

Abstract
The retinoblastoma gene Rb was the first tumor suppressor gene cloned, and it is well known as a negative regulator of the cell cycle through its ability to bind the transcription factor E2Fand repress transcription of genes required for S phase. Although over 100 other proteins have been reported to interact with Rb, in most cases these interactions are much less well characterized. Therefore, this review will primarily focus on Rb and E2F interactions. In addition to cell cycle regulation, studies of Rb and E2Fproteins in animal models have revealed important roles for these proteins in apoptosis and differentiation. Recent screens of Rb/E2Ftarget genes have identified new targets in all these areas. In addition, the mechanisms determining how different subsets of target genes are regulated under different conditions have only begun to be addressed and offer exciting possibilities for future research.
Keywords: Rb, E2F, development, tumorigenesis, cell proliferation and differentiation
The Rb and E2F families of protein
Mammalian Rb family of proteins
Rb, p107 (RBL1) and p130 (RB2) are members of a family of closely related proteins (Figure 1). Together, these proteins are often referred to as the ‘pocket proteins’ because their main sequence similarity resides in a domain, the pocket domain, which mediates interactions with viral oncoproteins as well as cellular proteins to exert the biological functions of this family. A spacer region that is not conserved separates the pocket domain into the A and B pockets. The spacers of p107 and p130 but not Rb contain binding sites for cyclin/cdk complexes (Zhu et al., 1995). The Rb family of proteins also contains numerous phosphorylation sites which can be phosphorylated by the G1phase cyclinD/cdk4 complexes and by the G1/S phase cyclinE/cdk2 and cyclinA/cdk2 complexes (Hinds et al., 1992; Ewen et al., 1993; Kato et al., 1993; Resnitzky et al., 1995; Du et al., 1996a). In general, the hyper-phosphorylated forms of Rb exhibit a decreased ability to interact with their target proteins and to exert their biological functions. As growthstimulating and growth-inhibitory factors generally affect the transcription, translation and stabilities of the D and E type cyclins as well as their inhibitors, these growth-signaling pathways regulate cell proliferation at least in part through regulating the phosphorylation of the Rb family of proteins, with the hypo-phosphorylated Rb being active and preventing transition into S phase (Chellappan et al., 1991).
Figure 1.

The mammalian and Drosophila pocket protein families. The pocket protein family in mammals consists of Rb, p107 and p130, and in Drosophila contains RBF and RBF2. The pocket domain, responsible for most protein–protein interactions, consists of two conserved sequences, A and B. The spacer region between A and B is conserved between p107 and p130 (yellow box), and can bind to cyclin/cdk complexes. The activity of Rb proteins is controlled by phosphorylation at numerous phosphorylation sites (indicated for Rb and RBF by *).
The biological functions of Rb include tumor suppression, regulation of the cell cycle, differentiation and apoptosis. These functions of Rb are mediated by its interaction with a large number of cellular proteins. Currently, over 100 proteins have been reported to interact with the Rb protein (Morris and Dyson, 2001), and most, if not all, of these interactions also involve the pocket domain. The best-studied binding partners of Rb are the E2F transcription factors.
E2F transcription factors in mammals
The E2F transcription factors function as heterodimers that are composed of a subunit of the E2F gene family and a subunit of the DP gene family. In mammalian systems, there are eight E2F family members and two DP family members (for reviews, see Dyson, 1998; Attwooll et al., 2004a). All the E2F and DP proteins, with the exception of the newly identified E2Fs 7 and 8, have both a conserved DNA-binding domain (DBD) and a dimerization domain (Dim, see Figure 2). The E2F family members can be divided into four subgroups. The first subgroup includes E2Fs 1–3 and is often referred to as the ‘activating E2Fs.’ In addition to the DNA-binding and dimerization sequences, these E2F proteins also contain a cdk-binding domain and a nuclear localization sequence near the N-terminus, and a transcriptional activation domain and an Rb-binding sequence near the C-terminus (Figure 2). This subgroup of E2F proteins generally has a role in the activation of E2F target genes at the G1/S transition and is important for proper cell cycle progression. The second E2F subgroup includes E2Fs 4 and 5 and is often referred to as the ‘repressive E2Fs.’ This subgroup of E2F proteins still contains the Rb-binding sequence at the C-terminus but does not have the cdk binding or the nuclear localization sequences in the N-terminus and has nuclear export sequences instead (Dynlacht et al., 1994b; Muller et al., 1997; Gaubatz et al., 2001; Apostolova et al., 2002). Consistent with these structural features, this subgroup of E2Fs functions mainly as repressors of E2F target gene expression, and they translocate into the nucleus when in complex with the Rb family of proteins during the G0/G1 phases of the cell cycle (Muller et al., 1997; Verona et al., 1997). E2Fs 6–8, representing the remaining two subgroups, also function as transcriptional repressors (Trimarchi et al., 1998, 2001; de Bruin et al., 2003a; Di Stefano et al., 2003; Attwooll et al., 2004b; Maiti et al., 2005). However, in contrast to E2Fs 1–5, these E2F proteins do not have the Rb-binding sequence at the C-terminus, and therefore their function and regulation are independent of the Rb family of proteins. These two subgroups of E2F will not be further addressed in this review.
Figure 2.
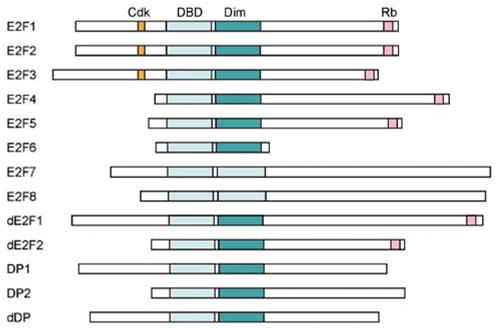
The mammalian and Drosophila E2F protein families. In mammals the E2F family is composed of E2Fs 1–8, DP1 and DP2, while in Drosophila it consists of dE2F1, dE2F2 and dDP. All E2Fs have a conserved DNA-binding domain (DBD) and (except for E2Fs 7 and 8) a dimerization domain for binding to DP (Dim). The mammalian-activating E2Fs 1–3 contain a cyclin/cdk-binding domain (cdk) at the N-terminus and a strong activation domain at the C-terminus. This activation domain overlaps with the Rb-binding domain (Rb) so that binding by Rb masks the activation domain. The repressive E2Fs 4 and 5 also bind the pocket proteins but lack the cyclin/cdk binding domain. E2F 6 shares only the DBD and dimerization domain with the rest of the family, while E2Fs 7 and 8 have two conserved DBDs, allowing them to function without DP to form a heterodimer. The DP proteins are distantly related members of the family that share the DBD and dimerization domain, allowing them to form heterodimers with E2F proteins.
Interestingly, despite the sequence similarities among the Rb family members and among the conserved C-terminal Rb-binding domains of E2F, specific members of the Rb family preferentially interact with specific members of the E2F family. As shown in Figure 3, while Rb preferentially binds to E2Fs 1–4, p107 and p130 predominantly bind to E2Fs 4 and 5 (Classon and Harlow, 2002). The preferential binding of activating E2Fs by Rb but not by p107 or p130 potentially underlies the observation that only Rb mutations are frequently detected in cancers.
Figure 3.
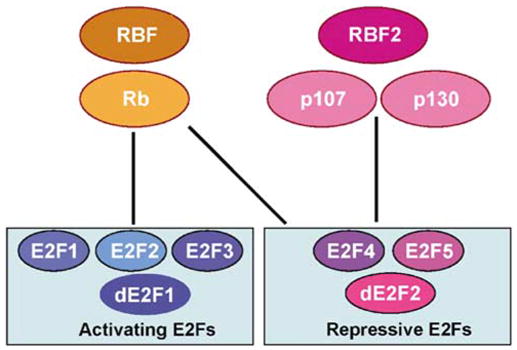
Interactions between the Rb and E2F proteins in mammals and Drosophila. The mammalian activating E2Fs, E2Fs 1–3, interact primarily with Rb. Similarly, the Drosophilaactivating E2F, dE2F1, interacts only with RBF. The mammalianrepressive E2Fs, E2Fs 4 and 5, interact with p107 and p130. In addition, E2F 4 can also interact with Rb. Similarly, the Drosophila-repressive E2F, dE2F2, interacts with both RBF and RBF2.
