

Abstract
Recent studies revealed that the lymphotoxin/lymphotoxin beta receptor (LT)/LTβR system activates the noncanonical nuclear factor-κB (NF-κB) signaling pathway involving I kappa B kinase 1/I kappa B kinase α (IKK1/IKKα) and NF-κB-inducing kinase (NIK) to direct processing of the nfκb2 protein p100 to yield RelB:p52 complexes. Despite the biochemical evidence, LT-, RelB-, p52-deficient mice show discrepant phenotypes. We now demonstrate that p105/p50 also constitutes an important pathway for LTβR signaling. Our studies revealed that mice deficient in either p50 or p52 have defects in the formation of inguinal lymph nodes (LNs), but that the complete defect in lymph node formation and splenic microarchitecture seen in LT-deficient mice is recapitulated only in mice deficient in both p50 and p52. Biochemically, we find not only that both p50- and p52-containing NF-κB activities are induced by LTβR signaling, but that the induction of NF-κB-containing complexes by LTβR engagement is perturbed in single knockouts. Importantly, the LTβR can additionally activate the less well-characterized p52:RelA and p50:RelB pathways, which play pivotal roles in vivo for the development and organization of lymphoid structures. Our genetic, cellular, and molecular evidence points toward a model of LT-mediated NF-κB regulation in which p105/p50 and p100/p52 have distinct and coordinating molecular specificities but differ in the upstream signaling pathways that regulate them.
Introduction
One of the primary hallmarks of the immune system is the ability to mount rapid and highly active responses to specific pathogens. Organization is a critical feature for immunity. Lymph nodes (LNs) are situated at strategic areas throughout the body to monitor the fluids that drain and cells that migrate there for pathologic changes. The development of LNs can be traced back to embryonic day 12.5 (E12.5) in the murine system, with the identification of lymphoid tissue inducer cells (LTICs) in the LN anlagen.1,2 The hematopoietically derived LTICs are CD4+CD3-IL-7Rα+ and express membrane-bound lymphotoxin (LTα1β2). The LTICs then provide this critical LT signal via the lymphotoxin beta receptor (LTβR) on the mesenchymal cells of the LN anlagen to orchestrate the complex steps of LN development.2-4 LTα- and LTβR-deficient mice thus fail to receive this signal and develop sans LNs.5,6 Beyond its early role in LN organogenesis, LT signaling is required for a number of processes pertaining to the organization of secondary lymphoid tissues in the adult mouse.3,7 Some of the critical elements controlled by LT include expression of homeostatic chemokines, follicular dendritic cells (FDCs), and marginal zone development.8,9 The homeostatic lymphoid chemokines SLC/CCL21, CXCL13/BLC, and their receptors have also been demonstrated to play a role in LN organogenesis.10-12 Additionally, CCL21 and CXCL13 are expressed by different subsets of stromal cells in the T- and B-cell zones, respectively, of secondary lymphoid organs. The differential localization of these 2 chemokines parallels and supports T- and B-cell compartmentalization within lymphoid tissues.13 Furthermore, primary and secondary immune responses for T and B lymphocytes as well as immunoglobulin switching and germinal-center formation are impaired in LT-deficient mice.14
Nuclear factor-κB (NF-κB)/Rel proteins are a family of pleiotropic transcription factors that regulates the expression of genes in numerous basic biological processes such as development, proliferation, survival, and differentiation.15 The impacts of NF-κB are pervasive as its effects extend across multiple systems in immunology, oncology, neurology, angiogenesis, and skeletal development. RelA (p65), RelB, c-Rel, NF-κB1 (p50, which is processed from p105), and NF-κB2 (p52, which is processed from p100) share the Rel homology domain (RHD) and make up the NF-κB family in mammals.16,17 NF-κB/Rel proteins can function as either homo- or heterodimers with many possible complexes. NF-κB activity is in part controlled by its associations with the inhibitory IκB family of proteins which include the p105 and p100 precursors. IκB activity in turn is regulated by the IκB kinase (IKK) complex, which through phosphorylation and subsequent ubiquitination targets the IκB family of proteins for degradation/processing.18 The liberated NF-κB complexes rapidly accumulate in the nucleus and are capable of binding cognate κB-sites and direct transcription of NF-κB target genes. Posttranslational modification by phosphorylation, nuclear-cytoplasmic shuttling, and association with histone deacetylases are some of the additional regulatory steps in NF-κB activation.15,18
Recently, another major evolutionarily conserved NF-κB pathway was identified in mammals. Studies of LT/LTβR signaling were instrumental in defining this so-called noncanonical pathway through the kinase activity of IKK1/IKKα.19 Secondary lymphoid organ deficiencies in mice harboring kinase-inactive IKK1 were correlated with an absence of LTβR-inducible proteolytic processing of Nfkb2-encoded p100 to p52.19,20 p52 was subsequently shown to function as a dimer with RelB, and the NF-κB-inducing kinase (NIK) was implicated as an upstream regulator of IKK1/IKKα activity and a required LT pathway signal transducer.21,22 Thus, our current understanding is that the noncanonical NF-κB pathway uses the NIK/IKK1 axis to induce RelB:p52 heterodimers that are hypothesized to regulate the expression of lymphoid chemokines, such as stromal cell-derived factor 1α (SDF-1α)/CXC12, B lymphoblastoid cell (BLC)/CXCL13, secondary lymphoid tissue chemokine (SLC)/CCL21, and EBI1 ligand chemokine (ELC)/CCL19. While inflammatory stimuli such as tumor necrosis factor (TNF), interleukin-1 (IL-1), and lipopolysaccharide (LPS) are potent inducers of the “canonical” pathway, in which the regulation of IκB-α, -β, and -ε proteins is sufficient to model its dynamic functionality,23 ligands such as LTβ, CD40L, and B-cell-activating factor of the TNF family (BAFF) have been intimately linked with the noncanonical pathway,20 whose mechanistic regulation and correlation with physiologic function is not yet as fully understood.
Previous work implied a critical role for p100/p52 in LT signaling and secondary lymphoid organ development; however, p100/p52-deficient mice were reported to exhibit only the limited phenotype characteristic of LT- or LTβR-deficient mice.4 These mice seem to have impaired development of LNs and the smaller size of inguinal LNs may be associated with stromal defect.24 In contrast, mice doubly deficient in RelA and TNFRI lacked secondary lymphoid organs, implicating the role of the classical RelA:p50 heterodimer in physiologic LT signaling.25 The role of p50, however, in the LTβR pathway has remained unclear because p50-deficient mice did not present with any major lymphoid organization phenotypes typically found in LT-deficient mice.4,26 Since RelA and RelB are known to form heterodimers with p50 and p52, respectively, we considered the possibility that p50 and p52 may provide obligate and parallel functions within the NF-κB signaling network in response to LTβR stimulation. To elucidate the relationship between p50- and p52-mediated signaling in lymph node organogenesis, we have examined phenotypes in p50 and p52 doubly mutant mice and undertook a biochemical characterization of p50 and p52 in LT signaling. In particular, we find significant deficits in lymph node formation, recapitulating the major phenotype of LT-deficient mice, and partial defects in both single mutants. Furthermore, we show that both p50 and p52 are activated by LTβR signaling and that their regulation is remarkably interdependent. Our study highlights the previously unappreciated role of p50 in the LT pathway and reveals critical, but asymmetric crosstalk between p50 and p52 in LTβR-mediated development and organization of secondary lymphoid tissues.
