

Abstract
A series of conformationally restricted inhibitors of human soluble epoxide hydrolase (sEH) has been developed. Inhibition potency of the described compounds ranges from 4.2 μM to 1.1 nM against recombinant sEH. N-(1-Acetylpiperidin-4-yl)-N′-(adamant-1-yl) urea (5a) was found to be a potent inhibitor (IC50 = 7.0 nM) that was also orally bioavailable in canines.
Keywords: Soluble epoxide hydrolase, Enzyme inhibition, Structure-activity relationships
The soluble epoxide hydrolase (sEH, E.C. 3.3.2.3) is a member of the α/β-hydrolase fold family of enzymes.1 A major function of sEH is the conversion of epoxides to vicinal diols through the catalytic addition of a water molecule.2 The endogenous substrates for the sEH include the cytochrome P450 metabolites of arachidonic acid (epoxyeicosatrienoic acids, EETs).3,4 EETs are known modulators of blood pressure and inflammation, and their conversion to dihydroxyeicosatrienoic acids (DHETs) by sEH reduces the beneficial activity of EETs. It has been shown that in vivo inhibition of sEH with highly selective inhibitors results in an increase in the concentration of EETs and is accompanied by a reduction in blood pressure in rodent models, thereby suggesting that sEH is a compelling target for the treatment of hypertension.5–8
Through the course of our research we have found N,N′-disubstituted ureas to be our most successful inhibitors of sEH (Fig. 1).9–13 The inhibitors shown in Figure 1 can be divided into three basic categories: those that are small, rigid, and contain non-polar groups (e.g., DCU), those that contain both rigid and flexible non-polar groups (e.g., CDU) and those that contain both a rigid non-polar group and flexible polar chain (e.g., 950, AUDA, and AUDA-BE).
Figure 1.
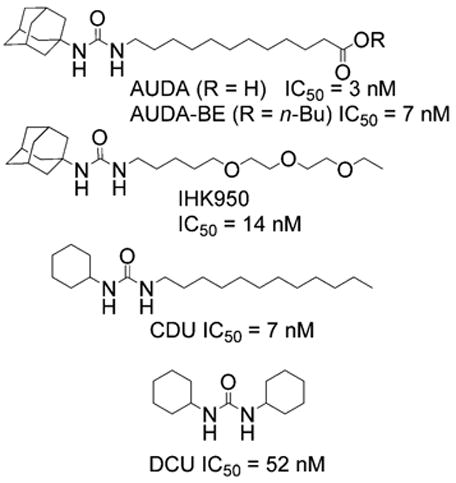
Common inhibitors of sEH. IC50 is for in vitro inhibition against recombinant human sEH.
We have found that while those compounds possessing a flexible side chain do show biological effects when assayed in vivo, they are rapidly metabolized and excreted, limiting their utility. In contrast, compounds that lack a flexible side chain (e.g., DCU) typically have physical properties that are so poor that they show almost no biological effects in vivo. Therefore, we have initiated a study aimed at examining the utility of conformationally restricted sEH inhibitors that also have a polar secondary pharmacophore. Not only did we wish to expand our current SAR by examining a broader range of structures, but we also aimed to synthesize inhibitors that were more ‘drug-like’ in structure.14–16 For the purposes of this study, we utilized the scaffolds shown in Figure 2 (compounds I and II) as platforms for inhibitor development. These scaffolds were designed to not only test the effectiveness of nitrogen containing secondary pharmacophores, but also to compare the more rigid scaffold I to the more flexible scaffold II.
Figure 2.
