

Abstract
In vivo, bromide (Br–), nitrite (NO2–), and thiocyanate (SCN–) compete for oxidation by eosinophil peroxidase (EPO) and H2O2, yielding, respectively, HOBr, NO2·, and HOSCN. We have recently shown that SCN– is the strongly preferred substrate for EPO in vivo and that HOSCN, in contrast with other EPO-generated oxidants and HOCl, is a relatively weak, cell-permeant, sulfhydryl (SH)–reactive oxidant. We here show that HOSCN is a uniquely potent (up to 100-fold) phagocyte oxidant inducer of tissue factor (TF) activity in human umbilical vein endothelial cells (HUVECs). This induction is attributable to transcriptional up-regulation of TF gene expression dependent upon both activation of the p65/c-Rel TF-κB transcription factor and activity of the ERK1/2 kinase pathway upstream of Egr-1 and was markedly further enhanced in the presence of wortmannin, an inhibitor of the PI3 kinase/Akt pathway. HOSCN also markedly activates the proinflammatory p65/p50 NF-κB pathway. Based on these findings we hypothesize that HOSCN generated by adherent and infiltrating eosinophils may provoke the development of a prothrombotic and proinflammatory endothelial/endocardial phenotype that promotes the pronounced thrombotic diathesis characteristic of the hypereosinophilic syndrome.
Introduction
Eosinophils (EOs) are specialized phagocytes that function to protect the host against metazoan parasites but can also mediate pathologic tissue damage in allergic inflammatory states. Perhaps the most striking example of EO-mediated pathology is the hypereosinophilic syndrome (HES), a systemic hematologic order characterized by multiorgan system involvement, most notably a characteristic, often lethal, form of endocarditis.1,2 Eosinophilic, or Loeffler, endocarditis is characterized by massive infiltration into the endocardium and myocardium of activated degranulating EOs and mural thrombosis leading to both pulmonary and systemic embolism. HES is also accompanied by a prominent systemic thrombotic diathesis that can manifest as hepatic vein thrombosis, cerebral sinus thrombosis, disseminated intravascular coagulation (DIC), skin microvessel thrombosis, and deep venous thrombosis.3-6 The etiology of this thrombotic diathesis is unclear.
EO-specific granule proteins are exocytosed and accumulate at sites of EO inflammation. These include the unique eosinophil peroxidase (EPO), a protein that accounts for 40% by weight of the EO-specific granule weight and shares 70% amino acid homology with the better-characterized neutrophil myeloperoxidase (MPO).7 In addition, EOs are endowed with a highly active NADPH oxidase system capable of sustaining up to 10 times the superoxide anion and H2O2 generation capacity of neutrophils.8 While it is clear that MPO functions predominantly to utilize H2O2 to oxidize chloride to hypochlorous acid (HOCl), the preferred physiologic substrate for EPO is less certain. Three unusual substrates—bromide (Br–), nitrite (NO –2), and thiocyanate (SCN–)—compete for oxidation by EPO in physiologic fluids in the presence of H2O2 to yield, respectively, hypobromous acid (HOBr), nitrogen dioxide (NO2 ·), or hypothiocyanous acid (HOSCN).9-11 These oxidant products have strikingly different reactivities: HOBr and NO2 · are potent, widely reactive membrane-lytic oxidants, whereas HOSCN is a weak, sulfhydryl (SH)–specific oxidant that penetrates into cells and imposes an intracellular oxidant stress.12-14 We have previously shown that in fluids of physiologic composition SCN– is the strongly preferred substrate for EPO, and therefore HOSCN is its predominant oxidant product.11 Despite the high potential for peroxidative damage attributable to the simultaneous presence of copious amounts of H2O2 and EPO deposition in tissue, however, little is known about the contribution of EPO-mediated oxidative damage to the pathology of eosinophilic inflammatory states.
Tissue factor (TF) plays a pivotal role in the pathology of thrombosis in vivo. Phagocyte oxidants including H2O2 and HOCl stimulate TF expression modestly (maximally 2- to 3-fold) in endothelial cells15,16 and monocytes.17 Oxidants have also been implicated in activation of the NF-κB transcription factor,18 and the TF promoter has an NF-κB–like “TF-κB” binding site.19 We hypothesized that EPO-derived oxidant, especially HOSCN, might also stimulate endothelial cell TF expression and thereby contribute to the pathogenesis of thrombosis in hypereosinophilic states. Using human umbilical vein endothelial cells (HUVECs) as an in vitro model of EO-mediated endothelial and endocardial toxicity, we here show that HOSCN is a uniquely potent phagocyte oxidant activator of TF expression. HOSCN also stimulates the p65/p50 NF-κB pathway, raising the possibility that HOSCN also provokes expression of a variety of proinflammatory gene products relevant to EO-mediated tissue damage.
Materials and methods
Reagents
Lipopolysaccharide (LPS) (Escherichia coli 055:B5), NaSCN, NaBr, NaNO2, HOCl, andrographolide, lactoperoxidase, and protease inhibitor cocktail were from Sigma Aldrich (St Louis, MO). Ionophore A23187 was from EMD Biosciences (San Diego, CA). Hanks balanced salt solution (HBSS) was from Invitrogen (Carlsbad, CA). Rabbit polyclonal antibodies against Egr-1, p65, p50, c-Rel, SP1, IκB-α, β-actin, and alkaline phosphatase (AP)–conjugated goat antirabbit and antimouse IgG and donkey antigoat IgG-AP were from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit polyclonal antibodies against phospho-AKT (Ser473), AKT, and p44/p42 MAPK (ERK1/2) and mouse monoclonal antibody against phospho-p44/p42 MAPK (Thr202/Tyr204, ERK1/2), U0126, and wortmannin were from Cell Signaling Technology (Beverly, MA). Mouse monoclonal HTF-1 and rabbit polyclonal antibodies against human TF20 were kindly provided by Dr Ronald Bach (Veterans Administration Medical Center [VAMC], Minneapolis, MN). Human EPO was prepared21 and kindly supplied by Dr Gerald J. Gleich, University of Utah, Salt Lake City. Nuclear Extraction kit (AY2002) was purchased from Panomics (Redwood City, CA).
Human umbilical vein endothelial cell (HUVEC) culture
HUVECs were isolated from umbilical cords and cultured as previously described.22 HUVEC monolayers were used for experiments at passages 2 or 3.
Oxidant modulation of HUVEC TF activity
HUVEC monolayers were exposed to oxidants using 3 different protocols: (1) by adding preformed reagent oxidants in a low-serum (1%) system (Figure 1A-B); (2) by an enzymatic reagent H2O2/EPO system in serum-free buffer supplemented with one of 3 different potential EPO substrates (Figure 1C); and (3) by adding preformed reagent oxidants in a high-serum (10%) system (Figures 2-3).
Figure 1.