RB and E2F proteins in Drosophila
There are two E2F (dE2F1 and dE2F2), one DP (dDP), and two Rb family (RBF and RBF2) genes in the Drosophila genome (Dynlacht et al., 1994a; Ohtani and Nevins, 1994; Du et al., 1996a; Sawado et al., 1998; Stevaux et al., 2002). Interestingly, the two Drosophila E2F proteins behave like the first two subgroups of the mammalian E2F proteins: dE2F1 mainly functions as a transcription activator (Du, 2000; Frolov et al., 2001), comparable to the mammalian-activating E2Fs 1–3, while dE2F2 primarily mediates active repression, similar to the mammalian-repressive E2Fs 4 and 5 (Frolov et al., 2001). Furthermore, similar to the mammalian Rb protein that can bind to both the activating and the repressive E2F proteins, RBF can bind to both dE2F1 and dE2F2 proteins in Drosophila (Frolov et al., 2001). In contrast, RBF2 can only bind dE2F2 (Stevaux et al., 2002) analogous to the mammalian p107/p130 proteins that bind preferentially to the repressive E2F proteins (see Figure 3). Thus, the Rb-E2F pathway is well conserved and is much simpler in Drosophila than in the mammalian systems. The advantages of this simplified model system have been exploited in examining the in vivo functions of the different classes of E2F and Rb family members as seen in many of the experiments described below.
Biological functions of the Rb and E2F family of proteins
Studies of the Rb and E2F proteins using the Drosophila model system
As a result of the simplicity of the Drosophila Rb/E2F protein families, significant insights into the biological functions of the E2F/Rb proteins have been derived from studies of this model system. Although there are two RBF genes in Drosophila, RBF2 is not an essential gene, and rbf2 mutant flies show no obvious phenotypes (Stevaux et al., 2005). In contrast, RBF appears to fulfill all of the cell cycle-related function of the Rb family of proteins in Drosophila. Characterization of the phenotypes of embryos devoid of RBF revealed that RBF is required for the repression of E2F target gene expression and for maintaining the first G1 cell cycle arrest during embryonic development (Du and Dyson, 1999). This establishment of the first G1 cell cycle arrest in the developing embryos requires Dacapo, the only member of the p21/p27 family of cdk inhibitors in Drosophila (de Nooij et al., 1996; Lane et al., 1996). Therefore, Rb and the p21/p27 family of cdk inhibitors function cooperatively to achieve a stable G1 cell cycle arrest during fly embryonic development. However, the exact functional relationship between the two families of cell cycle regulators varies in different cell types. For example, in the developing photoreceptor neurons RBF and Dacapo have redundant roles in maintaining G1 cell cycle arrest. Interestingly, in the absence of both RBF and Dacapo, the developing photoreceptor neurons fail to exit the cell cycle and continue to incorporate 5-bromodeoxyuridine (BrdU), in spite of the fact that these cells begin to differentiate and to express post-mitotic markers (Firth and Baker, 2005).
Analysis of the phenotype of flies with mutations of de2f1, the only activating class of E2F in Drosophila, showed that dE2F1 plays a critical role in the expression of the classical E2F target genes, such as RNR2 and proliferative cell nuclear antigen (PCNA), that are important for DNA replication (Duronio et al., 1995). However, in de2f1 mutants, although the rate of BrdU incorporation was significantly reduced, it was not completely blocked (Royzman et al., 1997). This effect is in contrast to that of cyclin E mutant, which showed an all or none effect on BrdU incorporation (Knoblich et al., 1994; Duronio et al., 1998; Follette et al., 1998). Therefore, during normal development dE2F1 is limiting for the expression of replication factors but is not limiting for the expression of cell cycle regulators such as cyclin E. Interestingly, flies with an allele of de2f1 (de2f1i2), which retains the DNA binding and dimerization domain but lacks the transcriptional activation and Rb-binding domain, are viable, but female are sterile due to increased gene amplification in follicle cells (Royzman et al., 1999). As this allele of dE2F1 cannot activate transcription, it was proposed that E2F and Rb proteins may regulate DNA replication directly at the DNA replication origins. Indeed, RBF, dE2F1 and dDP were found to be in a complex with ORC proteins and were bound to the chorion replication origin in vivo (Bosco et al., 2001). Therefore, the E2F/RB complex may have a role in regulating DNA replication at the replication origin in addition to a role in regulating transcription.
In contrast to de2f1, de2f2 mutant flies are viable (Cayirlioglu et al., 2001). Unexpectedly, reducing the gene dosage of rbf, de2f2, or dDP all substantially suppress the phenotype of de2f1 null mutants (Du, 2000; Frolov et al., 2001), suggesting that the phenotype of de2f1 mutants is at least in part due to the presence of an RBF/dE2F2 repressor complex. Characterization of the de2f1 and de2f2 double mutant or dDP mutant flies revealed slower progression through S phase and a defect in the G2/M transition (Frolov et al., 2001). Combining the de2f2 mutant with a novel allele of dE2F1 (dE2F1su89), which retains the transcription activation function but disrupts interaction with RBF, allowed the characterization of the active repression function of Rb/E2F complexes. Interestingly, the tissues that are most affected by the complete removal of Rb/E2F complexes are tissues with extensive endoreplication (Weng et al., 2003). This is apparently due to a requirement for Rb/E2F complexes to downregulate cyclin E levels after each round of S phase for the initiation of additional rounds of endoreplication.
In summary, it appears that the Rb proteins normally function to maintain G1 arrest in conjunction with the p21/p27 family of cdk inhibitors and that active repression by the Rb/E2F complex is required in continuously endoreplicating tissues. Although E2F does not seem to be absolutely required for S phase entry, it is required for the normal progression through S phase and for the G2/M transition. Furthermore, inappropriate activation of E2F, either by ectopic expression of dE2F1 or by mutation of rbf, does lead to ectopic S phase entry and increased apoptosis. These effects of E2F on the cell cycle and apoptosis are likely due to the fact that Rb/E2F regulates expression of multiple cell cycle and apoptotic targets as described in more detail below.
Studies of Rb family proteins in mice
The in vivo roles of Rb and E2F in mammalian systems have been addressed by knockout mice of individual family member. Because of the extensive functional overlap between the family members, such studies generally revealed functions for individual Rb or E2F members in regulating proliferation, apoptosis and differentiation in specific tissues. The best characterized of the pocket protein knockouts are Rb-deficient mice. Rb null mice die at embryonic day 13.5 and demonstrate ectopic S phases and extensive neuronal apoptosis in the central nervous system (CNS) and lens, as well as defects in the differentiation of muscles and red blood cells (Jacks et al., 1992). Some of these defects appear to be due to the unrestrained activity of activating E2Fs, since removal of any of E2F 1, 2 or 3 could decrease ectopic S phase in the CNS, lens and retina (Tsai et al., 1998; Ziebold et al., 2001; Saavedra et al., 2002). In contrast, only loss of E2F 1 could suppress apoptosis in all these tissues, while E2F 3 loss could suppress apoptosis only in the CNS. However, recent reports indicate that many of these Rb−/− defects are not cell autonomous but instead are secondary to placental abnormalities. Development of many tissues in Rb−/− embryos was remarkably restored when provided with wild-type placenta. In particular, although ectopic proliferation continued in the CNS, apoptosis was suppressed, and erythrocyte developmental delays were almost completely suppressed. In contrast, both ectopic proliferation and apoptosis in the lens were unaffected, and the skeletal muscle defect was not restored (de Bruin et al., 2003b; Wu et al., 2003). In light of these findings, the suppression effects of the Rb−/− phenotype by loss of the activating E2Fs will need to be revisited: does the removal of E2F 1 or 3 reduce CNS apoptosis through an indirect effect of restoring placental development? It should be pointed out that a recent study does show a cell intrinsic role of Rb in erythropoiesis under stress conditions (Spike et al., 2004).
Consistent with its role as a tumor suppressor, Rb+/− mice develop pituitary and thyroid tumors (Jacks et al., 1992). In agreement with the idea that the Rb and the p21/p27 family of cdk inhibitors cooperate in cell cycle regulation, removal of p21 or p27 in an Rb+/− background increases tumor development (Brugarolas et al., 1998; Park et al., 1999). Conversely, the incidence of pituitary tumors was decreased by eliminating E2F 1 (Yamasaki et al., 1998) or E2F 3 (Ziebold et al., 2003), although removal of E2F 3 actually increased the incidence of thyroid cancer in Rb+/− mice. This suggests that in at least some tumor types the ability of Rb to suppress tumorigenesis is at least in part due to its inhibition of the activating E2Fs. Surprisingly, removing the repressive E2F 4 also suppressed tumorigenesis in Rb+/− mice (Lee et al., 2002). It was suggested that removal of E2F 4 in Rb−/− tumor cells allowed p107 and p130 to associate with E2Fs 1 and 3, thereby replacing Rb’s ability to repress transcription. Consistent with these results, loss of E2F 4 could repress inappropriate proliferation and E2F target gene expression in Rb−/− cells. These results suggest that although E2F and Rb family members are classified based on their biochemical activities as shown in Figure 3, their biological function may vary under specific in vivo conditions.
