

Abstract
Angiogenesis contributes to a wide range of neoplastic, ischemic, and inflammatory disorders. Definition of the intrinsic molecular controls in angiogenic vessel growth promises novel therapeutic approaches for angiogenesis-related diseases. CD148 (also named DEP-1/PTPη) is a receptor-like protein tyrosine phosphatase that is abundantly expressed in vascular endothelial cells. To explore a role of CD148 in endothelial vessel formation, we generated a monoclonal antibody, Ab1, against the ectodomain sequence of CD148 and examined its effects on endothelial-cell growth and vessel formation. Here we report that a bivalent, but not a monovalent, form of the Ab1 antibody inhibits endothelial-cell growth and blocks angiogenesis in mouse cornea in vivo. We further demonstrate that (1) bivalent Ab1 arrests cell-cycle progression of CD148-transfected CHO cells at G0/G1 phase, (2) coexpression of catalytically inactive CD148 mutants attenuates the Ab1-cell growth inhibition, and (3) bivalent Ab1 suppresses phosphorylation of ERK1/2 kinases and Met tyrosine kinase as activated CD148 does, with an increase in CD148-associated tyrosine phosphatase activity. Taken together, these findings demonstrate that Ab1-induced ectodomain oligomerization arrests endothelial-cell growth through catalytic activity of the CD148 cytoplasmic domain. The present study defines CD148 as a valuable molecular target for antiangiogenesis therapy.
Introduction
Angiogenesis is a fundamental process in organogenesis and tissue regeneration. On the other hand, deregulated angiogenesis induced by pathologic stimuli contributes to numerous diseases, including cancer, cardiovascular disease, arthritis, and diabetes.1 Definition of the intrinsic molecular controls in angiogenic vessel growth promises better treatment strategies for angiogenesis-associated diseases.
Blood-vessel formation is tightly controlled through a balance between proangiogenic and antiangiogenic factors.2 Studies in recent decades have indicated a critical role for endothelial receptor protein tyrosine kinases (RPTKs) and their activating ligands to promote and coordinate vessel formation.3 These include receptors for vascular endothelial growth factor (VEGF), angiopoietins, ephrins, fibroblast growth factor (FGF), and hepatocyte growth factor (HGF). In contrast, the role of receptor-like protein tyrosine phosphatases (RPTPs) in this process is largely unknown, although coupled and counterbalanced functions of RPTKs and RPTPs have been well defined in neural targeting and differentiation.4
CD148 (also named DEP-1/PTPη) is a type III RPTP that is composed of an extracellular region containing 8 fibronectin type III–like repeats, a membrane-spanning region, and a single intracellular phosphatase domain.5 It is abundantly expressed in vascular endothelial cells,6,7 hematopoietic-cell lineages,8 and duct epithelia of thyroid, mammary, and gastrointestinal tissues.9-12 CD148 was initially shown to increase in abundance with high cell density in WI38 cells, prompting the name DEP-1 (density-enhanced phosphatase-1).5 The finding suggested a role for CD148 to convey density-mediated growth arrest signals. Subsequent studies further supported a role of CD148 in cell-growth control. First, CD148 expression is down-regulated in tumor cells or transformed cell lines, correlated with their malignant phenotype.11,12 Second, overexpression of CD148 suppresses tumor-cell growth in vitro and in vivo, concomitant with reduction in MAP kinase (ERK1/2) activity and PLCγ1 phosphorylation.10,11,13 Third, Ptprj (CD148) has been identified as a gene candidate for mouse colon-cancer susceptibility locus Scc1,14 and loss of heterozygosity (LOH) at PTPRJ locus was frequently found in human cancers.14 Finally, we have shown that mutant mice lacking catalytic activity of CD148 die at midgestation due to vascularization failure accompanied by increased endothelial-cell proliferation and vessel growth.15 In aggregate, these findings indicate an important role for CD148 in negative regulation of cell proliferation.
Consistent with these findings, recent studies have demonstrated CD148 inhibition of growth factor signaling. CD148 overexpression promotes site-selective dephosphorylation of the activated PDGF-beta receptor,16,17 and suppresses PDGF-mediated ERK1/2 activation and inositol trisphosphate (IP3) production.18 CD148 dephosphorylates VEGF receptor-2 on endothelial cell-cell contacts.19 Further, “substrate trapping” approaches have identified Met tyrosine kinase, HGF receptor, as a relevant substrate for CD148.20 The study also demonstrated site-selective dephosphorylation of Met tyrosine kinase by CD148.20 Further, studies on T lymphocytes have shown that CD148 suppresses PLCγ1 and ERK1/2 activities induced by T-cell receptor activation, suggesting that CD148 may regulate signaling of several receptors by acting on downstream targets of the receptors.21 Indeed, a more recent study has suggested direct interaction between CD148 and ERK1/2 kinases.22 Although these studies have identified the intracellular signaling pathways that CD148 may control, the mechanisms regulating CD148 activity remain undefined.
It is well known that RPTKs transduce signals by ligand-initiated ectodomain oligomerization to promote intermolecular, in trans cytoplasmic domain tyrosine phosphorylation and subsequent assembly of multicomponent signaling complexes.23 RPTPs are also anticipated to be regulated by ectodomain-binding events24; however, strong evidence for such coupling has not yet been provided. Data addressing this mechanism have been obtained only for RPTPα and CD45. The crystal structure of the RPTPα D1 domain revealed catalytic site occlusion by a helix-turn-helix segment of an opposing dyad partner.25 Forced dimerization of RPTPα inhibited catalytic activity in intact cells.26 Further, EGF-induced dimerization of EGFR-CD45 chimera suppressed CD45 catalytic activity.27 Mice mutant for the predicted inhibitory wedge in CD45 exhibited lymphoproliferative disorders consistent with increased activity of CD45.28 These findings prompted a model of dimerization-induced RPTP inhibition, which has been challenged by additional data. First, although RPTPα dimers have been detected after chemical cross-linking, differences in specific activities of monomeric and dimeric forms of RPTPα have not been demonstrated.29 Second, the residues that occlude the RPTPα active site are poorly conserved among other RPTPs, and the crystal structures of PTPμ and LAR catalytic domains failed to show structural support for dimer-induced inhibition.30,31 These data argue that dimerization-induced inhibition is not a universal mechanism regulating RPTP activity. To date, extracellular ligand interactions that have clearly demonstrated effects on phosphatase activity against specific tyrosine-phosphorylated substrates have not been identified in most RPTPs.32
To explore a potential role for CD148 in endothelial vessel formation, we have generated a monoclonal antibody, Ab1, against the ectodomain sequences of human CD148.5 We have shown that Ab1 antibody binds to CD148 on surfaces of endothelial cells with high affinity and specificity.6 In the present study, we examined the biologic effects of monovalent (Fab fragment) and bivalent (intact) Ab1 on endothelial-cell growth and corneal angiogenic responses in vivo. Our data demonstrate that bivalent, but not monovalent, Ab1 inhibits endothelial-cell growth and angiogenesis. These findings suggest that CD148 activity is potentially regulated by ectodomain-mediated oligomerization and that CD148 is a molecular target for antiangiogenesis therapy.