Materials and methods
Mice and LN detection
Nfkb1-/- and Nfkb2-/- mice were intercrossed as previously described.27 Nfkb1-/- mice were also purchased from Jackson Laboratories (Bar Harbor, ME). Animal care and use were in accordance with institutional and National Institutes of Health guidelines. Mice received intraperitoneal injections of 200 μL 0.33% Chicago Sky Blue and were scored for the presence or absence of LNs 7 to 10 days later as previously described.11
Cells and reagents
Mouse embryo fibroblasts (MEFs) were derived from embryonic day (E) 12.5-E13.5 embryos, grown in Dulbecco modified Eagle medium (DMEM) + 10% bovine calf serum, and used for experiments up to passage 5 or following immortalization by repeated passage according to the 3T3 procedure. LTβR-deficient fibroblasts were derived from splenic or lung isolations as previously described.28 Antibodies used in Western blots and electrophoretic mobility shift assay (EMSA) “supershifts” were purchased from Santa Cruz Biotechnology (Santa Cruz, CA): anti-p50/p105 (sc-114); anti-p52/p100 (sc-298); anti-p65 (sc-109 and sc-372); anti-cRel (sc-70); and anti-RelB (sc-226).
Gene expression analysis
Immunoblot and EMSA were performed as described.29 Quantitative real-time polymerase chain reaction (PCR) was performed with previously published primers20 and Applied Biosystems GeneAmp 5700 Sequence Detection System using Applied Biosystems' Sybr green kit (Applied Biosystems Foster City, CA). GAPDH (primer sequences available upon request) was used as normalization standard with 106 transcript units being defined as equal to the GAPDH transcript level, and cycle number differences were converted assuming 2-fold amplification with every cycle.
Flow cytometry and immunohistochemistry
FITC-conjugated B220, biotin-conjugated vascular cell adhesion molecule 1 (VCAM-1), intercellular adhesion molecule 1 (ICAM-1), and Thy1.1 antibodies were from Pharmingen (San Diego, CA). PE-conjugated streptavidin was obtained from Immunotech (Marseille, France). Cells were stained and analyzed by flow cytometry on a FACScan or FACScalibur using CellQuest (BD Biosciences, Palo Alto, CA) or FlowJo (Tree Star, Ashland, OR) softwares. Frozen sections of spleen were prepared, fixed, and stained as previously described.28 Biotin-conjugated CCL21 (R&D Systems, Minneapolis, MN), Thy1.2 (Pharmingen) and FITC-conjugated B220 (Pharmingen) antibodies were used. Images were acquired using an Olympus BX41 microscope (Olympus America, Melville, NY) with a 20×/0.50 NA objective and a 10×/0.22 NA eyepiece, for a total magnification of 200×, and an Axiocam camera and AxioVision 3.0 software (Carl Zeiss Microimaging, Thornwood, NY).
Bone marrow reconstitution
Mice were lethally irradiated with 900 to 1000 rads and adoptively transferred intravenously with 2 to 3 × 106 bone marrow cells. Bactrim was added to the drinking water for 2 weeks following irradiation. Spleen sections from the mice were analyzed 2 to 3 months following transfer.
Results
LTβR signaling activates specific NF-κB complexes
Studies of LTβR signaling were instrumental in defining the noncanonical NF-κB activation pathway. Its hallmark is IKK1/IKKα kinase activity-dependent induction of proteolytic processing of Nfkb2-encoded p100 to p52.19 However, p52-deficient mice were reported to demonstrate impaired stromal structures inside lymph nodes that prevent its full development.24 In contrast, LT/LTβR-deficient mice showed no LNs, raising the possibility that other NF-κB units may be critical in LTβR signaling. To determine which other NF-κB family members are also downstream of the LTβR, we examined the composition of NF-κB complexes activated in response to LTβR signaling. Wild-type MEFs were stimulated with an agonistic anti-LTβR antibody in a time course that elicited multiple strong NF-κB DNA-binding activities, which peaked by 12 to 24 hours (Figure 1A). Supershift assays with antibodies against specific NF-κB subunits revealed these to be primarily RelA:p50, RelB:p52, and p50:p50 dimers, with similar results seen in 24-hour stimulated extracts (Figure 1B). The abundance of p50-containing dimers was somewhat surprising, as most reports had focused on RelB, p52, and to a lesser extent RelA as mediators of the LTβR signaling pathway.20,30,31 A previous report did note the induction of p50-containing dimers following LT stimulation.31 However, the importance or functional significance of p50 to the LTβR NF-κB pathway has not been well defined. The prominence of p50-containing dimers by pathways emanating from the LTβR prompted us to further examine their physiologic role in LT signaling.
Figure 1.
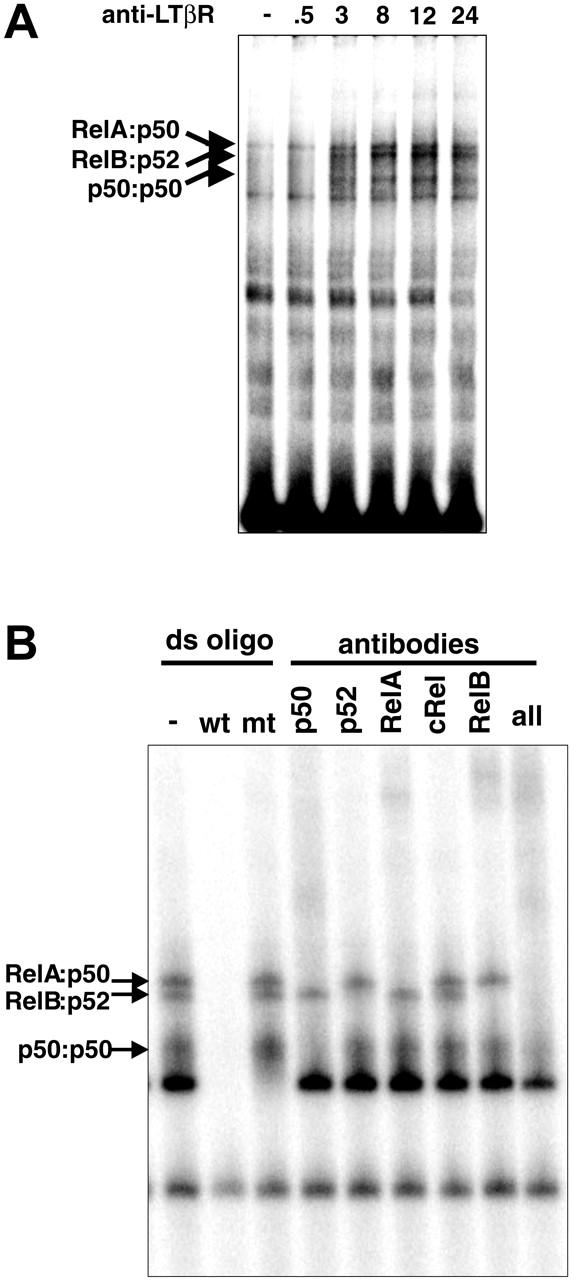
LTβR signaling activates multiple NF-κB pathways. (A) LTβR activates distinct NF-κB complexes. MEFs from wild-type mice were stimulated with agonistic anti-LTβR antibody for the indicated times (hours) and subjected to a NF-κB EMSA. (B) LTβR signaling activates multiple NF-κB complexes. Extracts from 24-hour-stimulated WT MEFs were subject to supershift analysis with the indicated NF-κB antibodies.
