

Abstract
A single HO endonuclease-induced double-strand break (DSB) is sufficient to activate the DNA damage checkpoint and cause Saccharomyces cells to arrest at G2/M for 12–14 h, after which cells adapt to the presence of the DSB and resume cell cycle progression. The checkpoint signal leading to G2/M arrest was previously shown to be nuclear-limited. Cells lacking ATR-like Mec1 exhibit no DSB-induced cell cycle delay; however, cells lacking Mec1's downstream protein kinase targets, Rad53 or Chk1, still have substantial G2/M delay, as do cells lacking securin, Pds1. This delay is eliminated only in the triple mutant chk1Δ rad53Δ pds1Δ, suggesting that Rad53 and Chk1 control targets other than the stability of securin in enforcing checkpoint-mediated cell cycle arrest. The G2/M arrest in rad53Δ and chk1Δ revealed a unique cytoplasmic phenotype in which there are frequent dynein-dependent excursions of the nucleus through the bud neck, without entering anaphase. Such excursions are infrequent in wild-type arrested cells, but have been observed in cells defective in mitotic exit, including the semidominant cdc5-ad mutation. We suggest that Mec1-dependent checkpoint signaling through Rad53 and Chk1 includes the repression of nuclear movements that are normally associated with the execution of anaphase.
Keywords: dynein, mitotic arrest, nucleus positioning, metaphase–anaphase transition
A single unrepaired or slowly repaired double-strand break (DSB) in the budding yeast Saccharomyces cerevisiae triggers the Mec1–Ddc2-dependent DNA damage checkpoint, causing G2/M cell cycle arrest through the activation of a protein kinase cascade (1–9). The trigger for the DNA damage checkpoint appears to be the formation of single-stranded DNA by 5′ to 3′ resection of the DSB, a step that depends on Cdk1 (10). Mec1–Ddc2 binding to ssDNA appears to occur both by intrinsic DNA binding of Ddc2 but also by association of Mec1–Ddc2 with the ssDNA-binding protein complex, RPA (11, 12). Checkpoint activation depends also on the binding of the PCNA-like complex of Rad17–Ddc1–Mec3 at the ssDNA/dsDNA junction and depends only on the RFC-like complex containing Rad24 (13–18). The DNA damage checkpoint has at least two branches, one controlled by Rad53 (Chk2/Cds1 in mammals and Saccharomyces pombe, respectively) and the other by Chk1. In addition, checkpoint activation depends on Rad9, a BRCT domain-containing protein that is associated with a DSB and plays a key role in the autophosphorylation of Rad53 and the phosphorylation of Chk1 (19–21). A primary objective of checkpoint activation appears to be the prevention of degradation of securin, Pds1, by the Cdc20-activated anaphase-promoting complex. Without securin, the separase enzyme can cleave cohesins linking sister chromatids and allow mitosis to proceed; thus stabilization of Pds1 results in cells remaining arrested in metaphase, before anaphase (22, 23).
Once the DNA checkpoint is triggered, cells remain arrested for as long as six cell doubling times (12–15 h). However, even in the absence of repair, damage-arrested wild-type cells will eventually turn off the checkpoint and resume proliferation by a process termed “adaptation” (1, 24–26). A number of adaptation-defective mutations have been identified that appear to prolong arrest in several ways. Deletion of the Ku proteins increases the rate of 5′ to 3′ DNA end resection and thus increases the level of damage signaling (2), whereas deletion of the Ptc2 and Ptc3 phosphatases prevents dephosphorylation of Rad53 and other targets and thus prolongs the arrest signal (27). The defects in several other adaptation-defective mutations are not yet known. Some adaptation-defective mutations, most notably those that cannot turn off the damage signal, also prevent cells from resuming cell cycle progression even after DNA damage is repaired (termed recovery).
The protein kinase cascade generates an arrest signal that is apparently nuclear-limited. In a heterokaryon in which one nucleus suffers an unrepairable HO-induced DSB and arrests before anaphase, a second, undamaged nucleus is unimpeded in carrying out mitosis (28). This result suggests that the DNA damage checkpoint does not send a signal transmitted through the cytoplasm that would affect the second nucleus. In this article, we show that there is a cytoplasmic aspect to the DNA checkpoint but one that affects nuclear positioning and not the segregation of chromosomes.
Results
Most Single-Checkpoint Mutants Still Delay Cell Cycle Progression in Response to a Single Unrepaired DSB.
We examined the way in which deletion of different checkpoint genes affects the extent of G2/M arrest after experiencing a single unrepairable DSB. Previous studies have suggested that mec1 mutants abolish any delay in cell cycle progression after DNA damage, whereas other checkpoint mutants, including rad53 and chk1, show a partial checkpoint deficiency (29–31). Not all of these previous comparisons have been carried out in response to a single type of DNA damage, and, in some cases, the DNA damage may interact with other checkpoints such as the DNA replication checkpoint and the spindle assembly checkpoint (32).