Materials and methods
Antibodies
A mouse IgG1 monoclonal antibody, Ab1, was raised against ectodomain sequences (aa 175-536) of human CD148 (DEP-1).6 The Fab fragments of Ab1 antibody, Ab1.Fab, were prepared by digesting 5 mg Ab1 in 100 mM sodium acetate/pH 5.5, 50 mm l-cysteine, 1 mM EDTA with 10 μg papain (Sigma, St Louis, MO) for 8 hours at 37°C and incubated with 75 mM iodoacetamide for 30 minutes at RT, followed by Protein-A sepharose (Sigma) affinity chromatography to remove free Fc domains. Fluorescein isothiocyanate (FITC)–conjugated Ab1 antibody was prepared as previously described.6 Anti–hemagglutinin (HA) epitope monoclonal antibody (clone 12CA5), in either unconjugated or horseradish peroxidase (HRP)–conjugated forms, was from Roche Applied Science (Indianapolis, IN); anti–VE cadherin goat antibody was from Santa Cruz Biotechnology (Santa Cruz, CA); rhodamine-conjugated rabbit anti–goat IgG was from Jackson Immunoresearch Lab (Baltimore, MD).
Expression plasmids
The full-length cDNA of human CD148 (kindly provided by Nicholas Tonks)5 was c-terminally tagged with 3xHA sequences and subcloned into the pSRα vector (pSRα CD148/HA).6 The expression plasmid for a catalytically inactive CD148 mutant, pSRα CD148 C-S/HA (C1239S), was constructed by oligonucleotide-specified overlap extension polymerase chain reaction (PCR). The cytoplasmic deletion mutant, pSRα CD148ΔCyto (terminated at aa1051) was produced using a HindIII site in the CD148 sequence. Plasmid constructions were confirmed by DNA sequencing.
Cell culture and transfection
Primary human renal microvascular endothelial cells (HRMECs) at the third passage were cultured as previously described.6 CHO cells were cultured and transfected by cationic liposome (Lipofectamine Plus; GIBCO Life Technologies, Grand Island, NY) according to the manufacturer's protocol.
Proliferation assay
HRMEC proliferation (Figure 1) was assayed by plating the cells in 12-well plates at the density of 3 × 104/well = 4.0 cm2 on day 0, with replacement of medium supplemented with either Ab1 (67 nM), Ab1.Fab (67 nM), or IgG1 control (67 nM) every 2 days subsequently. Cell number was evaluated by staining 2% glutaraldehyde-fixed cells with 0.5% crystal violet and measuring A570 of dye extracted by 1% SDS as previously described.33 For CHO-cell proliferation experiments in Figure 2, cells grown in 100-mm dishes were transfected with 4.0 μg of the expression plasmids and replated in 12-well plates at 16 hours (2.4 × 104 cells/well), and antibodies (67 nM) were added at 30 hours. Cell number was assayed at 72 hours after transfection by crystal violet assay. Proliferation assays were also conducted with stably transfected CHO cells (Figures 4, 5). The cells were plated in 12-well plates (2.0 × 104 cells/well) and synchronized by 48-hour serum starvation. Cells were then preincubated with Ab1, Ab1.Fab, or control IgG1 (67 nM) for 60 minutes, and growth medium supplemented with the indicated agents was replaced on day 0 and day 2. Cells were harvested for counting on day 2 and day 4. Cell number was evaluated by a crystal violet assay.33 In both HRMEC and CHO proliferation assays, FBS in the medium was reduced to 7.5% (HRMEC) or 2.5% (CHO). The crystal violet assay was validated to correlate with cell numbers by independent counting in a hemocytometer.
Figure 1.

Bivalent Ab1 remarkably inhibits proliferation of HRMECs. (A) HRMECs were plated in a 12-well plate at the density of 3 × 104 cells/4.0 cm2 (day 0). The medium was replaced every 2 days subsequently (arrows), supplemented with 67 nM (10 μg/mL) Ab1, Ab1.Fab, or mouse IgG1 (isotype-matched control antibody), and the cell number (means ± SEM, n = 6) was evaluated as described in “Materials and methods.” Bivalent Ab1, but not monovalent Ab1.Fab, markedly inhibited proliferation of HRMECs. (B) Cell numbers at day 6 cultured in the growth medium supplemented with indicated concentrations of agents were determined. Means ± SEM of quadruplicate samples are shown. Cell numbers (○) cultured in serum-free medium are also displayed.
Figure 2.

Expression of catalytically active CD148 confers Ab1-sensitive growth inhibition on CHO cells and the Ab1 activity is abrogated by coexpression of inactive CD148 mutants. (A) CHO cells were transiently transfected with CD148 expression plasmids (mock, WT, and C-S and ΔCyto forms) as indicated. The cells were replated at 16 hours and incubated from 30 hours after transfection in growth medium supplemented with either Ab1, Ab1.Fab, or control IgG1 (67 nM). Bars represent mean A570 determination values (± SEM) of 12 independent samplings for each group at 3 days (72 hours) after transfection, expressed as percent of control (the cells grown without agents). The protein levels of transfected genes were evaluated by Ab1 immunoblot on 5.0 μg lysate protein (insert). (B) CHO cells were cotransfected with 2.5 μg WT plasmid in combination with 2.5 μg plasmid expressing C-S (WT + C-S) or ΔCy (WT +ΔCy) form of CD148. The cells were treated with Ab1, Ab1.Fab, or control IgG1 (67 nM), and cell proliferation and CD148 protein levels were examined as described in panel A. The arrowhead (in insert) indicates the ΔCy mutant.
Figure 4.

Determination of Ab1 epitope sequence and peptide competition study. (A) Ab1 immunoblot on a display of overlapping 8 amino acid synthetic peptides derived from the CD148 ectodomain sequence used for immunization. Ab1 specifically reacted to the sequence QSRDTEVL. (B-C) Stable CHO cell line was plated, serum-depleted, and cultured in growth medium supplemented with either no addition (□) or with Ab1 (67 nM, •) in combination with the indicated molar ratios of peptide, biotin-SGSGQSRDTEVL (synthesized by Chiron Technologies, Clayton, Australia) (B) or Ab1.Fab (C). Cell numbers were assessed at day 2 and day 4 as described in “Materials and methods.” Cell numbers at day 4 are shown. Both peptide and Ab1.Fab effectively antagonized growth inhibitory activity of Ab1 at molar ratios exceeding 3-fold that of Ab1. The data in panels B and C are means ± SEM of quadruplicate determinations.
Figure 5.

Bivalent Ab1 inhibits cell-cycle progression at G0/G1 phase. (A) Stable and control CHO cell lines (CD148-CHO, control-CHO) were prepared as described in “Materials and methods.” Cells were plated, serum-depleted for 48 hours, and pretreated for 60 minutes with serum-free medium containing Ab1, Ab1.Fab, and control IgG1 (67 nM). Then, the medium was replaced to growth medium containing 67 nM of each agent (day 0). Cell number was evaluated at days 1, 3, and 5. An upper panel displays anti-HA immunoblot on cell lysates (25 μg protein) prepared from the CHO lines. (B) The CHO cells, CD148-CHO and control-CHO, were plated in 100-mm dishes (1 × 105 cells/dish), synchronized by 72-hour serum depletion, and pretreated for 60 minutes with serum-free medium containing Ab1 or Ab1.Fab (67 nM), and the medium was replaced with growth medium supplemented with Ab1 or Ab1.Fab (67 nM) (time 0 hours). Cell-cycle progression was analyzed by FACS as described in “Materials and methods.” The blue indicates S phase; red, G0/G1 and G2/M phases. Bivalent Ab1 inhibits S-phase entry in CD148-CHO cells, while Ab1.Fab does not.