p50 and p52 are required for the development of lymph nodes
One of the major defects in LT/LTβR-deficient mice is the failure to develop secondary lymphoid tissues such as LNs and Peyer patches (PPs). The deficiency in lymphoid organogenesis in either p50 (Nfkb1-/-) or p52 (Nfkb2-/-) mice is rather limited.4 Moreover, p52- but not p50-deficient mice were impaired in PP formation.30 However, analysis of mice deficient in p50 unexpectedly revealed a discriminating loss of inguinal LNs with all other nodes present at wild-type frequencies (Table 1). Similarly, p52-deficient mice exhibited a selective but incomplete deficiency in the formation of inguinal LNs (Table 1). In addition, complete Freund adjuvant (CFA) immunization did not recover inguinal LNs in the Nfkb2-/- mice, indicating a failure in LN genesis rather than maturation (data not shown). Our findings implicating p50 activation along with the current dogma of p100 processing to p52 in LT signaling led us to a new hypothesis that p50 and p52 may have their distinct roles, but also cooperate in the formation of lymph nodes. To test this idea, we intercrossed Nfkb1-/- mice with Nfkb2-/- mice to generate Nfkb1-/-Nfkb2+/-, Nfkb1+/-Nfkb2-/-, and Nfkb1-/-Nfkb2-/- mice. We analyzed LN development in the p50, p52 doubly mutant mice. Wild-type and Nfkb1+/- Nfkb2+/- mice developed a full set of LNs (Table 1). Nfkb1-/- Nfkb2+/- mice, in which 3 of the 4 combined p50 and p52 alleles are disrupted, revealed an additional loss of other LN types. Nfkb1+/-Nfkb2-/- mice displayed a dramatic loss in all peripheral LNs examined with the development of occasional peripheral LNs (Table 1). These mice also demonstrated a defect in mucosal LNs with development of a single rather than a chain of mesenteric LNs (Table 1). Overall, the LN phenotype of Nfkb1+/-Nfkb2-/- mice closely mirrors that of LTβ-deficient mice, further supporting the notion that p50 and p52 cooperate in transducing membrane LT signals in LN genesis. Mice in which 3 of the 4 p50 and p52 alleles were disrupted still retained mesenteric LNs. To determine if the single remaining p50 or p52 allele was responsible for the genesis of mesenteric LNs in Nfkb1+/-Nfkb2-/- and Nfkb1-/-Nfkb2+/- mice, we analyzed Nfkb1-/-Nfkb2-/- mice for development of the outstanding mesenteric LNs. Impressively, Nfkb1-/-Nfkb2-/- mice exhibited a complete deficiency in LN formation (Table 1). These data, therefore, confirm our hypothesis that p50 and p52 cooperate in and together are essential for the development of LNs.
Table 1.
Cooperation between p50 and p52 is essential for the development of lymph nodes
| Genotype | No. | Inguinal | Brachial | Axillary | Facial | Cervical | Periaortic | Mesenteric |
|---|---|---|---|---|---|---|---|---|
| WT | 6 | 12/12 | 12/12 | 12/12 | 12/12 | 12/12 | 18/18 | 18/18 |
| Nfkb1+/–Nfkb2+/– | 6 | 12/12 | 12/12 | 12/12 | 12/12 | 12/12 | 18/18 | 18/18 |
| Nfkb1–/– | 10 | 4/20 | 20/20 | 20/20 | 20/20 | 20/20 | 29/30 | 30/30 |
| Nfkb2–/– | 9 | 4/18 | 18/18 | 18/18 | 18/18 | 18/18 | 26/27 | 27/27 |
| Nfkb1–/–Nfkb2+/– | 5 | 0/10 | 1/10 | 8/10 | 8/10 | 4/10 | 14/15 | 15/15 |
| Nfkb1+/–Nfkb2–/– | 9 | 0/18 | 0/18 | 0/18 | 1/18 | 0/18 | 1/27 | 16/27 |
| Nfkb1–/–Nfkb2–/– | 6 | 0/12 | 0/12 | 0/12 | 0/12 | 0/12 | 0/18 | 0/18 |
Denominators denote the normal number of lymph nodes of each type counted and found in a wild-type mouse.
Distinct roles of p50 and p52 in LTβR-mediated NF-κB pathway
A previous study has suggested the complicated role of p50 and p52 in the development of PP.30 The development of most or all LNs in Nfkb1-/- and Nfkb2-/- mice prompted us to examine the cellular and molecular mechanisms for LTβR-induced NF-κB activity in the absence of either p50 or p52. The formation of LNs in Nfkb1-/- and Nfkb2-/- mice suggested that sufficient LTβR signals were transduced in the absence of p50 or p52. To test this, p50- or p52-deficient MEFs were stimulated with agonistic anti-LTβR and subjected to an NF-κB EMSA. Significant NF-κB DNA binding activity was detected after LTβR ligation in both Nfkb1-/- and Nfkb2-/- MEFs, indicating that neither p50 nor p52 is absolutely required for LTβR-induced NF-κB activity, though the kinetics of activation were aberrant in the knockouts (Figure 2A). We examined the composition of NF-κB complexes activated by the LTβR in Nfkb1-/- and Nfkb2-/- cells by probing κB-site-protein complexes with antibodies whose specificity is established through the use of gene-knockout cells (Figure 2B). These combined biochemical and genetic analyses demonstrate that the LTβR activates both RelA- and RelB-containing dimers in the absence of p50 or p52. In the p50-deficient MEFs, p52:RelB complexes continued to form while p52:RelA replaced p50:RelA complexes. Similarly, stimulation of p52-deficient MEFs resulted in the activation of p50:p50 homodimers and p50:RelA heterodimers as in wild type (WT) but with p50:RelB dimers substituting for p52:RelB complexes (Figure 2B). These results have revealed the underappreciated roles of p50:RelB and p52:RelA complexes in transducing LTβR signals, and further demonstrates that they are sufficient for LTβR-mediated signal transduction of the development of most LNs as witnessed in both of the singly deficient mice.