In addition to E2F, Rb has been shown to interact genetically with the inhibitor of differentiation protein Id2. The Id family of basic helix–loop–helix proteins lack DNA-binding domains and serve as dominant negative regulators of the bHLH transcription factors involved in differentiation. With the exception of the skeletal muscle defects, removal of Id2 corrected many of the defects of Rb−/− mice (Lasorella et al., 2000). However, given the placental effects recently reported, it is possible that some of these effects could be accounted for by a rescue of the Rb−/− placenta. Although the exact mechanism is not clear, this genetic observation is consistent with the notion that Rb normally can hold Id2 activity in check. In support of this idea, unrestricted Id2 appears to prevent terminal differentiation in fetal liver macrophages from Rb−/− embryos, at least in part by preventing association of the transcription factor PU.1 with its binding sites (Iavarone et al., 2004). Although Rb has no effect on PU.1-binding alone, it is able to reverse Id2 repression. An additional genetic interaction has been shown in tumor suppression: Rb+/− Id2−/− pituitary glands show decreased proliferation, premature differentiation, and decreased pituitary tumor incidence relative to Rb+/− glands, presumably due to loss of Id2’s inhibitory effect on transcription factors involved in differentiation (Lasorella et al., 2005). However, this observed effect may not reflect a direct physical interaction between Rb and Id2 since these two proteins may control different events in pituitary development.
In contrast to Rb−/− mice, p107 or p130 null animals appear to develop normally, although p107−/− ;p130−/− animals are not viable, apparently due to defects in chondrocytes leading to bone abnormalities (Cobrinik et al., 1996), suggesting some essential, tissue-specific roles for these proteins. In addition, although these proteins are less frequently found to be mutated in tumors, they do play a role in tumor suppression in the absence of Rb. For example, the additional loss of p107 in Rb−/− chimaeric mice is sufficient for development of retinoblastoma (Robanus-Maandag et al., 1998). More recently Rb+/−;p107−/− chimaeras were shown to develop a broad spectrum of tumors, most of which showed loss of the remaining Rb allele, indicating that p107 is an important tumor suppressor in the absence of Rb (Dannenberg et al., 2004). Similarly, Rb +/−;p130−/− and Rb−/− ;p130−/− chimaeras were also tumor prone. In particular, Rb−/−;p130−/− mice showed consistent development of retinoblastomas, pheochromocytomas and neuroendocrine hyperplasia in the lungs, suggesting that p130 is particularly important in restraining tumor development in these tissues (Dannenberg et al., 2004).
In summary, studies in the mouse models have shed light on the role of the Rb family of proteins in normal development and tumorigenesis. These phenotypes are likely the consequences of the roles of the Rb family of proteins in the control of cell cycle, apoptosis, and differentiation as discussed in detail below.
Cell cycle roles of the Rb family of proteins
The Rb family of proteins are best known for their ability to regulate the cell cycle, and most studies point to E2F as the key target that mediates the cell cycle role of Rb proteins. Compound knockout of E2Fs 1–3 leads to cell cycle arrest of mouse embryonic fibroblasts (MEFs) with an accumulation of the cdk inhibitor p21 (Wu et al., 2001). This is similar to RNAi knock-down of dE2F1 in Drosophila SL2 cells (Frolov et al., 2003), supporting the notion of a conserved role for the activating E2F proteins. Although simplified models suggest that the Rb/E2F complexes present during quiescence or G0 are replaced with free E2Fs at the G1/S transition, the actual composition of these complexes appears to vary with the cell cycle (Classon and Harlow, 2002; Bracken et al., 2004). In quiescent cells, p130 is the main pocket protein in complex with the inhibitory E2Fs, while in cycling cells it is replaced by p107/E2F 4 in G0/G1 phases and by Rb/E2F 1–3 complexes in S phase (Shirodkar et al., 1992). Although MEFs deficient in any one of the inhibitory E2Fs or in both p107 and p130 proliferate normally, they show defects in response to growth inhibitory signals. Triple knockout MEFs deficient in all three pocket proteins show increased entry into S phase, loss of contact inhibition, and resistance to senescence and DNA-damage-induced G1 arrest (Dannenberg et al., 2000; Sage et al., 2000).
In addition to control of the G1/S transition, Rb has also been implicated in regulating other phase of the cell cycle as well as cell cycle checkpoints. For example, overexpression of dE2F1 in the Drosophila developing wing accelerates both G1/S and G2/M transitions, while overexpression of RBF slowed all phase of the cell cycle, with the greatest effect on S-phase duration (Neufeld et al., 1998). These cell cycle effects are likely due to the ability of RBF and dE2F1 to control the expression of targets such as cyclin E and string (Drosophila cdc25), as well as multiple DNA replication factors that regulate the progression of different cell cycle phases. Recent screens of Rb/E2F target genes by microarray have also identified cell cycle checkpoint genes (Polager et al., 2002; Ren et al., 2002). For example, Mad2, a spindle checkpoint gene, was recently identified as an E2F target. Removal of Rb or overexpression of E2F 1 was sufficient to upregulate expression of Mad2 throughout the cell cycle. Although increased expression of a spindle checkpoint gene might be expected to inhibit tumorigenesis, removal of Rb or overexpression of Mad2 was sufficient for abnormal chromosome accumulation and delayed exit from mitosis. Significantly, partial suppression of Mad2 levels was sufficient to reverse the chromosomal abnormalities associated with removal of Rb (Hernando et al., 2004).
Rb is also involved in senescence, a permanent, nonreversible exit from the cell cycle. While pocket protein members may compensate for one another to some extent, Rb appears to be particularly important for senescence, and MEFs that are acutely inactivated for Rb are able to re-enter the cell cycle, even from a senescent state (Sage et al., 2003), in spite of intact p107 and p130. Senescent cells show nuclear reorganization with foci of DNA accumulation that correlate with transcriptionally inactive genes (Narita et al., 2003). These regions exhibit characteristics of heterochromatin: histone H3 methylated on lysine 9 and HP1 protein accumulation. These modifications are associated with the promoters of E2F target genes only during senescence and not in merely quiescent cells. Interestingly, although p107 and p130 were found at the promoters of E2F target genes in quiescent cells, Rb was only detected in senescent cells, suggesting a particular role for Rb in inducing senescence (Narita et al., 2003). Interestingly, these E2F targets could not be upregulated in senescent cells even with overexpression of E2F 1, while removal of Rb or p16 with siRNA decreased heterochromatin foci and allowed expression of E2F target genes.
The role of Rb in apoptosis
As indicated by the apoptosis in various tissues in Rb−/− mice, the Rb family of proteins has been found to suppress apoptosis. Some of the ability of Rb to limit apoptosis may be due to its ability to control the cell cycle: in response to DNA-damaging agents quiescent cells are more resistant to apoptosis than dividing cells, and overexpression of Rb is similar in effect to other modifications that arrest the cell cycle (Chau and Wang, 2003; Masselli and Wang, 2006). Similarly, models have proposed that the induction of apoptosis in Rb−/− cells is indirectly due to their continued proliferation as they initiate differentiation, and that this conflict signals for apoptosis.
However, other studies indicate that Rb has a direct role in suppressing apoptosis. For example, Rb can be cleaved by caspases, and mice engineered to express a caspase-resistant form of Rb show defects in tumor necrosis factor (TNF)-induced apoptosis in several tissues (Chau et al., 2002). Unlike wild-type Rb, caspase-resistant Rb could inhibit TNF-induced apoptosis in fibroblasts treated with cycloheximide, even though both populations arrested in G1. These results further suggest that Rb has antiapoptotic effects independent of its ability to regulate gene expression. As proteins reported to interact with Rb such as c-Jun N-terminal kinase and c-ABL (Morris and Dyson, 2001; Chau and Wang, 2003) can also induce apoptosis, it is possible that their interaction with Rb might also be able to inhibit their pro-apoptotic signaling. Interestingly, the caspase-resistant form of Rb is unable to prevent DNA-damage-induced apoptosis, suggesting that in addition to cell type, the role of Rb in suppressing apoptosis may depend on the apoptotic stimulus.
Regulation of E2F transcription does appear to be relevant to Rb’s antiapoptotic effect in some tissues. Many genes involved in apoptosis are direct targets of E2F, overexpression of E2F 1 has been shown to induce apoptosis, and E2F 1−/− thymocytes are defective in undergoing apoptosis (Field et al., 1996). Whether this ability to induce apoptosis is restricted to E2F 1 or is shared by other activating E2Fs is still unclear. E2F targets related to apoptosis include APAF-1, caspases 3 and 7 and p73 (Chau and Wang, 2003). Although some apoptosis appears to be independent of p53, E2F can activate the p53 pathway through upregulation of Arf and Pin-1, both of which contribute to p53 stabilization. The ability of E2F to affect apoptosis would be expected to inhibit tumor formation by leading to apoptosis of cells with excessive E2F activity. Consistent with this, E2F 1−/− mice are surprisingly tumor prone (Yamasaki et al., 1996), although they show a different tumor spectrum from that of Rb+/− mice. Tumor incidence of E2F 1−/− mice can be further increased by removing E2F 2 (Zhu et al., 2001), but not E2F 3, suggesting that tumor suppression is a characteristic only of E2Fs 1 and 2 (Cloud et al., 2002).