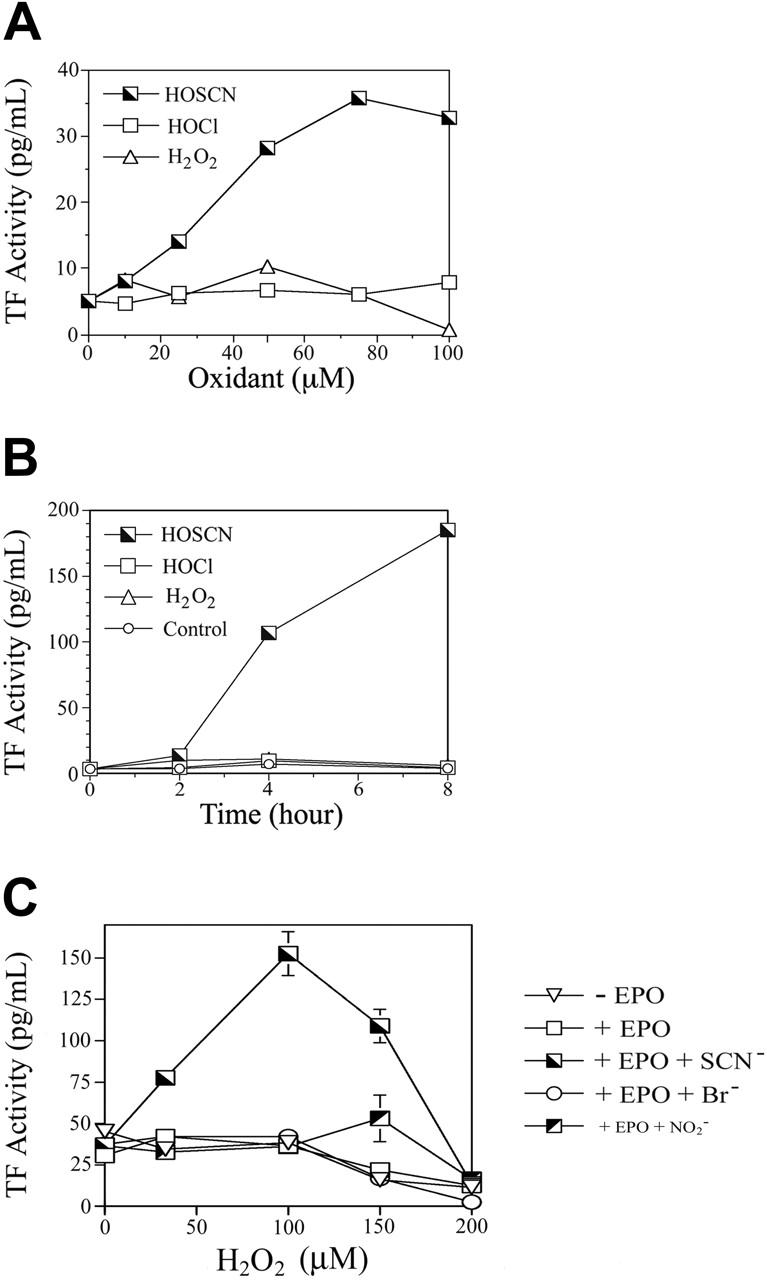
Comparative induction of HUVEC TF activity by phagocyte oxidants. (A) Dose response analysis of phagocyte oxidants. HUVEC monolayers were exposed to 0 to 100 μM reagent HOCl, HOSCN, or H2O2 for 4 hours in M199 medium with 1% FBS, medium was replaced with HBSA with 10 μM ionophore A23187, and scraped freeze-thawed lysates were assayed for TF activity using a 2-stage clotting assay. (B) Time course of phagocyte oxidant induction of TF activity. HUVEC monolayers were exposed to either buffer, 50 μM reagents HOCl, HOSCN, or H2O2, for 0 to 8 hours as described for panel A, and assayed for TF activity at the indicated time points. (C) Substrate-dependent TF activity induction by EPO. HUVEC monolayers were overlaid with 100 nM EPO in HEPES/Hanks buffer (+ EPO) or the same buffer to which 1 mM NaSCN (+ EPO + SCN–), 1 mM NaBr (+ EPO + Br–), or 1 mM NaNO2 (+ EPO + NO2– was added. The indicated final concentrations of H2O2 were added, monolayers incubated 30 minutes, supernatant buffer replaced with M199 with 10% FBS, and freeze-thawed cell lysates assayed for TF activity by one-stage clotting assay after a further 5 hours of incubation. All data are shown ± SD.
Figure 2.
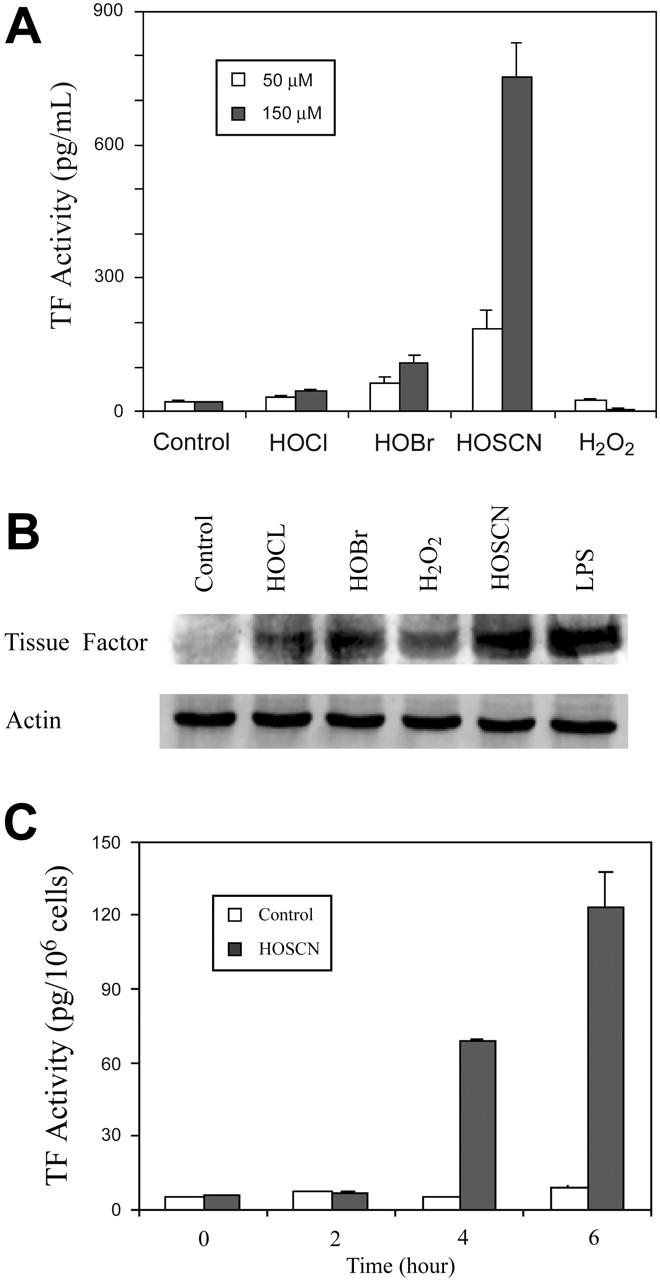
TF induction by phagocyte oxidants in the presence of 10% serum. (A) Total cell lysate TF activity. Monolayers of HUVECs were supplemented with buffer (control), or 50 μM(□), or 150 μM(▪) of reagent HOCl, HOBr, HOSCN, and H2O2 in M199 medium with 10% serum and incubated at 37°C for 4 hours prior to assay of TF activity in ionophore-treated cell lysates. (B) TF Western blot. HUVECs were treated as in panel A with 150 μM of the designated oxidant or 10 ng/mL LPS and TF was detected by Western blot. (C) Cell surface TF activity. HUVECs were exposed to either buffer control (□) or 150 μM HOSCN (▪) for the indicated times and harvested into single-cell suspension by trypsinization. A total of 6 × 105 cells were pelleted, washed with HBSA buffer, and then incubated with 6 nM factor VIIa, 600 nM human FX, and 10 mM CaCl2 at 37°C for 15 minutes. Supernatant fluid Xa activity was quantitated by clotting assay and TF activity derived from a standard curve using recombinant TF in the first stage. All data are shown ± SD.
Figure 3.

HOSCN induces oxidant-dependent transcriptional up-regulation of TF activity. (A) HUVECs were supplemented with either buffer (▵, control), 10 ng/mL LPS (□), or 150 μM HOSCN (▪) in M199 medium with 10% FBS, incubated at 37°C for the indicated times, and lysates assayed for TF activity. (B) HUVECs were exposed in the high serum system to either buffer (control), 150 μM HOSCN, or 10 ng/mL LPS, and in the presence or absence of 450 μM 5-thio-2-nitrobenzoic acid (TNB), and then incubated 5 hours prior to assay of TF activity. (C) HUVECs were exposed to buffer, 150 μM HOSCN, or 10 ng/mL LPS for the indicated time. Total cellular RNA was extracted and analyzed by RT-PCR using either TF-(252 bp) or 18S rRNA-(324 bp) specific primers. Gels were stained with SYBR Green I and imaged using ultraviolet transillumination.
