

Abstract
The nature of the immunopathogenic relationship underlying the very strong association of coeliac disease (CD) to the HLA-DQ (A1*0501, B1*0201) genotype is not known, but probably relates to binding of gluten-derived epitopes to the HLA-DQ (α1*0501, β1*0201) heterodimer (DQ2). These epitopes have not yet been defined. In this study we have tested the binding of various gluten-derived peptides to DQ2 in a cellular assay using Epstein–Barr virus (EBV)-transformed B lymphocytes and murine fibroblast transfectants. One of these peptides (peptide A), which has previously been shown to exacerbate the CD lesion in vitro and in vivo, was found to bind to DQ2, albeit only moderately, lending further credence to its possible role in the pathogenesis of CD. The nature of peptide A's binding to DQ2 was explored with truncated and conservative point substituted analogues and compared with the published DQ2 binding motif, the results of which explain the observed level of binding.
Keywords: HLA, DQ2, binding, coeliac disease
INTRODUCTION
Coeliac disease (CD), or gluten sensitive enteropathy, is associated in 95% of affected individuals with the HLA locus DQ2 (A1*0501, B1*0201). This can be inherited in either cis (in HLA-DR3 individuals) or trans (in DR5,7 heterozygous individuals) arrangement [1, 2]. HLA-DQ8 (A1*0301, B1*0302) also confers a susceptibility in HLA-DR4 (DRB1*0401/2) southern Europeans [3, 4]. The association of coeliac disease to HLA-DR3 [5] is thought to be through linkage disequilibrium with DQ2.
The characteristic small bowel morphological changes leading to malabsorption in this disorder are reversed on eliminating gluten from the diet. Current evidence suggests that gluten-sensitive small intestinal T cells recognize gluten-derived peptides when presented in association with DQ2 [6], with their subsequent activation leading to the observed mucosal damage [7–9]. The exact toxic epitope within gluten is, however, still not known, although recent evidence suggests that small intestinal derived T cells from CD patients may be able to recognize different epitopes from different gliadins [10].
It has been demonstrated, however, that a 19mer peptide from the N-terminal region of A-gliadin (corresponding to amino acids 31–49) can exacerbate the histological features of CD by in vitro organ culture studies [11] and in challenge studies on treated patient volunteers [12]. Furthemore, this peptide (referred to in this study as peptide A) may be recognized by T cells from the peripheral blood of CD patients, when presented by DQ2 [13].
In this study we examine the binding of various gluten-derived peptides, including peptide A, to HLA-DQ2 using a cellular assay, which is in contrast to work from other authors [14], who used a soluble assay with affinity-purified HLA molecules. These, and other workers, have elucidated the peptide binding motif of DQ2 [15–17]. We compare our findings, using truncations and conservative point substitutions of peptide A, with that work and speculate on the implications for the molecular pathogenesis of CD.
MATERIALS AND METHODS
Peptides
Peptides were synthesized using a solid-phase peptide synthesizer (Model 431 A; Applied Biosystems Inc, Foster City, CA) on a preloaded Wang resin, and 9-fluorenylmethoxy-carbonyl for temporary α-amino group protection (Calbiochem Novabiochem, Nottingham, UK), cleaved from the resin and lyophilized. Peptide analysis was performed by reverse phase high performance liquid chromatography (HPLC) and matrix-assisted laser desorption ionizing mass spectrometry (Bioton Spectrometer; Applied Biosystems). Purification to > 95% homogeneity was performed on a gel exclusion column (Sephadex G15; Pharmacia LKB Biotechnology, Uppsala, Sweden) and the fractions were collected using a UV-1 single path monitor (Pharmacia) and FRAC-100 fraction collector (Pharmacia). The resulting purified peptides were frozen using liquid nitrogen, double-lyophilized and stored at −20°C. N-terminal long chain biotinylation was performed on the peptidyl resin before cleavage using sulfosuccinimidyl-6-biotinamidohexanoate chemistry (Pierce & Warriner, Chester, UK) and deprotection. Stock solutions of peptides were prepared in water at 2 mg/ml and stored at −20°C. The peptides were sonicated to ensure solubility.