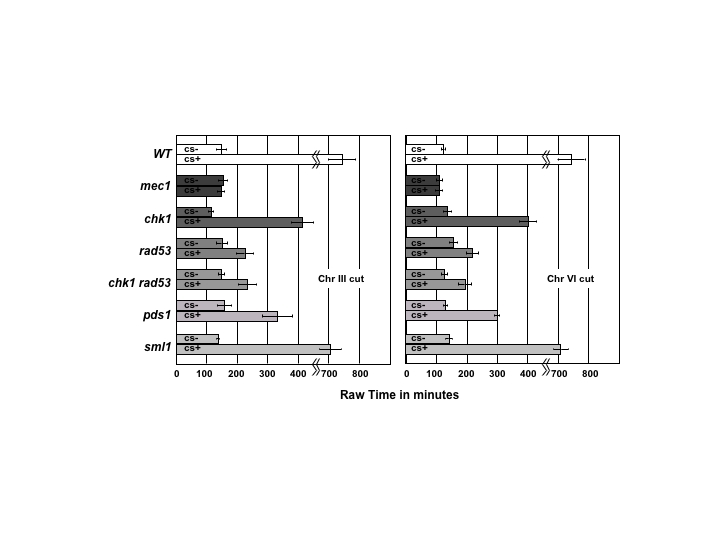
In strain JKM179, induction of a galactose-inducible HO endonuclease gene creates a single unrepairable DSB at the MAT locus (25), and cells experience a DNA-damage checkpoint-mediated G2/M arrest for an extended period before adapting and resuming cell division. To better characterize the checkpoint, we precisely measured the length of cell cycle arrest in individual cells. Unbudded G1 cells were micromanipulated into an ordered array on an agar nutrient plate containing galactose (YEP-galactose) to induce the DSB. We noted the time when each G1 cell first budded (i.e., entered the two-cell stage) and the time when the cell entered the next cycle by the appearance of another bud. In most cases, 100 cells were monitored every 30 min (Fig. 1C). To correct for the possibility that checkpoint mutations would affect the length of the cell cycle, each mutant was compared with an isogenic strain lacking the HO cut site. To ensure that there were no special features of the DSB made at MAT on chromosome III (Chr III), strains lacking the cleavage site at MAT were transformed with an HPH-marked construct that introduced a 117-bp HO cleavage site into the middle of chromosome VI (Chr VI), and a similar analysis was performed. The results of these analyses are shown in Fig. 1A and the unnormalized data are shown in supporting information (SI) Fig. 4. Because both mec1Δ and rad53Δ are lethal, we created strains with a sml1 deletion, which suppresses this lethality (33). By itself, sml1Δ has no effect on the length of checkpoint arrest compared with wild-type cells.
Fig. 1.

Duration of a single cell cycle after an HO-induced DSB is generated in G1 cells. (A) At least 50 cells were examined microscopically every 30 min, as described in Experimental Procedures, to determine the length of a single cell cycle (see C). In each case, the length of the cell cycle was normalized to that of an isogenic control, treated identically but lacking an HO cleavage site. Data are shown for a DSB at the MAT locus on Chr III or for a site on Chr VI. Statistical significance of cell cycle length is shown relative to mec1Δ sml1Δ, which shows no difference with or without induction of a DSB. (B) Cell cycle delay in pds1Δ and in several mutant derivatives. Statistical significance of delays are shown relative to mec1Δ sml1Δ. A pds1Δ rad53Δ chk1Δ mutant behaves comparable to mec1Δ. (C) Measurement of cell cycle delay. G1 cells are plated on galactose-containing plates and observed at 30-min intervals and their morphology recorded. A single cell cycle is considered to be from the time a tiny bud appears on the single cell until the dumbbell G2 cell completes mitosis and rebuds. Control cells of identical genotypes but lacking the HO cleavage site were treated in the same fashion.
For a DSB on both Chr III or on Chr VI, only the mec1Δ sml1Δ strain progresses through the cell cycle as fast as an isogenic strain lacking a DSB. All of the other single mutations exhibit a statistically significant delay. In the case of chk1Δ, this delay persists for many hours, but even rad53Δ sml1Δ shows a delay of >1 h. These data suggest that Mec1 is central in the establishment of the checkpoint, activating several independent downstream pathways that are, to varying degrees, required for the maintenance of the checkpoint. Activation of one of these downstream targets seems to be sufficient to establish at least a brief arrest.