Low-serum system. Reagent HOSCN was synthesized and quantitated as previously described using lactoperoxidase.11,12 HOCl was quantitated spectrophotometrically using its molar extinction coefficient 350 M–1 cm–1 at 292 nm and pH more than 11.23 Reagent HOBr was prepared by quantitative conversion of HOCl to HOBr by adding 20-fold excess sodium bromide, as previously described.24 For experiments comparing the dose response of phagocyte oxidants upon HUVEC TF activity (Figure 1A), growth medium was aspirated from HUVEC monolayer wells and replaced with prewarmed M199 medium with 1% fetal bovine serum (FBS) and 1.5 mM l-glutamine with 0 to 100 μM preformed HOCl, HOSCN, or H2O2 and incubated 4 hours at 37°C in a CO2 incubator. Medium was aspirated and wells overlaid with 1 mL PBS. Aspirated medium was centrifuged at 1000g for 10 minutes to pellet nonadherent cells. Pellets were resuspended in 0.5 mL ice-cold HBSA (137 mM NaCl, 5.38 mM KCl, 5.55 mM glucose, 10 mM HEPES, 12.5 mM CaCl2, 0.1% bovine serum albumin) buffer with 10 μM A23187 and monolayers frozen at –80°C until being thawed, scraped, and assayed for TF activity in a 2-stage clotting assay.25 The time course of oxidant induction of HUVEC TF activity (Figure 1B) was assessed by adding 50 μM HOCl, HOSCN, or H2O2 for 0 to 8 hours prior to processing for TF activity using the 2-stage assay.
Serum-free system. Confluent monolayers of HUVECs in 12-well plates were washed and overlaid with 1 mL per well of HEPES/Hanks buffer (HBSS with 10 mM HEPES, pH 7.4, 1.2 mM CaCl2, 0.492 mM MgCl2, 0.406 mM MgSO4) containing 100 nM EPO and 1 mM of either NaSCN, NaBr, or NaNO2. Plates were incubated 15 minutes at 37°C whereupon 0 to 200 μM boluses (final concentration) of H2O2 were added. Plates were incubated 30 minutes and then “rescued” by aspiration of supernatant buffer and replacement with 2 mL per well of M199 with 10% FBS and 1.5 mM l-glutamine. Plates were incubated 5 hours further and then processed for assay of tissue factor (Figure 1C) using a one-stage clotting assay.
High-serum system. HUVEC monolayers were aspirated of growth medium and replaced with M199 with 10% FBS and 1.5 mM l-glutamine, incubated 1 hour at 37°C in a CO2 incubator to allow equilibration, then either 50 μM or 150 μM (final concentration) H2O2, HOCl, HOBr, HOSCN, or a PBS buffer dilution control was added, the cells incubated 4 hours at 37°C, and assayed for TF activity by one-stage clotting assay (Figure 2A). Alternatively (Figure 3), either 150 μM HOSCN or 10 ng/mL LPS was added in the same high-serum system and monolayers processed for assay of tissue factor at 1, 2, 3, 4, 6, 8, and 24 hours by one-stage clotting assay.
Assay of tissue factor activity of intact HUVECs. HUVEC monolayers were exposed to either buffer control or 150 μM HOSCN at 37°C for 0, 2, 4, or 6 hours in the high-serum system (Figure 2C). Monolayers were washed with PBS plus 0.5 mM EDTA for 2 minutes, supernatants were removed, and the monolayers were harvested into single-cell suspension with 0.5 mM EDTA/0.017% trypsin for 1 minute and washed once with ice-cold PBS plus 10% (vol/vol) FBS, pH 7.4. HUVECs (6 × 105) were pelleted, washed with HBSA, and then incubated with 6 nM human factor VIIa and 600 nM human factor X (both from Enzyme Research, South Bend, IN) and 10 mM CaCl2 in HBSA at 37°C for 15 minutes. The supernatant was collected and assayed for TF activity using the Xa-based clotting times as previously described.25
Whole-cell lysate TF activity: one-stage assay. Two-hundred microliter aliquots of HUVEC ionophore-treated lysates were incubated at 37°C for 3 minutes prior to the addition of 100 μL citrated human platelet-poor plasma and measuring clotting time after recalcification in an ST4 coaguolometer (Diagnostica Stago, Parsippany, NJ). Relative TF activity was estimated by comparing clot times with a standard curve generated by assaying serial dilutions of Innovin (recombinant, relipidated human TF; Dade International, Deerfield, IL). To confirm TF dependence of the clotting assay, representative lysates were preincubated with 4 μg/mL mouse monoclonal antibody HTF-1 against human TF at 37°C for 30 minutes prior to initiating coagulation. In all cases resulting clotting times were equivalent to less than 2 pg/mL TF (not shown). For some experiments HUVEC lysates were assayed for TF activity using our previously described25 2-stage assay.
Reverse transcriptase–polymerase chain reaction (RT-PCR) analysis of TF mRNA
Confluent monolayers of HUVECs in T-75 flasks were exposed in the high-serum system to either buffer control, 150 μM HOSCN, or 10 ng/mL LPS. Flasks were washed with prewarmed HBSS without Ca2+ or Mg2+, and total cellular RNA was extracted using acid guanidium thiocyanate-phenol-chloroform.26 RNA was treated with DNase I and purified using Rneasy Mini Columns according to the manufacturer's protocol (Qiagen, Valencia, CA). Two micrograms of RNA was reverse transcribed with random decamers according to manufacturer's instructions using the RETROscript First Strand Synthesis Kit (Ambion, Austin, TX). Aliquots of 3 μL cDNA were subjected to PCR in the presence of a 1:9 primer-to-competimer ratio specific for 18S rRNA (QuantumRNA 18S Internal Standards; Ambion). TF-specific primers used were as follows: sense (5′-CCCGTCAATCAAGTCTACACTGTTC-3′) and antisense (5′TCCGAGGTTTGCTCCAGGTAAG-3′). Amplification was performed using 0.875 units per reaction of Taq and Tgo DNA polymerases (Roche Applied Science, Indianapolis, IN). The following parameters were used on a PTC-100 thermal cycler (MJ Research, Reno, NV): 94°C for 3 minutes; 26 cycles of 94°C for 45 seconds, 56.4°C for 30 seconds, 72°C for 45 seconds; followed by a 7-minute extension at 72°C. Aliquots were analyzed by submarine electrophoresis by loading 10 μL PCR product on 1.5% agarose gels. Gels were stained using a 1:10 000 dilution of SYBR Green I (Molecular Probes, Eugene, OR) in 1 × TBE buffer for 25 minutes at room temperature and imaged using ultraviolet transillumination.
Extraction of nuclear proteins for electrophoretic mobility shift assay (EMSA) and Western blot analysis
Nuclear extracts from 107 HUVECs were prepared with a Nuclear Extract Kit (Panomics) according to the manufacturer's instructions and stored at –80°C until use. Protein concentrations in these nuclear extracts were 3 to 5 mg/mL.
EMSA
Oligonucleotides for the TF-κB–like consensus site (5′-GTCCCGGAGTTTCCTACCGGG-3-3′) and TF-κB–like mutant control (5′-GTCCCGGAGTTAGATACCGGG-3′); proximal TF AP-1 consensus site (5′-CTGGGGTGAGTCATCCCTT-3′) and AP-1 mutant control (5′-CTGGGGTGAGTTGTCCCTT-3′); and Egr-1 binding consensus site (5′-CCCGGCGCGGGGGCGATTTCGAGTCA-3′) and mutant control (5′-CCCGGCGCTAGGGCGATTTCGAGTCA-3′) were obtained from Integrated DNA Technologies (Skokie, IL). NF-κB consensus (5′-AGTTGAGGGGACTTTCCCAGGC-3′) and mutant (5′-AGTTGAGGCGACTTTCCCAGGC-3′) oligonucleotides were purchased from Santa Cruz Biotechnology. The oligonucleotide was end labeled with [γ-32P]-ATP (Amersham Pharmacia Biotech, Piscat-away, NJ) and T4 polynucleotide kinase (Amersham Pharmacia Biotech). Nuclear extracts (5 μg for NF-κB and Egr-1 probes, 2 μg for AP-1 probe, 10 μg for TF-κB–like probe) were incubated with radiolabeled DNA probes (50 fmol) for 30 minutes at room temperature in a total 20 μL binding reaction containing 20 mM HEPES, pH 7.9, 50 mM KCl, 0.5 mM EDTA, 5% glycerol, 1 mM dithiothreitol, 0.5 mM PMSF, 1 mg/mL BSA, 0.1% NP-40, and 50 μg/mL poly(dI-dC). Protein-DNA complexes were separated from free DNA probe through 5% nondenaturing polyacrylamide gel in 0.5 × TBE buffer. The gels were dried and analyzed by autoradiography. For competition and antibody supershift experiments, binding reactions were preincubated with unlabeled consensus and mutant double-stranded oligonucleotides or supershift antibodies for 30 minutes on ice before the addition of the radiolabeled probes.