Johansen et al. [18] have demonstrated that the mycobacterial heat shock protein peptide MB243-255 (KPLLIIAEDVEGEY) binds strongly to DQ2, and another, MT3-13 (KTIAYDEEARR), binds strongly to DR3. Although there is some cross-binding between these two peptides and class II molecules [14], as we were using transfectants expressing single HLA heterodimers this is not important. We thus used these two peptides as positive controls, for comparison of strength of binding, in the assays using transfectants.
Apart from peptide A (LGQQQPFPPQQPYPQPQPF), corresponding to residues 31–49 of A-gliadin, two other gliadin-derived peptides were tested. Peptide B (QQYPLGQGSFRPSQQNPQA), corresponding to 202–220 of A-gliadin, was chosen because it includes sequence homology with the 54-kD E1b protein of adenovirus 12. This was suggested by Kagnoff et al. [19] as a model of molecular mimicry of possible significance to the aetiology of CD. Peptide C (VPVPQLQPQNPSQQQPQEQ), corresponding to 3–21 of A-gliadin, has PSQQ and QQQP tetra peptide motifs with a β-reverse turn secondary structure, again of suggested significance to the pathogenesis of CD [20].
B cell lines
B cell lines from the peripheral blood of CD patients were immortalized using Epstein–Barr virus (EBV) transformation as described elsewhere [21]. These lines were homozygous for HLA-DR3 and DQ2 by serological and polymerase chain reaction-sequence-specific probe (PCR-SSP) genomic HLA typing. Tissue culture was carried out in CO2-independent medium (Life Technologies Ltd, Paisley, UK), 1000 U/1000 mg/ml penicillin/streptomycin (Life Technologies), 5 mg/ml gentamicin (Sigma Chemical Co., Poole, UK), and 200 mml-glutamine (Life Technologies) in 10% heat-inactivated fetal calf serum (FCS; Life Technologies). The cells were tested for HLA class II expression by staining with murine monomorphic anti-HLA-DR (L243 clone; Becton Dickinson, Oxford, UK) and murine anti-DQ (SPV-L3 clone; Serotec, Oxford, UK) and conjugated goat anti-mouse-FITC MoAbs (Becton Dickinson) before FACS analysis (Fig. 1). The HLA-negative K562 erythroleukaemic cell line served as a negative control.
Fig. 1.
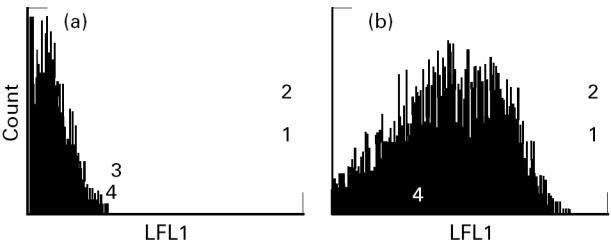
Measurement of HLA-DQ2 surface expression on HLA-negative K562 erythroleukaemic cell line (a) (mean fluorescence 1.248) and B cells from a coeliac disease patient homozygous for HLA-DR3, -DQ2 (b) (mean fluorescence 14.14) using anti-DQ (SPV-L3; Serotec), and FITC-conjugated goat anti-mouse (Becton Dickinson) MoAbs, by FACS analysis.
Transfectants
Murine L-cell (DAP-3) transfectants expressing HLA-DR or -DQ alleles were used to establish the binding affinities of the various peptides investigated, in comparison with two known strong binders—MB243-255 in the case of DQ2 and MT3-13 in the case of DR3 [18]. They were also used in the alanine point substitution experiments involving peptide A.