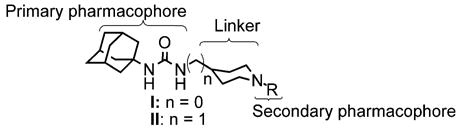
Piperidine-based scaffolds for rigid sEH inhibitors. Urea, amide, and carbamate groups as the central pharmacophore have yielded potent inhibitors with 1,3-substitution with aryl, alkyl, cycloalkyl or as shown, adamantyl groups.9,10
The synthesis of scaffold I is illustrated in Scheme 1. We began by reacting the commercially available piperidine 1 and benzaldehyde to form the corresponding Schiff base. Treatment with Boc anhydride followed by acid hydrolysis afforded the protected piperidine 2.17 Reaction of compound 2 with 1-ada-mantylisocyanate followed by removal of the Boc group with methanolic HCl afforded scaffold I with a high overall yield. Scaffold II was synthesized in an analogous manner.
Scheme 1.
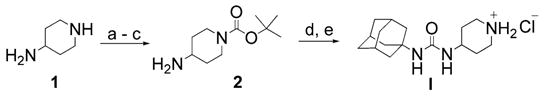
Reagents and conditions: (a) toluene, benzaldehyde (1 equiv), reflux, Dean–Stark trap; (b) Boc anhydride, 0–25 °C, 12 h; (c) KHSO4(aq) (1 equiv); (d) adamant-1-yl isocyanate, THF, rt; (e) HCl/MeOH (4 equiv H+), 74% (overall).
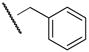
We found that the piperidine nucleus was readily alkylated as shown in Scheme 2 via reaction with an alkylbromide in the presence of catalytic KI to give the products in modest yield. Interestingly, we found that the alkylpiperidines listed in Table 1 were, in general, poor inhibitors. It is clear from these results that a positively charged piperidine is not tolerated by the enzyme. The exceptions to this trend are benzyl-substituted compounds 3d and 4d. These results suggest that the destabilizing effects of a cation may be compensated for by aryl–aryl interactions within the active site. It is also interesting to note that those compounds, which are based on scaffold II show an SAR that varies logically with substitution, whereas those which are based on scaffold I do not.
Scheme 2.
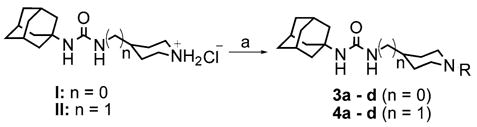
Reagents and conditions: (a) bromoalkane, KI, DMF, K2CO3, 50 °C, 12 h, 42–60%.
Table 1.
IC50 values for alkylpiperidine-based sEH inhibitors
|
n = 0
|
n = 1
|
|||
|---|---|---|---|---|
| Compound | IC50 (μM)a | Compound | IC50 (μM)a | |
| R:H | I | 0.30 | II | 4.2 |
|
|
3a | 3.8 | 4a | 3.9 |
|
|
3b | 0.81 | 4b | 2.6 |
|
|
3c | 1.2 | 4c | 0.61 |
|
3d | 0.01 | 4d | 0.11 |
As determined via a kinetic fluorescent assay.22
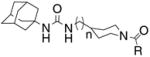
From the results described above, we postulated that eliminating the possibility of nitrogen protonation would result in an increase in potency. With this in mind, we turned our attention to acylated piperidines. We found that while acylation of scaffolds I and II can be accomplished through the reaction of scaffolds I or II with the requisite acid chloride, using carbo-diimide mediated coupling chemistry gave the desired products in consistently higher yield (Scheme 3).18 As can be seen in Table 2, conversion of the piperidine nitrogen from an amine to an amide resulted in a dramatic increase in potency. Inhibitors 5a–h show, in general, better potency than inhibitors 6a–h. This suggests that scaffold I may facilitate the formation of a beneficial polar interaction between the piperidinyl amide and a residue in the active site. In addition, the potency of inhibitors 5a–h and 6a–h seems to rely more on the presence of the amide functionality rather than the actual identity of the amide fragment.
Scheme 3.
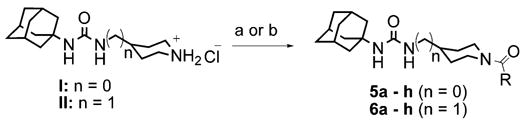
Reagents and conditions: (a) RCOCl, TEA, DCM, 6 h, 20–73%; (b) RCOOH, TEA, DMAP, EDCI, dichloromethane, 0 °C to rt, 12 h, 75–99%.