Immunofluorescence staining of p65 subunit of NF-κB in HUVECs
HUVECs were grown to confluence on glass coverslips and then exposed to M199 medium with 5% FBS (control), 10 μg/mL LPS, or 50 μM HOSCN for 4 hours. HUVECs were washed and fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100. Specimens were then overlaid with 1 μg/mL rabbit anti-p65 polyclonal antibody and incubated at room temperature for 4 hours, washed 3 times, exposed to FITC-conjugated goat antirabbit IgG, and analyzed by immunofluorescence microscopy (magnification, × 400). Images were obtained using an Olympus IX70 microscope, an Olympus Plan APO 100×/1.0 numeric aperture lens (oil), and an Olympus DP70 camera (Olympus, Melville, NY). Images were acquired using DP Manager software and were processed using Adobe Photoshop (Adobe Systems, San Jose, CA).
Kinase and transcription factor inhibitor studies
For kinase pathway inhibition studies, 10 μM U0126 and both 10 and 100 nM wortmannin were used as specific inhibitors of the ERK1/2 and PI3 kinase/Akt pathways, respectively. Andrographolide, 10 μg/mL, was used as an inhibitor of the NF-κB pathway.27 Monolayers of HUVECs in T-75 flasks were pretreated 1 hour with inhibitors and then exposed in 10% serum-containing medium with fresh inhibitors to either buffer control or 150 μM HOSCN. At various time points, flasks were either processed for TF activity assay or rinsed with HBSS without Ca2+ or Mg2+ and then extracted with 300 μL ice-cold lysis buffer (300 mM NaCl, 1.5 mM MgCl2, 200 μM EDTA, 0.1% Triton X-100, 25 mM HEPES, pH 7.6) with 5 mM dithiothreitol, 100 μM sodium orthovanadate, 20 mM β-glycerophosphate, 20 μg/mL leupeptin hydrochloride, and 1 mM PMSF for 10 minutes. After vigorous scraping the lysate was collected and incubated on ice for 10 minutes further and then centrifuged 10 minutes at 13 000g. Total cell extracts and nuclear protein extracts were separated on 10% SDS-polyacrylamide gels and transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA). Membranes were blocked for 1 hour at room temperature with 5% nonfat dry milk in Tris-buffered saline (10 mM Tris, 150 mM NaCl, pH 7.5) with 0.01% Tween 20 (TBS/T). Primary antibodies were diluted in blocking buffer and incubated overnight at 4°C. After washing 3 times with TBS/T at room temperature, membranes were blocked for 1 hour at room temperature before adding alkaline phosphatase–conjugated goat antirabbit or antimouse IgG antibodies (1:10 000 dilution) and incubating 1 hour at room temperature. After washing, membranes were exposed to 24 μL/cm2 ECF substrate (Amersham Pharmacia Biotech) for 10 minutes at room temperature and scanned on a Storm 860 Phosphoimager (Molecular Devices, Sunnyvale, CA).
Results
Comparative induction of TF activity in endothelial cells by phagocyte-derived oxidants
The principal stable oxidants generated by neutrophils are H2O2 and HOCl while EOs generate primarily H2O2 and HOSCN.11,12 We compared the relative capacity of each of these oxidants to induce TF activity in HUVEC monolayers (Figure 1). Increasing concentrations of reagent oxidants were added to tissue culture wells containing M199 medium with low (1%) concentrations of FBS and incubated for 4 hours, and scraped freeze-thawed lysates were assayed for TF activity (Figure 1A). Neither HOCl nor H2O2 influenced TF expression, but as little as 10 μM HOSCN increased TF activity, peaking at approximately 75 μM at 7-fold over baseline.
To determine the time course of TF activity induction by HOSCN and assess the possibility that our failure to detect stimulation by H2O2 and HOCl was due to assaying at a single time point, we added 50 μM of each oxidant and assayed monolayers at various time points thereafter (Figure 1B). HOSCN induces TF activity between 2 and 4 hours with a further increase through 8 hours. Subsequent experiments showed that 8 hours is the maximum point of TF activity, which wanes over the next 16 hours. This delay in TF induction suggests the possibility of a transcriptional regulatory mechanism.
We compared the ability of a purified EPO system composed of H2O2, EPO, and each of its 3 preferred substrates—thiocyanate, bromide, or nitrite—to induce HUVEC TF expression (Figure 1C). To avoid the potential artifact of serum components scavenging the more reactive oxidants generated in the presence of bromide and nitrite, these experiments were performed in a serum-free system. Thirty minutes after adding increasing concentrations of H2O2 to fuel the reactions, supernatant liquid was aspirated and the monolayers “rescued” by adding medium with 10% FBS before being assayed for TF activity after a further 4 hours of incubation. Only in the presence of thiocyanate was significant induction of TF activity detectable. Collectively the experiments in Figure 1 show that HOSCN is a uniquely potent phagocyte oxidant inducer of TF activity in endothelial cells.
The potent oxidants HOCl and HOBr form less reactive but more stable chloroamine and bromoamine adducts with primary amine moieties, including taurine, present in serum. These less reactive intermediates, unlike their parent oxidants, might induce TF expression by imposing intracellular oxidant stress, as does HOSCN. We therefore performed experiments in the presence of 10% FBS (Figure 2A). Indeed, under these conditions some weak stimulatory activity by HOBr and HOCl at both 50 and 150 μM is seen, but enhancement by HOSCN is strikingly enhanced to as much as 30-fold over baseline. H2O2 had no activity under these conditions. TF up-regulation was confirmed by Western blot analysis (Figure 2B). Because TF exists on cell membranes predominantly in an “encrypted” form that is inactive, we assayed the capacity of HOSCN to induce TF activity on intact cellular surfaces rather than whole cell lysates, where TF is maximally deencrypted.28 As shown in Figure 2C, HOSCN also markedly stimulates TF activity on intact endothelial surfaces. In experiments not shown we have found that about 15% of total lysate HUVEC TF activity is expressed on the intact cell surface (ie, “de-encrypted”).
HOSCN up-regulation of TF gene transcription: comparison with LPS
To gauge the capacity of HOSCN to stimulate TF activity in endothelium, we compared it with that of the potent in vivo agonist, LPS. As shown in Figure 3A, 150 μM HOSCN induction rivals that of 10 ng/mL LPS and has a similar time course, peaking at 8 hours and declining thereafter as well as exhibiting a lag time of 2 to 3 hours prior to induction. To show that the activity of our HOSCN preparation was attributable to its oxidant character and not some contaminant, such as LPS, we examined the influence of TNB, which rapidly reduces or “scavanges” HOSCN, upon this activity. As shown in Figure 3B, TNB nearly totally reverses the capacity of our HOSCN preparation to induce TF activity without affecting LPS induction. To determine whether HOSCN, as does LPS, stimulates TF expression through transcriptional up-regulation, we performed semiquantitative RT-PCR analysis on RNA extracted from HUVEC monolayers at various time points after exposure to these agonists. As shown in Figure 3C, both HOSCN and LPS strikingly up-regulate TF mRNA levels that are first detectable by 2 hours and peak at 3 to 4 hours.
