

Abstract
Killer immunoglobulin–like receptors (KIRs) are a family of regulatory cell-surface molecules expressed on natural killer (NK) cells and memory T-cell subsets. Their ability to prevent the formation of an activation platform and to inhibit NK cell activation is the basis of the missing self model of NK cell function. The benefits of KIR expression for T-cell biology are unclear. We studied how KIR2DL2 regulates T-cell function. Engagement of KIR2DL2 by the ligand human leukocyte antigen (HLA)–Cw3 did not affect conjugate formation between CD4+KIR2DL2+ T cells and superantigen-pulsed target cells or the development of mature immune synapses with lipid rafts. KIR2DL2 and the corresponding HLA-C ligand were initially recruited to the peripheral supramolecular activation cluster (pSMAC). Consequently, KIR2DL2 engagement did not inhibit the phosphorylation of early signaling proteins and T-cell–receptor (TCR)–mediated cytotoxicity or granule exocytosis. After 15-30 minutes, KIR2DL2 moved to the central supramolecular activation cluster (cSMAC), colocalizing with CD3. TCR synapses dissociated, and phosphorylated phospholipase C (PLC)–γ1, Vav1, and extracellular signal–regulated kinase 1/2 (ERK1/2) were reduced 90 minutes after stimulation. Gene array studies documented that the inhibition of late signaling events by KIR2DL2 affected transcriptional gene activation. We propose that KIRs on memory T cells operate to uncouple effector functions by modifying the transcriptional profile while leaving granule exocytosis unabated.
Introduction
Immune homeostasis is tightly regulated by negative regulatory signals providing a counterbalance to activating stimuli.1,2 Negative regulatory receptors have been described on all hematopoietic cells; a prime example that illustrates their importance is natural killer (NK) cell function. Self tolerance of NK cells is ensured by a set of inhibitory cell surface receptors that bind to self–major histocompatibility (MHC) class I ligands.3 In humans, these receptors include the family of killer immunoglobulin-like receptors (KIRs).
Inhibitory KIRs have long cytoplasmic tails with 2 immunoreceptor tyrosine-based inhibitory motifs (ITIMs). Upon ligation of an inhibitory KIR, the ITIMs are phosphorylated and bind to the src homology 2 (SH2) domains of the phosphatase SHP-1. As a result of this binding, the catalytic site of SHP-1 is released from autoinhibition.4 In NK cells, cognate binding of inhibitory KIRs to their human leukocyte antigen (HLA) ligands is enough to facilitate receptor clustering, ITIM phosphorylation, and SHP-1 activation.5 The direct target of SHP-1 appears to be the guanine nucleotide exchange factor Vav1.6 Dephosphorylation of Vav1 leads to inhibition of Rac1,6 thereby preventing cytoskeletal rearrangement. One consequence is complete inhibition of NK cell activation. In fact, the formation of an activation platform between NK cells and target cells is completely prevented.7
In addition to NK cells, KIRs are also expressed on T cells.8,9 Naive and memory T cells are equipped to support their transcription;10 however, expression is only found on senescent or enddifferentiated CD4+ and CD8+ T cells that have lost the expression of CD28.11 The biologic function of inhibitory KIR expression on T cells is difficult to envision and likely different from NK cells. In NK cells, they are the basis for the missing self hypothesis12 (ie, NK cells only respond to cells that have lost MHC class I expression). Otherwise, the inhibitory receptors keep NK cells completely unresponsive. Such a model would not be meaningful for T cells, the activation of which is dependent on MHC-restricted T-cell–receptor (TCR) triggering. Not surprisingly, functional studies of inhibitory receptors on T cells have come to conflicting results.8,13,14 Inhibitory KIRs on selected tumor-specific CD8+ T cells in patients with melanoma15 and renal carcinoma were shown to inhibit tyrosine phosphorylation of early signaling proteins, lipid rafts, TCR/CD3 clustering, and the reorganization of the actin cytoskeleton; they also completely shut down T-cell activation. Consequently, the tumor-specific immune response was dampened, supporting a model of KIR expression on T cells as a cause of defective immunosurveillance.8 Other studies of inhibitory receptors, such as immunoglobulin-like transcript 2 (ILT-2) on human CD8 T cells and GP49B1 on murine CD8 T cells, have only reported an effect on interferon-γ (IFN-γ) production but not on cytotoxicity.14,16 In transgenic mice coexpressing KIR2DL3 and its ligand HLA-Cw3, KIRs even appeared to favor clonal expansion and T-cell survival in vivo.13
Since T cells are uniquely equipped to transcribe KIRs, their expression on T cells must be evolutionarily selected. However, the model that the biologic function of KIRs lies in the accumulation of functionally inert cells that with time increasingly compete for space is unappealing. We have hypothesized that the function of KIR expression on T cells lies in the modification of activation-dependent T-cell function and not their global inhibition. This hypothesis is consistent with the observation that different T-cell functions require different activation thresholds and that attenuation of T-cell receptor signaling has not only quantitative but also qualitative consequences.17
Materials and methods
Cells and materials
CD4+CD28– T cells were cloned and maintained as previously described.9,18 KIR2DL3+ NK cells were isolated by AutoMACS (Miltenyi Biotec, Bergisch Gladbach, Germany). 721.221 and 721.221 cells transfected with HLA-Cw3 were kind gifts from Dr Eric Long (National Institutes of Health, Rockville, MD). Antibodies and reagents used: anti-CD3 (OKT3 Ortho Diagnostics, Raritan, NJ); anti-KIR2DL3/2DL2/2DS2 monoclonal antibodies (mAbs) GL183 (Beckman Coulter, Fullerton, CA) and CH-L (Pharmingen, San Diego, CA), anti-CD16 mAb (3G8), anti–HLA-ABC (Pharmingen), anti–HLA-DR (L243; American Type Culture Collection, Rockville, MD); staphylococcal enterotoxin B (SEB; Toxin Technology, Sarasota, Florida); Nα-benzyloxycarbonyl-L-lysine thiobenzyl ester hydrochloride (BLT), and 5,5′-dithio-bis-(2-nitrobenzoic acid) (DTNB; Sigma, St Louis, MO); fluorescently labeled cholera toxin B subunit (CTXβ; Molecular Probes, Eugene, OR); anti-ZAP70 polyclonal antibody (Dr Paul Leibson, Mayo Clinic, Rochester, MN); anti–PLC-γ mAb, anti-Vav1 mAb, and antiphosphotyrosine antibody 4G10–conjugated agarose beads (Upstate Biotechnology, Lake Placid, NY); and antiphosphotyrosine antibody P-Tyr-100 (Cell Signaling Technology, Beverly, MA). For confocal imaging experiments, anti-CD3, anti-CD11a, anti–HLA-ABC, and anti-KIR2DL2 (CH-L) were fluorescently labeled with Zenon antibody-labeling kits (Molecular Probes). The protocol was approved by the Mayo Clinic and the Emory University Institutional Review Board.
Conjugate formation assay
T cells were labeled with Alexa488-CTXβ (Molecular Probes) and target cells 721.221 or 721.221/HLA-Cw3 with Alexa647-CTXβ. Target cells were incubated with 2 ng/mL SEB for 2 hours. T cells and target cells (10 000 and 20 000, respectively) were mixed in a final volume of 200 μL, centrifuged at 25g (300 rpm) at 4°C for 3 minutes, and kept at 37°C for various times. Samples were promptly analyzed on a FACSort (Becton Dickinson, Franklin Lakes, NJ). Results are expressed as the percentage of T cells that formed conjugates with target cells.