Immunoprecipitation and immunoblot analysis
CD148 protein level was assessed by lysis of the cells in SDS buffer (0.1 M Tris/pH 6.8, 2% SDS, 4% glycerol), SDS–polyacrylamide gel separation, and immunoblotting using HRP-conjugated Ab1 or anti-HA antibody as previously described.6 Phosphorylation of Met kinase, a receptor for HGF, was examined as below. HRMECs were plated in 100-mm dishes at 30% confluence and serum depleted in DMEM/0.1% FBS for 18 hours. The cells were preincubated with 67 nM Ab1 antibodies (monovalent or bivalent) for one hour at 37°C, and medium was replaced with DMEM containing 10 ng/mL HGF (R & D Systems, Minneapolis, MN) supplemented with Ab1 (67 nM) or Ab1.Fab (67 nM). The cells were lysed in lysis buffer (10 mM Tris/pH 8.0, 150 mM NaCl, 10% glycerol, 1% NP-40, 2 mM sodium ortho-vanadate, 5 mM EDTA, 5 mM NaF) containing protease inhibitor cocktail (Roche Applied Science) at indicated time points, and clarified lysate protein (100 μg) was subjected to immunoprecipitation with anti-Met polyclonal antibody (Santa Cruz Biotechnology). The immunoprecipitates were separated by SDS electrophoresis on 7.5% polyacrylamide gels and transferred to PVDF membranes (Amersham Biosciences, Piscataway, NJ), and immunodetection was carried out using phospho-specific Met antibody (BD Biosciences, Transduction, San Diego, CA) as previously described.20 The membrane was reblotted with anti-Met polyclonal antibody (Santa Cruz Biotechnology). Immunoblots of active ERK1/2 and Akt kinases were carried out as follows. HRMECs and stably transfected CHO cells were plated at 30% confluence, serum depleted (HRMECs: 24 hours; CHO: 72 hours), and pretreated with Ab1 (67 nM, monovalent or bivalent) for 60 minutes. Then, the cells were incubated with DMEM containing 2.5% FBS and 67 nM Ab1 (monovalent or bivalent) and lysed in the buffer (50 mM Tris/pH 6.8, 2% SDS, 10% glycerol, 10 mM sodium pyrophosphate, 100 mM NaF, 1 mM Na3VO4, protease inhibitor cocktail). For ERK1/2 immunoblotting, 20 μg proteins was subjected to SDS electrophoresis on 12% polyacrylamide gels and immunoblotted with phospho-peptide–specific ERK1/2 antibody (New England Biolabs, Beverly, MA). Total ERK1/2 level was assessed by reblotting the membrane with anti-ERK1/2 antibody (Upstate Biotechnology, Lake Placid, NY). For Akt immunodetection, 200 μg protein was subjected to immunoprecipitation with anti-Akt monoclonal antibody (Cell Signaling Technology, Beverly, MA), separated by SDS-PAGE, and immunoblotted using phosphopeptide–specific Akt (phospho-ser473) polyclonal antibody (Cell Signaling Technology). Total Akt was assessed using anti-Akt polyclonal antibody (Cell Signaling Technology).
PTPase activity assay
HRMECs were plated at 30% confluence, serum depleted for 24 hours, and pretreated with Ab1 (67 nM, monovalent or bivalent) for 60 minutes. Then, the cells were incubated with DMEM containing 2.5% FBS and 67 nM Ab1 (monovalent or bivalent) for 15 minutes and lysed in buffer (50 mM HEPES/pH 7.5, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 1 mM DTT, 5 μg/mL aprotinin, 1 μg/mL leupeptin, 1 mM PMSF), and clarified protein lysate (150 μg) was subjected to immunoprecipitation with affinity-purified CD148 rabbit antibody6 for 4 hours at 4°C. The washed immunocomplexes were incubated with 100 μM synthetic phosphotyrosine peptide (RRLIEDAEpYAAR) in Tris buffer (pH 7.5) containing 0.5 mg/mL BSA and 0.5 mM DTT for 15 minutes at 37°C, and released phosphates were calculated by the malachite green development according to the manufacturer's instruction (Upstate Biotechnology).
Cell cycle analysis
Stably transfected CHO lines (CD148-CHO) were prepared by cotransfection with the plasmids pSRα CD148/HA6 and pZeoSV2 (Invitrogen, Carlsbad, CA) at a 10:1 ratio, followed by Zeocin (800 μg/mL; Invitrogen) selection. The CHO cell lines that express CD148 at the level comparable with that in HRMECs were selected. The CHO cells (control-CHO) that lack CD148/HA were also selected and used as a control. The cells were plated at 1 × 105 cells/100-mm dish and synchronized by 72-hour serum depletion. Cells were then preincubated (t = 0) with either Ab1 or Ab1.Fab (67 nM) for 60 minutes, and then medium was replaced with growth medium supplemented with either Ab1 or Ab1.Fab (67 nM). Cells were harvested at indicated time points, fixed with cold 70% ethanol for 30 minutes, washed with PBS, and incubated with 50 μg/mL RNAase (Roche Applied Science) for 30 minutes at 37°C. Propidium iodide was added (25 μg/mL) and incubated for 30 minutes at RT in the dark. Cells were analyzed by flow cytometry using a FACS Caliber (Becton Dickinson, San Jose, CA) instrument using an argon laser at 531 nm with a 585-nm band pass filter. DNA analysis was conducted using ModFitLT V2.0 software (Verify Software House, Topsham, ME).
Immunohistochemistry
Acetone-fixed cryostat sections of mouse tissues were blocked, incubated with FITC-conjugated Ab1, mounted, and analyzed by confocal microscopy (Zeiss LSM410; Carl Zeiss, Heidelberg, Germany) as previously described.6 Double immunolabeling for CD148 and VE-cadherin was performed by sequential addition of goat anti–VE cadherin antibody and rhodamine-conjugated rabbit anti–goat IgG as described previously.6
Mouse corneal angiogenesis assay
Hydron pellets incorporating sucralfate with either vehicle alone, 90 ng basic FGF (3 pM/pellet; gift from Scios), or basic FGF in combination with control IgG1 (11 μg), Ab1 (11 μg), or Ab1.Fab (3.8 μg, molar equivalent) in equivalent stoichiometric amounts (74 pM/pellet) were prepared as previously described.34 Pellets were surgically implanted into corneal stromal micropockets created 1-mm medial to the lateral corneal limbus of C57BL/6 male mice (7-9 weeks old). At day 5, corneas were photographed at an incipient angle of 35° to 50° from the polar axis in the meridian containing the pellet, using a Zeiss slit lamp. Images were digitalized and processed by subtractive color filters (Adobe Photoshop; Adobe Systems, San Jose, CA); the fraction of the total corneal image that was vascularized and the ratio of pixels marking neovascular capillaries, both within the vascularized region and within the total corneal image, were calculated using Bioquant software (Bioquant, Nashville, TN).