HOSCN influence upon AP-1, Egr-1, NF-κB, and TF-κB transcription factor activity
Because TF transcription is regulated by upstream binding sites for the AP-1, Egr-1, and TF-κB–like transcription factors,17,29-31 we assessed which, if any, of these factors was activated by HOSCN. First, because there is precedent for oxidant activation of the p65/p50 heterodimeric form of NF-κB18 that is typically the most abundant isoform of the NF-κB family, we assayed the effect of LPS and HOSCN upon p65/p50 activation using an EMSA analysis of HUVEC nuclear extracts prepared after 1 hour of exposure, reasoning that transcription factor activation would significantly precede transcriptional activation. As shown in Figure 4A, both LPS and HOSCN—but not H2O2, HOCl, or HOBr—markedly increase expression of a p65/p50-specific retardation band. That this band was composed of both p65 and p50 was confirmed by “supershift assays” using specific polyclonal antibodies. Specificity of the interaction was confirmed by the finding that excess unlabeled but not excess mutant unlabeled probe totally eliminated the p65/p50-specific band. However, human TF has a TF-κB–like upstream sequence that is specific for the p65/c-Rel heterodimer, not p65/p50 (NF-κB). As shown in Figure 4B, both LPS and HOSCN activate p65/c-Rel, though, as expected, this heterodimer is much less abundant than the p65/p50 isoform. p65-specific antibodies produced a supershift, and c-Rel–specific antibodies diminished the intensity of the parent band without a convincing supershift. These results suggest that this complex is at least in part p65/c-Rel heterodimer.
Figure 4.

HOSCN activates the p65/p50 and p65/c-Rel heterodimeric forms of NF-κB. (A) EMSA analysis of NF-κB activation. HUVECs were stimulated with 10 μg/mL LPS or 150 μM of the designated oxidant as described in the legend to Figure 2 for 1 hour prior to extraction of nuclear proteins. Five-microgram aliquots of nuclear extracts were incubated with radiolabeled NF-κB consensus binding sequence oligonucleotide and separated on 5% nondenaturing polyacrylamide gels. The mobility of the shifted NF-κB consensus probe is shown by the arrow; the slower migration of supershifted bands is shown in brackets. The lanes designated “consensus” show the effect of adding 10-fold excess unlabeled probe; “mutant,” the effect of adding 10-fold excess unlabeled mutant NF-κB consensus oligonucleotide; and “p65 Ab,” “p50 Ab,” and “c-Rel Ab,” the effects of adding polyclonal antibodies to the designated proteins. (B) As in panel A, except HUVECs were incubated with the stimulant for 2 hours, the loading of 10 μg nuclear extract protein/lane, and use of a radiolabeled oligonucleotide probe, derived from the authentic TF-κB–like sequence. (C) Immunofluorescent microscopy localization of p65 in HUVEC monolayers exposed to 10 μg/mL LPS or 50 μM HOSCN in M199 medium with 5% FBS for 4 hours, fixed with 4% paraformaldehyde, and permeabilized with 0.1% Triton X-100. p65 was visualized using a rabbit anti-p65 polyclonal primary antibody and an FITC-conjugated goat antirabbit IgG secondary antibody by immunofluorescence microscopy (magnification, × 400). (D) Western blot analysis of the nuclear extracts used for 1 hour (left panel) and 2 hours (right panel) using polyclonal antibodies specific for the designated proteins with SP1 as a loading control. For IκB-α only, cytosolic rather than nuclear extracts were analyzed with actin as a loading control.
Because activation of p65/p50 NF-κB is accompanied by translocation of subunits from the cytoplasm to the nucleus, we localized p65 in LPS- and HOSCN-exposed HUVEC monolayers by immunofluorescence microscopy. Both LPS and HOSCN effected nuclear translocation of the p65 subunit (Figure 4C). Nuclear translocation of c-Rel, p65, and p50 was confirmed using Western blot assays of nuclear extracts (Figure 4D). Degradation of cytoplasmatic IκB-α was also confirmed. These results show HOSCN activates both p65/p50 (NF-κB) and p65/c-Rel (TF-κB) to an extent comparable to that of LPS.
EMSA analysis of AP-1 transcription factor activation (Figure 5A) shows that neither LPS nor HOSCN provoked detectable AP-1 activation, but baseline expression was prominent. An anti–c-Jun antibody produced a supershift, whereas anti–c-Fos diminished intensity of the retarded band without a clear supershift. These results suggest that AP-1 is constitutively activated and not significantly further stimulated by either LPS or HOSCN. We also assessed whether HOSCN could activate Egr-1 as well as its upstream ERK1/2 kinase activator. Using an Egr-1–specific probe and PMA as a positive control we found modest up-regulation by HOSCN (Figure 5B). This was confirmed by Western blot analysis of nuclear extracts (Figure 5C). HOSCN rapidly (within minutes) stimulates phosphorylation and activation of the p44/p42 components of ERK1/2 (Figure 5D).
Figure 5.
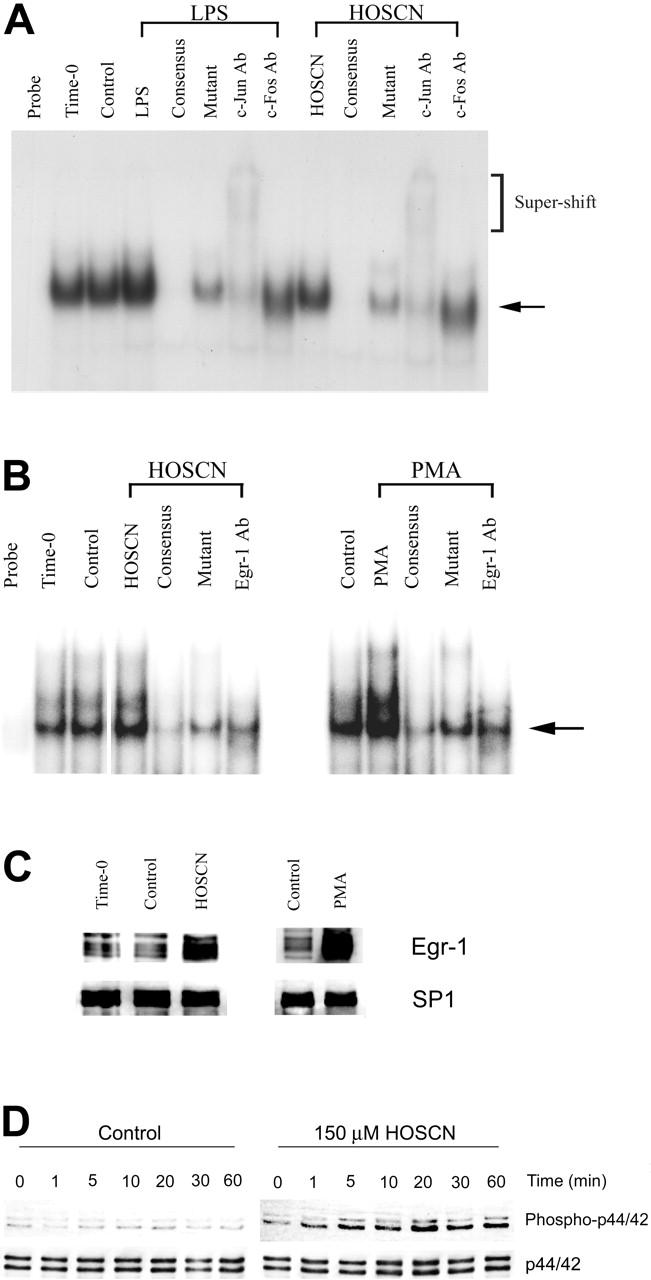
HOSCN modulation of the AP-1 and Egr-1 pathways. (A) EMSA analysis of AP-1. HUVECs were stimulated with 10 μg/mL LPS or 150 μM HOSCN for 1 hour in M199 medium with 10% FBS. Two microgram aliquots of nuclear extracts were incubated with a radiolabeled, TF promoter–derived proximal AP-1 binding site oligonucleotide and analyzed as described for Figure 4. The arrow shows the mobility of the shifted probe and brackets the mobility of supershifted probe. Lanes designated “consensus” show the effect of adding 10-fold excess unlabeled AP-1 probe; “mutant,” the effect of adding 10-fold excess unlabeled mutant AP-1 probe; and “c-Jun” and “c-Fos,” the effect of adding monoclonal antibodies to the designated proteins. (B-D) EMSA analysis of Egr-1 and demonstration of upstream ERK1/2 kinase activation by HOSCN. (B) HUVECs were stimulated with 150 μM HOSCN (left panel) or 50 ng/mL PMA (right panel) for 1 hour in M199 medium with 10% FBS and nuclear extracts prepared. Five-microgram nuclear protein aliquots were incubated with a radiolabeled Egr-1 binding oligonucleotide probe and analyzed as described for Figure 4. The arrow shows the mobility of Egr-1–probe complex. Lanes designated “consensus” show the effect of adding 10-fold excess unlabeled Egr-1 probe; “mutant,” the effect of adding 10-fold excess unlabeled mutant Egr-1; and “Egr-1 Ab,” the effect of adding a polyclonal antibody for Egr-1. (C) Western blots of the nuclear extracts shown in panel B probed with an antibody for Egr-1 with SP1 as a loading control. (D) HUVECs were exposed to buffer (left panel) or 150 μM HOSCN (right panel) in complete medium for the indicated times, whole cell lysates prepared, and Western blots probed with an antibody specific for the phospho forms of the p44 and p42 components of Erk1/2, here designated “phospo-p44/42” (top row). p44/42 (Erk1/2) was probed as a protein loading control (bottom row).