If the DNA damage checkpoint centers its action on the prevention of sister-chromatid separation by preventing separase-mediated cleavage of Pds1, then a pds1Δ mutant might display no arrest. However, this is not the case (Fig. 1B). In fact, a complete lack of HO-induced arrest is seen only in a chk1Δ rad53Δ pds1Δ triple mutant. We note that Pds1 is not only necessary to inhibit separase, it also aids in the transport of separase into the yeast nucleus (23). It is therefore possible that Rad53 and/or Chk1 could play a role in the transport of separase into the nucleus (34) in addition to regulating the stability of Pds1.
A Cytoplasmic Target of the DNA Damage Checkpoint.
DAPI staining of checkpoint-arrested cells revealed that preanaphase arrest is not uniform. Although all cells have a short mitotic spindle that one expects for such arrest; not all cells exhibit a compact nucleus located in the mother cell (Class I, Fig. 2A). Some cells exhibit a protrusion at least some of the DNA through the bud neck (Class II) and a significant number of cells exhibit DAPI staining as a “bow-tie” in which the DNA is about equally distributed on both sides of the bud neck (Class III). Both rad53Δ and chk1Δ cells show a statistically significant increase in the distribution of these three classes of arrest compared with DSB-arrested wild-type cells examined after the same delay (Fig. 2B). These results suggested that there could be differences between wild-type and chk1Δ cells in the degree of chromosome segregation or in nuclear positioning.
Fig. 2.

Nuclear positioning in DNA-damaged cells. (A) DAPI staining of wild-type cells undergoing extended G2/M arrest induced by a single DSB. Three classes of DAPI images are shown: Class I with all of the DAPI staining in a compact nucleus; Class II with a small protrusion of DAPI stain through the bud neck; Class III, “bow-tie” staining with substantial DAPI staining in both mother and daughter cells. (B) Distribution of DAPI staining classes in wild-type (WT), chk1Δ, and rad53Δ sml1Δ cells. (C) Wild-type cells grown on glucose (Glu) for 4 h, with Nuf2-GFP-labeled centromeres and Spc29-RFP-labeled SPBs, show an average centromere separation of 1.11 μm with a standard deviation of 0.426 μm (n = 8). Wild-type cells grown on galactose (Gal) for 4 h show an average centromere separation of 0.948 μm with a standard deviation of 0.299 μm (n = 10). These differences are not statistically significant (P = 0.679).
To examine the nature of the difference in DAPI staining, we asked whether chk1Δ cells had already executed at least part of mitosis, in which chromosomes would segregate toward the spindle poles, and the distance between spindle poles would elongate. We examined both spindle pole bodies (with RFP-tagged Spc29) and centromeres (using Nuf2-GFP) (35), as shown in Fig. 2B. In undamaged wild-type cells before anaphase, sister centromeres are separated by 0.94–1.11 μm, as expected for chromosomes under tension, and, similarly, spindle pole bodies (SPBs) are separated by 1.5–2.0 μm (36), but there is clearly no large separation of centromeres or SPB that is seen when cells enter anaphase. The distance separating fluorescently tagged sister centromeres is not different in wild-type cells with or without a broken chromosome and is only slightly increased in HO-damaged cells in cells lacking chk1Δ (Fig. 2C). We note that these results demonstrate that cells arrested by the Mec1-dependent DNA damage checkpoint are arrested such that sister centromeres are constantly under tension; only in this way can one see two masses of separated GFP-labeled sister centromeres (37–40). This state is maintained for as long as 12–15 h before adaptation occurs, and sister chromatids separate.
The bow-tie DAPI staining could be the result of frequent excursions of the nucleus through the narrow bud neck or by other deformations of the arrested nucleus. By following the movement of GFP-marked SPB in living cells, we examined excursions of the paired SPB back and forth through the bud neck (Fig. 3A and SI Movie 1). The mobility of the paired SPB can be expressed as the total distance traversed, relative to the midpoint of the spindle at T = 0, as a function of time. As shown in the kymographs in Fig. 3B and in SI Fig. 5 and in the plots of the distances traveled (Fig. 3B), in wild-type cells, there is not much difference in the total spindle pole movement in cells traversing G2 (YEP–Glu) and in checkpoint arrested (YEP–Gal) cells. Mitotic spindles remain constant in length and traverse a total of 3.31 ± 0.93 μm over 8.5 min of the ≈20-min period representing the duration of these mitotic spindles (the time of spindle pole separation to spindle pole elongation in wild-type cells). After induction of a DSB, the spindles traverse 3.72 ± 0.65 μm over 8.5 min. Thus, the wild-type response to a DSB is a prolonged duration of G2, without loss of the regulatory processes that maintain spindle length and position.
Fig. 3.