Figure 2.
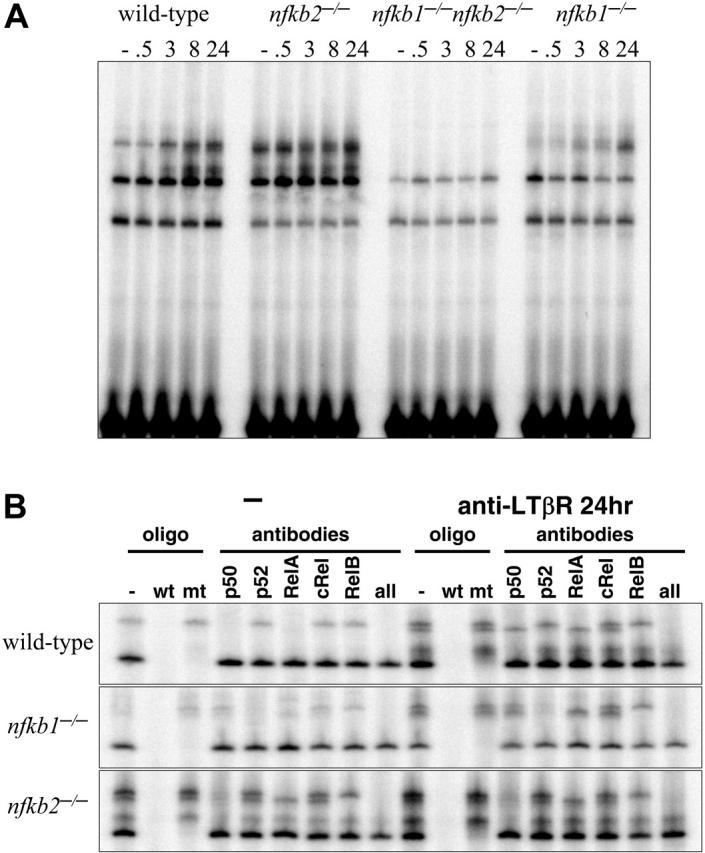
p50/105 and p52/p100 are required for LTβR-induced NF-κB activities. (A) LTβR-induced NF-κB activation is completely abolished in p50 and p52 double mutants and perturbed in p50 and p52 single-mutant MEFs. MEFs of the indicated genotype were stimulated with agonistic anti-LTβR antibody for the indicated times and subject to a NF-κB EMSA. Three specific activities are indicated by arrows as in Figure 1A. (B) Identification of p50- and p52-containing NF-κB activities. Extracts from wild-type, Nfkb1-/-, and Nfkb2-/- MEFs, untreated or stimulated for 24 hours with agonistic LTβR antibody, were subject to supershift analyses with the indicated NF-κB antibodies and competitor oligonucleotides.
Both p50 and p52 are required for LTβR-mediated NF-κB activation
Earlier, we had shown that p50 in conjunction with p52 are essential for LN development. Here we tested the idea that p50 and p52 together are essential for LTβR-induced NF-κB activation. To determine if the residual NF-κB activation in response to LTβR stimulation in p50- and p52-deficient MEFs was dependent on the remaining p50 or p52 genes, we analyzed p50 and p52 doubly deficient MEFs. Impressively, the agonist LTβR antibody failed to stimulate any detectable NF-κB DNA binding activity in Nfkb1-/- Nfkb2-/- fibroblasts (Figure 2A). Taken together, these results demonstrate that expression of both p50 and p52 is required for NF-κB activity elicited downstream of the LTβR. These findings further support our hypothesis that the LTβR signaling pathway uses both p50 and p52 to transduce signals essential for the development of all LNs.
p50 and p52 are essential for LTβR-induced adhesion molecules
LT signals provide essential molecular cues that regulate the development and organization of secondary lymphoid tissues. Adhesion molecules such as ICAM-1 and VCAM-1 are highly expressed in clusters that correspond to LN and PP anlagen in the developing embryo.1 These adhesion molecule signals are LT dependent because LT-deficient embryos fail to develop these clusters.32 We and others have previously shown that LTβR signaling can recapitulate adhesion molecule induction in vitro with fibroblasts or stromal cells.22,28 This provided us with a good model to test the individual and joint requirements of p50 and p52 in several key LTβR target genes important for lymphoid tissue development and organization.
We started by testing whether the LTβR-induced adhesion molecules are dependent on NF-κB. Fibroblasts were transduced to express a mutant form of IκBα, which acts as a superrepressor of NF-κB activity. MIGR1 vector-transduced cells responded to LTβR stimulation by inducing ICAM-1 and VCAM-1 expression. However, the ICAM-1 and VCAM-1 induction in response to LTβR signaling was abolished in cells transduced with IκBα, demonstrating that this response is dependent on NF-κB (data not shown). Agonistic antibody to LTβR is also specific since it can stimulate wild-type cells but not LTβR-deficient cells. This now allowed us to test the roles of specific NF-κB subunits, p50 and p52, in LTβR-induced adhesion molecules.
The LN deficiencies observed in LT-deficient and p50 and p52 mutant mice suggested that p50 and p52 play essential roles in the LTβR signaling pathway. To address this issue directly, we tested the requirements of p50 and p52 in LT-induced adhesion molecules. Wild-type, p50-, and p52-deficient MEFs were stimulated with agonist LTβR antibody and adhesion molecule expression was determined by flow cytometry. Although wild-type MEFs strongly induced ICAM-1 and VCAM-1 expression after LTβR stimulation, Nfkb1-/- MEFs responded with a very weak shift (Figure 3). Moreover, Nfkb2-/- MEFs failed to induce ICAM-1 and VCAM-1 expression in response to LTβR ligation (Figure 3). These genetic and cellular data stress the requirement of both p50 and p52 in LTβR-induced gene expression.
Figure 3.