The relative importance of loss of Rb-mediated repression versus activation by E2Fs is still not clear and may vary with the cell type or apoptotic stimulus. In human fibroblasts, overexpression of an E2F DNAbinding domain construct can induce cell death regardless of the phase of the cell cycle, indicating that Rb’s active repression of target genes is important to inhibit apoptosis (Young and Longmore, 2004). In contrast to E2F-dependent cell cycle genes, Rb was found at the promoter of E2F apoptotic targets at all stages of the cell cycle in human fibroblasts. This was distinct from mouse cells, where Rb was never detected at E2F apoptotic targets, although the repressive E2F 4 and other pocket proteins were present. These results may also reflect the fact that human fibroblasts showed significantly higher levels of nuclear Rb than did mouse fibroblasts (Young and Longmore, 2004). In addition, different pocket proteins appear to be important in inhibiting apoptosis in different tissues. In post-mitotic neurons, p130 is a key player in suppressing expression of E2F target genes by binding to E2F 4. In fact, overexpression of p130 is sufficient to protect neurons from apoptotic stimuli, and removal of p130 with siRNA is sufficient to induce apoptosis. This suppressive activity of p130 requires its interaction with corepressors SUV39H1 and histone deacetylases (HDACs) (Liu et al., 2005).
A role for Rb and E2F in regulating apoptosis was also observed in Drosophila, where overexpression of dE2F1 or removal of RBF all led to increased apoptosis (Asano et al., 1996; Du et al., 1996b; Du and Dyson, 1999). dE2F1 was shown to be required for UV irradiation-induced expression of the pro-apoptotic gene hac-1 (the Drosophila homolog of APAF-1) (Zhou and Steller, 2003). Although this pro-apoptotic role of activating E2Fs has been firmly established in both Drosophila and mammalian systems, recent evidence indicates that dE2F1 can also play a protective role in inhibiting UV induced apoptosis. Specifically, developing wing discs show UV-dependent apoptosis in the intervein area but not at the dorsal/ventral boundary. Wing discs lacking dDP or deficient in dE2F1 show a reversal of this phenotype, with decreased apoptosis in the intervein area and increased apoptosis at the D/V boundary. These apoptotic effects are apparently due to a repression of the apoptotic regulator hid by RBF and the activating E2F dE2F1 (Moon et al., 2005).
The role of Rb in differentiation
Similar to indirect models of apoptosis, loss of Rb could indirectly prevent differentiation if cells unable to exit the cell cycle could not differentiate. In some hematopoetic cell types, Rb null cells cannot reach the final stage of differentiation, and in mice these cells may contribute to myeloproliferative disorders. It has been speculated that this is due to the increase in the pool of precursor cells that can give rise to tumors (Spike et al., 2004). In other cases, the loss of Rb appears to disrupt the coordination between cell cycle exit and differentiation. In Rb−/− skin for example, suprabasal keratinocytes continued to divide even though they lost expression of basal keratins and induced expression of more differentiated keratin (Ruiz et al., 2004). In sensory hair cells of the ear, Rb is normally upregulated during terminal differentiation, but when Rb is removed there is increased proliferation not only of the proliferating progenitors but also of the normally postmitotic, differentiated cells (Sage et al., 2005). Although these mature cells showed abnormal orientation, they were functional even while expressing proliferation markers such as PCNA, and acute loss of Rb in these mature cells was sufficient for them to re-enter the cell cycle. These examples demonstrate that, at least in some tissues, Rb plays a critical role in coordinating cell cycle exit with differentiation and in maintaining the quiescent state of the differentiated cells, and loss of Rb does not prevent differentiation even when cell cycle exit is blocked.
Recent studies showed that Rb and E2F proteins may have a more direct role in differentiation. The importance of E2F and pocket protein activity has been shown in confluent (non-dividing) MEFs that are induced to differentiate into adipocytes. Under these conditions, removal of E2F 4 results in spontaneous differentiation even though the cell cycle is not affected, suggesting that E2F 4 suppresses differentiation independently of proliferation (Landsberg et al., 2003). Recently, lung-specific deletion of Rb has been reported. Not unexpectedly, these mice showed both increased proliferation and increased apoptosis in the lung. More interestingly, these lungs demonstrated a specific effect on the neuroendocrine cells, which are normally scattered at airway branch points. Not only were these clusters comprised of more cells when Rb was ablated, but there was also an increased number of individual aggregates, suggesting that Rb normally prevents cells from adopting the neuroendocrine fate (Wikenheiser-Brokamp, 2004). Although this ability to restrict differentiation has not been genetically separated from the ability to restrict proliferation, it is potentially of particular interest in tumorigenesis since small cell lung cancer, which is derived from neuroendocrine cells, is specifically associated with loss of Rb function.
In addition to regulating E2F, Rb has been shown to interact with numerous other proteins. The best characterized of these are involved in differentiation and include basic helix–loop–helix transcription factors such as Id and MyoD. The muscle defect of Rb−/− embryos appears to be due to an inability of muscle cells to complete differentiation (Rb family-mediated muscle differentiation is reviewed by De Falco and Simone in this issue). Muscle cells lacking Rb can growth arrest, but they show delayed upregulation of late differentiation markers and are capable of re-entering the cell cycle when stimulated with mitogens. Rb can cooperate with the transcription factor MyoD to induce transcription of muscle target genes, and MyoD was shown to bind to Rb (Gu et al., 1993), although the physiological significance of these interactions is unclear. An alternative model suggests that Rb may promote differentiation indirectly by sequestering inhibitors of MyoD (Nguyen and McCance, 2005). Recently, specific posttranslational modifications have been demonstrated to affect the ability of Rb to mediate differentiation. Differentiated myotubes showed an increase in acetylated Rb, and an acetylation site mutant of Rb, although able to acutely arrest the cell cycle, was not able to cooperate with MyoD to induce differentiation and maintain permanent growth arrest (Nguyen et al., 2004). One model for Rb activation of MyoD involves its ability to sequester inhibitors such as HDACS or E1alike inhibitor of differentiation (EID-1) (MacLellan et al., 2000; Miyake et al., 2000). Non-actetylated Rb appears less able to bind MDM2 and therefore has a decreased ability to target EID-1 for degradation (Nguyen et al., 2004). Verification of this role of Rb in differentiation in vivo will require further use of mouse models.
Transcriptional targets of Rb and E2F proteins
Since Rb and E2F proteins are transcriptional regulators, identification of their targets is critical to our understanding of their biological functions. Rb and E2F proteins were originally known for their ability to regulate the G1/S transition, so E2F target genes were initially identified by examining the promoters of genes known to be turned on at the G1/S transition for E2Fbinding sites. These early studies identified E2F target genes that are either cell cycle regulators (such as cyclins E, A and B) or DNA replication factors (such as PCNA, dihydrofolate reductase (DHFR) and thymidine kinase).
More recently, microarray technology has allowed identification of E2F target genes at the genome level. Several different approaches have been carried out. Initial microarray screens focused on the overexpression of E2F or Rb family members (Muller et al., 2001; Polager et al., 2002). While these studies identified large arrays of altered genes, they cannot distinguish genes directly regulated by Rb or E2F from genes that changed expression due to secondary effects of Rb or E2F expression (such as alterations in the cell cycle). Furthermore, overexpression of E2Fs or Rb might also result in non-physiological binding and activation or repression.
To identify potential direct targets of Rb and E2F proteins, a second generation of screens used the so called chromosomal immunoprecipitation (ChIP) on CHIP approach to identify genes which have promoters bound to endogenous E2F/Rb (Ren et al., 2002). Specifically, small DNA fragments that are bound by the endogenous Rb and E2F proteins are enriched by Chromosomal IP and are then amplified and hybridized to a promoter array. However, as the complete human promoter microarray is not currently available, the current ChIP on CHIP screens have used either a promoter library of genes known to be regulated in a cell–cycle-dependent manner (Ren et al., 2002) or a library of CpG islands (Wells et al., 2003), as GC rich regions are often found at promoters and origins of replication. The ChIP on CHIP screen with the cell–cycle-regulated promoter array found that in quiescent cells, antibodies against the repressive E2F 4 precipitated the promoters of genes involved not only in the cell cycle but also in DNA replication, DNA damage and repair, chromosome condensation and separation, the G2/M checkpoint, and mitosis. Many components of multimeric complexes appeared in the screen, suggesting that they are transcriptionally co-regulated. The importance of pocket protein repression in controlling these genes was confirmed by the finding that in p107−/−;p130−/− MEFs many of these genes were deregulated, as expected if repression by E2F 4 complexes controlled their expression. In S-phase cells, the activating E2F 1 precipitated primarily genes involved in DNA replication and repair, suggesting that these genes require not only de-repression but also activation. In the ChIP on CHIP screen of CpG islands, targets other than cell cycleregulated genes were found, including genes involved in embryogenesis, differentiation and development. As the binding of E2F or Rb to a given promoter does not necessary mean that E2F or Rb will be the rate-limiting determinant of expression for that gene, combining the microarray gene expression data and the CHIP on CHIP data should allow a better identification of Rb/E2F target genes. In addition, these newly identified genes can be studied to determine whether they are important to the biological roles of Rb/E2F described above.