Effect of DNA damage checkpoint deficiencies on nuclear movements. (A) Images from a time course showing the positions of Spc72-RFP-labeled SPBs (arrows) in wild-type and chk1Δ cells. (B) Time course of movements every 30 sec seen by the distance migrated in two dimensions by each of the SPBs (one black, one gray) in a representative wild-type, chk1Δ, and chk1Δ dyn1Δ cell and for rad53Δ sml1Δ and rad53Δ sml1Δ dyn1Δ cells. The damage-independent movement of cdc5-ad and its suppression by dyn1Δ is also shown. A kymograph is shown in the upper right or upper left corner. The plot is a two-dimensional projection of the data. The three (or five) images corresponding to a single time-lapse point were projected to a single image by using only the brightest pixel at any one location in all five image planes. Live cell images can be seen in SI Movie 1. The distances in x and y are in micrometers.
In contrast, 6 h after induction of the DSB, chk1Δ cells showed a statistically significant difference in the motion of the spindle poles (5.07 ± 1.5 μm) compared with undamaged isogenic cells (3.37 ± 1.4 μm), P < 0.05, P = 0.0437. rad53Δ sml1Δ showed a similar behavior, with much greater movement of the SPB in DNA damaged arrest (5.21 ± 1.58). To quantitatively distinguish spindle dynamics in wild-type vs. chk1Δ or rad53Δ, we summed the distance (d) each spindle pole traveled as a function of time and determined the mean. The mean distance traveled for spindle poles in chk1Δ = 0.403 ± 0.105 μm/min and wild type = 0.316 ± 0.014 μm/min. The increase in mean movement confirms that the spindle poles, and hence the spindle, is significantly more motile in cells lacking chk1Δ relative to wild-type cells. The increased frequency of bow-tie DAPI staining can be attributed to movements of the nucleus through the very narrow bud neck without any separation of sister centromeres or spindle elongation. In contrast, a pds1Δ mutant, monitored at the same time, did not show any change in nuclear migrations before and after induction of the DSB (3.61 ± 1.79 vs. 3.58 ± 1.08).
The excursions of the SPB across the bud neck depend on dynein, encoded by DYN1, which we have previously shown is responsible for the movements that orient and move the nucleus through the bud neck before and during anaphase (41–43). A dyn1Δ chk1Δ strain shows the same very limited nuclear migration that is seen in wild-type cells before mitosis (Fig. 3B and SI Fig. 5B). In the dyn1Δ chk1Δ strain, total movement over 8.5 min was reduced to 2.74 ± 1.18 μm, compared with 5.07 ± 1.58 μm in the chk1Δ strain (P < 0.05; P = 0.0195). The absence of dynein also suppressed the movements seen in rad53Δ, from 5.21 (±1.58) μm to 2.22 μm (±0.52).
We carried out a similar analysis on pds1Δ cells during their arrest. There was no significant difference in the movement of the SPBs in glucose (3.58 ± 1.09 μm) vs. galactose (3.05 ± 1.27 μm). Thus, even though the nuclear constraints on mitosis are significantly weakened, there is no correlation with nuclear movements, which remain constrained, presumably because both Chk1 and Rad53 kinases are still active.
In the course of this work, we also examined an adaptation-defective mutation, cdc5-ad, in which a single DSB provokes permanent cell cycle arrest (2, 26). We found that the cdc5-ad mutant exhibited frequent nuclear excursions after DNA damage, but unlike the other mutants we have looked at, there were equivalently large movements even in the absence of DNA damage (SI Fig. 5). Again, these movements were dynein-dependent (Fig. 3B). Because Cdc5 is an essential protein, it was clear that the movements in glucose as well as galactose (4.49 ± 0.75 vs. 4.58 ± 1.76 μm, respectively) represent a gain-of-function phenotype that might reflect a defect in activating the mitotic exit network (see below). The results for cdc5-ad in glucose are significantly different from those in wild-type cells (P = 0.03). In contrast, when we examined a temperature-sensitive cdc5-td mutation that is presumably null at the restrictive temperature, we did not observe the types of nuclear excursions we found for cdc5-ad, at 24°C, 30°C, or 37°C (data not shown). A further indication that cdc5-ad is a gain-of-function mutation is that it is semidominant in preventing adaptation after induction of unrepaired DSBs in a diploid in which both MATa and MATα are cleaved, and HML and HMR are absent (SI Fig. 6).
Discussion
The response of budding yeast cells to a single unrepaired DSB is remarkably complex. Cells experience a Mec1-dependent delay before anaphase, but there is no delay in S phase or activation of an S phase-dependent checkpoint (C. Zierhut and J. F. X. Diffley, personal communication). Recently, Javaheri et al. (44) showed that an HO-induced DSB can cause a very short delay in the G1–S transition, but it is on the order of 15 min and cannot account for the much longer delays seen in rad53Δ and chk1Δ cells. Thus, nearly all of the delay in completing one cell cycle is caused by arrest before anaphase.