Results
Bivalent Ab1 inhibits endothelial-cell growth
A growing body of evidence demonstrates an important role of CD148 in negative regulation of cell growth.10,11,13,14 In the present study, we asked first whether Ab1 might affect endothelial-cell growth through its interaction with CD148 as either an antagonist or an agonist of CD148-mediated responses. Shown in Figure 1A, Ab1 suppressed serum-stimulated endothelial-cell growth, effectively reducing proliferation to levels comparable with those seen in the absence of serum. Maximal growth inhibitory effects were observed at 67 nM (10 μg/mL) and the IC50 was at 3 to 5 nM (Figure 1B). In contrast to bivalent Ab1, monovalent Ab1 (the Fab fragment of Ab1 antibody, Ab1.Fab) was inactive at equivalent and higher concentrations (up to 335 nM), as was a bivalent class-matched mouse IgG1 (Figure 1B). Bivalent Ab1 did not induce apoptosis in endothelial cells, as scored by a TUNEL assay, annexin V binding, or nuclear fragmentation (data not shown). Cell-growth inhibition by Ab1 was not restricted to HRMECs, but was also observed on other endothelial cell lines expressing CD148, including HMEC-16 and EAhy926 (data not shown), and in vivo in mouse tissues, as shown in a corneal angiogenesis assay (or a corneal pocket model).
Expression of CD148 confers Ab1-sensitive growth inhibition on CHO cells, and coexpression of inactive CD148 mutants abrogates the Ab1 activity
To verify the specificity and the mechanism of Ab1-cell growth inhibition, we expressed wild-type and mutant forms of CD148 in CHO cells, a line lacking endogenous CD148, and examined the effects of Ab1 on their cell growth. CHO populations transiently transfected with wild-type (WT) CD148/HA showed modest reduction in proliferation in growth medium (8%-14% reduction in cell numbers at 5 days) when the cells were plated at low cell density (20% confluence). In contrast, growth inhibition was markedly accentuated in the presence of bivalent Ab1, while monovalent Ab1 and class-matched mouse IgG1 were inactive (Figure 2A). Moreover, Ab1 was inactive as a growth suppressor for the CHO cells expressing comparable levels of either a catalytically inactive (C-S mutant) or a cytoplasmic domain-deleted (ΔCy) CD148/HA (Figure 2A). Independent biotin surface labeling and anti-CD148 cell-staining experiments revealed that Ab1 binding has negligible effects to reduce cell-surface CD148 level over a period of up to 16 hours (data not shown). These results indicate that both CD148 expression and intact catalytic activity are required to confer Ab1-sensitive growth inhibition on CHO cells.
These findings further suggest that Ab1-induced ectodomain oligomerization may increase CD148 activity and inhibit cell proliferation, as depicted in Figure 2A. To test this model, we coexpressed catalytically inactive forms of CD148 with wild-type CD148/HA, anticipating that these mutant forms would function as dominant-negative antagonists. Shown in Figure 2B, transient coexpression of CD148 mutants (C-S or ΔCyto) in stoichiometric excess of CD148/HA abrogated the activity of bivalent Ab1 to inhibit cell growth (Figure 2B).
Bivalent Ab1 suppresses phosphorylation of ERK1/2 and Met kinases accompanied by an increase in CD148-associated PTP activity
To determine whether Ab1 increases CD148-mediated dephosphorylation activity, we investigated the effects of Ab1 on downstream targets of CD148 signaling using HRMECs and CHO cells. Since CD148 was shown to bind to and dephosphorylate ERK1/2 kinases,22 we first investigated the activity of Ab1 to suppress ERK1/2 phosphorylation. Shown in Figure 3A, bivalent Ab1 suppressed the magnitude and duration of serum-stimulated ERK1/2 activation in HRMECs and stable CHO cell lines, which express physiologic levels of CD148, while serum-stimulated Akt activation was not affected by bivalent Ab1. Monovalent Ab1 was inactive in suppressing ERK1/2 phosphorylation. Second, we evaluated the effects of bivalent Ab1 to reduce the state of tyrosine phosphorylation of Met tyrosine kinase (HGF receptor), a recognized substrate of CD148,20 in HRMECs that abundantly express Met kinase. Shown in Figure 3B, bivalent Ab1 treatment suppressed Met tyrosine phosphorylation over 30 minutes, while monovalent Ab1 was inactive. These findings demonstrate that Ab1 binding to CD148 dephosphorylates downstream targets of CD148 and inhibits cell proliferation as active CD148 does.
Figure 3.

Bivalent Ab1 suppresses phosphorylation of ERK1/2 and Met kinases with an increase in CD148-associated PTP activity. (A) HRMECs and the CHO cell line (CHO-CD148), which is stably expressing CD148, were serum starved for 24 hours and 72 hours, respectively; pretreated with 67 nM Ab1 (bivalent) or Ab1.Fab (monovalent); and stimulated with medium supplemented with 2.5% FBS in the presence of Ab1 or Ab1.Fab (67 nM). The cell lysates were subjected to immunoblots using phospho-peptide–specific ERK1/2 (p-ERK) and Akt (p-Akt) antibodies. Blots were stripped and reprobed for total ERK1/2 (ERK) and Akt (Akt). (B) Serum-starved subconfluent HRMECs were pretreated with Ab1 or Ab1.Fab (67 nM), and then stimulated with 10 ng/mL HGF in the presence of Ab1 or Ab1.Fab (67 nM). Met kinase (HGF receptor) was immunoprecipitated from the cleared lysates and its phosphorylation level was examined by immunoblots using a phospho-peptide–specific Met antibody (p-Met) as described in “Materials and methods.” Blots were stripped and reprobed for total Met (Met). (C) HRMECs were plated on 100-mm dishes, serum starved, and incubated with either Ab1 or Ab1.Fab or control IgG1 (67 nM) for 15 minutes. The cells were lysed in buffer and CD148 was immunoprecipitated using affinity-purified CD148 rabbit antibody or control rabbit IgG. The washed immunocomplexes were assayed for PTP activity with or without 1 mM sodium orthovanadate (VO4) as described in “Materials and methods.” The data are presented as means ± SEM of quadruplicate determinations.
Finally, we assessed whether CD148-associated PTP activity was increased following exposure of the cells to bivalent Ab1, by assaying immunoprecipitated CD148 for pervanadate-sensitive phosphatase activity using a phosphotyrosine peptide as substrate. As shown in Figure 3C, the results exhibited a moderate increase (∼ 55%) in CD148-associated PTP activity in the Ab1-treated HRMECs, although Ab1 treatment did not alter CD148 abundance in immunocomplexes (data not shown). In aggregate, these findings indicate increased CD148 activity by Ab1 treatment. Although CD148-associated PTP activity was consistently increased by exposure to bivalent Ab1, the increase was not prominent when assayed in this manner. This is possibly because immunoprecipitation itself oligomerizes and activates solubilized CD148, or alternatively, bivalent Ab1–treated CD148 may not completely be solubilized or recovered by the immunoprecipitation.