p50 and p52 are essential for LTβR-induced adhesion molecules. Loss of LTβR-induced expression of adhesion molecules in p50-and p52-deficient MEFs. WT, Nfkb1-/-, and Nfkb2-/- MEFs were stimulated for 2 to 3 days with a control (gray lines) or agonistic LTβR (black lines) antibody, stained with VCAM-1 or ICAM-1 antibodies, and analyzed by flow cytometry.
p50 and p52 are required for LTβR-mediated expression of lymphoid chemokines
The LT/LTβR signaling pathway is also required for expression of secondary lymphoid tissue chemokines such as CCL21 and CXCL13 in lymphoid but not nonlymphoid tissues.9,28 In addition to their role in maintaining proper compartmentalization of T and B lymphocytes within lymphoid follicles, the chemokines CCL21 and CXCL13 and their receptors have also been implicated in LN formation.10-12 We therefore hypothesized that LTβR signaling induces the expression of secondary lymphoid tissue chemokines in an NF-κB-dependent manner. In order to address this issue, we tested whether LT signals can stimulate the transcription of CCL21 in vitro. Treatment of wild-type MEFs with agonist LTβR antibody augmented the expression of CCL21 transcripts (Figure 4A) and Cxcl13 transcripts (Figure 4B) as assessed by real-time PCR. We next interrogated the contributions of p50 and p52 in this process. MEFs deficient in p50 or p52 were stimulated with the agonist LTβR antibody and tested for CCL21 and CXCL13 induction. In contrast to WT MEFs, Nfkb1-/- MEFs induced suboptimal levels of CXCL13 and weakly induced CCL21, demonstrating the critical role of p50 in lymphoid chemokine expression downstream of the LTβR. Nfkb2-/- MEFs showed negligible induction of CCL21 and CXCL13 in response to LT but with a higher baseline transcription of CCL21 and CXCL13. We hypothesized that the weak responses to LT in the p50 and p52 singly deficient MEFs were the result of compensation by p52 and p50, respectively. To directly address this issue, we assayed Nfkb1-/-Nfkb2-/- MEFs. The weak CCL21 and CXCL13 responses in response to LT stimulation observed in the Nfkb1-/- and Nfkb2-/- cells were completely abrogated in the Nfkb1-/-Nfkb2-/- fibroblasts (Figure 4A-B), confirming our hypothesis that p50 and p52 can provide some residual activities in the absence of the other, which may be sufficient for the formation of most lymphoid tissues. Furthermore, the Nfkb1-/-Nfkb2-/- MEFs did not exhibit the elevated basal levels of CCL21 and CXCL13 shown by the Nfkb2-/- MEFs. These results reveal that p50 and p52 are required for LTβR-mediated induction of CCL21 and CXCL13 for the formation and organization of lymphoid tissues. Together these data indicate that p50 and p52 both play essential roles in LTβR-induced adhesion molecule and chemokine expression and suggest that the impairment of LT signaling to fully induce adhesion molecules and chemokines in the LN anlagen of p50 and p52 doubly mutant mice results in LN aplasia.
Figure 4.

p50 and p52 are essential for LTβR-induced expression of lymphoid tissue chemokines CCL21 and CXCL13. (A) Distorted CCL21 induction in p50 and p52 singly and doubly deficient MEFs in response to LTβR signaling. WT, Nfkb1-/-, Nfkb2-/-, and Nfkb1-/- Nfkb2-/- MEFs were stimulated for 24 hours with agonistic LTβR antibody and assessed for transcription of CCL21. (B) Distorted CXCL13 induction in p50 and p52 singly and doubly deficient MEFs in response to LTβR signaling. WT, Nfkb1-/-, Nfkb2-/-, and Nfkb1-/- Nfkb2-/- MEFs were stimulated for 24 hours with agonistic LTβR antibody and assessed for transcription of CXCL13. The RNA was isolated from the cells and converted to cDNA for analysis of CCL21 and CXCL13 transcription by real-time PCR. Error bars indicate a single standard deviation in the data of 3 biological replicates. Each biological replicate was done in 3 technical Q-PCR triplicates (though some reactions failed).
p50 and p52 expression on radiation-resistant stromal cells is required for T/B lymphocyte segregation
The critical roles of p50 and p52 in LT induction of the lymphoid chemokines CCL21 and CXCL13 led us to investigate whether loss of p50 and p52 in vivo would perturb T/B lymphocyte segregation. We and others had previously observed abnormalities in lymphocyte maturation, but rather normal T/B cell segregation in the spleens from mice deficient in either p50 or p52.26,33 Likewise, T/B lymphocyte segregation was maintained in lymph nodes of p50 and p52 singly deficient mice while altered in the spleens of LTβR-/- and RelB-/- mice (data not shown). However, the cooperative roles of p50 and p52 in the formation of LNs further prompted us to investigate T- and B-cell segregation in Nfkb1+/-Nfkb2-/- mutant mice. The paucity of mature lymphocytes, especially B cells, in the Nfkb1+/-Nfkb2-/- mice did not allow us to adequately assess T/B segregation directly in these mice. To circumvent this problem, Nfkb1+/-Nfkb2-/- mice were lethally irradiated and reconstituted with bone marrow from WT mice. In contrast to the WT to WT chimera, WT to Nfkb1+/-Nfkb2-/- chimeric mice lacked well-defined T-cell zones and demonstrated perturbed T-/B-cell segregation in the spleen (Figure 5). Consistent with a role for the import of p50 and p52 expression in radiation-resistant stromal cells and not hematopoietic cells in lymphocyte compartmentalization, Nfkb1+/-Nfkb2-/- to WT chimeras did not exhibit a defect in T/B lymphocyte segregation (Figure 5). Sections from LNs revealed similar T/B segregation defects in the WT to Nfkb1+/-Nfkb2-/- chimeric mice (data not shown). Overall, we observed a severe degree of defective T- and B-cell compartmentalization in the WT to Nfkb1+/-Nfkb2-/- chimeras that was similar but less severe than WT to LTβR-/- chimeras and LT- and LTβR-deficient mice. In line with the requirement for LTβR expression on radioresistant stromal cells, T/B lymphocyte segregation was not restored in the WT to LTβR-/- chimeras.34 We predict that mice completely deficient in both p50 and p52 would demonstrate T/B disorganization on par with LT-deficient mice, though the experiment would be logistically difficult given the block in lymphocyte development and the runted phenotype to survive bone marrow reconstitution. These data indicate that expression of both p50 and p52 on radiation-resistant stromal cells is required for T-/B-cell segregation. These findings support the notion that both p50 and p52 transduce critical signals downstream of the LTβR to effectively organize secondary lymphoid tissues.
Figure 5.