To assess the functional importance of the ERK1/2/Egr-1, PI3 kinase/Akt, and NF-κB pathways, we assessed the effects upon HOSCN-dependent TF induction of U0126, a specific ERK1/2 inhibitor, wortmannin, a potent inhibitor of the PI3K/Akt pathway, and andrographolide, a recently described specific inhibitor of the NF-κB pathway.27 As expected, U0126 blocked phospho-p44/42 (ERK1/2) and wortmannin inhibited phospho-Akt expression (Figure 6A). HOSCN activates ERK1/2 and slightly stimulates phosphor-Akt expression (Figure 6A). TF detected by Western blot was increased by HOSCN but decreased in the presence of HOSCN and U0126. Wortmannin, even in the absence of HOSCN, significantly increases TF expression, which was greatly further enhanced by addition of HOSCN (Figure 6A). The effect of these kinase inhibitors was quantitated by assaying TF activity induction in whole cell lysates (Figure 6B). Both U0126 and andrographolide inhibit stimulation by HOSCN, whereas wortmannin stimulates activity. Thus, both the ERK1/2/Egr-1 and NF-κB pathways participate importantly in HOSCN induction of endothelial cell TF activity, whereas the PI3 kinase/Akt pathway inhibits both constituitive and HOSCN-stimulated TF expression.
Figure 6.
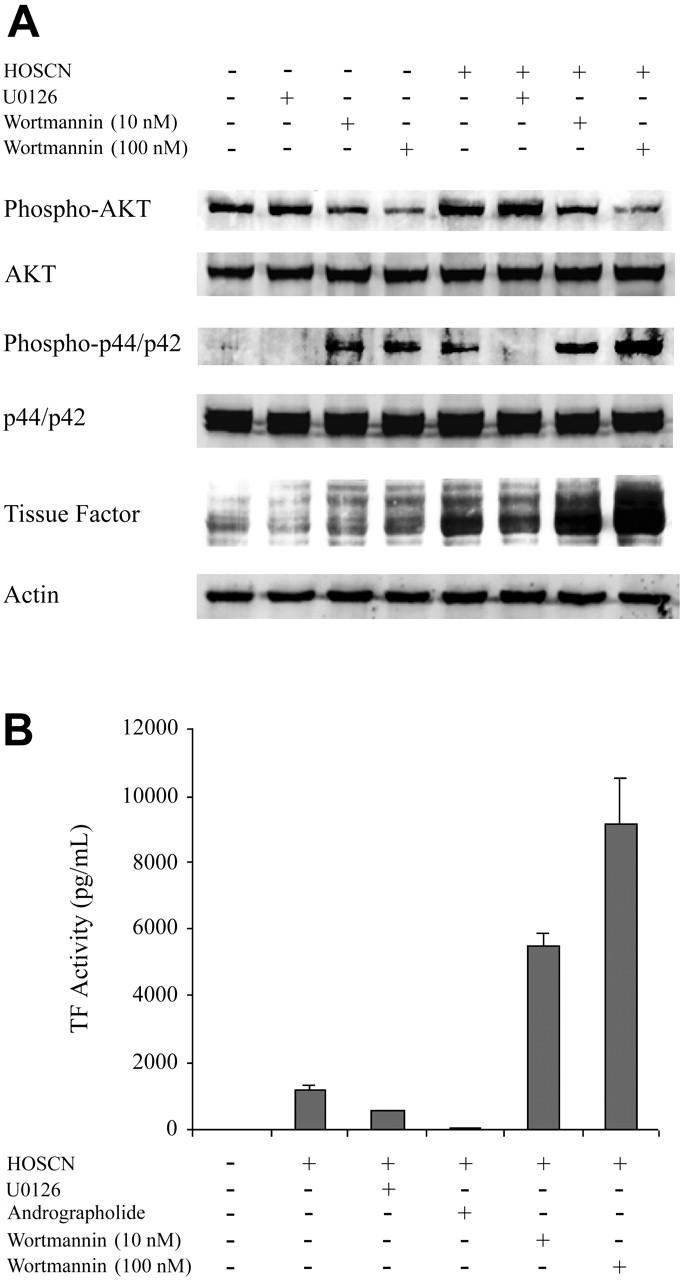
Kinase inhibitor studies and Western blot analysis of kinase pathways in HUVECs exposed to HOSCN. (A) Monolayers of HUVECs were pretreated 1 hour with U0126 (10 μM) or wortmannin (10 nM and 100 nM) and then exposed to buffer or 150 μM HOSCN in M199 medium containing 10% FBS and fresh inhibitors for 4 hours. Western blots of whole cell lysates were probed for phospho-p44/42 (Erk1/2), p44/42 (Erk1/2), phospho-AKT, AKT, and TF. (B) HUVECs were pretreated with 10 μM U0126, 10 μg/mL andrographolide, or wortmannin (10 nM and 100 nM) for 1 hour and then exposed to 150 μM HOSCN in M199 medium containing 10% FBS and fresh inhibitors for 4 hours. TF activity in lysates was determined by one-stage clotting assay. All data are shown ± SD.
Discussion
Although phagocyte oxidants are typically considered in the context of pathogen killing and host tissue cytotoxicity, recent work reveals their modulation of an unexpectedly wide variety of intracellular signaling pathways (reviewed by Soberman32 and Nathan33). Oxidants typically inhibit phophatases but can activate several kinase signaling pathways that have the capacity to promote gene transcriptional activation.32,33 Previous studies have shown that high (millimolar) concentrations of H2O2 stimulate TF activity up to 3-fold in HUVECs,15 monocytes,17 and vascular smooth muscle cells.34 Exposure of microvascular endothelial cells to HOCl stimulates TF maximally 2.5-fold.16 We hypothesized that HOSCN, by virtue of its SH specificity, capacity to impose intracellular oxidant stress,12 and lack of reactivity with catalase, might be an especially effective transcriptional inducer of TF activity. We therefore directly compared the capacity of the major phagocyte oxidants to influence TF expression in HUVECs.
We show that HOSCN is a uniquely potent (up to 100-fold) phagocyte oxidant inducer of HUVEC TF activity whose efficacy rivals that of the powerful (patho)physiologic TF inducer LPS (Figure 3A). TF induction was much more pronounced in the presence of complete media with 10% serum (Figure 3) than in a simple buffer system (Figure 1C), a finding that may reflect the formation of HOSCN-derived stable sulfenyl thiocyanate (R-S-SCN) adducts on cysteinyl or other serum SH compounds such as glutathione.13 In the former, arguably more physiologic conditions, the other phagocyte oxidants provoked less than 3-fold stimulation. HOSCN-induced TF activity is expressed not only in whole cell lysates but also on intact HUVEC cell surfaces (Figure 2C) and is attributable to pronounced transcriptional activation of TF gene expression (Figure 3C). Importantly, because endothelial cell apoptosis induces TF expression,35 HOSCN-stimulated TF expression occurs at concentrations of HOSCN that have no effect, even 24 hours later, on viability and consistently induce levels of apoptosis below those of medium controls (data not shown). The concentrations (more than 20 μM) of HOSCN required to induce TF activity in HUVECs are within the range that accumulates in supernatant buffer in the presence of activated EOs.12,13 Moreover, when EOs infiltrate endothelium/endocardium in HES, activated EOs come in close apposition to endothelial cells and are therefore likely exposed to much higher concentrations of locally generated HOSCN.