IFN-γ analysis
Resting CD4+CD28– T-cell clones were stimulated with irradiated P815 cells coated with mouse IgG, anti-CD3 and mouse IgG, or anti-CD3 and anti-KIR2DL2 mAbs. In other experiments, the T cells were stimulated with irradiated SEB-coated 721.221 or 721.221/HLA-Cw3 cells. In each case, 50 000 CD4+CD28–KIR2DL2+ T cells were coincubated with 25 000 target cells. The supernatants were harvested after 48 hours, and IFN-γ was determined by enzyme-linked immunosorbent assay (ELISA; BD Pharmingen, San Diego, CA).
Cell proliferation
Carboxy-fluorescein diacetate succinimidyl ester (CFSE)–labeled CD4+CD28–KIR2DL2+ T cells (50 000) were incubated with 25 000 SEB-coated target cells (721.221 or 721.221/HLA-Cw3). On day 5, the cells were analyzed on a FACSort.
Cytotoxicity and degranulation assay
Standard 4-hour 51Cr-release or CytoTox 96 Non-Radioactive Assays (Promega, Madison, WI) were carried out with P815 cells coated with mouse IgG, anti-CD3 and mouse IgG, or anti-CD3 and anti-KIR2DL2 mAbs as target cells, and either KIR2DL3+ NK cells or CD4+CD28–KIR2DL2+ T cells as effector cells. BLT esterase assays were used to assess degranulation.19 Effector cells were incubated with either antibody-coated P815 cells or with SEB-coated 721.221 or 721.221/HLA-Cw3 target cells; supernatants were collected after 4 hours.
Confocal microscopy image analysis
721.221 and 721.221/HLA-Cw3 cells were incubated at 37°C with 2 ng/mL SEB for 2 hours. For experiments involving the visualization of lipid rafts, T cells were stained with either Alexa488 or with Alexa555-conjugated CTXβ. For visualizing LFA-1, CD3, HLA-Cw3, and KIR2DL2, T cells were labeled with fluorescently conjugated anti-CD11a, anti-CD3, anti–HLA-A, HLA-B, HLA-C, and anti-KIR2DL2 (CH-L) antibodies. The nonblocking function of anti–HLA-A, HLA-B, and HLA-C antibody has been shown previously.20 The anti-KIR2DL2 antibody (CH-L), used under the same conditions as in the imaging experiments, did not affect the inhibitory function of KIR2DL2 on IFN-γ production (Figure S1; see the Supplemental Figure link at the top of the online article, at the Blood website). Labeled T cells and target cells mixed at a ratio of 1:2 were centrifuged at 25g (300 rpm) at 4°C for 3 minutes. The conjugates were gently resuspended in medium RPMI 1640 without phenol red plus 5% FCS (Invitrogen, GIBCO, Grand Island, NY), incubated at 37°C for the indicated times, and transferred to collagen-coated glass-bottom optical dishes (MatTek Corporation, Ashland, MA) just before imaging.
Cell conjugates were visualized using a Zeiss LSM 510 META laser-scanning confocal microscope equipped with argon/krypton and helium/neon lasers (Zeiss, Thornwood, NY). Images were collected using a 100 ×/1.4 numeric aperture (NA) Plan-Apochromat or a 63 ×/1.4 NA Plan-Neofluor oil immersion objective by an investigator who was blinded to the experimental protocol. A second independent investigator determined the number of conjugates and presence or absence of raft clustering at the T-cell–target-cell interphase. Three-dimensional analysis images were acquired through the z-axis at 0.5 to 1.0 μM increments. Images were analyzed with the LSM510 Examiner 3.2 software package (Carl Zeiss, Thornwood, NY).
Microarrays and data analysis
T cells were incubated with SEB-coated 721.221 or 721.221/HLA-Cw3 target cells for 0, 4, and 24 hours. Total RNA was extracted using an RNeasy Mini Kit (Qiagen, Valencia, CA). Total RNA (1 μg) was used to make antisense RNA according to manufacturer's instructions (Affymetrix, Santa Clara, CA; http://www.affymetrix.com/support/technical/manual/expression_manual.affx). Affymetrix GeneChip U133A hybridization was performed by Genomics Core Laboratory (Medical College of Georgia, Augusta, GA). Gene expression signal was summarized by GC robust multi-array average (GC-RMA) (Affymetrix), which uses sequence information for background correction and a quantile algorithm for normalization.21,22 Genes were identified that fulfilled the following criteria: (1) expression signal greater than 45 (3-fold higher than background/noise values); (2) a “present” detection call; and (3) a 3-fold higher signal at 4 or 24 hours compared to 0 hour. A total of 107 genes passed this filter.
Transmembrane signaling
T cells were coincubated with superantigen-coated (1 ng/mL SEB) 721.221 or 721.221/HLA-Cw3 target cells for various time points. Cells were lysed, and 500 μg of each cell lysate was subjected to immunoprecipitation followed by Western blotting as previously described.23 In other experiments, CD4+CD28–KIR2DL2+ T cells were stimulated with 1 μg/mL mouse IgG, 1 μg/mL mouse IgG + 1 μg/mL anti-CD3, or 1 μg/mL mouse IgG + 1 μg/mL anti-KIR2DL2 (GL183) at 37°C. The reaction was stopped, the cells were lysed, and the amount of total and phosphorylated ERK1/2 was measured using total and phosphospecific ERK1/2 ELISA (BioSource International, Camarillo, CA). Results are shown as the amount of phosphorylated ERK1/2 relative to total ERK1/2.
Results
Unrestrained cytotoxic activity of CD4+CD28– T cells despite expression of inhibitory KIRs
KIRs are expressed on CD4+CD28– and CD8+CD28– T cells at equal expression levels as on NK cells (Figure 1A). Expression of KIRs on T cells is stochastic, and individual T-cell clones can therefore coexpress stimulatory KIR2DS2 and inhibitory KIR2DL2 and KIR2DL3,9 all of which are recognized by the same antibody, GL183. To examine the function of inhibitory KIRs, we selected CD4+CD28– clones that only had transcripts for KIR2DL2 (Figure 1B). The ability of KIRs to inhibit cytotoxicity on NK cells is well accepted,3 and we therefore first examined the effect of KIR2DL2 activity on TCR-mediated cytotoxicity of CD4+CD28– T cells in a 4-hour reverse cytotoxicity assay using antibody-coated 51Cr-labeled P815 mouse mastocytoma cells. TCR triggering by anti-CD3 induced considerable cytotoxicity, which is consistent with the previous finding that these cells are cytotoxic effector cells expressing perforin and granzyme B (Figure 1C).24-27 To our surprise, we observed no inhibitory effect on the ability of the T cell to kill the target cells, when KIR2DL2 was coengaged with the anti-KIR2DL2 antibody. In contrast, the CD16-induced cytotoxic activity of purified KIR2DL3+ NK cells was inhibited in the very same experimental system upon cross-linking with anti-KIR2DL2 (Figure 1C). The NK cell donor was selected to lack the KIR2DS2 gene; T-cell clones tested were negative for KIR2DS2 transcripts. Inhibition of NK cell activity, as well as the lack of inhibition in T cells, was seen over a large range of concentrations of the stimulatory antibodies (1-300 ng/mL), excluding coexpression of KIR2DS2 or different activation thresholds for the distinct effects in T and NK cells.
Figure 1.