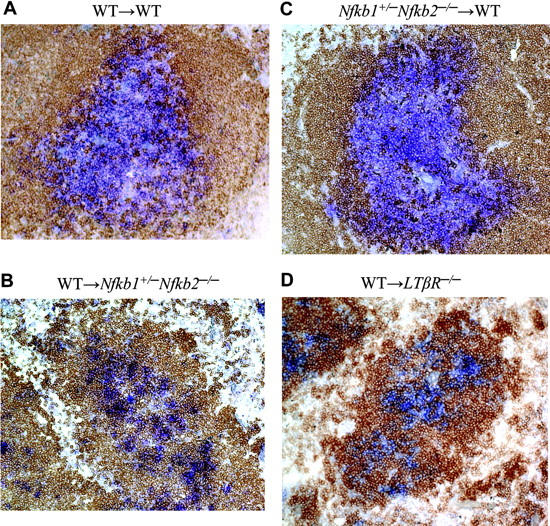
p50 and p52 expression in radiation-resistant stromal cells is required for T/B lymphocyte segregation. WT, Nfkb1+/- Nfkb2-/-, and LTβR-/- mice were lethally irradiated and reconstituted with WT or Nfkb1+/- Nfkb2-/- bone marrow cells. Spleens from the indicated bone marrow chimeric mice were stained for T cells with Thy1.1 (blue) and B cells with B220 (brown) antibodies 2 to 3 months after reconstitution. At least 20 follicles from each spleen were scored for degree of T/B segregation (complete, partial, and disorganized). WT→WT (A) and Nfkb1+/- Nfkb2-/-→WT (C) chimeras showed 100% complete segregation. WT→Nfkb1+/- Nfkb2-/- (B) and WT→LTβR-/- (D) chimeras exhibited 100% disorganized T/B segregation. The difference is very significant (P < .001). Data shown are representative sections from 3 spleens per group.
Discussion
LT developmental signaling through the LTβR activates several signaling pathways that play critical roles in the development, maturation, and continued organization of secondary lymphoid organs. We show that LT activates multiple NF-κB complexes, including p50:RelA, p52:RelB, p50:p50, and the poorly characterized p50:RelB and p52:RelA dimers to effect the activation of genes critical for LN development and organization of secondary lymphoid organs. Our study demonstrates that the regulation of both p50 and p52 activities is critical to every aspect of LT signal transduction and physiology we examined, and that their distinct proteolytic processing mechanisms are key to proper LT signal transduction (Table 2). While earlier studies had focused on describing different NF-κB dimers activated by LTβR and specifying a unique function for each in terms of gene transcription,20,30,31 our data from combined genetic and biochemical perturbation experiments further reveal a high degree of functional overlap as well as critical crosstalk between the NF-κB pathways. In fact, here we show that adhesion molecule induction by the LTβR is controlled by both p50 and p52, demonstrating the complexity in the system. Rather than 2 distinct linear pathways, the inter-dependent regulation of components of the NF-κB/IκB system describes a signaling network that produces multiple NF-κB activation complexes.
Table 2.
Summary of NF-κB requirements in LTβR signaling
|
Gene expression
|
In vivo
|
|||||
|---|---|---|---|---|---|---|
| Genotype | NF-κB complexes activated by LTβR | VCAM ICAM | Ccl21 | Cxcl13 | LN development | T/B zone |
| WT | p50:RelA, p52:RelB, p50:p50 | +++ | +++ | +++ | +++ | + |
| Nfkb1+/–Nfkb2+/– | ND | ND | ND | ND | +++ | + |
| Nfkb1–/– | p52:RelA, p52:RelB | + | + | ++ | ++ | + |
| Nfkb2–/– | p50:RelA, p50:RelB, p50:p50 | – | + | +/– | ++ | + |
| Nfkb1+/–Nfkb2–/– | ND | ND | ND | ND | + | – |
| Nfkb1–/–Nfkb2–/– | – | ND | – | – | – | ND |
ND indicates not detected; +++, full response; ++, intermediate response; +, weak response; –, no response; and +/–, very weak response and elevated basal levels.
Lymphoid organ phenotypes in NF-κB-deficient mice
Mice deficient in either p50 or p52 display LT-characteristic phenotypes, indicating the necessary roles of each molecule in LT signaling and lymphoid organogenesis and organization; however, their incomplete penetrance relative to doubly deficient mice also speaks to the importance of residual LTβR-induced NF-κB signals. The discriminatory loss of inguinal LNs in Nfkb1-/- and Nfkb2-/- mice and retention of mesenteric LNs in Nfkb1+/-Nfkb2-/- and Nfkb1-/-Nfkb2+/- mice suggests there is a hierarchy in LN development. To put it another way, each set of LNs may have different developmental requirements. Furthermore, there is a gene dosage effect on LN development that starts to manifest itself as 2 of the 4 combined Nfkb1 and Nfkb2 alleles are lost. Interestingly, more severe defects in both T/B lymphocyte segregation in the spleen and LN organogenesis are revealed as 3 of the 4 p50 and p52 alleles are lost. The gene dosage effect does not appear to be a simple linear relationship but instead indicates a complex nonlinear cooperation between p50 and p52 for lymphoid tissue development and organization. Although Nfkb1-/- and Nfkb2-/- mice have similar deficiencies in LNs, Nfkb1+/-Nfkb2-/- and Nfkb1-/- Nfkb2+/- mice show altered patterns in LN genesis, demonstrating the functional similarities and differences between p50 and p52. The increased sensitivity of inguinal LNs also bore out in the CCR7-/- mice.12 On the other end of the spectrum, the robustness of mesenteric LN development was also found in LTβ-/- mice, which lack all peripheral LNs but selectively retain mucosal LNs.35 In line with our hypothesis, minimal LTβR signaling through LIGHT, another ligand for LTβR, was sufficient for the formation of mesenteric LNs. The outstanding mesenteric LNs were lost in Ltb-/- Light-/- mice, recapitulating the LN phenotype expected of LTβR-/- mice.6,36 Our genetic data clearly demonstrate the essential cooperative roles of p50 and p52 in lymphoid tissue organogenesis and organization, yet the contributions of p50 and p52 and how they fit with other NF-κB pathways in unmanipulated wild-type mice remain to be determined. These genetic systems provide biologically relevant consequences of varying signal doses from full, to intermediate, to low, to nil.
Our data suggest that both p50 and p52 need to be expressed on the same cell for full LTβR activation. The combined deficiency in p50 and p52 that results in the in vivo loss of LNs supports this notion. An alternative explanation is that p50 and p52 may be expressed on different cells. For example, the loss of p50 on a hematopoietic cell combined with the loss of p52 on a stromal cell may synergize to produce the observed phenotype. This alternative theory is, however, not supported by our experimental data showing further decreased LTβR-induced NF-κB activation in fibroblasts lacking both p50 and p52, compared with single knockout cells. Furthermore, gene expression of the RelB target genes Ccl21 and Cxcl13 is blunted in Nfkb1-/- and Nfkb2-/- cells, and completely ablated in nfkb-/-Nfkb2-/- fibroblasts. Interestingly, baseline Cxcl13 and to a lesser extent Ccl21 transcription is elevated in Nfkb2-/- cells and is unresponsive to further LT stimulation. This finding parallels an earlier observation that pulmonary CCL21 expression is enhanced in Nfkb2-/- mice.28 These data strongly suggest that the constitutive activation of NF-κB dimers seen in the Nfkb2-/- cells disrupts the homeostatic regulation of NF-κB, resulting in enhanced chemokine expression both in vitro and in vivo. Lastly, our data indicate that radiation-resistant stromal cells, but not hematopoietic cells, need to express both p50 and p52 to orchestrate proper T/B lymphocyte compartmentalization in secondary lymphoid organs. These results point to the requirement of p50 and p52 expression on the stromal cell responding to membrane LT signals.