Only a deletion of Mec1 completely abolishes the checkpoint response, whereas even the double-mutant combination of rad53Δ chk1Δ shows a significant delay in cell cycle progression. There is also a significant delay in the absence of Pds1. The most striking discovery is that both chk1Δ cells and rad53Δ sml1Δ cells exhibit frequent excursions of the preanaphase spindle back and forth through the narrow bud neck, leading to the spreading out of the DNA and/or distortions of the nucleus (as seen in the bow-tie DAPI staining) without changing sister-centromere or SPB separation from a preanaphase distance. We suggest that the excursion of the nucleus is best explained by the idea that normally there are two components to checkpoint-mediated arrest of mitosis, a constraint on sister-chromatid separation but also a prevention of the movement of the spindle into the bud, as normally occurs in anaphase. In the absence of the complete DNA damage checkpoint, the cytoplasmic microtubule apparatus becomes active in orienting and driving the nucleus toward the end of the bud. In normal mitosis, when one spindle pole interacts with the bud cortex, it activates the mitotic exit network (45, 46), including the phosphatase Cdc14 that triggers telophase and progression into the next cell cycle. If the SPB reaches the bud cortex but the intranuclear DNA damage checkpoint is still in force, Cdc14 may not be activated and preanaphase will continue, resulting in continuing spindle migrations may continue back and forth across the neck. Indeed nuclear excursions such as those we describe have been previously seen in mutants defective in anaphase and later stages of the mitosis (47). Our observation of similar movements in the cdc5-ad mutation of the Polo kinase, but here even in the absence of an induced DSB, could imply that this mutant fails to activate properly the mitotic exit network. One way that this phenotype might arise would be if a key component of anaphase, e.g., the action of the separase Esp1 to destroy sister-chromatid cohesion, was prevented whereas the rest of the anaphase program was executed. Esp1 is sequestered in the cytoplasm until anaphase, and its entry into the nucleus is facilitated by Pds1 (23), but there are likely to be other controls on its transport, which may require the Rad53 and/or Chk1 in the transport of separase into the nucleus (34). Separase also plays a key role in regulating Cdc55 PP2A phosphatase to initiate mitotic exit (48).
We note that the chk1Δ and pds1Δ strains do not carry a sml1Δ deletion, which is carried in the rad53Δ strain (see SI Table 1). Given that sml1Δ does not confer wild-type nuclear migration on the rad53Δ strain, we think it unlikely that sml1Δ would change the behavior of chk1Δ; similarly, given that sml1Δ by itself is not different from wild type in terms of nuclear positioning, we think it unlikely that sml1Δ would change the behavior of pds1Δ.
We had previously shown that the primary DNA damage arrest signals are nuclear limited, so that one damaged nucleus in a common cytoplasm with an undamaged nucleus would not prevent the undamaged nucleus from carrying out mitosis (28). Here, we show that there is a cytoplasmic aspect to the DNA damage checkpoint that prevents a premature initiation of dynein-dependent nuclear orientation and movement. Whether the checkpoint kinases shuttle out and into the nucleus or, more likely, phosphorylate an exported protein will be important to learn. We note that Rad53 has been shown to play another cytoplasmic role, in the regulation of morphogenetic events under replication stress, including the phosphorylation of septins and the timing of degradation of the Swe1 protein kinase (21, 49, 50).
Experimental Procedures
Strains.
All strains are derivatives of JKM139 (MATa) and JKM179 (MATα) that have the genotype hmlΔ::ADE1 hmrΔ::ADE1 leu2-3, 112 ade3::GAL::HO ade1 lys5 ura3–52 (51). A list of strains is given in SI Table 1. Deletions including rad9Δ::KAN, rad53Δ::NAT, sml1Δ::KAN, mec1Δ::NAT, were constructed by PCR-based gene disruptions (52, 53) by using plasmids pKAN-MX2, pAG25, and pAG32 (a gift from John McCusker, Duke University, Durham, NC). Details are available upon request. Multiple mutants were made either by transformation or by obtaining meiotic segregants from dissected tetrads. All mec1Δ and rad53Δ deletions also carry sml1Δ to ensure viability; sml1Δ does not affect checkpoint arrest. A cdc5-ad derivative was constructed by using a pop in/pop out plasmid described by Tozcyski et al. (26). Strain Y63, generously provided by Achille Pellicioli (Universitá degli Studi di Milano), is a JKM179 derivative containing degron-modified CDC5 (cdc5-td), which was constructed by the method of Dohmen et al. (54).
Microscopic Analysis of Length of Arrest.