Epitope mapping and peptide competition support
As a final test of whether Ab1-cell growth inhibition resides in its capacity to engage ectodomain sequences of CD148, we mapped the epitope to which Ab1 binds within the 361–amino acid immunogen (Figure 4). Peptides spanning the immunization sequences were synthesized and displayed on nitrocellulose (Research Genetics, Huntsville, AL). Ab1 bound to the 8–amino acid sequence QRSDTEVL with high avidity as depicted in Figure 4A. This peptide sequence competed effectively for Ab1 binding to the immunized protein in Biacore competition assays, where Ab1 displays an apparent Kd of 5.4 nM (Ergang Shi and T.O.D., unpublished results, January 2001). Further, a peptide containing the QSRDTEVL sequence effectively antagonized Ab1-mediated cell-growth inhibition in a stable CHO cell line (Figure 4B). Shown in Figure 4C, the growth inhibitory effect of Ab1 on stably transfected CHO cells was also antagonized by preincubation of the cells with 3-fold and greater molar excesses of Ab1.Fab. This finding shows that Ab1.Fab has similar affinity for CD148 expressed on CHO cells as bivalent antibody does, and is consistent with its effect to occupy surface epitope sites as a monomer and to antagonize the capacity for bivalent Ab1 to inhibit cell growth. Both findings are consistent with the conclusion that Ab1 activity is dependent upon its interaction with the exposed surface epitope.
Bivalent Ab1 inhibits S-phase entry
To determine the point at which Ab1 inhibits cell proliferation, we evaluated the effects of Ab1 on cell-cycle progression using the CHO stable lines and fluorescence-activated cell sorter (FACS) analysis. Figure 5 depicts a representative analysis of 5 independent stable CD148 (CD148-CHO) and control (control-CHO) CHO transfectants. Shown in Figure 5A, bivalent Ab1 inhibited proliferation of the CD148-CHO cells, while it was inactive upon control CHO cells lacking CD148 expression. It is noteworthy that the growth profile of CD148-CHO cells was almost comparable with that of control-CHO cells in the absence of Ab1. Figure 5B demonstrates a representative FACS analysis of cell-cycle progression in those stable lines. Cell-cycle synchronization at G0/G1 phase was achieved by serum withdrawal for 72 hours, followed by serum replacement in the presence of either Ab1 or Ab1.Fab (67 nM). Shown in Figure 5B, bivalent Ab1 blocked G1/S-phase transition in CD148-CHO cells but not in control-CHO cells, while Ab1.Fab was inactive. Notably, and like the primary endothelial-cell results, Ab1 did not cause apoptosis with nuclear fragmentation. These results indicate that bivalent Ab1 blocks cell-cycle progression at G0/G1 phase.
Bivalent Ab1 blocks angiogenesis in a corneal pocket model
Finally, we explored whether locally administered bivalent Ab1 has antiangiogenic activity, using a well-defined in vivo angiogenesis system, the mouse cornea. Shown in Figure 6A, Ab1 immunoreactivity was detected by immunofluorescence microscopy on microvascular and arterial endothelial cells in various mouse tissues with a similar pattern observed in human tissues,6 including kidney (Figure 6Ai), brain (Figure 6Aii), heart (Figure 6Aiii) and corneal angiogenic vessels induced by FGF (Figure 6Aiv). A double-immunolabeling study for CD148 and VE-cadherin confirmed CD148 distribution to endothelial cells, and high magnification views showed CD148 accumulation at sites of endothelial cell-cell contacts where VE-cadherin localizes (Figure 6Av), correlating with the findings in cultured endothelial cells.6 Further, Ab1 bound a 220-kDa protein in crude detergent extracts from mouse kidney as it did in cultured human endothelial cells.6 The immunized CD148 ectodomain protein (Ec) and the Ab1 epitope peptide (pep) abolished the Ab1 immunoreactivity to mouse kidney tissue lysates, while an irrelevant cytoplasmic domain protein (Cy) did not. These findings demonstrate Ab1 recognition of CD148 on mouse endothelial cells.
Figure 6.

Ab1 labels CD148 on murine endothelium and inhibits corneal angiogenesis in mice. (Ai-v) Mouse cryostat tissue sections were immunostained with FITC-conjugated Ab1. Ab1 labels arterial and capillary endothelium (arrows) of various tissues, including kidney (i), cerebellar cortex (ii), endocardium (iii), and corneal angiogenic vessels (iv) following stimulation with a basic FGF–impregnated hydron pellet (ppt). Highly magnified image of renal artery exhibits endothelial localization of CD148, which overlaps VE-cadherin (arrowheads, v). Art indicates renal artery; glom, renal glomerulus; and peri, peritubular capillary. Images were taken using a Zeiss LSM 410 laser-scanning microscope (Carl Zeiss) with Zeiss 10×/0.3 numerical aperture (NA), 40×/1.3 NA, and 63×/1.3 NA objective lenses, and with Image Browser 5 LSM ver3.2 (Carl Zeiss). Figure panels were prepared with Adobe Photoshop 7.0 (Adobe Systems). Original magnifications: 100× (i), 400× (ii), 400× (iii), 400× (iv), and 1200× (v). (vi) Mouse kidney tissue lysates (300 μg) were subjected to immunoblotting using HRP-conjugated Ab1 preabsorbed with CD148 recombinant proteins (50 μg) (CD148ec indicates ectodomain; CD148cy, cytoplasmic domain) or epitope peptide (10 μg) (pep). (B) Hydron pellets impregnated with (i) vehicle (PBS), (ii) bFGF (3.0 pM), (iii) bFGF + control mouse IgG1 (74 pM), (iv-v) bFGF + Ab1 (74 pM), or (vi) bFGF + Ab1 Fab fragment (74 pM) were implanted and photographed at 5 days after implantation. (C) The graph displays means ± SEM (n = 8, excluding IgG1 controls [n = 6]) for the fractional areas vascularized, the vascular density within that region, and the vessel density as a fraction of the total corneal image area, as indicated in the figure.
To determine the antiangiogenic activity of Ab1 in vivo, we evaluated corneal angiogenic responses to implantation of inert pellets impregnated with basic FGF in combination with vehicle, control mouse IgG1, Ab1, or Ab1.Fab. Shown in Figure 6B, locally administered bivalent, but not monovalent, Ab1 markedly attenuated the angiogenic response to FGF in the cornea (iv-v), where endothelial cells express CD148 (Figure 6Aiv). The antiangiogenic effect was prominent, represented graphically by analysis of the corneal area vascularized, the density of vessels within the vascularized area, and the overall vascular density of the corneal image as indicated (Figure 6C). Antiangiogenesis activity was also detected in similar assays where Ab1 was injected intraperitoneally (data not shown).