Although p52-deficient mice display deficits in splenic CCL21 and CXCL13 expression, p50-deficient mice express comparable levels of CCL21 and CXCL13 as wild-type mice.37 Our current study, however, identifies a role for p50 in the expression of CCL21 and CXCL13. The in vitro stimulation of MEFs may be more sensitive in detecting more subtle differences than in vivo chemokine expression. Moreover, the in vitro stimulations allowed us to directly address the responses specific to anti-LTβR stimulation. There are likely many more factors that can contribute to chemokine expression in vivo that may mask the defects seen with the p50-deficient fibroblasts. Continual stimulation of the LTβR along with other NF-κB-activating stimuli may thus allow for more permissive conditions in vivo.
Our analyses of p50 and p52 in LTβR signaling are supported by our use of MEFs for a number of in vitro assays. Although MEFs represent an excellent cellular and genetic model of p50 and/or p52 deficiency, they do not necessarily reflect the complex cell types likely required for lymphoid tissue development and organization found in vivo. The cellular heterogeneity may result from different states of maturation, activation, or microenvironments, leading to disparate responses from cell to cell. We believe the MEFs provide a reasonably good model to study LTβR signal transduction, as our analysis of stromal cells derived from the spleen revealed similar dependencies on NF-κB for induction of adhesion molecules in response to LTβR stimulation (data not shown). In addition, we show that the MEFs are capable of inducing the lymphoid chemokine genes Ccl21 and Cxcl13 following LTβR ligation.
NF-κB dimer specificity and compensatory pathways
As described, mice deficient in either p50 or p52 do not have a global defect in the formation of LNs, despite key contributions made by p50 or p52 in LTβR signaling. Nfkb1-/- and Nfkb2-/- cells contain both RelA and RelB complexes, but their regulation is perturbed. The substitution of p50:RelA with p52:RelA complexes in the absence of p50 may permit for some functional compensation, as does the switch from p52:RelB to p50:RelB complexes in Nfkb2-/- fibroblasts. In fact, only the reduction or removal of p50 in the Nfkb2-/-, p52 in the Nfkb1-/-, and hence the residual and alternate NF-κB dimers resulted in more pronounced phenotypes found in vivo, such as T/B segregation and LN development. Furthermore, p50:RelB dimers have been observed to be activated by LTβR in wild-type cells but its role in the development of LNs or lymphoid microenvironment has not been presented,38 even if our experimental methodology failed to detect this dimer. The abundance and variety of p50-containing dimers elicited by LTβR signals in wild-type and Nfkb2-/- cells further highlights the complexity and importance of p50 in the LT signaling cascade. The respective roles of p50:p50 homodimers and p52:RelB and p50: RelB heterodimers remain to be determined. It is not known whether the latter 2 dimers are functionally equivalent in target gene activation while being differentially regulated, and whether p50:p50 homodimers induced by LTβR will repress or enhance transcription of genes.
The genetic ablation of p52 in Nfkb2-/- fibroblasts resulted in constitutive NF-κB activation and increased basal transcription of the lymphoid chemokines CCL21 and especially CXCL13, revealing a counterregulatory role for the Nfkb2 gene products in the NF-κB network. Exactly how p100/p52 modulates the different NF-κB pathways elicited by the LTβR and other upstream signaling pathways remains an active area of research. p100 may exert its effects at multiple levels such as by inhibiting the nuclear translocation of p52 or other NF-κB subunits to repress NF-κB activation.39 We had previously shown that the C-terminal domain of p100 can repress RelB binding to DNA.30 Additionally, the transcriptional targets of p52 may act to modulate later phases of NF-κB activity.
The LTβR signals myriad biological responses pertaining to the development of lymphoid tissues. Here we reveal a novel and previously unappreciated role for p50 in the LTβR signal transduction pathway and a critical role in a signaling network that allows for LT-responsive p50 and p52 regulation in the development of LNs. We found that the LTβR activates a far more diverse set of NF-κB dimers than previously appreciated, leading us to propose that signaling by other cytokine receptors should also be examined in terms of an NF-κB signaling network to produce a multitude of factors rather than engaging distinct, linea,r canonical and noncanonical NF-κB signaling cascades. Perturbation of the homeostatic signaling network by either genetic ablation (described here), allele variants in the population, random mutations in cancer cells, or possibly directed or inadvertent pharmacologic intervention can have severe consequences not only on reversible signal transduction responses as in inflammation, but also irreversibly during development of splenic and lymphoid microarchitectures.
Acknowledgments
A.H. thanks T. H. Leung and the S. Hedrick lab for help with Q-PCR experiments, R. Ryseck for sharing knockout mice, B. Kennedy for outstanding mouse husbandry services, J. Browning for anti-LTβR antibody, Santa Cruz Biotechnology for antibodies, D. Baltimore for continued support, and R. Rickert and M. Karin for discussions. Y.-X.F. thanks Dave Palucki for his assistance with mice.
Prepublished online as Blood First Edition Paper, September 29, 2005; DOI 10.1182/blood-2005-06-2452.
Supported by a Medical Scientist National Research Service Award (5 T32 GM07281) to J.C.L. and by National Institutes of Health grant nos. AI062026 (Y.-X.F.), DK58891 (Y.-X.F.), and GM071573 (A.H.).