Co-transfection of HLA-DRα and -DRβ chain cDNA cloned into the RSV.5neo [22] and RSV.3 [23] expression vectors, respectively, was accomplished using calcium phosphate precipitation. Full length DQA and DQB cDNA clones were generated by reverse transcriptase (RT)-PCR amplification of mRNA isolated from a homozygous DR3, DQ2 B cell line. Following complete DNA sequencing of the cDNA clones, co-transfection of the HLA DQA1*0501 and DQB1*0201 cDNA cloned into the RSV.5neo and RSV.5gpt [22] expression vectors was accomplished using Lipofectamine-mediated transfection (Life Technologies). Transfected cell lines were grown in Ex-cell 320 (JRH Biosciences, Lenexa, KS) supplemented with 10% FCS (Life Technologies). HLA-DR transfectants were selected in medium containing 0·5 mg/ml G418, 10 mg/ml mycophenolic acid (Life Technologies) and 100 mg/ml xanthine (Sigma). Cells expressing the highest levels of HLA-DR or -DQ were selected by repetitive FACS sorting and cloning using HLA-DR or -DQ-specific, FITC-labelled MoAbs.
Binding assays
The binding assay used was the previously described signal amplification method [24, 25]. Briefly, cells (3 × 105/well) were incubated with 50 μl biotinylated peptide and 50 μl PBS/0·1% bovine serum albumin (BSA; Sigma) in 96-well round-bottomed plates for 18 h at 37°C. Each cell line/peptide combination was tested in duplicate and each peptide was tested at multiple concentrations. When transfected cell lines were used for assays, duplicate wells without peptide were included for each transfected line and were stained using the appropriate anti-HLA-DR (L243; Becton Dickinson) or -DQ (SPV-L3; Serotec) specific and conjugated goat anti-mouse-FITC MoAbs, in order to measure the level of HLA expression (Fig. 2). Binding of the biotinylated peptides was detected by staining twice with 1:100 avidin D-FITC (Vector Labs, Burlingame, CA) and once with 1:100 biotinylated anti-avidin D (Vector Labs) in a sandwich amplification technique at 4°C. Binding was measured as FITC fluorescence of 5000 events gated on size and propidium iodide fluorescence—to exclude dead cells—using an EPICS Profile II Fluoresence Activated Cell Sorter (Coulter, Luton, UK) (Fig. 3, transfectants; Fig. 4, B cell line).
Fig. 2.
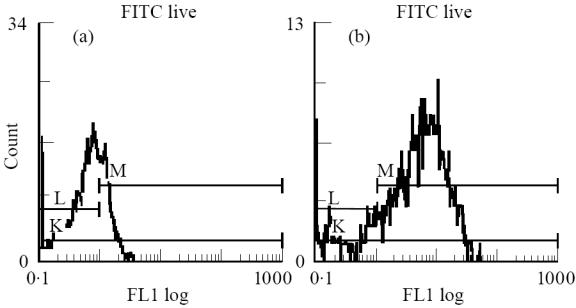
Measurement of HLA-DQ2 surface expression on HLA-negative DAP-3 parent line (a) (mean fluorescence 0.689) and DQ2-expressing transfectants (b) (mean fluorescence 4.37), using murine anti-DQ (SPV-L3; Serotec) and FITC-conjugated goat anti-mouse (Becton Dickinson) MoAbs, by FACS analysis.
Fig. 3.