The relative simplicity of the Drosophila Rb and E2F proteins has made it an ideal system for the genomewide identification of Rb/E2F targets. Specifically, RNAi has been used to deplete the activating E2F (dE2F1), the inhibitory E2F (dE2F2), dDP, RBF or RBF2 from Drosophila cell lines (Dimova et al., 2003) or specific tissues (Stevaux et al., 2005). Although it is possible that secondary changes in gene expression could have occurred, the screen did detect actual changes in expression, suggesting that the E2F/Rb interactions were rate limiting under these conditions. A surprising result of this screen was that very few genes showed both a decrease in expression in the absence of dE2F1 and an increase in expression in the absence of dE2F2, suggesting that most genes are more influenced either by repression or by activation rather than by both activities equally (Dimova et al., 2003). Most of the genes found to be regulated largely by dE2F1 were involved in processes similar to those found in the ChIP on CHIP screens, including the cell cycle, DNA replication and repair, mitosis, chromosome segregation, and checkpoints. However, the genes regulated primarily by dE2F2 were not involved in DNA repair or S-phase processes. While the functions of many genes in this category are unknown, a surprising number appear to be unrelated to the cell cycle and instead are involved in development, including male and female specific genes. The importance of dE2F2 repression was confirmed by finding that expression of many of these genes was deregulated in de2f2 null flies.
Further examination of repression of these differentiation genes has highlighted the complexity of Rb/E2F control of transcription and the importance of interactions with other factors. In comparing the requirement for dE2F2 and RBF2 to repress expression in Drosophila cell lines, ovaries and embryos, microarray data showed that different genes were deregulated in each tissue with surprisingly little overlap, suggesting cell-type specific requirements for expression (Stevaux et al., 2005). Comparison of dE2F2 and RBF2 binding to the promoters of these genes showed that in some cases the differences in expression correlated with dE2F2/RBF2 occupancy of the promoter, suggesting that cell type-specific factors determine dE2F2/RBF2 distribution. In contrast, at other promoters dE2F2 and RBF2 were always present, suggesting that the functional relevance of promoter occupancy also varies with cell type. These findings emphasize the need to further understand the mechanisms determining Rb and E2F functions in specific contexts.
Mechanisms of transcriptional control
E2F and Rb control transcription through a variety of mechanisms. While E2F alone can activate transcription, binding by Rb not only blocks transcriptional activation but also leads to active repression. Inhibition of Rb function would be predicted to both remove active repression and to allow transactivation by E2F. The relative importance of de-repression versus activation may depend on the specific target. For example, mutation of the E2F-binding site upstream of B-myb and cyclin E results in premature expression, reflecting a requirement for active suppression. In contrast, similar mutations result in loss of appropriate upregulation of DHFR and thymidine kinase, indicating a requirement for activation (Mundle and Saberwal, 2003).
A role for chromatin remodeling by Rb as a part of its repressive activity was first identified by the interaction of Rb with BRG1, a component of the human SWI/SNF complex. BRG1 interacts with the unphosphorylated pocket domain, and in BRG1-deficient cells its re-expression is capable of conferring a growth arrested, flat cell phenotype (Dunaief et al., 1994). However, it has since been shown that the Rb-binding site of BRG1 is not conserved in the Drosophila BRG homolog, and mutation of human BRG1 so that it could no longer bind Rb did not affect its ability to arrest cells in G1, suggesting that the genetic interaction between BRG1 and Rb does not reflect a direct biochemical interaction. Instead, it appears that BRG1 induces expression of a cyclinE/cdk2 inhibitor, resulting in decreased phosphorylation of Rb, thus allowing it to remain in an active state (Kang et al., 2004).
One mechanism of Rb repression is through recruitment of co-repressors. Rb was first shown to interact with HDAC 1 through its pocket domain (Brehm et al., 1998). Although HDAC and E2F both bind to Rb’s pocket domain, they can simultaneously interact with hypophosphorylated Rb, allowing HDAC to be recruited to E2F sites to repress transcription. Rb that has been phosphorylated by cyclinD/cdk4 cannot bind to HDACs, allowing the expression of targets such as cyclin E (Zhang et al., 2000). This mode of repression by Rb is in direct contrast to activation by E2F, which can bind to the histone acetylases (HATs) p300/CBP and P/CAF, making the DNA more accessible to transcription factors. RB/E2F-binding status, acetylation of E2F-dependent promoters and induction of transcription have been shown to be coordinated in a number of studies examining the proteins localized to the promoters of E2F-dependent genes at different stages of the cell cycle (Takahashi et al., 2000; Ferreira et al., 2001; Rayman et al., 2002; Caretti et al., 2003; Taubert et al., 2004). As compared to quiescent cells, in late G1 there is a loss of the pocket proteins and repressive E2Fs (E2F 4/p130), which are replaced by activating E2Fs. At the same time, there is a switch from HDACs to HATs. This is accompanied by hyperacetylation of histones H3 and H4 and the initiation of E2F-dependent gene expression (Frolov and Dyson, 2004).
Rb may also repress transcription through its ability to recruit histone methylase activity. Endogenous Rb associates with SUV39H1, an enzyme which methylates lysine 9 on histone H3. Additionally, HP1, a protein which binds methylated lysine 9 and is associated with transcriptionally silent regions of chromatin, can bind Rb (Nielsen et al., 2001). Furthermore, Rb interacts with the DNA methyltransferase enzyme DNMT1, which cooperates to repress reporter genes (Robertson et al., 2000); however, DNA methylation was not detected at the repressed promoters, suggesting that the effect of DNMT1 may not be through its enzymatic activity but instead might help recruit other co-repressors.
While general mechanisms of repression and activation have been well studied, more recent efforts have been directed toward elucidating the mechanisms by which various E2F and Rb family members carry out their distinct functions. Since the crystallization of an E2F 4/DP DNA-bound heterodimer suggested that all the DNA-contacting residues are conserved among E2F family members (Zheng et al., 1999), it is unlikely that specificity is conferred by variations in the DNA sequence of the E2F-binding sites. An alternative hypothesis is that specificity of E2F family members is determined by their interaction with other transcription factors (Figure 4). The marked box domain of E2F family members may play a role in this specificity. This domain, directly downstream of the DBD, was first found to be important in the interaction of E2F 1 with the adenoviral protein E4, and it allowed for a synergistic effect of these two proteins on the activation of the E2 promoter, leading to the hypothesis that this region might also bind to cellular transcription factors (Jost et al., 1996). This was confirmed by studies examining the specificity of E2F 1 induced apoptosis. Construction of chimaeric proteins containing E2Fs 1 and 3 sequences indicated that the marked box domain and adjacent regions of E2F 1 were critically involved in the ability of activating E2Fs to induce apoptosis (Hallstrom and Nevins, 2003).
Figure 4.
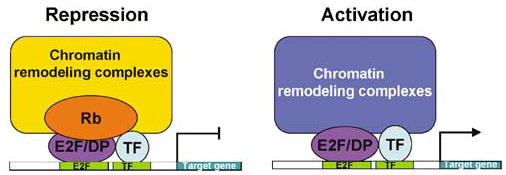
Combinatorial control of gene expression by Rb/E2F with other transcription factors. Owing to specific interactions with other transcription factors, individual members of the Rb and E2F families can differentially regulate specific target genes. As shown in the repression model, specific Rb/E2F complexes may be preferentially recruited to an E2F-binding site based on the ability of Rb and/or E2F to bind to another transcription factor with an adjacent binding site. Similarly, in the activation model, the ability of individual E2F family members to bind to other transcription factors may preferentially recruit both activators to promoters containing adjacent binding sites, synergistically activating transcription.
Indeed, the ubiquitous E box-binding transcription factor TFE-3 was identified in a yeast two-hybrid screen using the marked box domain of E2F 3 as bait to identify interacting proteins (Giangrande et al., 2003, 2004). Although this marked box domain shows 55% homology between E2Fs 1 and 3, TEF-3 specifically bound only to E2F 3. E2F 3 and TFE-3 could synergistically activate p68, a polymerase α subunit with both an E-box and E2F sites proximal to the promoter, and they could directly bind to one another through the E2F 3 marked box domain. In fact, the synergistic activation required this interaction, since an E2F 3 chimaera with the E2F 1 marked box domain could not activate transcription, and in the absence of E2F 3, TFE-3 was unable to bind to the p68 promoter. These findings suggest that the specificity of transcriptional activation by individual E2F proteins is determined at least in part by the transcription factors that bind synergistically with the E2F transcription factor (Figure 4).