Table 2.
IC50 values for acylpiperidine-based sEH inhibitors
 |
n = 0
|
n = 1
|
||
|---|---|---|---|---|
| Compound | IC50 (nM)a | Compound | IC50 (nM)a | |
|
|
5a | 7.0 | 6a | 5.0 |
|
|
5b | 3.2 | 6b | 8.7 |
|
|
5c | 2.6 | 6c | 6.7 |
|
|
5d | 1.1 | 6d | 1.8 |
|
|
5e | 1.3 | 6e | 3.2 |
|
|
5f | 1.2 | 6f | 7.6 |
|
5g | 1.7 | 6g | 5.4 |
|
|
5h | 2.1 | 6h | 7.3 |
As determined via a kinetic fluorescent assay.22
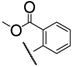
Having firmly established the importance of the amide functionality, we then investigated the effects of additional polar functionality on the potency of these inhibitors. Using the chemistry outlined in Scheme 3, a series of methyl ester based inhibitors was generated (Table 3, 7a–e and 8a–e). As with the other amide-based inhibitors described in this study, esters 7a–e and 8a–e do not show a large variation in potency. There does seem to be a small advantage in having an aryl linker instead of an alkyl one. Curiously, there is very little difference in whether the methyl ester is in the ortho-, meta-, or para-position, indicating that the active site is quite tolerant of increased steric bulk.
Table 3.
IC50 values for piperidine-based sEH inhibitors containing ester and acid functionalized amides
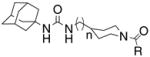 |
n = 0
|
n = 1
|
||
|---|---|---|---|---|
| Compound | IC50 (nM)a | Compound | IC50 (nM)a | |
|
|
7a | 9.0 | 8a | 6.2 |
|
|
7b | 2.7 | 8b | 3.4 |
|
7c | 1.7 | 8c | 1.8 |
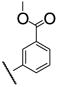
|
7d | 1.1 | 8d | 4.1 |
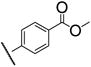
|
7e | 1.1 | 8e | 1.5 |
|
|
7f | 2.5 × 102 | 8f | 1.7 × 102 |
|
|
7g | 72 | 8g | 41 |
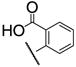
|
7h | 1.6 × 102 | 8h | 4.0 × 102 |
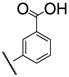
|
7i | 10 | 8i | 43 |
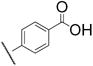
|
7j | 3.3 | 8j | 11.8 |
As determined via a kinetic fluorescent assay.22
Esters 7a–e and 8a–e were smoothly converted to the corresponding acids (7f–j and 8f–j, respectively) by reaction with methanolic KOH. The potency of the resultant acids showed a remarkable dependence on the position of the carboxylate, as shown by data in Table 3. Conformational analysis (using CONFLEX, as implemented in CAChe Workstation Pro 6.1, Fujit-su Inc.) suggested that the carboxylate in compounds 7f could hydrogen bond to the urea in an intramolecular fashion (data not shown). This implies that the observed SAR trend for compound 7f–j and 8f–j may be due, in part, to the ability of the inhibitor to form intramolecular hydrogen bonds, thereby destabilizing any interactions that would support binding in the active site.