Discussion
Interaction of monoclonal antibody Ab1 with CD148 has defined both biologic responses and mechanistic information about the process through which CD148 regulates cellular events. Bivalent Ab1 decreased proliferation in serum-stimulated endothelial cells and CD148-expressing CHO cells. Bivalent Ab1 arrested cell-cycle progression at G0/G1 phase in CHO stable lines and inhibited angiogenic vessel growth in mouse cornea in vivo. Previous work has implicated CD148 in negative regulation of cell proliferation. CD148 inhibits cell proliferation in various types of cells when it is overexpressed.10,11,35,36 Further, CD148 has been shown to be increased and activated in cells as they become confluent, suggesting its role to convey density-mediated growth arrest signals.5 These findings provide evidence that bivalent, but not monovalent, Ab1 acts to enhance CD148′s tyrosine phosphatase–mediated signals.
This model is further supported by additional evidence. First, Ab1 inhibits cell-cycle progression at G0/G1 phase at the cell-cycle point where p27 mediates cell-cycle arrest, consistent with previous work demonstrating CD148-mediated p27 up-regulation.10 Second, bivalent Ab1 prominently suppresses phosphorylation of Met and ERK1/2 kinases, substrates of CD148, as activated CD148 does.20-22 In addition, a CD148 immunocomplex PTP assay showed that Ab1 treatment increases CD148-associated PTP activity. Although CD148 was shown to reduce PLCγ1 phosphorylation21 when it is overexpressed, this activity was indistinguishable in response to Ab1 exposure (data not shown). This finding suggests that Ab1 may not fully transduce CD148 signals, explaining why serum starvation or reduction is required for Ab1 to completely arrest cell proliferation. Alternatively, very high levels of CD148 expression may be required to suppress PLCγ1 phosphorylation. Identification of a natural ligand for CD148 and further comparison of these agonists may be required to determine whether Ab1 fully mimics natural regulation of CD148 activity. It is noteworthy that the growth arrest imposed on the CHO-cell system by combined CD148 expression and Ab1 exposure persists for days in the presence of antibody, and is more effective when serum-starved cells are pre-exposed to Ab1 prior to replacement of serum. In addition, bivalent Ab1 exposure did not cause prominent down-regulation of cell-surface CD148 (data not shown). This timing effect suggests that Ab1-mediated CD148 activation provides a tonic, persistent, and dominant inhibitory effect upon cell-growth signals.
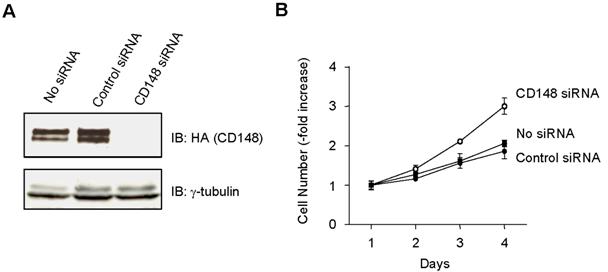
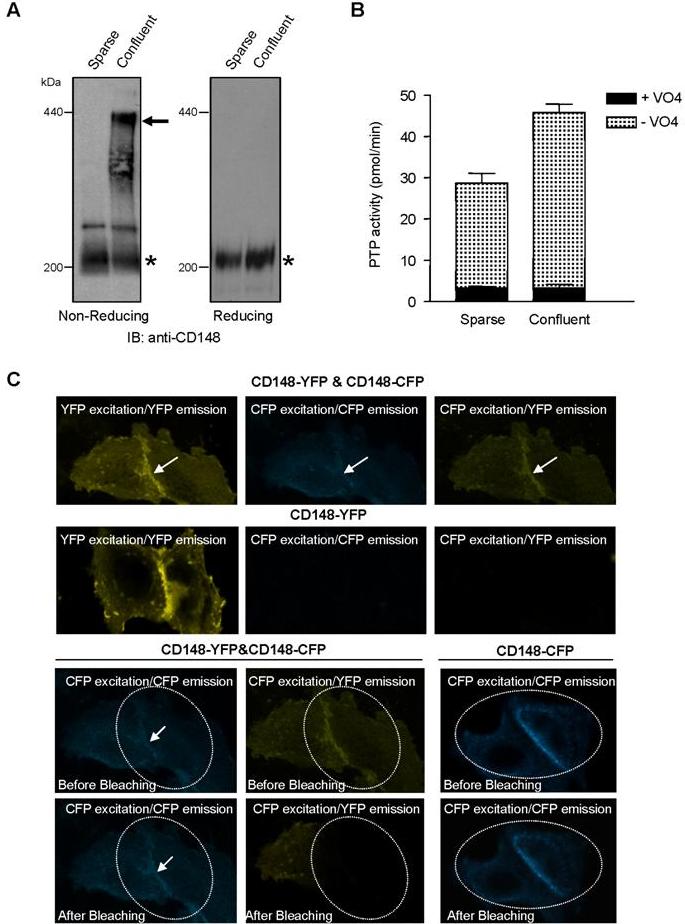
Our observations suggest that ectodomain-induced oligomerization is a potential mechanism of CD148 activation to evoke biologic responses. First, monovalent Ab1.Fab is inactive in imposing growth arrest in cultured cell assays and is remarkably less angiostatic in FGF-induced corneal angiogenesis assays, while it retains high affinity for the defined recognition epitope and abrogates responses to bivalent Ab1. Second, cotransfection of catalytically inactivate forms of CD148, either point mutant or truncation proteins, abrogates the capacity for bivalent Ab1 to inhibit proliferation in cells expressing wild-type CD148. This antagonistic effect of these dominant-negative proteins also argues for a primary role of CD148 to inhibit cell growth. Indeed, CD148 gene knockdown increased cell proliferation in CHO-CD148 cells (Figure S1, available on the Blood website; see the Supplemental Figures link at the top of the online article). This model is also supported by an additional experiment demonstrating that EGF-induced dimerization of the epidermal growth factor receptor (EGFR)/CD148 chimera inhibits 32D-cell proliferation (Figure S2). Finally, supplemental data demonstrate potential CD148 oligomerization in confluent (resting) endothelial cells (Figure S3).
A model for receptor PTP inhibition by dimerization has been advanced by the studies of RPTPα and CD45, RTPs with 2 catalytic domains. Access to the active site was occluded by a helix-turn-helix loop from adjacent partners within homodimeric dyad pairs of the RPTPα D1-catalytic domain.25 Consistent with RPTPα crystal structures, forced dimerization of disulphide-bonded cysteine-containing mutant RPTPα inhibited its catalytic activity in transfected cells.26 Similarly, the catalytic function of CD45 was inhibited by dimerization forced by EGF binding to chimeric EGFR/CD45 fusion proteins.27 On the other hand, more recent studies have shown that RPTPα constitutively forms a dimer in the membrane, and its activity is defined by rotational coupling of the RPTPα dimers.37 Although RPTPs with 2 catalytic domains have been well studied, very little is known about oligomerization of receptor PTPs that have a single catalytic domain, such as CD148. Our findings demonstrate that activity of CD148 is potentially regulated by ectodomain oligomerization. Consistent with our results, a recent study has shown that Sap-1 RPTP, a member of CD148 RPTP family, potentially forms dimers through its ectodomains.38 It is noteworthy that ligation of CD148 with a different CD148 monoclonal antibody induced proliferation of anti-CD3–activated T cells,39 whereas transient transfection of intact CD148 negatively regulates T-cell activation.40 This suggests that CD148 ectodomain antibodies may act as inhibitors or activators, and that antibody-mediated rotational constraints may positively or negatively regulate CD148 activity. Also, it is possible that serum depletion may cause structural changes of CD148 and increase accessibility of Ab1 to CD148, resulting in prominent growth inhibition. Further investigation is required to determine the mechanism of Ab1-mediated CD148 activation.