Similarly, the repressive E2Fs, E2Fs 4 and 5 were found to specifically interact with the SMAD transcription factors (Chen et al., 2002). In response to transforming growth factor-β (TGF-β) signaling, SMAD2 and 3 are phosphorylated, allowing them to bind to SMAD4, and this complex can either activate or repress transcription of target genes. The TGF β response element of c-myc contains a consensus E2F site adjacent to the SMAD-binding site, and mutation of either site led to decreased SMAD binding and loss of TGF-β responsiveness. Chromatin immunoprecipitation experiments demonstrated that SMADs 2–4, as well as E2Fs 4 and 5 and p107 were found at the TGF-β response element, but that the other RB family members and activating E2Fs were absent. In fact, SMAD3 could directly bind to both E2Fs 4 and 5 as well as p107. This interaction was required for repression of c-myc in response to TGF-β since c-myc expression did not decrease in E2F 4−/−;E2F 5−/− or p107−/− cells. This repressive ability of E2Fs 4 and 5 may therefore, in some cases, be due to their ability to be recruited to the promoter by their specific interaction with other transcription factors (Figure 4).
Post-translational modifications of E2F 1 may also regulate its ability to associate with different E2F promoters, although specific co-factors have not been defined. In response to DNA damage, E2F 1 was shown to re-localize from cell cycle promoters to some apoptotic targets, such as p73. E2F 1 was acetylated in response to DNA damage, and expression from the p73 promoter but not the DHFR promoter depended both on E2F 1 acetylation and on the acetyltransferase p300/CBP-associated factor (PCAF). This model suggests that acetylation of E2F 1 may regulate its ability to interact with other transcription factors, adapting its localization under different circumstances. Further, these other factors may specifically require the recruitment of PCAF rather than other acetyltransferases such as p300 that can also activate E2F-dependent transcription (Pediconi et al., 2003).
Finally, recent work in Drosophila has also contributed to further understanding of the differences between activating and repressive E2Fs. In an attempt to understand the specific function of dE2F2, the repressive E2F, native complexes were purified from embryo extracts (Korenjak et al., 2004). In addition to either RBF or RBF2, these complexes contained components of the dMyb complex, a known transcriptional regulator. The dMyb complex showed no co-staining with actively transcribed regions of chromatin, consistent with its association with repressive E2Fs. Depletion of these dMyb subunits by RNAi resulted in upregulation of genes normally controlled exclusively by dE2F2 repression, demonstrating that dMyb is required for dE2F2/RB repression. Rather than playing a role in the cell cycle, these E2F target genes are involved in differentiation, often in expression of sex-specific genes. Interestingly, the Myb/Rb/E2F interaction also appears to be evolutionarily conserved: Rb and components of the Myb complex form part of the synMuv class of genes in Caenorhabditis elegans that control vulval development, and, although less studied, the human Myb homologs also appear to bind to Rb directly.
Concluding remarks
In summary, regulation of specific target genes by Rb/E2F depends not only on the presence of specific Rb and E2F family members but also on their post-translational modifications and other interacting factors. Although Rb has been extensively studied since its identification as the first tumor suppressor gene, much remains to be discovered about both Rb and E2F’s physiological roles and the specific mechanisms of action that account for their roles. Recent molecular discoveries have begun to provide a mechanistic explanation for the observation that some target genes are controlled by specific E2F and Rb family members, and further understanding of the specific interactions between E2F/Rb family members and other transcription factors may also explain complex patterns of timing and levels of induction. The mechanisms and targets found in biochemical and microarray experiments will need to be examined in vivo to determine which interactions and targets are crucial for determining the physiological outcome.
Acknowledgments
We thank Dr Kay Macleod for reading of this paper. This work is supported by grants from NIH and ACS to WD. WD is a Scholar of the Leukemia and Lymphoma Society.
References
- Apostolova MD, Ivanova IA, Dagnino C, D’Souza SJ, Dagnino L. J Biol Chem. 2002;277:34471–34479. doi: 10.1074/jbc.M205827200. [DOI] [PubMed] [Google Scholar]
- Asano M, Nevins JR, Wharton RP. Genes Dev. 1996;10:1422–1432. doi: 10.1101/gad.10.11.1422. [DOI] [PubMed] [Google Scholar]
- Attwooll C, Denchi EL, Helin K. EMBO J. 2004a;23:4709–4716. doi: 10.1038/sj.emboj.7600481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwooll C, Oddi S, Cartwright P, Prosperini E, Agger K, Steensgaard P, et al. J Biol Chem. 2004b;280:1199–1208. doi: 10.1074/jbc.M412509200. [DOI] [PubMed] [Google Scholar]
- Bosco G, Du W, Orr-Weaver TL. Nat Cell Biol. 2001;3:289–295. doi: 10.1038/35060086. [DOI] [PubMed] [Google Scholar]
- Bracken AP, Ciro M, Cocito A, Helin K. Trends Biochem Sci. 2004;29:409–417. doi: 10.1016/j.tibs.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. Nature. 1998;391:597–601. doi: 10.1038/35404. [DOI] [PubMed] [Google Scholar]
- Brugarolas J, Bronson RT, Jacks T. J Cell Biol. 1998;141:503–514. doi: 10.1083/jcb.141.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caretti G, Salsi V, Vecchi C, Imbriano C, Mantovani R. J Biol Chem. 2003;278:30435–30440. doi: 10.1074/jbc.M304606200. [DOI] [PubMed] [Google Scholar]
- Cayirlioglu P, Bonnette PC, Dickson MR, Duronio RJ. Development. 2001;128:5085–5098. doi: 10.1242/dev.128.24.5085. [DOI] [PubMed] [Google Scholar]
- Chau BN, Borges HL, Chen TT, Masselli A, Hunton IC, Wang JY. Nat Cell Biol. 2002;4:757–765. doi: 10.1038/ncb853. [DOI] [PubMed] [Google Scholar]
- Chau BN, Wang JY. Nat Rev Cancer. 2003;3:130–138. doi: 10.1038/nrc993. [DOI] [PubMed] [Google Scholar]
- Chellappan SP, Hiebert S, Mudryj M, Horowitz JM, Nevins JR. Cell. 1991;65:1053–1061. doi: 10.1016/0092-8674(91)90557-f. [DOI] [PubMed] [Google Scholar]
- Chen CR, Kang Y, Siegel PM, Massague J. Cell. 2002;110:19–32. doi: 10.1016/s0092-8674(02)00801-2. [DOI] [PubMed] [Google Scholar]
- Classon M, Harlow E. Nat Rev Cancer. 2002;2:910–917. doi: 10.1038/nrc950. [DOI] [PubMed] [Google Scholar]
- Cloud JE, Rogers C, Reza TL, Ziebold U, Stone JR, Picard MH, et al. Mol Cell Biol. 2002;22:2663–2672. doi: 10.1128/MCB.22.8.2663-2672.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobrinik D, Lee MH, Hannon G, Mulligan G, Bronson RT, Dyson N, et al. Genes Dev. 1996;10:1633–1644. doi: 10.1101/gad.10.13.1633. [DOI] [PubMed] [Google Scholar]
- Dannenberg JH, Schuijff L, Dekker M, van der Valk M, te Riele H. Genes Dev. 2004;18:2952–2962. doi: 10.1101/gad.322004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannenberg JH, van Rossum A, Schuijff L, te Riele H. Genes Dev. 2000;14:3051–3064. doi: 10.1101/gad.847700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin A, Maiti B, Jakoi L, Timmers C, Buerki R, Leone G. J Biol Chem. 2003a;278:42041–42049. doi: 10.1074/jbc.M308105200. [DOI] [PubMed] [Google Scholar]