We were intrigued by the observation that trifluoroacetamides 5d and 6d showed a dramatic increase in potency over acetamides 5a and 6a. Using the published crystal structure of human sEH with a bound urea-based ligand (N-(4-iodophenyl)-N′-cyclohexyl urea, CIU, PDB accession number 1VJ5),19 inhibitor 5d was manually docked into the active site in order to further understand the observed benefit of the trifluoroacetamide functionality. As can be seen in Figure 3, compound 5d is bound primarily through interactions with Tyr381, Tyr465, and Asp333 with the urea pharmacophore. In addition, the trifluoroacetamide functionality of compound 5d can hydrogen bond with Gln382 via the carbonyl and one of the fluorine atoms. It is reasonable to postulate that the observed increase in potency of trifluoroacetamides 5d and 6d over acetamides 5a and 6a is mainly due to the presence of the additional interactions between the –CF3 and Gln382. Docking inhibitor 5d into the active site in the opposite orientation resulted in unfavorable steric interactions between the adamantane and Met337, and removed any opportunity for the trifluoroacetamide to participate in productive hydrogen bonding.
Figure 3.
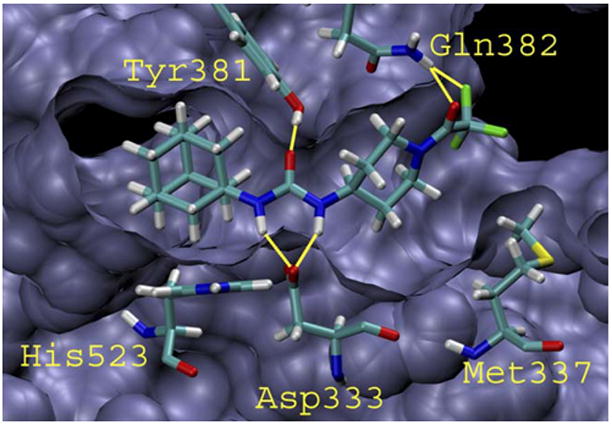
Compound 5d docked into the active site of human sEH. Hydrogen bonds are indicated by the yellow lines. Tyr465 has been removed for the sake of clarity.21
At this point, we selected a small number of compounds and screened them for oral bioavailability in dogs.20 As can be seen in Table 4, not only do these compounds have appreciable blood levels, but compound 5a shows an almost 10-fold increase in AUC as compared to AUDA. In addition, the observation that blood level is clearly dependent on inhibitor structure indicates that we may be able to optimize a subset of the compounds reported herein for improved oral availability.
Table 4.
Pharmacokinetic profile data for selected compounds as obtained via oral dosing in a canine model
| Compound | AUCa (×104 nM min) |
|---|---|
| 5a | 3.7 |
| 6a | 0.55 |
| 5b | 0.65 |
| 5c | 0.25 |
| 5e | 0.061 |
| 6e | 0.033 |
| 5d | 0.33 |
| 5f | 0.47 |
| AUDA | 0.31 |
Area under the curve, estimated from a plot of inhibitor plasma concentration (nM) versus time (minutes) following an oral dose of 0.3 mg/kg of the indicated compounds in tristerate.20
In conclusion, we have reported a series of sEH inhibitors that use a piperidine moiety to rigidify their structure. A preliminary screen of inhibitor potency against recombinant sEH reveals that simple amide-based inhibitors are well tolerated. In contrast, acid functionalized inhibitors show a distinct SAR, which is consistently less potent than the corresponding esters across both scaffolds. The data presented clearly indicate the potential value of these and other heterocyclic compounds as effective in vivo inhibitors of sEH. We currently have detailed experiments underway with the aim of determining the efficacy and pharmacokinetic properties of these compounds. The results of these studies will be reported in due course.
Acknowledgments
The authors thank Dr. James Sanborn for many helpful discussions. We also thank Dr. William Jewel and the staff of the UC Davis Mass Spectrometry Center for the use of their mass spectrometer. P.D.J. was supported by an NIH/NHLBI Ruth L. Kirchstein NRSA Grant (F32 HL078096). This work was supported in part by an NIEHS Grant (ES02710), an NIEHS Superfund Grant (P42 ES04699), an NIH/NHLBI grant (R01 HL59699-06A1), and a Translational Technology Grant from the UC Davis Medical Center.
Footnotes
Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bmcl.2006. 07.009.