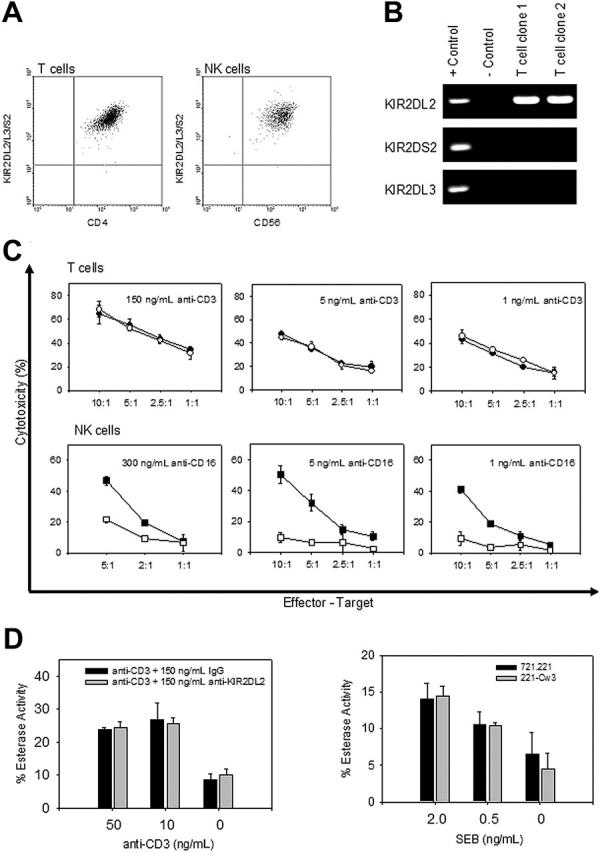
KIR2DL2 does not inhibit TCR-mediated cytotoxicity. (A) The level of KIR2DL2/2DL3/2DS2 expression on CD4+CD28– T cells and CD56+ NK clones were examined by FACS analysis. (B) KIR+ CD4+CD28– T-cell clones were tested for the presence of KIR2DL2, KIR2DL3, and KIR2DS2 mRNA by PCR. (C) The ability of KIR2DL2 to inhibit either T-cell–mediated (top panel) or NK cell–mediated (bottom panel) cytotoxicity was examined in a 4-hour cytotoxicity assay against anti-CD3 (•) and anti-CD3 + anti-KIR2DL2/2DL3/2DS2 (○) or anti-CD16 (▪) and anti-CD16 + anti-KIR2DL2/2DL3/2DS2 (□)–labeled P815 cells. Both anti-CD3 and anti-CD16 were tested over a wide dose range, and selected concentrations are shown. (D) The effect of KIR2DL2 on CD4+CD28– T cells on granule exocytosis was measured in 4-hour BLT release assay against either anti-CD3 and anti-CD3 + anti-KIR2DL2-coated P815 cells (left panel) or SEB-coated 721.221 and 721.221/HLA-Cw3 cells (right panel). Results are shown as mean ± SD of triplicate cultures.
Similar results were obtained when a granule exocytosis assay was used (Figure 1D). Anti-CD3–coated P815 cells induced granule release in CD4+CD28– T cells as indicated by granzyme A release.19 Cross-linking of KIR2DL2 did not have an inhibitory effect. The granule exocytosis allowed for study of the effect of KIR2DL2 in a natural ligand system. In pilot studies, T-cell clones were selected that were responsive to the superantigen SEB presented by 721.221 cells. 721.221 cells express MHC class II molecules but lack cell surface expression of MHC class I molecules. 721.221 cells transfected with HLA-Cw3, a ligand for KIR2DL2, induced release of cytotoxic granules from KIR2DL2+CD4+ T cells to the same extent as were released by stimulation with untransfected target cells.
Mature immune synapse forms between KIR2DL2+ CD4+CD28– T cells and HLA-Cw3–expressing 721.221 cells
The initial studies of granule exocytosis and cytotoxicity did not document an inhibitory effect of KIR2DL2 expressed on T cells, while inhibitory effects were seen when KIR2DL3+ NK cells were used in the same experimental systems. To explore whether KIR2DL2 on T cells, in contrast to NK cells, would allow conjugate formation between T cells and target cells, T cells were stained with Alexa488-conjugated CTXβ, and target cells were stained with Alexa647-conjugated CTXβ. T cells were incubated with SEB-coated 721.221 or 721.221/HLA-Cw3 cells for the indicated time periods, and conjugate formation was monitored by fluorescence-activated cell sorting (FACS) analysis. As shown in Figure 2A, KIR2DL2–HLA-Cw3 receptor-ligand interaction did not affect the percentage of T cells that were in conjugates with target cells.
Figure 2.

KIR2DL2 does not affect the formation of a mature activating immune synapse. (A) Alexa488-conjugated CTXβ-labeled T cells and Alexa647-conjugated CTXβ target cells (721.221 [left panel] or 721.221/HLA-Cw3 [right panel]) were coincubated at a ratio of 1:2 in the absence or presence of the superantigen SEB (2 ng/mL) in the absence of IL-2. At the indicated time points, the percentage of T cells in conjugates was determined by FACS analysis. Results shown as means of triplicate measurements are representative of 3 experiments. (B) T cells labeled with Alexa555-conjugated CTXβ were coincubated with HLA-Cw3–positive and –negative target cells and SEB as described in panel A. Images of multiple fields were obtained by a blinded observer using a Zeiss LSM 510 confocal microscope equipped with a 63 ×/1.4 oil-immersion objective lens after 30 minutes of incubation, and conjugate formation and lipid raft clustering were analyzed by counting at least 100 T cells in different fields. Results are expressed as the percentage of T cells that were in conjugates with target cells (left panel) and the percentage of conjugates that showed lipid raft clustering at the T-cell–target-cell contact area (right panel). Results are representative of 3 experiments. (C) T cells were labeled with Alexa555-conjugated CTXβ (red; top panel) or with Alexa488-labeled CD3 mAbs (green; bottom panel). T cells and SEB-coated 721.221/HLA-Cw3 target cells were mixed at a 1:2 ratio and centrifuged for optimal cell-cell contact formation. Cells were examined at room temperature using a Zeiss LSM 510 confocal microscope equipped with a 100 ×/1.4 oil-immersion objective lens 15 minutes after initial cell contact. (D) HLA-Cw3 was visualized with Alexa488-labeled anti–HLA-ABC antibody (green), and CD3 was detected with Alexa555-labeled anti-CD3 mAb (red). Conjugates were formed and examined as described for panel C. (E) Lipid rafts (top panel) and CD3 (bottom panel) were visualized with Alexa555 (red). LFA-1 was detected by Alexa488-conjugated CD11a mAb (green). Conjugates were formed and examined as described in panel C. Taken together, the images demonstrate synapse formation with rafts and CD3 in the cSMAC; and KIR2DL2, HLA-Cw3, and LFA-1 in the pSMAC after 15 minutes. All confocal pictures are representative of at least 3 experiments with at least 5 contact areas analyzed in each experiment. Results are shown as mean ± SD of triplicate experiments.
Conjugate formation was associated with the classic redistribution of lipid rafts to the contact area between T cells and target cells. T cells labeled with Alexa555-CTXβ and SEB-pulsed target cells were coincubated for 15 minutes. At that time point, approximately 80% of the KIR2DL2-expressing T cells were found in conjugates with 721.221 target cells, irrespective of the expression of the ligand HLA-Cw3 (Figure 2B, left panel). Lipid raft redistribution to the T-cell–target-cell interphase was observed in 80% of the T-cell–721.221 conjugates compared with 78% when HLA-Cw3–expressing 721.221 cells were used (Figure 2B, right panel). CD3 colocalized with the lipid rafts (Figure 2C) in the contact area between the T cells and either type of target cell. HLA-Cw3 was clustered peripherally to the lipid raft and CD3 cluster at 15 minutes, documenting that KIR2DL2 recognized its ligands and was recruited (Figure 2D).