G1 cells were micromanipulated on grids on YEP–galactose media. Number of cells or buds was counted every 30 min. The time from first budding (to two-celled stage) to next budding (to three- or four-celled stage) was used as a measure of length of arrest.
Cytological Analysis of DNA and Spindles.
Cells were induced with galactose. Five-milliliter aliquots were removed at appropriate times and fixed with 70% ethanol for 1 h at room temperature. Spheroplasts were prepared by treating with 40 μg/ml zymolyase (ICN Pharmaceuticals, Costa Mesa, CA) for 30 min at 37°C. DNA was stained by DAPI (Boeringer, Mannheim, Germany). Cells were visualized under a fluorescent microscope.
Live Imaging of Spindle Pole and Sister-Centromere Separations and Movements.
Cells were grown to logarithmic growth phase in complete media with glucose (YPD). To induce HO endonuclease, cells were harvested by centrifugation, washed once with sterile water, and diluted to an OD of 0.2 in complete media with galactose (YPG) for 4 h. Cells were harvested by centrifugation, washed with sterile water, and resuspended in 50 μl of sterile water. Eight microliters of the cell suspension was applied to a galactose-rich gelatin slab. Image acquisition was carried out as described (55) on a TE2000 microscope (Nikon, East Rutherford, NJ) with a 1.4 N.A., ×100 differential interference contrast (DIC) oil-immersion lens (56). Large budded cells were imaged at 30-s intervals over 8.5 min. Images were acquired with an ORCA II ER cooled CCD camera. Cells were pipetted onto slabs of 25% gelatin containing minimal media ±2% glucose as described by Yeh et al. (57). The microscope was modified for automated switching between fluorescence and DIC by replacement of the camera mount with a filter wheel (BioPoint 99B100; Ludl Electronic Products, Hawthorne, NY) containing the analyzer component of the DIC optics. The computer-controlled (MetaMorph 4.6 software) microscope executed an acquisition protocol taking fluorescence images at 0.75-μm axial steps and a single DIC image corresponding to the central fluorescence image. Fluorescence excitation through a 490/10-nm filter was normally attenuated to 1–10% of the available light from a 100 W mercury arc lamp. Fluorescence emission was collected through a 530/15-nm band-pass filter. DIC images were made by rotating the analyzer into the light path and taking a 0.6-s exposure. The images corresponding to a single time-lapse point were projected to a single image by using only the brightest pixel at any one location. Registration of DIC and fluorescence images was verified by imaging of 1-μm fluorescent beads in DIC and fluorescence modes. After determining the spindle midpoint, the absolute distance traveled by the midpoint over time was measured by using the midpoint position at T0 as a starting point. This value, referred to as total movement, was used to quantify the dynamicity of the spindle in three dimensions. Alternatively, the position of each spindle pole was determined in three dimensions. The change in distance was plotted over successive time points for each pole (d). To estimate the average movement, the sum of the distance traveled at each time point was plotted (Δ of d) vs. time. The slope of this line represents spindle dynamicity. The data from this analysis was indistinguishable from the three-dimensional midpoint analysis.
Supplementary Material
Acknowledgments
We thank Xiaohu Wan (University of North Carolina) for expert technical assistance with data analysis. Achille Pellicioli generously provided the cdc5-td strain. We are grateful to Jake Harrison for his insightful comments and suggestions. This research was supported by National Institutes of Health Grants GM61799 (to J.E.H.) and GM32238 (to K.B.).
Abbreviations
- DIC
differential interference contrast
- DSB
double-strand break
- SPB
spindle pole body.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0609636104/DC1.