- de Bruin A, Wu L, Saavedra HI, Wilson P, Yang Y, Rosol TJ, et al. Proc Natl Acad Sci USA. 2003b;100:6546–6551. doi: 10.1073/pnas.1031853100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Nooij JC, Letendre MA, Hariharan IK. Cell. 1996;87:1237–1247. doi: 10.1016/s0092-8674(00)81819-x. [DOI] [PubMed] [Google Scholar]
- Di Stefano L, Jensen MR, Helin K. EMBO J. 2003;22:6289–6298. doi: 10.1093/emboj/cdg613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimova DK, Stevaux O, Frolov MV, Dyson NJ. Genes Dev. 2003;17:2308–2320. doi: 10.1101/gad.1116703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W. Development. 2000;127:367–379. doi: 10.1242/dev.127.2.367. [DOI] [PubMed] [Google Scholar]
- Du W, Dyson N. EMBO J. 1999;18:916–925. doi: 10.1093/emboj/18.4.916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W, Vidal M, Xie JE, Dyson N. Genes Dev. 1996a;10:1206–1218. doi: 10.1101/gad.10.10.1206. [DOI] [PubMed] [Google Scholar]
- Du W, Xie JE, Dyson N. EMBO J. 1996b;15:3684–3692. [PMC free article] [PubMed] [Google Scholar]
- Dunaief JL, Strober BE, Guha S, Khavari PA, Alin K, Luban J, et al. Cell. 1994;79:119–130. doi: 10.1016/0092-8674(94)90405-7. [DOI] [PubMed] [Google Scholar]
- Duronio RJ, Bonnette PC, O’Farrell PH. Mol Cell Biol. 1998;18:141–151. doi: 10.1128/mcb.18.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duronio RJ, O’Farrell PH, Xie JE, Brook A, Dyson N. Genes Dev. 1995;9:1445–1455. doi: 10.1101/gad.9.12.1445. [DOI] [PubMed] [Google Scholar]
- Dynlacht BD, Brook A, Dembski M, Yenush L, Dyson N. Proc Natl Acad Sci USA. 1994a;91:6359–6363. doi: 10.1073/pnas.91.14.6359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dynlacht BD, Flores O, Lees JA, Harlow E. Genes Dev. 1994b;8:1772–1786. doi: 10.1101/gad.8.15.1772. [DOI] [PubMed] [Google Scholar]
- Dyson N. Genes Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- Ewen ME, Sluss HK, Sherr CJ, Matsushime H, Kato J, Livingston DM. Cell. 1993;73:487–497. doi: 10.1016/0092-8674(93)90136-e. [DOI] [PubMed] [Google Scholar]
- Ferreira R, Naguibneva I, Mathieu M, Ait-Si-Ali S, Robin P, Pritchard LL, et al. EMBO Rep. 2001;2:794–799. doi: 10.1093/embo-reports/kve173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field SJ, Tsai FY, Kuo F, Zubiaga AM, Kaelin WG, Jr, Livingston DM, et al. Cell. 1996;85:549–561. doi: 10.1016/s0092-8674(00)81255-6. [DOI] [PubMed] [Google Scholar]
- Firth LC, Baker NE. Dev Cell. 2005;8:541–551. doi: 10.1016/j.devcel.2005.01.017. [DOI] [PubMed] [Google Scholar]
- Follette PJ, Duronio RJ, O’Farrell PH. Curr Biol. 1998;8:235–238. doi: 10.1016/s0960-9822(98)70089-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolov MV, Dyson NJ. J Cell Sci. 2004;117:2173–2181. doi: 10.1242/jcs.01227. [DOI] [PubMed] [Google Scholar]
- Frolov MV, Huen DS, Stevaux O, Dimova D, Balczarek-Strang K, Elsdon M, et al. Genes Dev. 2001;15:2146–2160. doi: 10.1101/gad.903901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolov MV, Stevaux O, Moon NS, Dimova D, Kwon EJ, Morris EJ, et al. Genes Dev. 2003;17:723–728. doi: 10.1101/gad.1031803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaubatz S, Lees JA, Lindeman GJ, Livingston DM. Mol Cell Biol. 2001;21:1384–1392. doi: 10.1128/MCB.21.4.1384-1392.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giangrande PH, Hallstrom TC, Tunyaplin C, Calame K, Nevins JR. Mol Cell Biol. 2003;23:3707–3720. doi: 10.1128/MCB.23.11.3707-3720.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giangrande PH, Zhu W, Rempel RE, Laakso N, Nevins JR. EMBO J. 2004;23:1336–1347. doi: 10.1038/sj.emboj.7600134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Schneider JW, Condorelli G, Kaushal S, Mahdavi V, Nadal-Ginard B. Cell. 1993;72:309–324. doi: 10.1016/0092-8674(93)90110-c. [DOI] [PubMed] [Google Scholar]
- Hallstrom TC, Nevins JR. Proc Natl Acad Sci USA. 2003;100:10848–10853. doi: 10.1073/pnas.1831408100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernando E, Nahle Z, Juan G, Diaz-Rodriguez E, Alaminos M, Hemann M, et al. Nature. 2004;430:797–802. doi: 10.1038/nature02820. [DOI] [PubMed] [Google Scholar]
- Hinds PW, Mittnacht S, Dulic V, Arnold A, Reed SI, Weinberg RA. Cell. 1992;70:993–1006. doi: 10.1016/0092-8674(92)90249-c. [DOI] [PubMed] [Google Scholar]
- Iavarone A, King ER, Dai XM, Leone G, Stanley ER, Lasorella A. Nature. 2004;432:1040–1045. doi: 10.1038/nature03068. [DOI] [PubMed] [Google Scholar]
- Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- Jost CA, Ginsberg D, Kaelin WG., Jr Virology. 1996;220:78–90. doi: 10.1006/viro.1996.0288. [DOI] [PubMed] [Google Scholar]
- Kang H, Cui K, Zhao K. Mol Cell Biol. 2004;24:1188–1199. doi: 10.1128/MCB.24.3.1188-1199.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato J, Matsushime H, Hiebert SW, Ewen ME, Sherr CJ. Genes Dev. 1993;7:331–342. doi: 10.1101/gad.7.3.331. [DOI] [PubMed] [Google Scholar]
- Knoblich JA, Sauer K, Jones L, Richardson H, Saint R, Lehner CF. Cell. 1994;77:107–120. doi: 10.1016/0092-8674(94)90239-9. [DOI] [PubMed] [Google Scholar]
- Korenjak M, Taylor-Harding B, Binne UK, Satterlee JS, Stevaux O, Aasland R, et al. Cell. 2004;119:181–193. doi: 10.1016/j.cell.2004.09.034. [DOI] [PubMed] [Google Scholar]
- Landsberg RL, Sero JE, Danielian PS, Yuan TL, Lee EY, Lees JA. Proc Natl Acad Sci USA. 2003;100:2456–2461. doi: 10.1073/pnas.0138064100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane ME, Sauer K, Wallace K, Jan YN, Lehner CF, Vaessin H. Cell. 1996;87:1225–1235. doi: 10.1016/s0092-8674(00)81818-8. [DOI] [PubMed] [Google Scholar]
- Lasorella A, Noseda M, Beyna M, Yokota Y, Iavarone A. Nature. 2000;407:592–598. doi: 10.1038/35036504. [DOI] [PubMed] [Google Scholar]
- Lasorella A, Rothschild G, Yokota Y, Russell RG, Iavarone A. Mol Cell Biol. 2005;25:3563–3574. doi: 10.1128/MCB.25.9.3563-3574.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EY, Cam H, Ziebold U, Rayman JB, Lees JA, Dynlacht BD. Cancer Cell. 2002;2:463–472. doi: 10.1016/s1535-6108(02)00207-6. [DOI] [PubMed] [Google Scholar]
- Liu DX, Nath N, Chellappan SP, Greene LA. Genes Dev. 2005;19:719–732. doi: 10.1101/gad.1296405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLellan WR, Xiao G, Abdellatif M, Schneider MD. Mol Cell Biol. 2000;20:8903–8915. doi: 10.1128/mcb.20.23.8903-8915.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti B, Li J, de Bruin A, Gordon F, Timmers C, Opavsky R, et al. J Biol Chem. 2005;280:18211–18220. doi: 10.1074/jbc.M501410200. [DOI] [PubMed] [Google Scholar]
- Masselli A, Wang JY. Oncogene. 2006;25:1290–1298. doi: 10.1038/sj.onc.1209161. [DOI] [PubMed] [Google Scholar]
- Miyake S, Sellers WR, Safran M, Li X, Zhao W, Grossman SR, et al. Mol Cell Biol. 2000;20:8889–8902. doi: 10.1128/mcb.20.23.8889-8902.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon NS, Frolov MV, Kwon EJ, Di Stefano L, Dimova DK, Morris EJ, et al. Dev Cell. 2005;9:463–475. doi: 10.1016/j.devcel.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Morris EJ, Dyson NJ. Adv Cancer Res. 2001;82:1–54. doi: 10.1016/s0065-230x(01)82001-7. [DOI] [PubMed] [Google Scholar]