Further imaging studies documented that the transient interaction platform formed between KIR2DL2+ T cells and HLA-Cw3–expressing target cells is characteristic of an activating immune synapse.28 Cells were stained with Alexa488-labeled anti–LFA-1 and Alexa555-labeled anti-CD3 mAb, and lipid rafts were visualized by Alexa555-labeled CTXβ. Representative confocal images of a mature immune synapse with lipid raft and CD3 accumulation in the central supramolecular activation cluster (cSMAC) and LFA-1 accumulation in the peripheral SMAC (pSMAC) are shown in Figure 2E.
KIR2DL2 initially recruited to the pSMAC moves with delay into the cSMAC
Initial studies had shown that KIR2DL2 does not form an inhibitory synapse such as in NK cells,7 but locates to the pSMAC within the first 10 to 15 minutes after T-cell activation. Serial time points after initial cell contact were examined to obtain information on the kinetics of KIR2DL2-HLA-Cw3 redistribution and the relative localizations of KIR2DL2, HLA-Cw3, and CD3. Antibodies used for the staining did not suppress the function of KIR2DL2 in functional assays (Figure S1).20 After 10 minutes of co-incubation, both KIR2DL2 and its ligand formed a ring in the pSMAC. By 20 minutes, they both had moved to the cSMAC, and by 30 minutes they both had been recruited to the center of the immune synapse (Figure 3A), where they colocalized with CD3 (Figure 3B-C).
Figure 3.

Recruitment of KIR2DL2 to the cSMAC is delayed. (A) KIR2DL2 was visualized by Alexa555-labeled CH-L, and HLA-Cw3 was detected by Alexa488-labeled anti–MHC class I mAb. KIR2DL2+ CD4+ T cells were pelleted with 721.221/HLA-Cw3 target cells in the presence of 2 ng/mL SEB. The cells were transferred to optical dishes, and the movement of KIR2DL2 and HLA-Cw3 was observed using a confocal microscope at room temperature. Representative x-y sections for KIR2DL2 (red) and HLA-Cw3 (green) are shown for 10, 20, and 30 minutes (left 2 columns). The middle 3 columns show z-axis image reconstruction in the plane of the contact site. Line plots show a quantitative image analysis in this plane, demonstrating that the green and red fluorescence colocalize in the pSMAC after 10 minutes and in the cSMAC after 20 minutes. (B) T cells were stained with Alexa488-labeled anti-CD3 mAb and Alexa555-labeled anti-KIR2DS2/2DL2/2DL3 mAb and then coincubated with HLA-Cw3–expressing target cells as described in panel A. Representative sections at 2 time points are shown. At 10 minutes, CD3 (green) localized to the cSMAC, while KIR2DL2 (red) was found in the pSMAC. At 30 minutes, both molecules were in the cSMAC. Representative x-y section images and z-axis–reconstructed images in the plane of contact site are shown for both time points. (C) T cells were stained with Alexa555-labeled anti-CD3 mAb, and target cells (721.221/HLA-Cw3) were stained with Alexa488-labeled anti–MHC class I mAb. At 10 minutes, CD3 (red) localized to the cSMAC, while HLA-Cw3 (green) was found in the pSMAC. At 30 minutes, both molecules were in the cSMAC. Representative x-y section images and z-axis–reconstructed images in the plane of contact site are shown for both timepoints. For all panels, conjugates were formed and examined as described for Figure 2C.
Early TCR signaling events are not inhibited by KIR2DL2
Since cell-surface expression of KIR2DL2 did not inhibit the formation of a mature immune synapse, we hypothesized that the early signaling events of T-cell activation were not inhibited. KIR2DL2+ T cells were stimulated with SEB-pulsed 721.221 or 721.221/HLA-Cw3 target cells. At time points indicated (Figure 4A), tyrosine-phosphorylated proteins were immunoprecipitated and analyzed. KIR2DL2 did not have any inhibitory effect on ZAP70, PLC-γ, and Vav1 phosphorylation within the first 25 minutes (Figure 4A).
Figure 4.

KIR2DL2 affects early and late TCR-mediated signaling events differently. (A) KIR2DL2+ T cell clones were stimulated with SEB-coated 721.221 or 721.221/HLA-Cw3 target cells for the indicated time points. Cell lysates were tested for phosphorylated ZAP70, phosphorylated PLC-γ1, and phosphorylated Vav1. (B) KIR2DL2+ T-cell clones were stimulated with 1 μg/mL control IgG, 1 μg/mL, anti-CD3 + 1 μg/mL IgG, or 1 μg/mL anti-CD3 + 1 μg/mL anti-KIR2DL2 for the indicated timepoints. The amount of total and phosphorylated ERK1/2 was determined by ELISA. The amount of phosphorylated ERK1/2 relative to the amount of total ERK1/2 is shown as mean of triplicate measurements. Results are representative of 3 experiments. (C) KIR2DL2+ T-cell clones were stimulated with SEB-coated 721.221 or 721.221/HLA-Cw3 target cells for 60, 90, and 120 minutes. Cell lysates were tested for phosphorylated PLC-γ1 and phosphorylated Vav1. (D) KIR2DL2+ T-cell clones were stimulated as in panel B and the level of ERK1/2 phosphorylation was established as in panel B. Results are shown as mean ± SD of triplicate measurements.
To examine more distal signaling events, we focused on ERK1/2 phosphorylation, which allows an integrative assessment of early signaling events. Since ERK1/2 is abundantly present in 721.221 cells, we used appropriate mAb to stimulate T cells instead of the natural ligand system. T cells were stimulated with control IgG, anti-CD3 and IgG, or anti-CD3 and anti-KIR2DL2 mAb for up to 30 minutes. Phosphorylated ERK1/2 was determined by ELISA. Results are expressed relative to total ERK1/2 (Figure 4B). KIR2DL2 did not have an effect on the generation of phosphorylated ERK1/2 in the first 30 minutes of T-cell activation.
KIR2DL2 inhibits ERK1/2 phosphorylation during later-phase T-cell activation
Since KIR2DL2 moved into the cSMAC 15 to 30 minutes after the initial contact between the T cells and the target cells, we examined the effects of KIR2DL2 engagement on TCR-mediated signaling at later stages. The phosphorylation of PLC-γ and Vav1 was investigated between 60 to 120 minutes after TCR stimulation. The phosphorylation levels of both molecules were reduced starting at 90 minutes by KIR2DL2 (Figure 4C). The ratio of phosphorylated to total ERK1/2 continued to increase up to 120 minutes after the start of T-cell activation. This increase was substantially less in the cultures where KIR2DL2 was co–cross-linked, which resulted in a significant inhibition of ERK phosphorylation 90 minutes after stimulation (Figure 4D). These data suggest that KIR2DL2 does not affect early signaling events but inhibits sustained signaling.
TCR-induced transcriptional activation is sensitive to KIR2DL2
To examine the effect of KIR2DL2 on transcriptional activation after TCR stimulation, KIR2DL2+ T cells were stimulated with SEB-coated 721.221 or 721.221/HLA-Cw3 target cells. Gene expression profiles were obtained at 0, 4, and 24 hours. Results after normalization are shown in Figure 5A for 107 genes that all showed at least a 3-fold increase between 0 and 4 hours. At 0 hours, expression levels were equal; all data points mapped to the diagonal in a 2-dimensional scatter plot. At 4 hours, gene expression of most genes increased more in the absence of HLA-Cw3; the cluster of data points moved to above the diagonal. Similar results were seen after 24 hours. The inhibition did not apply to all genes, and some genes were equally induced under both stimulation conditions. The results indicate that KIR2DL2 engagement blocked the majority of TCR-induced gene transcription. This finding is consistent with the hypothesis that induction of most gene transcription requires sustained signaling.