As a rigorous test of whether Ab1 growth inhibition resides in its capacity to react to ectodomain sequences of CD148, we determined the epitope to which Ab1 binds. Recently, 5 polymorphisms have been identified in extracellular sequences of human PTPRJ (CD148), resulting in amino-acid changes.14 Further, it has been shown that PTPRJ genotypes homozygous for the Gln276Pro and Arg326Gln, and the Asp872Glu allele, are more frequent in thyroid carcinoma patients than in healthy individuals.41 Of interest, the Ab1 epitope includes this Gln276, suggesting the importance of this region in CD148 function. It is noteworthy that while the predicted mouse sequence from this position diverges within this octomer peptide (DPSLTEIL mu vs QSRDTEVL hu),42 the human sequence–derived peptide antagonizes Ab1 recognition of the murine 220-kDa CD148 derived from kidney tissue on immunoblots. This finding suggests that alternatively spliced forms distinct from published cDNA sequences may be expressed on mouse endothelium.
Finally, we tested whether Ab1 blocks angiogenic vessel growth in vivo. Shown in Figure 6, the Ab1 effectively inhibited angiogenic vessel growth in mouse cornea. The high level of expression of CD148 in endothelial cells at a wide range of microvascular sites and its accumulation at points of interendothelial contact suggest it may function as an angiostatic regulator of endothelial proliferation, particularly if cell-cell interactions promote its oligomerization. It is noteworthy that bivalent Ab1 has no activity to disrupt contact inhibition in endothelial cells (data not shown). In this context, Ab1 may act as an antiangiogenesis agent to inhibit angiogenesis without side effects on endothelial permeability. Further evaluation of whether systemic injection of Ab1 effectively inhibits pathologic angiogenesis, such as tumor angiogenesis, without side effects, is required to support use of Ab1 in antiangiogenic therapy.
In conclusion, we demonstrate for the first time that catalytic activity associated with the CD148 cytoplasmic domain is increased by bivalent antibody against the ectodomain sequence, with effects demonstrable on specific substrates. The findings demonstrate that ectodomain oligomerization is a potential regulatory mechanism of CD148 activity. Finally, the present study implicates CD148 as a molecular target for antiangiogenesis therapy. Efforts to define natural ligands of CD148 or to generate synthetic agonists that oligomerize and activate CD148 offer a novel strategy for antiangiogenesis therapy.
Supplementary Material
Acknowledgments
The authors thank Nick Tonks for the human DEP-1 cDNA plasmid, David Smith and Ute Priglinger for preparation of plasmids and stable cell lines, Douglas Cerretti and William Fanslow (Immunex) for support, and Ray Harris for critical reading and input to the article.
Prepublished online as Blood First Edition Paper, April 4, 2006; DOI 10.1182/blood-2005-10-4296.
Supported by the National Institutes of Health grants DK38517, DK52483, and CA68485; the T. J. Martell Foundation; the National Kidney Foundation; grant JDF no. 2-2000-147 from the Juvenile Diabetes Research Foundation; Immunex Corporation; and the Molecular Recognition, DNA Sequencing, and Cell Imaging Shared Resources of the Vanderbilt-Ingram Cancer Center.
T.T. designed and performed research, analyzed data, and wrote the article; K.T., N.T., and H.L. performed research. R.L.M. prepared and provided vital new reagents; and T.O.D. supported research and reviewed data.
T.T., R.L.M., and T.O.D. hold a patent related to the work that is described in the present study.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6: 389-395. [DOI] [PubMed] [Google Scholar]
- 2.Daniel TO, Abrahamson D. Endothelial signal integration in vascular assembly [in process citation]. Annu Rev Physiol. 2000;62: 649-671. [DOI] [PubMed] [Google Scholar]
- 3.Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9: 685-693. [DOI] [PubMed] [Google Scholar]
- 4.Desai CJ, Sun Q, Zinn K. Tyrosine phosphorylation and axon guidance: of mice and flies. Curr Opin Neurobiol. 1997;7: 70-74. [DOI] [PubMed] [Google Scholar]
- 5.Ostman A, Yang Q, Tonks NK. Expression of DEP-1, a receptor-like protein-tyrosine-phosphatase, is enhanced with increasing cell density. Proc Natl Acad Sci U S A. 1994;91: 9680-9684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takahashi T, Takahashi K, Mernaugh R, et al. Endothelial localization of receptor tyrosine phosphatase, ECRTP/DEP-1, in developing and mature renal vasculature. J Am Soc Nephrol. 1999; 10: 2135-2145. [DOI] [PubMed] [Google Scholar]
- 7.Borges LG, Seifert RA, Grant FJ, et al. Cloning and characterization of rat density-enhanced phosphatase-1, a protein tyrosine phosphatase expressed by vascular cells. Circ Res. 1996;79: 570-580. [DOI] [PubMed] [Google Scholar]
- 8.de la Fuente-Garcia MA, Nicolas JM, Freed JH, et al. CD148 is a membrane protein tyrosine phosphatase present in all hematopoietic lineages and is involved in signal transduction on lymphocytes. Blood. 1998;91: 2800-2809. [PubMed] [Google Scholar]
- 9.Autschbach F, Palou E, Mechtersheimer G, et al. Expression of the membrane protein tyrosine phosphatase CD148 in human tissues. Tissue Antigens. 1999;54: 485-498. [DOI] [PubMed] [Google Scholar]
- 10.Trapasso F, Iuliano R, Boccia A, et al. Rat protein tyrosine phosphatase eta suppresses the neoplastic phenotype of retrovirally transformed thyroid cells through the stabilization of p27(Kip1). Mol Cell Biol. 2000;20: 9236-9246. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Keane MM, Lowrey GA, Ettenberg SA, Dayton MA, Lipkowitz S. The protein tyrosine phosphatase DEP-1 is induced during differentiation and inhibits growth of breast cancer cells. Cancer Res. 1996;56: 4236-4243. [PubMed] [Google Scholar]
- 12.Zhang L, Martelli ML, Battaglia C, et al. Thyroid cell transformation inhibits the expression of a novel rat protein tyrosine phosphatase. Exp Cell Res. 1997;235: 62-70. [DOI] [PubMed] [Google Scholar]
- 13.Iuliano R, Trapasso F, Le Pera I, et al. An adenovirus carrying the rat protein tyrosine phosphatase eta suppresses the growth of human thyroid carcinoma cell lines in vitro and in vivo. Cancer Res. 2003;63: 882-886. [PubMed] [Google Scholar]