- Muller H, Bracken AP, Vernell R, Moroni MC, Christians F, Grassilli E, et al. Genes Dev. 2001;15:267–285. doi: 10.1101/gad.864201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller H, Moroni MC, Vigo E, Petersen BO, Bartek J, Helin K. Mol Cell Biol. 1997;17:5508–5520. doi: 10.1128/mcb.17.9.5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundle SD, Saberwal G. FASEB J. 2003;17:569–574. doi: 10.1096/fj.02-0431rev. [DOI] [PubMed] [Google Scholar]
- Narita M, Nunez S, Heard E, Lin AW, Hearn SA, Spector DL, et al. Cell. 2003;113:703–716. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- Neufeld TP, de la Cruz AF, Johnston LA, Edgar BA. Cell. 1998;93:1183–1193. doi: 10.1016/s0092-8674(00)81462-2. [DOI] [PubMed] [Google Scholar]
- Nguyen DX, Baglia LA, Huang SM, Baker CM, McCance DJ. EMBO J. 2004;23:1609–1618. doi: 10.1038/sj.emboj.7600176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DX, McCance DJ. J Cell Biochem. 2005;94:870–879. doi: 10.1002/jcb.20375. [DOI] [PubMed] [Google Scholar]
- Nielsen SJ, Schneider R, Bauer UM, Bannister AJ, Morrison A, O’Carroll D, et al. Nature. 2001;412:561–565. doi: 10.1038/35087620. [DOI] [PubMed] [Google Scholar]
- Ohtani K, Nevins JR. Mol Cell Biol. 1994;14:1603–1612. doi: 10.1128/mcb.14.3.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MS, Rosai J, Nguyen HT, Capodieci P, Cordon-Cardo C, Koff A. Proc Natl Acad Sci USA. 1999;96:6382–6387. doi: 10.1073/pnas.96.11.6382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pediconi N, Ianari A, Costanzo A, Belloni L, Gallo R, Cimino L, et al. Nat Cell Biol. 2003;5:552–558. doi: 10.1038/ncb998. [DOI] [PubMed] [Google Scholar]
- Polager S, Kalma Y, Berkovich E, Ginsberg D. Oncogene. 2002;21:437–446. doi: 10.1038/sj.onc.1205102. [DOI] [PubMed] [Google Scholar]
- Rayman JB, Takahashi Y, Indjeian VB, Dannenberg JH, Catchpole S, Watson RJ, et al. Genes Dev. 2002;16:933–947. doi: 10.1101/gad.969202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, et al. Genes Dev. 2002;16:245–256. doi: 10.1101/gad.949802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnitzky D, Hengst L, Reed SI. Mol Cell Biol. 1995;15:4347–4352. doi: 10.1128/mcb.15.8.4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robanus-Maandag E, Dekker M, van der Valk M, Carrozza ML, Jeanny JC, Dannenberg JH, et al. Genes Dev. 1998;12:1599–1609. doi: 10.1101/gad.12.11.1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP. Nat Genet. 2000;25:338–342. doi: 10.1038/77124. [DOI] [PubMed] [Google Scholar]
- Royzman I, Austin RJ, Bosco G, Bell SP, Orr-Weaver TL. Genes Dev. 1999;13:827–840. doi: 10.1101/gad.13.7.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royzman I, Whittaker AJ, Orr-Weaver TL. Genes Dev. 1997;11:1999–2011. doi: 10.1101/gad.11.15.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz S, Santos M, Segrelles C, Leis H, Jorcano JL, Berns A, et al. Development. 2004;131:2737–2748. doi: 10.1242/dev.01148. [DOI] [PubMed] [Google Scholar]
- Saavedra HI, Wu L, de Bruin A, Timmers C, Rosol TJ, Weinstein M, et al. Cell Growth Differ. 2002;13:215–225. [PubMed] [Google Scholar]
- Sage C, Huang M, Karimi K, Gutierrez G, Vollrath MA, Zhang DS, et al. Science. 2005;307:1114–1118. doi: 10.1126/science.1106642. [DOI] [PubMed] [Google Scholar]
- Sage J, Miller AL, Perez-Mancera PA, Wysocki JM, Jacks T. Nature. 2003;424:223–228. doi: 10.1038/nature01764. [DOI] [PubMed] [Google Scholar]
- Sage J, Mulligan GJ, Attardi LD, Miller A, Chen S, Williams B, et al. Genes Dev. 2000;14:3037–3050. doi: 10.1101/gad.843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawado T, Yamaguchi M, Nishimoto Y, Ohno K, Sakaguchi K, Matsukage A. Biochem Biophys Res Commun. 1998;251:409–415. doi: 10.1006/bbrc.1998.9407. [DOI] [PubMed] [Google Scholar]
- Shirodkar S, Ewen M, DeCaprio JA, Morgan J, Livingston DM, Chittenden T. Cell. 1992;68:157–166. doi: 10.1016/0092-8674(92)90214-w. [DOI] [PubMed] [Google Scholar]
- Spike BT, Dirlam A, Dibling BC, Marvin J, Williams BO, Jacks T, et al. EMBO J. 2004;23:4319–4329. doi: 10.1038/sj.emboj.7600432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevaux O, Dimova D, Frolov MV, Taylor-Harding B, Morris E, Dyson N. EMBO J. 2002;21:4927–4937. doi: 10.1093/emboj/cdf501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevaux O, Dimova DK, Ji JY, Moon NS, Frolov MV, Dyson NJ. Cell Cycle. 2005;4:1272–1280. doi: 10.4161/cc.4.9.1982. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Rayman JB, Dynlacht BD. Genes Dev. 2000;14:804–816. [PMC free article] [PubMed] [Google Scholar]
- Taubert S, Gorrini C, Frank SR, Parisi T, Fuchs M, Chan HM, et al. Mol Cell Biol. 2004;24:4546–4556. doi: 10.1128/MCB.24.10.4546-4556.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimarchi JM, Fairchild B, Verona R, Moberg K, Andon N, Lees JA. Proc Natl Acad Sci USA. 1998;95:2850–2855. doi: 10.1073/pnas.95.6.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimarchi JM, Fairchild B, Wen J, Lees JA. Proc Natl Acad Sci USA. 2001;98:1519–1524. doi: 10.1073/pnas.041597698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai KY, Hu Y, Macleod KF, Crowley D, Yamasaki L, Jacks T. Mol Cell. 1998;2:293–304. doi: 10.1016/s1097-2765(00)80274-9. [DOI] [PubMed] [Google Scholar]
- Verona R, Moberg K, Estes S, Starz M, Vernon JP, Lees JA. Mol Cell Biol. 1997;17:7268–7282. doi: 10.1128/mcb.17.12.7268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells J, Yan PS, Cechvala M, Huang T, Farnham PJ. Oncogene. 2003;22:1445–1460. doi: 10.1038/sj.onc.1206264. [DOI] [PubMed] [Google Scholar]
- Weng L, Zhu C, Xu J, Du W. EMBO J. 2003;22:3865–3875. doi: 10.1093/emboj/cdg373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikenheiser-Brokamp KA. Development. 2004;131:4299–4310. doi: 10.1242/dev.01232. [DOI] [PubMed] [Google Scholar]
- Wu L, de Bruin A, Saavedra HI, Starovic M, Trimboli A, Yang Y, et al. Nature. 2003;421:942–947. doi: 10.1038/nature01417. [DOI] [PubMed] [Google Scholar]
- Wu L, Timmers C, Maiti B, Saavedra HI, Sang L, Chong GT, et al. Nature. 2001;414:457–462. doi: 10.1038/35106593. [DOI] [PubMed] [Google Scholar]
- Yamasaki L, Bronson R, Williams BO, Dyson NJ, Harlow E, Jacks T. Nat Genet. 1998;18:360–364. doi: 10.1038/ng0498-360. [DOI] [PubMed] [Google Scholar]
- Yamasaki L, Jacks T, Bronson R, Goillot E, Harlow E, Dyson NJ. Cell. 1996;85:537–548. doi: 10.1016/s0092-8674(00)81254-4. [DOI] [PubMed] [Google Scholar]
- Young AP, Longmore GD. Oncogene. 2004;23:814–823. doi: 10.1038/sj.onc.1207187. [DOI] [PubMed] [Google Scholar]
- Zhang HS, Gavin M, Dahiya A, Postigo AA, Ma D, Luo RX, et al. Cell. 2000;101:79–89. doi: 10.1016/S0092-8674(00)80625-X. [DOI] [PubMed] [Google Scholar]
- Zheng N, Fraenkel E, Pabo CO, Pavletich NP. Genes Dev. 1999;13:666–674. doi: 10.1101/gad.13.6.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Steller H. Dev Cell. 2003;4:599–605. doi: 10.1016/s1534-5807(03)00085-6. [DOI] [PubMed] [Google Scholar]
- Zhu JW, Field SJ, Gore L, Thompson M, Yang H, Fujiwara Y, et al. Mol Cell Biol. 2001;21:8547–8564. doi: 10.1128/MCB.21.24.8547-8564.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Harlow E, Dynlacht BD. Genes Dev. 1995;9:1740–1752. doi: 10.1101/gad.9.14.1740. [DOI] [PubMed] [Google Scholar]
- Ziebold U, Lee EY, Bronson RT, Lees JA. Mol Cell Biol. 2003;23:6542–6552. doi: 10.1128/MCB.23.18.6542-6552.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebold U, Reza T, Caron A, Lees JA. Genes Dev. 2001;15:386–391. doi: 10.1101/gad.858801. [DOI] [PMC free article] [PubMed] [Google Scholar]