Figure 5.
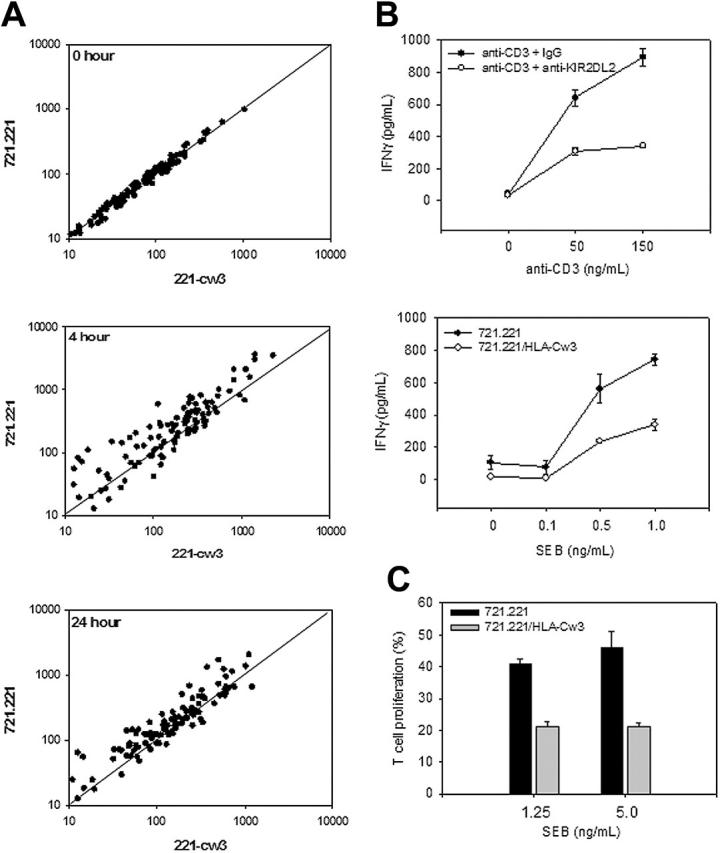
KIR2DL2 inhibits TCR-mediated IFN-γ production and cell proliferation. (A) KIR2DL2+ T cells were stimulated with SEB-coated 721.221 or 721.221/HLA-Cw3 target cells. Gene expression was profiled at 0, 4, and 24 hours. Only those genes that showed at least a 3-fold increase in expression after activation were selected, and the expression levels of these genes under the 2 culture conditions were compared. KIR2DL2 engagement by HLA-Cw3 reduced the stimulation-induced up-regulation of the majority of genes. (B) Induction of IFN-γ was found to be strongly inhibited by gene array. To confirm this result, KIR2DL2+ T cells were incubated with anti-CD3 + IgG (150 ng/mL) or anti-CD3 + anti-KIR2DL2 (150 ng/mL) coated P815 cells (top panel); or with SEB-coated 721.221 or 721.221/HLA-Cw3 target cells (bottom panel) for 48 hours. IFN-γ in the supernatant was quantified by ELISA. Results are shown as mean ± SD of triplicate cultures representative of 5 experiments. (C) CFSE-labeled KIR2DL2+ T cells were incubated with SEB-coated 721.221 or 721.221/HLA-Cw3 target cells for 5 days. The percentage of T-cell proliferating was determined by FACS analysis. Results are shown as mean and SD of triplicates representative of 3 experiments.
The gene array data were confirmed for selected candidate genes by conventional techniques. Results in Figure 5B show IFN-γ production by ELISA. SEB-coated 721.221 and 721.221/HLA-Cw3 were coincubated with KIR2DL2+CD4+CD28– T cells for 48 hours, and the amount of IFN-γ produced was quantified in the supernatant. Parallel experiments were performed using IgG, anti-CD3 + IgG, or anti-CD3 + anti-KIR2DL2–coated P815 cells as stimulators. Regardless of the experimental system, KIR2DL2 engagement reduced the amount of IFN-γ produced by T cells (Figure 5B).
To explore whether inhibition of sustained TCR-mediated signaling inhibits cell-cycle entry and progression, CFSE-labeled T cells were coincubated with SEB-pulsed 721.221 or 721.221/HLA-Cw3 target cells for 5 days. KIR2DL2 engagement with HLA-Cw3 resulted in reduced TCR-mediated proliferation (Figure 5C).
KIR2DL2 acts by prematurely terminating the immune synapse
Since KIR2DL2, although with substantial delay, is finally recruited to the cSMAC, it may have an effect on the sustained integrity of the immune synapse. To address this question, T cells incubated with SEB-pulsed 721.221 or 721.221/HLA-Cw3 cells were monitored for up to 90 minutes by confocal microscopy, and conjugates were examined for the presence of an immune synapse by using Alexa555-labeled CTXβ. At 30 minutes, there was no difference in raft formation regardless of whether the target cell expressed the KIR2DL2 ligand HLA-Cw3 or not (Figure 2B). At 90 minutes, approximately 72% of T cells were found in conjugates with 721.221 cells; 79% of the conjugates had lipid rafts localized to the center of the contact area between the T cells and these target cells (Figure 6). A similar percentage of 71% of the T cells were in conjugates with 721.221/HLA-Cw3 cells, but only 29% of these conjugates showed lipid rafts in the contact area (Figure 6).
Figure 6.
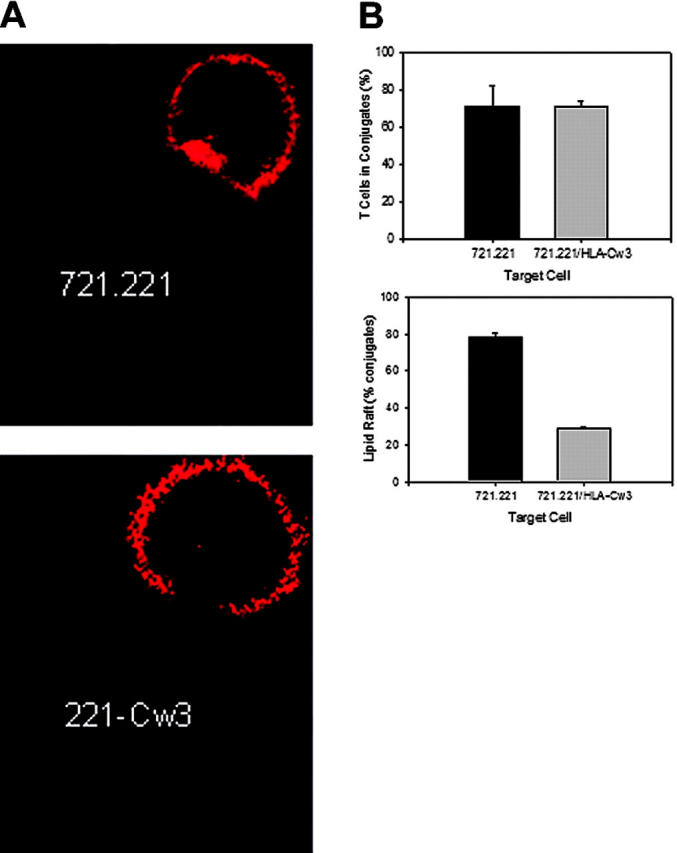
KIR2DL2 decreases immune synapse stability. (A) Lipid rafts were visualized with Alexa555-conjugated CTXβ (red) in KIR2DL2+ T cells pelleted with SEB-coated 721.221 or 721.221/HLA-Cw3 target cells. The conjugates were transferred to optical dishes, and the kinetics of rafts (red) disbanding in contact areas was monitored by confocal microscopy. A representative picture after 90 minutes is shown. At that time, the raft persisted in conjugates with MHC class I–negative cells (top panel) but had resolved in conjugates with HLA-Cw3–positive cells (bottom panel). (B) Cells were prepared and treated as described in panel A, and images of 10 fields were visualized with a 63 ×/1.4 oil-immersion objective lens. The percentage of T cells in conjugates with target cells (top panel) and the percentage of conjugates with lipid rafts at the contact zone (bottom panel) were determined. Results are shown as mean ± SD of 3 independent experiments.