TF gene transcription in monocytes and endothelial cells is regulated by 5′ regulatory sequences containing binding sites for the AP-1, Egr-1, and p65/c-rel transcription factors.19,28-30 To our knowledge, all described examples of TF gene transcription activation show participation of all 3 of these transcription factors simultaneously, suggesting that their coordinate action might be required. We find that exposure to HOSCN activates p65/c-Rel to an extent similar to that of LPS (Figure 4B,D). In contrast, both HOSCN and LPS minimally influence activation of AP-1 or Egr-1, both of which are constitutively expressed in resting cultured HUVECs (Figure 5). This suggests that both LPS and HOSCN induce TF predominantly through activation the p65/c-Rel in the setting of constitutive AP-1 and Egr-1 activation.
That simultaneous activation of these 3 pathways may be required for TF expression is further supported, though not conclusively shown, by our inhibitor studies (Figure 6). U0126, a specific inhibitor of the ERK1/2 kinase that is upstream of Egr-1 activation, attenuates TF induction by HOSCN. Therefore, Egr-1 expression may promote, though not itself be sufficient for, the HOSCN TF response. Andrographolide, a specific inhibitor of NF-κB, blocks HOSCN TF induction, confirming an essential regulatory role for this transcription factor. The precise mechanism by which HOSCN activates NF-κB (and p65/c-Rel) is not addressed by our studies. Both of the IκB kinases (α and β) that phosphorylate the NF-κB–inhibitory IκB-α to cause its ubiquination and subsequent degradation have redox-sensitive cysteines whose oxidation state regulates their activities.35-37 In addition, both p50 and c-Rel have a cysteine, Cys62, that forms a covalent conjugate with the NF-κB inhibitor, andrographolide,27 suggesting that redox modification of this amino acid influences NF-κB activation. These as well as various redox-sensitive kinase cascades38 are candidates for further investigation of NF-κB activation by HOSCN. The PI3 kinase/Akt inhibitor wortmannin stimulates both baseline and HOSCN-dependent TF expression (Figure 6A), implicating this pathway in constituitive down-regulation of TF in HUVECs. However, the downstream mechanisms of this effect are not elucidated by our studies but are likely complex in view of the potential indirect influences of Akt upon AP-1, Egr-1, and NF-κB.39 Notably, although PI3 kinase/Akt typically functions to stimulate protein synthesis, for TF expression it is clearly inhibitory (Figure 6A-B39-41).
Our findings raise the possibility that in eosinophilic endothelial and endocardial inflammatory states, such as HES, eosinophilic endocarditis, and certain vasculitides, infiltrating activated EOs might expose adjacent endothelial cells to high concentrations of HOSCN that could induce cell surface TF activity and thereby contribute to a thrombotic diathesis. HOSCN also strongly activates TF expression in peripheral blood mononuclear cells (data not shown), suggesting another mechanism for thrombosis. However, the etiology of this thrombotic diathesis is likely to involve other, nonoxidant factors as well—for example, nonenzymatic inactivation of thrombomodulin by electrostatic binding of EPO and other EO cationic-specific granule proteins.42
By this same mechanism, MPO might also promote thrombosis in neutrophil inflammatory states. Van Dalen et al have shown that in the presence of physiologic concentrations of SCN– (100 μM) and Cl– (100 mM) MPO generates approximately equimolar amounts of HOSCN and HOCl.43 Enzymatically active extracellular MPO is stable enough to accumulate in atherosclerotic plaques44 along with chlorotyrosine-modified protein,45 a specific biomarker of MPO-mediated HOCl oxidation,45 and blood MPO levels are a powerful independent marker of coronary artery disease.46 It is possible, especially in smokers, whose serum thiocyanate levels are several-fold normal, that MPO-catalyzed generation of HOSCN, whether from activated neutrophils or from extracellular MPO, might induce local endothelial cell TF expression and thereby help explain the long-described associations of smoking, inflammation, and thrombosis.
HOSCN activates the more abundant p65/p50 NF-κB heterodimer as well as p65/c-Rel TF-κB (Figure 4). Because p65/p50 NF-κB binding sites are important transcriptional regulatory elements of genes such as ICAM1, VCAM1, E-selectin, IL8, and TNF,47 HOSCN might also induce expression of these proinflammatory cell adhesion molecules and cytokines in addition to TF. We are currently testing this hypothesis.
In summary, we show that HOSCN, the principal physiologic oxidant product of EPO, is a uniquely potent phagocyte oxidant activator inducer of TF expression in endothelial cells through a mechanism that predominantly reflects activation of the p65/c-Rel/TF-κB pathway. In addition, HOSCN strongly stimulates the activation of the p65/50 NF-κB transcription factor that regulates expression of several critical proinflammatory cytokines and adhesion molecules. Thus, HOSCN generated by either EPO or MPO might stimulate prothrombotic and proinflammatory gene expression in vascular endothelium. This, in turn, raises the possibility that phagocyte peroxidases, through generation of HOSCN, function in part to promote and link thrombosis and inflammation.
Acknowledgments
We acknowledge the valuable contributions of Julia Nguyen for assistance with endothelial cell culture and Thomas E. Welch for assistance with the transcription factor EMSA assays.
Prepublished online as Blood First Edition Paper, September 15, 2005; DOI 10.1182/blood-2005-05-2152.
Supported by National Institutes of Health grants HL-70937 and HL-65578.
J.-G.W. and S.A.M. contributed equally to this work.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Weller PF, Bubley GJ. The idiopathic hypereosinophilic syndrome. Blood. 1994;83: 2759-2779. [PubMed] [Google Scholar]
- 2.Brito-Babapulle F. The eosinophilias, including the idiopathic hypereosinophilic syndrome. Br J Haematol. 2003;121: 203-223. [DOI] [PubMed] [Google Scholar]
- 3.Fitzpatrick JE, Johnson C, Simon P, Owenby J. Cutaneous microthrombi: a histologic clue to the diagnosis of hypereosinophilic syndrome. Am J Dermatopathol. 1987;9: 419-422. [PubMed] [Google Scholar]
- 4.Kojima K, Sasaki T. Veno-occlusive disease in hypereosinophilic syndrome. Intern Med. 1995;34: 1194-1197. [DOI] [PubMed] [Google Scholar]
- 5.Schulman H, Hertzog L, Zirkin H, Hertzanu Y. Cerebral sinovenous thrombosis in the idiopathic hypereosinophilic syndrome in childhood. Pediatr Radiol. 1999;29: 595-597. [DOI] [PubMed] [Google Scholar]
- 6.Valente O, Scarpinella-Bueno MA. Deep venous thrombosis in hypereosinophilic syndrome. Am Fam Physician. 1994;50: 921-922. [PubMed] [Google Scholar]
- 7.Gleich GJ. Mechanisms of eosinophil-associated inflammation. J Allergy Clin Immunol. 2000;105: 651-663. [DOI] [PubMed] [Google Scholar]
- 8.DeChatelet LR, Shirley PS, McPhail LC, et al. Oxidative metabolism of the human eosinophil. Blood. 1977;50: 525-535. [PubMed] [Google Scholar]
- 9.Weiss SJ, Test ST, Eckmann CM, Roos D, Regiani S. Brominating oxidants generated by human eosinophils. Science. 1986;234: 200-203. [DOI] [PubMed] [Google Scholar]
- 10.Wu W, Chen Y, Hazen SL. Eosinophil peroxidase nitrates protein tyrosyl residues: implications for oxidative damage by nitrating intermediates in eosinophilic inflammatory disorders. J Biol Chem. 1999;274: 25933-25944. [DOI] [PubMed] [Google Scholar]
- 11.Slungaard A, Mahoney JR Jr. Thiocyanate is the major substrate for eosinophil peroxidase in physiologic fluids: implications for cytotoxicity. J Biol Chem. 1991;266: 4903-4910. [PubMed] [Google Scholar]
- 12.Arlandson M, Decker T, Roongta VA, et al. Eosinophil peroxidase oxidation of thiocyanate: characterization of major reaction products and a potential sulfhydryl-targeted cytotoxicity system. J Biol Chem. 2001;276: 215-224. [DOI] [PubMed] [Google Scholar]
- 13.Thomas EL. Products of lactoperoxidase-catalyzed oxidation of thiocyanate and halides. In: Pruitt KM, Tenovuo JO, eds. The Lactoperoxidase System: Chemistry and Biological Significance. New York, NY: Marcel Dekker; 1985: 31-53.