References
- 1.Sandell LL, Zakian VA. Cell. 1993;75:729–739. doi: 10.1016/0092-8674(93)90493-a. [DOI] [PubMed] [Google Scholar]
- 2.Lee SE, Moore JK, Holmes A, Umezu K, Kolodner R, Haber JE. Cell. 1998;94:399–409. doi: 10.1016/s0092-8674(00)81482-8. [DOI] [PubMed] [Google Scholar]
- 3.Pellicioli A, Lee SE, Lucca C, Foiani M, Haber JE. Mol Cell. 2001;7:293–300. doi: 10.1016/s1097-2765(01)00177-0. [DOI] [PubMed] [Google Scholar]
- 4.Zhou BB, Elledge SJ. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 5.Lowndes NF, Murguia JR. Curr Opin Genet Dev. 2000;10:17–25. doi: 10.1016/s0959-437x(99)00050-7. [DOI] [PubMed] [Google Scholar]
- 6.McGowan CH, Russell P. Curr Opin Cell Biol. 2004;16:629–633. doi: 10.1016/j.ceb.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 7.Kastan MB. Biomed Pharmacother. 2004;58:72–73. [Google Scholar]
- 8.Chen Z, Xiao Z, Chen J, Ng SC, Sowin T, Sham H, Rosenberg S, Fesik S, Zhang H. Mol Cancer Ther. 2003;2:543–548. [PubMed] [Google Scholar]
- 9.Harrison JC, Haber JE. Annu Rev Genet. 2006;40:209–235. doi: 10.1146/annurev.genet.40.051206.105231. [DOI] [PubMed] [Google Scholar]
- 10.Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, Carotenuto W, Liberi G, Bressan D, Wan L, Hollingsworth NM, et al. Nature. 2004;431:1011–1017. doi: 10.1038/nature02964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rouse J, Jackson SP. Mol Cell. 2002;9:857–869. doi: 10.1016/s1097-2765(02)00507-5. [DOI] [PubMed] [Google Scholar]
- 12.Zou L, Elledge SJ. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 13.Melo JA, Cohen J, Toczyski DP. Genes Dev. 2001;15:2809–2821. doi: 10.1101/gad.903501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kondo T, Wakayama T, Naiki T, Matsumoto K, Sugimoto K. Science. 2001;294:867–870. doi: 10.1126/science.1063827. [DOI] [PubMed] [Google Scholar]
- 15.Zou L, Cortez D, Elledge SJ. Genes Dev. 2002;16:198–208. doi: 10.1101/gad.950302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Majka J, Burgers PM. DNA Repair. 2005;4:1189–1194. doi: 10.1016/j.dnarep.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 17.Majka J, Burgers PM. Proc Natl Acad Sci USA. 2003;100:2249–2254. doi: 10.1073/pnas.0437148100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shiomi Y, Shinozaki A, Nakada D, Sugimoto K, Usukura J, Obuse C, Tsurimoto T. Genes Cells. 2002;7:861–868. doi: 10.1046/j.1365-2443.2002.00566.x. [DOI] [PubMed] [Google Scholar]
- 19.van den Bosch M, Lowndes NF. Cell Cycle. 2004;3:119–122. [PubMed] [Google Scholar]
- 20.Toh GW, Lowndes NF. Biochem Soc Transact. 2003;31:242–246. doi: 10.1042/bst0310242. [DOI] [PubMed] [Google Scholar]
- 21.Smolka MB, Albuquerque CP, Chen SH, Schmidt KH, Wei XX, Kolodner RD, Zhou H. Mol Cell Proteomics. 2005;4:1358–1369. doi: 10.1074/mcp.M500115-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Gramont A, Cohen-Fix O. Trends Biochem Sci. 2005;30:559–568. doi: 10.1016/j.tibs.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 23.Agarwal R, Tang Z, Yu H, Cohen-Fix O. J Biol Chem. 2003;278:45027–45033. doi: 10.1074/jbc.M306783200. [DOI] [PubMed] [Google Scholar]
- 24.Pellicioli A, Lee SE, Lucca C, Foiani M, Haber JE. Mol Cell. 2001;7:293–300. doi: 10.1016/s1097-2765(01)00177-0. [DOI] [PubMed] [Google Scholar]
- 25.Lee S, Pellicioli A, Demeter J, Vaze M, Gasch AP, Malkova A, Brown P, Stearns T, Foiani M, Haber JE. Cold Spring Harbor Symp Quant Biol. 2000;65:303–314. doi: 10.1101/sqb.2000.65.303. [DOI] [PubMed] [Google Scholar]
- 26.Toczyski DP, Galgoczy DJ, Hartwell LH. Cell. 1997;90:1097–1106. doi: 10.1016/s0092-8674(00)80375-x. [DOI] [PubMed] [Google Scholar]
- 27.Leroy C, Lee SE, Vaze MB, Ochsenbein F, Guerois R, Haber JE, Marsolier-Kergoat MC. Mol Cell. 2003;11:827–835. doi: 10.1016/s1097-2765(03)00058-3. [DOI] [PubMed] [Google Scholar]
- 28.Demeter J, Lee SE, Haber JE, Stearns T. Mol Cell. 2000;6:487–492. doi: 10.1016/s1097-2765(00)00047-2. [DOI] [PubMed] [Google Scholar]
- 29.Gardner R, Putnam CW, Weinert T. EMBO J. 1999;18:3173–3185. doi: 10.1093/emboj/18.11.3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanchez Y, Bachant J, Wang H, Hu F, Liu D, Tetzlaff M, Elledge SJ. Science. 1999;286:1166–1171. doi: 10.1126/science.286.5442.1166. [DOI] [PubMed] [Google Scholar]