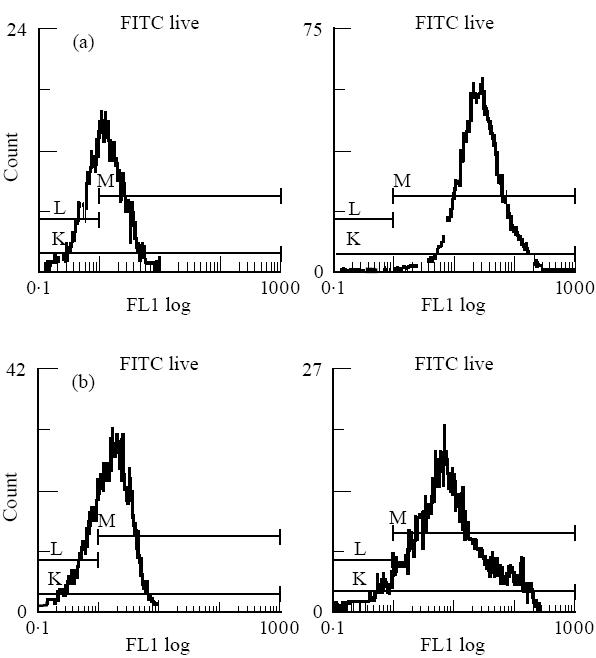
FACS histograms demonstrating binding of peptide A to DQ2-expressing transfectants and the HLA-negative DAP-3 parent line control. (a) DQ2 transfectants with protein A at 0 μmol/l (left, mean fluorescence 1.28) and 250 μmol/l (right, mean fluorescence 23.4). (b) DAP-3 control with protein A at 0 μmol/l (left, mean fluorescence 1.50) and 250 μmol/l (right, mean fluorescence 8.07).
Fig. 4.
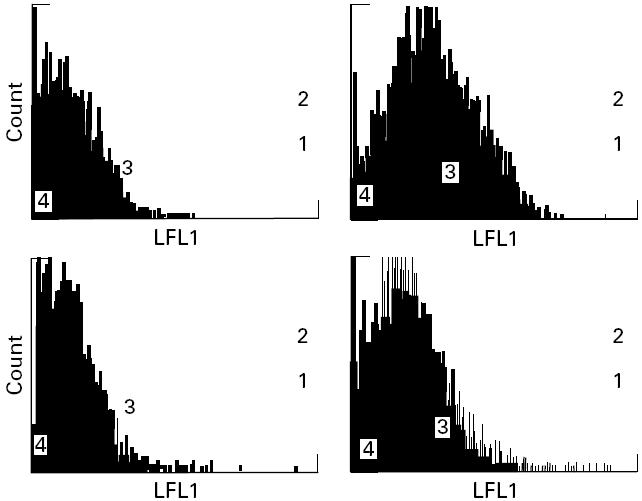
Binding of peptide A to B cells homozygous for HLA-DR3, -DQ2 and HLA-negative K562 controls by FACS analysis (see Materials and Methods). Top: B cells with protein A at 0 μmol/l (left, mean fluorescence 1.507) and 250 μmol/l (right, mean fluorescence 6.034). Bottom: K562 cells with protein A at 0 μmol/l (left, mean fluorescence 1.462) and 250 μmol/l (right, mean fluorescence 2.889).
For each transfected cell line tested, linear peptide binding means (mean channel fluorescence) were adjusted for background binding to the parental cell line (DAP-3), for each cell line's autofluorescence in the absence of peptide and for the level of HLA expression. Using this approach, an arbitrary unit of binding was calculated from the following formula:
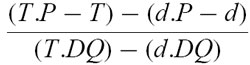 |
where T.P or T = mean fluorescence of transfectant with or without peptide, d.P or d = mean fluorescence of parent line with or without peptide, and T.DQ or d.DQ = mean fluorescence of transfectant or parent line with DR or DQ MoAb.
The results represent mean binding values from at least three sets of experiments for each set of data.
Computer modelling
The modelling method used was similar to that used for other HLA molecules [26, 27]. Briefly, the model of the HLA-DQ2 molecule was built from that of the HLA-DR1 molecule [28, 29], using the COMPOSER suite of programs contained within SYBYL (Tripos Associates, St Louis, MO) [30, 31]. The amino acid sequences of the α- and β-chains encoded by the DQA1*0501 and DQB1*0201 genes were built into the coordinates of the HLA class II α- and β-chains of the HLA-DR1:influenza HA 307–319 structure. Insertions and deletions relative to the DR1 sequence were built initially using the loop searching algorithms within COMPOSER and subjected to energy minimization. To optimize the position of peptide A within the HLA-DQ2 binding groove, candidate residues for the HLA-DQ2 P1 pocket were used to align the test sequence within the binding cleft. Alignments that gave unacceptable contacts were eliminated and remaining alignments were ranked by inspection of the peptide–side chain interactions with the HLA-DQ2 molecule, particularly those with the P1, P6 and P9 pockets, and by calculating interaction energies for those pockets. The optimal model was refined using a gradient torsional optimization algorithm [26]. Initially the main chain of the molecule and the Cβ carbons were fixed. Following the convergence of the side refinement, all atoms of the molecule were released and the optimization was performed on the entire model.