References and notes
- 1.Arand M, Grant DF, Beetham JK, Friedberg T, Oesch F, Hammock BD. FEBS Lett. 1994;338:251. doi: 10.1016/0014-5793(94)80278-5. [DOI] [PubMed] [Google Scholar]
- 2.Oesch F. Xenobiotica. 1973;3:305. doi: 10.3109/00498257309151525. [DOI] [PubMed] [Google Scholar]
- 3.Zeldin DC, Kobayashi J, Falck JR, Winder BS, Hammock BD, Snapper JR, Capdevila JH. J Biol Chem. 1993;268:6402. [PubMed] [Google Scholar]
- 4.Spector AA, Fang X, Snyder GD, Weintraub NL. Prog Lipid Res. 2004;43:55. doi: 10.1016/s0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- 5.Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, Zeldin DC, Kroetz DL. Circ Res. 2000;87:992. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
- 6.Imig JD, Zhao X, Capdevila JH, Morisseau C, Hammock BD. Hypertension. 2002;39:690. doi: 10.1161/hy0202.103788. [DOI] [PubMed] [Google Scholar]
- 7.Imig JD, Zhao X, Zaharis CZ, Olearczyk JJ, Pollock DM, Newman JW, Kim IH, Watanabe T, Hammock BD. Hypertension. 2005;46:975. doi: 10.1161/01.HYP.0000176237.74820.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spiecker M, Liao JK. Arch Biochem Biophys. 2005;433:420. doi: 10.1016/j.abb.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 9.Morisseau C, Goodrow MH, Dowdy D, Zheng J, Greene JF, Sanborn JR, Hammock BD. Proc Natl Acad Sci USA. 1999;96:8849. doi: 10.1073/pnas.96.16.8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McElroy NR, Jurs PC, Morisseau C, Hammock BD. J Med Chem. 2003;46:1066. doi: 10.1021/jm020269o. [DOI] [PubMed] [Google Scholar]
- 11.Kim IH, Morisseau C, Watanabe T, Hammock BD. J Med Chem. 2004;47:2110. doi: 10.1021/jm030514j. [DOI] [PubMed] [Google Scholar]
- 12.Kim IH, Heirtzler FR, Morisseau C, Nishi K, Tsai HJ, Hammock BD. J Med Chem. 2005;48:3621. doi: 10.1021/jm0500929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Proc Natl Acad Sci USA. 2005;102:9772. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Navia MA, Chaturvedi PR. Drug Discovery Today. 1996;1:179. [Google Scholar]
- 15.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv Drug Delivery Rev. 2001;46:3. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 16.Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. J Med Chem. 2002;45:2615. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 17.Sonda S, Kawahara T, Murozono T, Sato N, Asano K, Haga K. Bioorg Med Chem. 2003;11:4225. doi: 10.1016/s0968-0896(03)00412-7. [DOI] [PubMed] [Google Scholar]
- 18.Dhaon MK, Olsen RK, Ramasamy K. J Org Chem. 1982;47:1962. [Google Scholar]
- 19.Gomez GA, Morisseau C, Hammock BD, Christianson DW. Biochemistry. 2004;43:4716. doi: 10.1021/bi036189j. [DOI] [PubMed] [Google Scholar]
- 20.Selected soluble epoxide hydrolase inhibitors were prepared at 6 mg/mL in triglycerides. Each dog was dosed orally with three different inhibitors per experiment (dose = 0.3 mg/kg per inhibitor). Plasma samples were collected at 0, 15, 30, 60, 120, 180, 240, 300, 360, 480, and 1440 min, and analyzed by LC/MS/MS (Quattro Premier; Micromass, MA). The pharmacokinetic parameters were calculated with WinNonlin 5.0 (Pharsight, CA).
- 21.Humphrey W, Dalk A, Schulten K. J Mol Graph. 1996;14.1:33. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 22.Jones PD, Wolf NM, Morisseau C, Whetstone P, Hock B, Hammock BD. Anal Biochem. 2005;343:66. doi: 10.1016/j.ab.2005.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]