- 14.Ruivenkamp CA, van Wezel T, Zanon C, et al. Ptprj is a candidate for the mouse colon-cancer susceptibility locus Scc1 and is frequently deleted in human cancers. Nat Genet. 2002;31: 295-300. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi T, Takahashi K, St John PL, et al. A mutant receptor tyrosine phosphatase, CD148, causes defects in vascular development. Mol Cell Biol. 2003;23: 1817-1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kovalenko M, Denner K, Sandstrom J, et al. Site-selective dephosphorylation of the platelet-derived growth factor beta-receptor by the receptor-like protein-tyrosine phosphatase DEP-1. J Biol Chem. 2000;275: 16219-16226. [DOI] [PubMed] [Google Scholar]
- 17.Persson C, Engstrom U, Mowbray SL, Ostman A. Primary sequence determinants responsible for site-selective dephosphorylation of the PDGF beta-receptor by the receptor-like protein tyrosine phosphatase DEP-1. FEBS Lett. 2002;517: 27-31. [DOI] [PubMed] [Google Scholar]
- 18.Jandt E, Denner K, Kovalenko M, Ostman A, Bohmer FD. The protein-tyrosine phosphatase DEP-1 modulates growth factor-stimulated cell migration and cell-matrix adhesion. Oncogene. 2003;22: 4175-4185. [DOI] [PubMed] [Google Scholar]
- 19.Lampugnani MG, Zanetti A, Corada M, et al. Contact inhibition of VEGF-induced proliferation requires vascular endothelial cadherin, {beta}-catenin, and the phosphatase DEP-1/CD148. J Cell Biol. 2003;161: 793-804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palka HL, Park M, Tonks NK. Hepatocyte growth factor receptor tyrosine kinase met is a substrate of the receptor protein-tyrosine phosphatase DEP-1. J Biol Chem. 2003;278: 5728-5735. [DOI] [PubMed] [Google Scholar]
- 21.Baker JE, Majeti R, Tangye SG, Weiss A. Protein tyrosine phosphatase cd148-mediated inhibition of t-cell receptor signal transduction is associated with reduced lat and phospholipase cgamma1 phosphorylation. Mol Cell Biol. 2001;21: 2393 -2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Massa A, Barbieri F, Aiello C, et al. The expression of the phosphotyrosine phosphatase DEP-1/PTPeta dictates the responsivity of glioma cells to somatostatin inhibition of cell proliferation. J Biol Chem. 2004;279: 29004-29012. [DOI] [PubMed] [Google Scholar]
- 23.Lemmon MA, Schlessinger J. Transmembrane signaling by receptor oligomerization. Methods Mol Biol. 1998;84: 49-71. [DOI] [PubMed] [Google Scholar]
- 24.Weiss A, Schlessinger J. Switching signals on or off by receptor dimerization. Cell. 1998;94: 277-280. [DOI] [PubMed] [Google Scholar]
- 25.Bilwes AM, den Hertog J, Hunter T, Noel JP. Structural basis for inhibition of receptor protein-tyrosine phosphatase-alpha by dimerization. Nature. 1996;382: 555-559. [DOI] [PubMed] [Google Scholar]
- 26.Jiang G, den Hertog J, Su J, et al. Dimerization inhibits the activity of receptor-like protein-tyrosine phosphatase-alpha. Nature. 1999;401: 606-610. [DOI] [PubMed] [Google Scholar]
- 27.Majeti R, Bilwes AM, Noel JP, Hunter T, Weiss A. Dimerization-induced inhibition of receptor protein tyrosine phosphatase function through an inhibitory wedge. Science. 1998;279: 88-91. [DOI] [PubMed] [Google Scholar]
- 28.Majeti R, Xu Z, Parslow TG, et al. An inactivating point mutation in the inhibitory wedge of CD45 causes lymphoproliferation and autoimmunity. Cell. 2000;103: 1059-1070. [DOI] [PubMed] [Google Scholar]
- 29.Jiang G, den Hertog J, Hunter T. Receptor-like protein tyrosine phosphatase alpha homodimerizes on the cell surface. Mol Cell Biol. 2000;20: 5917-5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nam HJ, Poy F, Krueger NX, Saito H, Frederick CA. Crystal structure of the tandem phosphatase domains of RPTP LAR. Cell. 1999;97: 449-457. [DOI] [PubMed] [Google Scholar]
- 31.Hoffmann KM, Tonks NK, Barford D. The crystal structure of domain 1 of receptor protein-tyrosine phosphatase mu. J Biol Chem. 1997;272: 27505-27508. [DOI] [PubMed] [Google Scholar]
- 32.Beltran PJ, Bixby JL. Receptor protein tyrosine phosphatases as mediators of cellular adhesion. Front Biosci. 2003;8: d87-d99. [DOI] [PubMed] [Google Scholar]
- 33.Huynh-Do U, Stein E, Lane AA, et al. Surface densities of ephrin-B1 determine EphB1-coupled activation of cell attachment through alphavbeta3 and alpha5beta1 integrins. EMBO J. 1999;18: 2165-2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huynh-Do U, Vindis C, Liu H, et al. Ephrin-B1 transduces signals to activate integrin-mediated migration, attachment and angiogenesis. J Cell Sci. 2002;115: 3073-3081. [DOI] [PubMed] [Google Scholar]
- 35.Kellie S, Craggs G, Bird IN, Jones GE. The tyrosine phosphatase DEP-1 induces cytoskeletal rearrangements, aberrant cell-substratum interactions and a reduction in cell proliferation. J Cell Sci. 2004;117: 609-618. [DOI] [PubMed] [Google Scholar]
- 36.Trapasso F, Yendamuri S, Dumon KR, et al. Restoration of receptor-type protein tyrosine phosphatase eta function inhibits human pancreatic carcinoma cell growth in vitro and in vivo. Carcinogenesis. 2004;25: 2107-2114. [DOI] [PubMed] [Google Scholar]
- 37.van der WT, Blanchetot C, Overvoorde J, den Hertog J. Redox-regulated rotational coupling of receptor protein-tyrosine phosphatase alpha dimers. J Biol Chem. 2003;278: 13968-13974. [DOI] [PubMed] [Google Scholar]
- 38.Walchli S, Espanel X, Van Huijsduijnen RH. Sap-1/PTPRH activity is regulated by reversible dimerization. Biochem Biophys Res Commun. 2005;331: 497-502. [DOI] [PubMed] [Google Scholar]
- 39.Tangye SG, Phillips JH, Lanier LL, de Vries JE, Aversa G. CD148: a receptor-type protein tyrosine phosphatase involved in the regulation of human T cell activation. J Immunol. 1998;161: 3249-3255. [PubMed] [Google Scholar]
- 40.Tangye SG, Wu J, Aversa G, et al. Negative regulation of human T cell activation by the receptor-type protein tyrosine phosphatase CD148. J Immunol. 1998;161: 3803-3807. [PubMed] [Google Scholar]
- 41.Iuliano R, Le Pera I, Cristofaro C, et al. The tyrosine phosphatase PTPRJ/DEP-1 genotype affects thyroid carcinogenesis. Oncogene. 2004;23: 8432-8438. [DOI] [PubMed] [Google Scholar]
- 42.Kuramochi S, Matsuda S, Matsuda Y, et al. Molecular cloning and characterization of Byp, a murine receptor-type tyrosine phosphatase similar to human DEP-1. FEBS Lett. 1996;378: 7-14. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}