A.H. and Y.-X.F. share senior authorship.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Yoshida H, Naito A, Inoue J, et al. Different cytokines induce surface lymphotoxin-alphabeta on IL-7 receptor-alpha cells that differentially engender lymph nodes and Peyer's patches. Immunity. 2002;17: 823-833. [DOI] [PubMed] [Google Scholar]
- 2.Cupedo T, Kraal G, Mebius RE. The role of CD45+CD4+CD3- cells in lymphoid organ development. Immunol Rev. 2002;189: 41-50. [DOI] [PubMed] [Google Scholar]
- 3.Fu YX, Chaplin DD. Development and maturation of secondary lymphoid tissues. Annu Rev Immunol. 1999;17: 399-433. [DOI] [PubMed] [Google Scholar]
- 4.Weih F, Caamano J. Regulation of secondary lymphoid organ development by the nuclear factor-kappaB signal transduction pathway. Immunol Rev. 2003;195: 91-105. [DOI] [PubMed] [Google Scholar]
- 5.De Togni P, Goellner J, Ruddle NH, et al. Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science. 1994;264: 703-707. [DOI] [PubMed] [Google Scholar]
- 6.Futterer A, Mink K, Luz A, Kosco-Vilbois MH, Pfeffer K. The lymphotoxin β receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity. 1998;9: 59-70. [DOI] [PubMed] [Google Scholar]
- 7.Rennert PD, James D, Mackay F, Browning JL, Hochman PS. Lymph node genesis is induced by signaling through the lymphotoxin beta receptor. Immunity. 1998;9: 71-79. [DOI] [PubMed] [Google Scholar]
- 8.Fu Y-X, Huang G, Wang Y, Chaplin DD. Lymphotoxin-expressing B cells induce formation of splenic clusters of follicular dendritic cells. J Exp Med. 1998;187: 1009-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ngo VN, Korner H, Gunn MD, et al. Lymphotoxin alpha/beta and tumor necrosis factor are required for stromal cell expression of homing chemokines in B and T cell areas of the spleen. J Exp Med. 1999;189: 403-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ansel KM, Ngo VN, Hyman PL, et al. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature. 2000;406: 309-314. [DOI] [PubMed] [Google Scholar]
- 11.Luther SA, Ansel KM, Cyster JG. Overlapping roles of CXCL13, interleukin 7 receptor alpha, and CCR7 ligands in lymph node development. J Exp Med. 2003;197: 1191-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohl L, Henning G, Krautwald S, et al. Cooperating mechanisms of CXCR5 and CCR7 in development and organization of secondary lymphoid organs. J Exp Med. 2003;197: 1199-1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cyster JG. Chemokines and cell migration in secondary lymphoid organs. Science. 1999;286: 2098-2102. [DOI] [PubMed] [Google Scholar]
- 14.Fu YX, Huang GM, Matsumoto M, Molina H, Chaplin DD. Independent signals regulate development of primary and secondary follicle structure In spleen and mesenteric lymph node. Proc Natl Acad Sci U S A. 1997;94: 5739-5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109: S81-S96. [DOI] [PubMed] [Google Scholar]
- 16.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16: 225-260. [DOI] [PubMed] [Google Scholar]
- 17.Silverman N, Maniatis T. NF-kappaB signaling pathways in mammalian and insect innate immunity. Genes Dev. 2001;15: 2321-2342. [DOI] [PubMed] [Google Scholar]
- 18.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu Rev Immunol. 2000;18: 621-663. [DOI] [PubMed] [Google Scholar]
- 19.Senftleben U, Cao Y, Xiao G, et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293: 1495-1499. [DOI] [PubMed] [Google Scholar]
- 20.Dejardin E, Droin NM, Delhase M, et al. The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappaB pathways. Immunity. 2002;17: 525-535. [DOI] [PubMed] [Google Scholar]
- 21.Yin L, Wu L, Wesche H, et al. Defective lymphotoxin-beta receptor-induced NF-kappaB transcriptional activity in NIK-deficient mice. Science. 2001;291: 2162-2165. [DOI] [PubMed] [Google Scholar]
- 22.Matsushima A, Kaisho T, Rennert PD, et al. Essential role of nuclear factor (NF)-kappaB-inducing kinase and inhibitor of kappaB (IkappaB) kinase alpha in NF-kappaB activation through lymphotoxin beta receptor, but not through tumor necrosis factor receptor I. J Exp Med. 2001;193: 631-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. 2002;298: 1241-1245. [DOI] [PubMed] [Google Scholar]
- 24.Carragher D, Johal R, Button A, et al. A stroma-derived defect in NF-kappaB2-/- mice causes impaired lymph node development and lymphocyte recruitment. J Immunol. 2004;173: 2271-2279. [DOI] [PubMed] [Google Scholar]
- 25.Alcamo E, Hacohen N, Schulte LC, Rennert PD, Hynes RO, Baltimore D. Requirement for the NF-kappaB family member RelA in the development of secondary lymphoid organs. J Exp Med. 2002;195: 233-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Franzoso G, Carlson L, Poljack L, et al. Mice deficient in nuclear factor (NF)-kappa-B/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. J Exp Med. 1998;187: 147-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Franzoso G, Carlson L, Xing L, et al. Requirement for NF-kappaB in osteoclast and B-cell development. Genes Dev. 1997;11: 3482-3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lo JC, Chin RK, Lee Y, et al. Differential regulation of CCL21 in lymphoid/nonlymphoid tissues for effectively attracting T cells to peripheral tissues. J Clin Invest. 2003;112: 1495-1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoffmann A, Leung TH, Baltimore D. Genetic analysis of NF-kappaB/Rel transcription factors defines functional specificities. EMBO J. 2003;22: 5530-5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yilmaz ZB, Weih DS, Sivakumar V, Weih F. RelB is required for Peyer's patch development: differential regulation of p52-RelB by lymphotoxin and TNF. EMBO J. 2003;22: 121-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muller JR, Siebenlist U. Lymphotoxin beta receptor induces sequential activation of distinct NF-kappa B factors via separate signaling pathways. J Biol Chem. 2003;278: 12006-12012. [DOI] [PubMed] [Google Scholar]
- 32.Eberl G, Marmon S, Sunshine MJ, Rennert PD, Choi Y, Littman DR. An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat Immunol. 2004;5: 64-73. [DOI] [PubMed] [Google Scholar]
- 33.Caamano JH, Rizzo CA, Durham SK, et al. Nuclear factor (NF)-kappa B2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. J Exp Med. 1998;187: 185-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Endres R, Alimzhanov MB, Plitz T, et al. Mature follicular dendritic cell networks depend on expression of lymphotoxin beta receptor by radioresistant stromal cells and of lymphotoxin beta and tumor necrosis factor by B cells. J Exp Med. 1999;189: 159-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koni PA, Sacca R, Lawton P, Browning JL, Ruddle NH, Flavell RA. Distinct roles in lymphoid organogenesis for lymphotoxin α and β revealed in lymphotoxin β-deficient mice. Immunity. 1997;6: 491-500. [DOI] [PubMed] [Google Scholar]
- 36.Scheu S, Alferink J, Potzel T, Barchet W, Kalinke U, Pfeffer K. Targeted disruption of LIGHT causes defects in costimulatory T cell activation and reveals cooperation with lymphotoxin beta in mesenteric lymph node genesis. J Exp Med. 2002;195: 1613-1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weih DS, Yilmaz ZB, Weih F. Essential role of RelB in germinal center and marginal zone formation and proper expression of homing chemokines. J Immunol. 2001;167: 1909-1919. [DOI] [PubMed] [Google Scholar]
- 38.Derudder E, Dejardin E, Pritchard LL, Green DR, Korner M, Baud V. RelB/p50 dimers are differentially regulated by tumor necrosis factor-alpha and lymphotoxin-beta receptor activation: critical roles for p100. J Biol Chem. 2003;278: 23278-23284. [DOI] [PubMed] [Google Scholar]
- 39.Ishikawa H, Carrasco D, Claudio E, Ryseck RP, Bravo R. Gastric hyperplasia and increased proliferative responses of lymphocytes in mice lacking the COOH-terminal ankyrin domain of NF-kappaB2. J Exp Med. 1997;186: 999-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]