- 14.Grisham MB, Ryan EM. Cytotoxic properties of salivary oxidants. Am J Physiol. 1990;258: C115-C121. [DOI] [PubMed] [Google Scholar]
- 15.Schorer AE, Rick PD, Swaim WR, Moldow CF. Structural features of endotoxin required for stimulation of endothelial cell tissue factor production; exposure of preformed tissue factor after oxidant-mediated endothelial cell injury. J Lab Clin Med. 1985;106: 38-42. [PubMed] [Google Scholar]
- 16.Sugiyama S, Kugiyama K, Aikawa M, et al. Hypochlorous acid, a macrophage product, induces endothelial apoptosis and tissue factor expression: involvement of myeloperoxidase-mediated oxidant in plaque erosion and thrombogenesis. Arterioscler Thromb Vasc Biol. 2004;24: 1309-1314. [DOI] [PubMed] [Google Scholar]
- 17.Cadroy Y, Dupouy D, Boneu B, Plaisancie H. Polymorphonuclear leukocytes modulate tissue factor production by mononuclear cells: role of reactive oxygen species. J Immunol. 2000;164: 3822-3828. [DOI] [PubMed] [Google Scholar]
- 18.Kabe Y, Ando K, Hirao S, Yoshida M, Handa H. Redox regulation of NF-kappaB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid Redox Signal. 2005;7: 395-403. [DOI] [PubMed] [Google Scholar]
- 19.Oeth PA, Parry GC, Kunsch C, et al. Lipopolysaccharide induction of tissue factor gene expression in monocytic cells is mediated by binding of c-Rel/p65 heterodimers to a kappa B-like site. Mol Cell Biol. 1994;14: 3772-3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carson SD, Ross SE, Bach R, Guha A. An inhibitory monoclonal antibody against human tissue factor. Blood. 1987;70: 490-493. [PubMed] [Google Scholar]
- 21.Agosti JM, Altman LC, Ayars GH, et al. The injurious effect of eosinophil peroxidase, hydrogen peroxide, and halides on pneumocytes in vitro. J Allergy Clin Immunol. 1987;79: 496-504. [DOI] [PubMed] [Google Scholar]
- 22.Balla J, Jacob HS, Balla G, et al. Endothelial-cell heme uptake from heme proteins: induction of sensitization and desensitization to oxidant damage. Proc Natl Acad Sci U S A. 1993;90: 9285-9289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morris JC. The acid ionization constant of HOCl from 5 to 35°. J Phys Chem. 1966;70: 3798-3805. [Google Scholar]
- 24.Carr AC, Decker EA, Park Y, Frei B. Comparison of low-density lipoprotein modification by myeloperoxidase-derived hypochlorous and hypobromous acids. Free Radic Biol Med. 2001;31: 62-72. [DOI] [PubMed] [Google Scholar]
- 25.Key NS, Slungaard A, Dandelet L, et al. Whole blood tissue factor procoagulant activity is elevated in patients with sickle cell disease. Blood. 1998;91: 4216-4223. [PubMed] [Google Scholar]
- 26.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162: 156-159. [DOI] [PubMed] [Google Scholar]
- 27.Xia YF, Ye BQ, Li YD, et al. Andrographolide attenuates inflammation by inhibition of NF-kappa B activation through covalent modification of reduced cysteine 62 of p50. J Immunol. 2004;173: 4207-4217. [DOI] [PubMed] [Google Scholar]
- 28.Mackman N. Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler Thromb Vasc Biol. 2004;24: 1015-1022. [DOI] [PubMed] [Google Scholar]
- 29.Mackman N. Regulation of the tissue factor gene. Thromb Haemost. 1997;78: 747-754. [PubMed] [Google Scholar]
- 30.Guha M, O'Connell MA, Pawlinski R, et al. Lipopolysaccharide activation of the MEK-ERK1/2 pathway in human monocytic cells mediates tissue factor and tumor necrosis factor alpha expression by inducing Elk-1 phosphorylation and Egr-1 expression. Blood. 2001;98: 1429-1439. [DOI] [PubMed] [Google Scholar]
- 31.Hall AJ, Vos HL, Bertina RM. Lipopolysaccharide induction of tissue factor in THP-1 cells involves Jun protein phosphorylation and nuclear factor kappaB nuclear translocation. J Biol Chem. 1999;274: 376-383. [DOI] [PubMed] [Google Scholar]
- 32.Soberman RJ. The expanding network of redox signaling: new observations, complexities, and perspectives. J Clin Invest. 2003;111: 571-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nathan C. Specificity of a third kind: reactive oxygen and nitrogen intermediates in cell signaling. J Clin Invest. 2003;111: 769-778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Penn MS, Patel CV, Cui MZ, DiCorleto PE, Chisolm GM. LDL increases inactive tissue factor on vascular smooth muscle cell surfaces: hydrogen peroxide activates latent cell surface tissue factor. Circulation. 1999;99: 1753-1759. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Y, Chen F. Reactive oxygen species (ROS), troublemakers between nuclear factor-kB (NF-kB) and c-Jun NH2-terminal kinase (JNK). Cancer Res. 2004;64: 1902-1905. [DOI] [PubMed] [Google Scholar]
- 36.Rossi A, Kapahi P, Natoli G, et al. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IB kinase. Nature. 2000;403: 103-108. [DOI] [PubMed] [Google Scholar]
- 37.Chen F, Shi X. Signaling from toxic metals to NF-κB and beyond: not just a matter of reactive oxygen species. Environ Health Perspect. 2002;110(suppl 5): 807-811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279: L1005-L1028. [DOI] [PubMed] [Google Scholar]
- 39.Guha M, Mackman N. The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem. 2002;277: 32124-32132. [DOI] [PubMed] [Google Scholar]
- 40.Schabbauer G, Tencati M, Pedersen B, Pawlinski R, Mackman N. PI3K-Akt pathway suppresses coagulation and inflammation in endotoxemic mice. Arterioscler Thromb Vasc Biol. 2004;24: 1963-1969. [DOI] [PubMed] [Google Scholar]
- 41.Blum S, Issbruker K, Willuweit A, et al. An inhibitory role of the phosphatidylinositol 3-kinase-signaling pathway in vascular endothelial growth factor-induced tissue factor expression. J Biol Chem. 2001;276: 33428-33434. [DOI] [PubMed] [Google Scholar]
- 42.Slungaard A, Vercellotti GM, Tran T, Gleich GJ, Key NS. Eosinophil cationic granule proteins impair thrombomodulin function: a potential mechanism for thromboembolism in hypereosinophilic heart disease. J Clin Invest. 1993;91: 1721-1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Dalen CJ, Whitehouse MW, Winterbourn CC, Kettle AJ. Thiocyanate and chloride as competing substrates for myeloperoxidase. Biochem J. 1997;327(pt 2): 487-492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Daugherty A, Dunn JL, Rateri DL, Heinecke JW. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J Clin Invest. 1994;94: 437-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hazen SL, Heinecke JW. 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J Clin Invest. 1997;99: 2075-2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang R, Brennan ML, Fu X, et al. Association between myeloperoxidase levels and risk of coronary artery disease. JAMA. 2001;286: 2136-2142. [DOI] [PubMed] [Google Scholar]
- 47.Hanada T, Yoshimura A. Regulation of cytokine signaling and inflammation. Cytokine Growth Factor Rev. 2002;13: 413-421. [DOI] [PubMed] [Google Scholar]