The dissociation of the immune synapse, with the resolution of lipid rafts and its associated decompartmentalization of TCRs, costimulatory receptors, and signaling molecules, could result in the termination of signaling events and explain both the drop-off in ERK phosphorylation and the inhibition of transcriptional events. In naive CD4 T cells, TCR signaling needs to be maintained for 12 hours or longer to achieve full activation.29 It is likely that effector-memory T cells, such as KIR2DL2-expressing T cells, do not require such a sustained signal; however, the termination of the immune synapse in the first 1 to 2 hours after activation may still prevent transcriptional activation but be sufficient to induce granule release. To test this hypothesis, conjugates of KIR2DL2-expressing T cells and SEB-pulsed 721.221 cells lacking the HLA-Cw3 ligand cells were mechanically disrupted by adding anti–HLA-DR or control antibodies. The cells were cultured for a total of 48 hours, after which supernatants were harvested to assess the production of IFN-γ. The mechanical disruption of the recognition unit at 60, 90, and even 180 minutes inhibited IFN-γ production to a similar extent as observed when KIR2DL2 was triggered by its ligand HLA-Cw3 on the 721.221 target cells (Figure 7). This finding supports the interpretation that KIR2DL2 functions by prematurely terminating the immune synapse and that the delayed recruitment of KIR2DL2 to the cSMAC allows unrestrained cytotoxic activity but attenuates T-cell–activation events that depend on the induction of gene transcription.
Figure 7.

Sustained TCR-mediated signaling is required for the transcriptional activation of the IFN-γ gene. KIR2DL2+ T cells were incubated with SEB-coated 721.221 or 721.221/HLA-Cw3 target cells. Anti–MHC class II monoclonal antibodies (L243) were added to the cultures at the specified time points, and the cultures were continued for a total of 48 hours. Supernatants were harvested, and IFN-γ was quantified by ELISA. Results are shown as mean ± SD of triplicate cultures. TCR synapse dissociation within the first 90 minutes mimics the inhibitory effect of KIR2DL2.
Discussion
Homeostasis in the lymphoid system is achieved through a balance between stimulatory and inhibitory signals. Negative regulatory receptors raise the threshold for activation and prevent the induction of an immune cascade to irrelevant stimuli or minor variations in the microenvironment of antigen-presenting cells and self antigens. A classic example is negative regulatory receptors that recognize self–MHC class I molecules and control NK cell activation. Negative regulation also plays an important role in T-cell homeostasis. Here, regulatory receptors are usually induced upon T-cell activation and provide negative feedback signals. A prime example is cytotoxic T lymphocyte–associated antigen 4 (CTLA-4),30 which is expressed on the cell surface within 24 hours after stimulation and terminates T-cell activation by recruiting phosphatases.
KIR molecules on CD4 and CD8 memory T cells function fundamentally differently from other feedback mechanisms. Expression of KIRs is acquired during memory T-cell differentiation.9 While it is unknown how KIRs are induced and why they are restricted to subsets of memory T cells, their expression is constitutive at high levels, as is the case with NK cells, and is no longer subject to changes with T-cell activation. It is therefore assumed that they are not part of a feedback loop but set the activation threshold and prevent activation of T cells, as they do in NK cells. However, such a silencing mechanism is counterintuitive; T cells do not scan the environment for the absence of MHC class I molecules, and the activation of CD8 T cells actually depends on their presence. The biologic implication of KIR expression would therefore be a compromised memory response.
In the present study, we provide evidence for the model that KIRs change the T-cell effector profile rather than preventing T-cell activation, and that the main function of the KIR is to modify the consequences of T-cell activation. We document that KIR2DL2 on CD4 T cells differentially affects effector functions; granule release and cytotoxicity are fully competent, whereas TCR-induced gene transcription is inhibited. The likely mechanism for this finding is a delayed recruitment of KIR2DL2 into the cSMAC. Because of the delay, only late and not early signaling events are inhibited.
Cell-mediated cytotoxicity is only dependent on the release of preformed granules and therefore is a very rapid event. To prevent cytotoxicity, KIRs need to rapidly accumulate in the contact area between the T cell and the target cell. Inhibitory KIRs on NK cells that are binding to their HLA ligands on a target cell form an inhibitory NK cell–immune synapse within 1 minute. The inhibitory NK cell immune synapse is composed of a central and a peripheral supramolecular inhibition cluster,7 with inhibitory KIRs and SHP-1 localizing to the central cluster. One of the major roles of the inhibitory KIRs is the blocking of the lipid raft movement to the NK-cell–target-cell contact area.31,32 Since lipid rafts contain most of the activating receptors and activating signaling molecules, this active exclusion of lipid rafts from the cell contact area provides an important mechanism to block cell activation. KIRs do so by activating SHP-1, a direct target of which is Vav1.6 As Vav1 is the main conductor of actin cytoskeleton reorganization,33-35 the inhibition of Vav-1 activation by inhibitory KIRs is an efficacious mechanism to paralyze all effector functions in NK cells.
None of these observations in NK cells holds up for KIR2DL2 expressed in CD4 T cells. The ligation of KIR2DL2 to its ligand HLA-Cw3 did not result in the disruption of conjugate formation. Mature activating immune synapses were formed between KIR2DL2+CD4+ T cells and HLA-Cw3+ target cells. KIR2DL2 did not inhibit the movement of the lipid rafts and CD3 to the immune synapse. Consistent with this finding, phosphorylation of ZAP70, PLC-γ1, and Vav1 was induced.
While KIR2DL2 was not able to suppress early CD4 T-cell activation events, it did inhibit the transcription of most TCR-dependent genes; IFN-γ production was reduced and cell proliferation diminished. A possible explanation for this response pattern is the kinetics of KIR2DL translocation. KIR2DL2 is first recruited to the pSMAC, but eventually moves to the cSMAC. Inhibition of ERK phosphorylation was first observed approximately 90 minutes after TCR triggering. Transcription of IFN-γ and other activation markers may require sustained TCR signaling36 in analog to what has been shown for the proliferation of naive murine T cells37 and their IL-2 production.29 Indeed, the mechanical disruption of T-cell–target-cell conjugates up to 180 minutes after stimulation inhibited IFN-γ production.