RESULTS
Direct binding of gluten-derived peptides to DQ2 and DR3
The peptide MB243-255 bound well to DQ2 in our transfectant model (Fig. 5), correlating well with the different binding model of Johansen et al. [14, 18]. Peptide A bound moderately, peptide B weakly and peptide C showed no appreciable binding.
Fig. 5.
Direct binding of gluten-derived peptides A, B and C and the mycobacterial heat shock protein peptide MB243-255, used as a positive control, to HLA-DQ2-expressing L-cell transfectants, measured by FACS analysis. Net binding is expressed in arbitrary units, the calculation of which is described in Materials and Methods. (See Materials and Methods for amino acid sequences of the peptides.) ▵, Peptide A; ○, peptide B; □, peptide C; ▪, peptide MB243-255
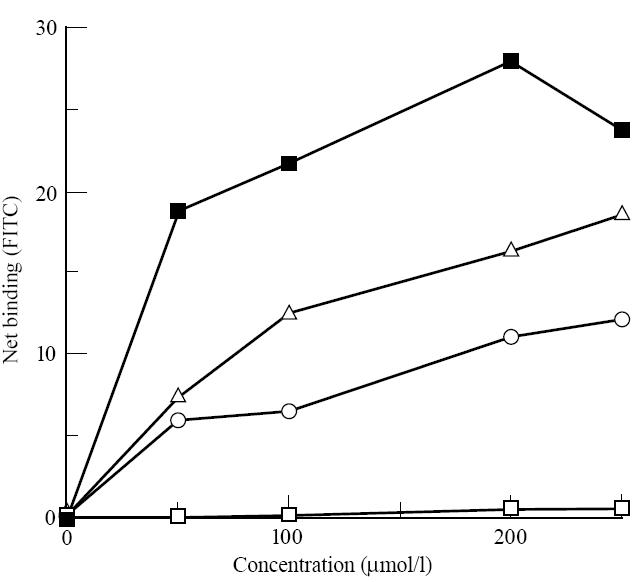
Having established that peptide A, a candidate toxic epitope from A-gliadin, bound to DQ2-expressing transfectants, we tested it in an assay against those expressing DR3 (Fig. 6). Binding did not occur, although peptide B, on the other hand, appeared to bind as well to DR3 as peptide A did to DQ2. Peptide C bound very weakly. The peptide MT3-13 bound strongly, as shown in soluble assays [14, 18], confirming the specificity of our model.
Fig. 6.
Direct binding of gluten-derived peptides and the mycobacterial heat shock protein peptide MT3-13, used as a positive control, to HLA-DR3-expressing L-cell transfectants, measured by FACS analysis. Net binding is expressed in arbitrary units, the calculation of which is described in Materials and Methods. ▴, Peptide MT3-13; ▵, peptide A; ○, peptide B; □, peptide C.
Truncations and point substitutions of peptide A
Since peptide A bound to the DQ2 transfectants, albeit only moderately, we attempted to define the residues within it that are important for binding.
For the truncation experiments we used lymphoblastoid B cell lines, derived from CD patients, homozygous for HLA-DR3, -DQ2, having established with the transfectants that peptide A bound to DQ2 but not to DR3.