- 31.Neecke H, Lucchini G, Longhese MP. EMBO J. 1999;18:4485–4497. doi: 10.1093/emboj/18.16.4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garber PM, Rine J. Genetics. 2002;161:521–534. doi: 10.1093/genetics/161.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao X, Rothstein R. Proc Natl Acad Sci USA. 2002;99:3746–3751. doi: 10.1073/pnas.062502299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jensen S, Segal M, Clarke DJ, Reed SI. J Cell Biol. 2001;152:27–40. doi: 10.1083/jcb.152.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Joglekar AP, Bouck DC, Molk JN, Bloom KS, Salmon ED. Nat Cell Biol. 2006;8:581–585. doi: 10.1038/ncb1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pearson CG, Maddox PS, Zarzar TR, Salmon ED, Bloom K. Mol Biol Cell. 2003;14:4181–4195. doi: 10.1091/mbc.E03-03-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He X, Asthana S, Sorger PK. Cell. 2000;101:763–775. doi: 10.1016/s0092-8674(00)80888-0. [DOI] [PubMed] [Google Scholar]
- 38.Goshima G, Yanagida M. Cell. 2000;100:619–633. doi: 10.1016/s0092-8674(00)80699-6. [DOI] [PubMed] [Google Scholar]
- 39.Pearson CG, Maddox PS, Salmon ED, Bloom K. J Cell Biol. 2001;152:1255–1266. doi: 10.1083/jcb.152.6.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tanaka T, Fuchs J, Loidl J, Nasmyth K. Nat Cell Biol. 2000;2:492–499. doi: 10.1038/35019529. [DOI] [PubMed] [Google Scholar]
- 41.Li YY, Yeh E, Hays T, Bloom K. Proc Natl Acad Sci USA. 1993;90:10096–10100. doi: 10.1073/pnas.90.21.10096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bloom K. Curr Biol. 2001;11:R326–R329. doi: 10.1016/s0960-9822(01)00176-2. [DOI] [PubMed] [Google Scholar]
- 43.Yeh E, Yang C, Chin E, Maddox P, Salmon ED, Lew DJ, Bloom K. Mol Biol Cell. 2000;11:3949–3961. doi: 10.1091/mbc.11.11.3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Javaheri A, Wysocki R, Jobin-Robitaille O, Altaf M, Cote J, Kron SJ. Proc Natl Acad Sci USA. 2006;103:13771–13776. doi: 10.1073/pnas.0511192103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Bettignies G, Johnston LH. Curr Biol. 2003;13:R301. doi: 10.1016/s0960-9822(03)00230-6. [DOI] [PubMed] [Google Scholar]
- 46.Smeets MF, Segal M. Cell Cycle. 2002;1:308–311. doi: 10.4161/cc.1.5.143. [DOI] [PubMed] [Google Scholar]
- 47.Palmer RE, Koval M, Koshland D. J Cell Biol. 1989;109:3355–3366. doi: 10.1083/jcb.109.6.3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Queralt E, Lehane C, Novak B, Uhlmann F. Cell. 2006;125:719–732. doi: 10.1016/j.cell.2006.03.038. [DOI] [PubMed] [Google Scholar]
- 49.Enserink JM, Smolka MB, Zhou H, Kolodner RD. J Cell Biol. 2006;175:729–741. doi: 10.1083/jcb.200605080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smolka MB, Chen SH, Maddox PS, Enserink JM, Albuquerque CP, Wei XX, Desai A, Kolodner RD, Zhou H. J Cell Biol. 2006;175:743–753. doi: 10.1083/jcb.200605081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moore JK, Haber JE. Mol Cell Biol. 1996;16:2164–2173. doi: 10.1128/mcb.16.5.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wach A, Brachat A, Pohlmann R, Philippsen P. Yeast. 1994;10:1793–1808. doi: 10.1002/yea.320101310. [DOI] [PubMed] [Google Scholar]
- 53.Goldstein AL, Pan X, McCusker JH. Yeast. 1999;15:507–511. doi: 10.1002/(SICI)1097-0061(199904)15:6<507::AID-YEA369>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 54.Dohmen RJ, Wu P, Varshavsky A. Science. 1994;263:1273–1276. doi: 10.1126/science.8122109. [DOI] [PubMed] [Google Scholar]
- 55.Shaw SL, Yeh E, Bloom K, Salmon ED. Curr Biol. 1997;7:701–704. doi: 10.1016/s0960-9822(06)00299-5. [DOI] [PubMed] [Google Scholar]
- 56.Salmon ED, Shaw SL, Waters J, Waterman-Storer CM, Maddox PS, Yeh E, Bloom K. Methods Cell Biol. 2003;56:185–215. [PMC free article] [PubMed] [Google Scholar]
- 57.Yeh E, Skibbens RV, Cheng JW, Salmon ED, Bloom K. J Cell Biol. 1995;130:687–700. doi: 10.1083/jcb.130.3.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}