Several authors in a variety of experimental systems have demonstrated an inhibitory effect of negative regulatory receptors in T cells similar to those in NK cells, with inhibition of early signaling events. CD94/NKG2A was inhibitory for γδ T cells,38 ILT-2 inhibited superantigen-induced cytoskeletal reorganization in CD8 T cell clones,39 and Guerra reported suppression of the activity of tumor-infiltrating KIR+ cells by HLA-C recognition.8 It cannot be excluded that our observations apply to CD4 and not to CD8 T cells. Indeed, KIR expressing CD4 and CD8 T cells differ in several respects. Both types of T cells have increased ability of granule-mediated cytotoxicity; however, CD4 T cells frequently express stimulatory KIR receptors and produce plenty of inflammatory cytokines, while cytokine production in CD8 T cells declines. Also, CD4 and CD8 T cells form different kinds of activation platforms.40 Our findings in CD4 T cells, however, are not without precedence; and the uncoupling of cytotoxic function from full T-cell activation has been observed for a variety of inhibitory receptors, including those in CD8 T cells. KIR2DL3 in the KIR2DL3 transgenic mice,13 CD85j on human CD8 T cells,14 and the murine inhibitory NK receptor GP49B1 on murine CD8 T cells16 all inhibited IFN-γ production while leaving the TCR-mediated cytotoxicity intact. The reasons for these different observations are unclear. Differences in experimental conditions are a possibility, and it would not be surprising if an antibody-mediated system favors global suppression. However, contradictory results have also been observed for natural ligand systems. Heterogeneity of T-cell populations from different individuals may be another explanation, and low affinity stimulation may be more susceptible to global suppression. However, we consistently found resistance to KIR-mediated suppression of cytotoxicity in all T-cell clones tested and over a wide range of stimulatory signals.
The kinetics of KIR recruitment is central to understanding the functional implications of KIR expression on T cells. There are contradicting reports about the involvement of cAMP, actin cytoskeleton, and tubulin in the movement of KIRs.32,41,42 It appears logical that an inhibitory receptor should not rely on the actin cytoskeleton to move in the plasma membrane. One of the major mechanisms of inhibitory receptor function is preventing the reorganization of the actin cytoskeleton, which obviously cannot be accomplished by an inhibitory receptor that relies on actin cytoskeleton to exert its inhibitory function. NKG2A, another inhibitory molecule, was also suggested not to depend on actin cytoskeleton for its lateral movement in the plasma membrane.43
Why inhibitory KIRs move with different dynamics in the cell membrane of CD4 T cells than in NK cells is intriguing, but, at this time, unresolved. Differences in plasma membrane compositions, cell-surface molecule sets, and adaptor molecule sets are possible mechanisms; however, these remain hypothetical since even for NK cells, the mechanisms governing KIR movements have not been defined.
Our studies provide evidence that granule release/cytotoxicity and de novo production of inflammatory mediators can be uncoupled after T-cell activation. While killing requires only a short activation time, inductions of gene transcription and proliferation require sustained signaling. The organization of the lytic machinery is easier to accomplish than the formation of a mature immune synapse. Stimulation conditions to induce cytotoxicity are less stringent; granule release is seen with lower antigen concentration than are needed for the induction of gene transcription. Betts et al have postulated that this uncoupling of major T-cell function has major biologic implications.44 With low antigen concentrations, such as with low viral load, the T-cell response is focused on killing the appropriate target cells, thereby avoiding inflammation and collateral damage. Only with higher viral load do antigen-specific T cells respond with the secretion of proinflammatory cytokines and the recruitment and activation of other effector mechanisms. A similar uncoupling of effector function after T-cell activation is seen with inhibitory KIRs. The expression of these negative regulatory receptors on subsets of effector memory T cells may therefore serve the purpose to impart strikes selectively. KIR expression may enable T cells to use cytotoxic mechanisms without causing collateral damage through the production of cytokines and without distorting T-cell homeostasis through clonal expansion.
Supplementary Material
Acknowledgments
The authors thank Tamela Yeargin for manuscript editing.
Prepublished online as Blood First Edition Paper, February 9, 2006; DOI 10.1182/blood-2005-06-2519.
Supported in part by grants from the National Institutes of Health (RO1 AR 41974, RO1 AR 42567, RO1 AG 15043, and UI9-AI 44142).
G.H. designed research, performed research, analyzed data, and wrote the paper; K.S. performed research and analyzed data; D.C. performed research and analyzed data; S.P. performed research; W.-W.L. performed research; C.M.W. designed research and analyzed data; and J.J.G. designed research, analyzed data, and wrote the paper.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Leibson PJ. The regulation of lymphocyte activation by inhibitory receptors. Curr Opin Immunol. 2004;16: 328-336. [DOI] [PubMed] [Google Scholar]
- 2.Dikic I, Giordano S. Negative receptor signalling. Curr Opin Cell Biol. 2003;15: 128-135. [DOI] [PubMed] [Google Scholar]
- 3.Moretta L, Moretta A. Killer immunoglobulin-like receptors. Curr Opin Immunol. 2004;16: 626-633. [DOI] [PubMed] [Google Scholar]
- 4.Barford D, Neel BG. Revealing mechanisms for SH2 domain mediated regulation of the protein tyrosine phosphatase SHP-2. Structure. 1998;6: 249-254. [DOI] [PubMed] [Google Scholar]
- 5.Faure M, Barber DF, Takahashi SM, Jin T, Long EO. Spontaneous clustering and tyrosine phosphorylation of NK cell inhibitory receptor induced by ligand binding. J Immunol. 2003;170: 6107-6114. [DOI] [PubMed] [Google Scholar]
- 6.Stebbins CC, Watzl C, Billadeau DD, Leibson PJ, Burshtyn DN, Long EO. Vav1 dephosphorylation by the tyrosine phosphatase SHP-1 as a mechanism for inhibition of cellular cytotoxicity. Mol Cell Biol. 2003;23: 6291-6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vyas YM, Maniar H, Lyddane CE, Sadelain M, Dupont B. Ligand binding to inhibitory killer cell Ig-like receptors induce colocalization with Src homology domain 2-containing protein tyrosine phosphatase 1 and interruption of ongoing activation signals. J Immunol. 2004;173: 1571-1578. [DOI] [PubMed] [Google Scholar]
- 8.Guerra N, Michel F, Gati A, et al. Engagement of the inhibitory receptor CD158a interrupts TCR signaling, preventing dynamic membrane reorganization in CTL/tumor cell interaction. Blood. 2002;100: 2874-2881. [DOI] [PubMed] [Google Scholar]
- 9.Snyder MR, Muegge LO, Offord C, et al. Formation of the killer Ig-like receptor repertoire on CD4+CD28null T cells. J Immunol. 2002;168: 3839-3846. [DOI] [PubMed] [Google Scholar]
- 10.Xu J, Vallejo AN, Jiang Y, Weyand CM, Goronzy JJ. Distinct transcriptional control mechanisms of killer immunoglobulin-like receptors in natural killer (NK) and in T cells. J Biol Chem. 2005;280: 24277-24285. [DOI] [PubMed] [Google Scholar]
