse. Nat Genet. 2000;24: 381-386. [DOI] [PubMed] [Google Scholar]
- 23.Wade CM, Kulbokas EJ III, Kirby AW, et al. The mosaic structure of variation in the laboratory mouse genome. Nature. 2002;420: 574-578. [DOI] [PubMed] [Google Scholar]
- 24.Cooper DN, Krawczak M. Cytosine methylation and the fate of CpG dinucleotides in vertebrate genomes. Hum Genet. 1989;83: 181-188. [DOI] [PubMed] [Google Scholar]
- 25.Nadeau JH. Modifier genes in mice and humans. Nat Rev Genet. 2001;2: 165-174. [DOI] [PubMed] [Google Scholar]
- 26.Harvey PJ, Keightley AM, Lam YM, Cameron C, Lillicrap D. A single nucleotide polymorphism at nucleotide-1793 in the von Willebrand factor (VWF) regulatory region is associated with plasma VWF:Ag levels. Br J Haematol. 2000;109: 349-353. [DOI] [PubMed] [Google Scholar]
- 27.Keightley AM, Lam YM, Brady JN, Cameron CL, Lillicrap D. Variation at the von Willebrand Factor (vWF) gene locus is associated with plasma vWF:Ag levels: identification of three novel single nucleotide polymorphisms in the vWF gene promoter. Blood. 1999;93: 4277-4283. [PubMed] [Google Scholar]
- 28.National Center for Biotechnology Information. dbSNP: SNP database. http://www.ncbi.nlm.nih.gov/SNP/index.html. Accessed January 5, 2006.
- 29.The International Society on Thrombosis and Haemostasis Scientific and Standardization Committee VWF Information Homepage. http://www.vwf.group.shef.ac.uk/. Accessed January 5, 2006.
- 30.Brown SA, Eldridge A, Collins PW, Bowen DJ. Increased clearance of von Willebrand factor antigen post-DDAVP in Type 1 von Willebrand disease: is it a potential pathogenic process? J Thromb Haemost. 2003;1: 1714-1717. [DOI] [PubMed] [Google Scholar]
- 31.van Schooten CJ, Tjernberg P, Westein E, et al. Cysteine-mutations in von Willebrand factor associated with increased clearance. J Thromb Haemost. 2005;3: 2228-2237. [DOI] [PubMed] [Google Scholar]
- 32.Ginsburg D, Bowie EJW. Molecular genetics of von Willebrand disease. Blood. 1992;79: 2507-2519. [PubMed] [Google Scholar]
- 33.Johnson CM, Bowie EJW. Pigs with von Willebrand disease may be resistant to experimental infective endocarditis. J Lab Clin Med. 1992;120: 553-558. [PubMed] [Google Scholar]
- 34.Sun H, Ringdahl U, Homeister JW, et al. Plasminogen is a critical host pathogenicity factor for group A streptococcal infection. Science. 2004;305: 1283-1286. [DOI] [PubMed] [Google Scholar]
- 35.Herwald H, Cramer H, Morgelin M, et al. M protein, a classical bacterial virulence determinant, forms complexes with fibrinogen that induce vascular leakage. Cell. 2004;116: 367-379. [DOI] [PubMed] [Google Scholar]
- 36.Goodeve AC, Eikenboom JC, Ginsburg D, et al. A standard nomenclature for von Willebrand factor gene mutations and polymorphisms. On behalf of the ISTH SSC Subcommittee on von Willebrand factor. Thromb Haemost. 2001;85: 929-931. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.