- 11.Schmidt D, Martens PB, Weyand CM, Goronzy JJ. The repertoire of CD4+ CD28– T cells in rheumatoid arthritis. Mol Med. 1996;2: 608-618. [PMC free article] [PubMed] [Google Scholar]
- 12.Ljunggren HG, Karre K. In search of the “missing self”: MHC molecules and NK cell recognition. Immunol Today. 1990;11: 237-244. [DOI] [PubMed] [Google Scholar]
- 13.Cambiaggi A, Darche S, Guia S, Kourilsky P, Abastado JP, Vivier E. Modulation of T-cell functions in KIR2DL3 (CD158b) transgenic mice. Blood. 1999;94: 2396-2402. [PubMed] [Google Scholar]
- 14.Ince MN, Harnisch B, Xu Z, et al. Increased expression of the natural killer cell inhibitory receptor CD85j/ILT2 on antigen-specific effector CD8 T cells and its impact on CD8 T-cell function. Immunology. 2004;112: 531-542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ikeda H, Lethe B, Lehmann F, et al. Characterization of an antigen that is recognized on a melanoma showing partial HLA loss by CTL expressing an NK inhibitory receptor. Immunity. 1997;6: 199-208. [DOI] [PubMed] [Google Scholar]
- 16.Gu X, Laouar A, Wan J, et al. The gp49B1 inhibitory receptor regulates the IFN-gamma responses of T cells and NK cells. J Immunol. 2003; 170: 4095-4101. [DOI] [PubMed] [Google Scholar]
- 17.Faroudi M, Utzny C, Salio M, et al. Lytic versus stimulatory synapse in cytotoxic T lymphocyte/target cell interaction: manifestation of a dual activation threshold. Proc Natl Acad Sci U S A. 2003; 100: 14145-14150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Snyder MR, Lucas M, Vivier E, Weyand CM, Goronzy JJ. Selective activation of the c-Jun NH2-terminal protein kinase signaling pathway by stimulatory KIR in the absence of KARAP/DAP12 in CD4+ T cells. J Exp Med. 2003;197: 437-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takayama H, Trenn G, Sitkovsky MV. A novel cytotoxic T lymphocyte activation assay: optimized conditions for antigen receptor triggered granule enzyme secretion. J Immunol Methods. 1987; 104: 183-190. [DOI] [PubMed] [Google Scholar]
- 20.Stewart CA, Laugier-Anfossi F, Vely F, et al. Recognition of peptide-MHC class I complexes by activating killer immunoglobulin-like receptors. Proc Natl Acad Sci U S A. 2005;102: 13224-13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19: 185-193. [DOI] [PubMed] [Google Scholar]
- 22.Wu Z, Irizarry R, Gentleman R, Murillo F, Spencer F. A model based background adjustment for oligonucleotide expression arrays. J Am Stat Assoc. 2005;99: 909-917. [Google Scholar]
- 23.Blagoev B, Ong SE, Kratchmarova I, Mann M. Temporal analysis of phosphotyrosine-dependent signaling networks by quantitative proteomics. Nat Biotechnol. 2004;22: 1139-1145. [DOI] [PubMed] [Google Scholar]
- 24.Park W, Weyand CM, Schmidt D, Goronzy JJ. Co-stimulatory pathways controlling activation and peripheral tolerance of human CD4+CD28– T cells. Eur J Immunol. 1997;27: 1082-1090. [DOI] [PubMed] [Google Scholar]
- 25.Namekawa T, Wagner UG, Goronzy JJ, Weyand CM. Functional subsets of CD4 T cells in rheumatoid synovitis. Arthritis Rheum. 1998;41: 2108-2116. [DOI] [PubMed] [Google Scholar]
- 26.Schirmer M, Vallejo AN, Weyand CM, Goronzy JJ. Resistance to apoptosis and elevated expression of Bcl-2 in clonally expanded CD4+CD28– T cells from rheumatoid arthritis patients. J Immunol. 1998;161: 1018-1025. [PubMed] [Google Scholar]
- 27.Vallejo AN, Schirmer M, Weyand CM, Goronzy JJ. Clonality and longevity of CD4+CD28null T cells are associated with defects in apoptotic pathways. J Immunol. 2000;165: 6301-6307. [DOI] [PubMed] [Google Scholar]
- 28.Monks CR, Freiberg BA, Kupfer H, Sciaky N, Kupfer A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. 1998;395: 82-86. [DOI] [PubMed] [Google Scholar]
- 29.Huppa JB, Gleimer M, Sumen C, Davis MM. Continuous T cell receptor signaling required for synapse maintenance and full effector potential. Nat Immunol. 2003;4: 749-755. [DOI] [PubMed] [Google Scholar]
- 30.Carter LL, Carreno BM. Cytotoxic T-lymphocyte antigen-4 and programmed death-1 function as negative regulators of lymphocyte activation. Immunol Res. 2003;28: 49-59. [DOI] [PubMed] [Google Scholar]
- 31.Lou Z, Jevremovic D, Billadeau DD, Leibson PJ. A balance between positive and negative signals in cytotoxic lymphocytes regulates the polarization of lipid rafts during the development of cell-mediated killing. J Exp Med. 2000;191: 347-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fassett MS, Davis DM, Valter MM, Cohen GB, Strominger JL. Signaling at the inhibitory natural killer cell immune synapse regulates lipid raft polarization but not class I MHC clustering. Proc Natl Acad Sci U S A. 2001;98: 14547-14552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hornstein I, Alcover A, Katzav S. Vav proteins, masters of the world of cytoskeleton organization. Cell Signal. 2004;16: 1-11. [DOI] [PubMed] [Google Scholar]
- 34.Tybulewicz V. Vav-family proteins in T-cell signalling. Curr Opin Immunol. 2005;17: 267-274. [DOI] [PubMed] [Google Scholar]
- 35.Villalba M, Bi K, Rodriguez F, Tanaka Y, Schoenberger S, Altman A. Vav1/Rac-dependent actin cytoskeleton reorganization is required for lipid raft clustering in T cells. J Cell Biol. 2001;155: 331-338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dustin ML. Coordination of T cell activation and migration through formation of the immunological synapse. Ann N Y Acad Sci. 2003;987: 51-59. [DOI] [PubMed] [Google Scholar]
- 37.Schrum AG, Turka LA. The proliferative capacity of individual naive CD4(+) T cells is amplified by prolonged T cell antigen receptor triggering. J Exp Med. 2002;196: 793-803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carena I, Shamshiev A, Donda A, Colonna M, Libero GD. Major histocompatibility complex class I molecules modulate activation threshold and early signaling of T cell antigen receptor-gamma/delta stimulated by nonpeptidic ligands. J Exp Med. 1997;186: 1769-1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dietrich J, Cella M, Colonna M. Ig-like transcript 2 (ILT2)/leukocyte Ig-like receptor 1 (LIR1) inhibits TCR signaling and actin cytoskeleton reorganization. J Immunol. 2001;166: 2514-2521. [DOI] [PubMed] [Google Scholar]
- 40.Anfossi N, Doisne JM, Peyrat MA, et al. Coordinated expression of Ig-like inhibitory MHC class I receptors and acquisition of cytotoxic function in human CD8+ T cells. J Immunol. 2004;173: 7223-7229. [DOI] [PubMed] [Google Scholar]
- 41.Davis DM, Chiu I, Fassett M, Cohen GB, Mandelboim O, Strominger JL. The human natural killer cell immune synapse. Proc Natl Acad Sci U S A. 1999;96: 15062-15067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Standeven LJ, Carlin LM, Borszcz P, Davis DM, Burshtyn DN. The actin cytoskeleton controls the efficiency of killer Ig-like receptor accumulation at inhibitory NK cell immune synapses. J Immunol. 2004;173: 5617-5625. [DOI] [PubMed] [Google Scholar]
- 43.Sanni TB, Masilamani M, Kabat J, Coligan JE, Borrego F. Exclusion of lipid rafts and decreased mobility of CD94/NKG2A receptors at the inhibitory NK cell synapse. Mol Biol Cell. 2004;15: 3210-3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Betts MR, Price DA, Brenchley JM, et al. The functional profile of primary human antiviral CD8+ T cell effector activity is dictated by cognate peptide concentration. J Immunol. 2004;172: 6407-6417. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.