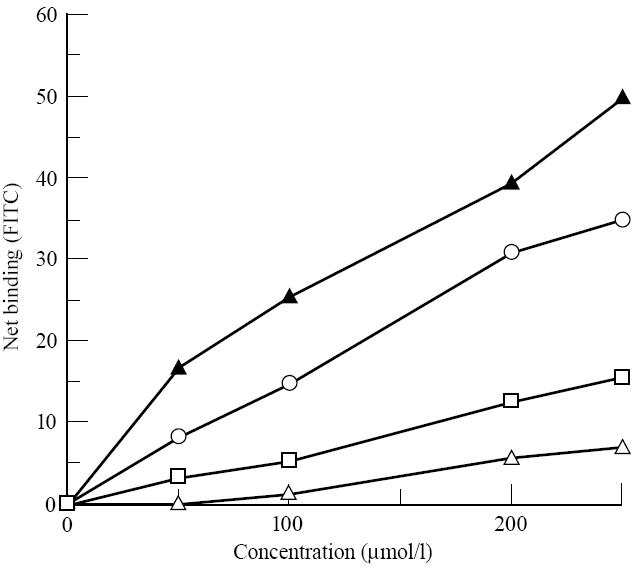
Truncations of one (F49) and then two amino acids (P48, F49) from the C-terminal end appeared to increase binding (Fig. 7), but loss of a third amino acid in addition (Q47) resulted in a dramatic reduction in binding.
Fig. 7.
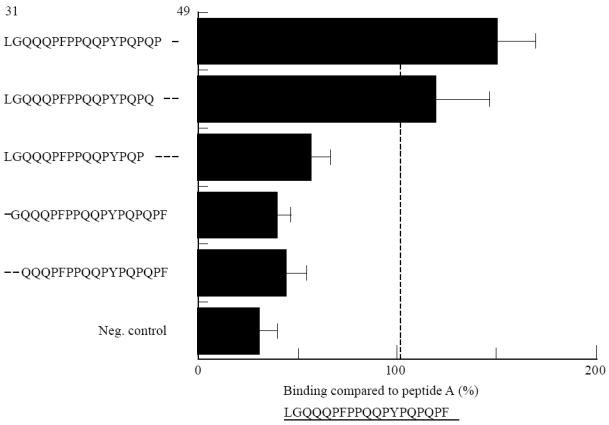
Direct binding of truncated peptide A analogues (250 μm) to B cell lines homozygous for HLA-DR3 and -DQ2, derived from coeliac disease patients. Binding is expressed as a percentage compared with binding of peptide A. The negative control is the HLA-negative K562 erythroleukaemic cell line.
Truncation of the N-terminal L31 or L31 and G32 also resulted in marked loss of binding. Thus, the minimum peptide derived from peptide A that was found to bind to DQ2, with anything nearing the affinity of peptide A, corresponded to residues 31–47.
DQ2 transfectants were used to assess the effect of alanine point substitutions on the binding of peptide A. Given the large representation of proline residues in the N-terminal region of A-gliadin, substitution of these residues was undertaken, in addition to some glutamine residues, which are also well represented. Alanine substitutions at P36 and P38 resulted in increased binding affinity (Fig. 8), whereas substitutions at other proline positions either had no effect (P46) or caused a reduction in binding (P39, P42, P44, P48). Substitution by alanine at Q33 and Q41 (Fig. 8) also resulted in a lowering of binding affinity.
Fig. 8.

Direct binding of alanine point substituted analogues of peptide A (250 μm) to HLA-DQ2-expressing L-cell transfectants. Binding is expressed as a percentage compared with binding of peptide A. The L-cell parent line, DAP-3, is the negative control.
Computer modelling
A model of the three-dimensional structure of the HLA-DQ2 peptide binding cleft, constructed from published sequences [32], allowed the establishment of a best fit model of peptide A within the binding groove. The P1 pocket of the DQ2 mole- cule is comprised of residues αHis26, αGln33, αPhe34, αTrp45, βAsn82 and βGlu86, of which αPhe34 and βAsn82 are conserved in all DQA and DQB alleles (Fig. 9, inset). The P1 pocket therefore has both a hydrophobic and hydrophilic character, with net negative charge contributed by βGlu86, suggesting that the pocket would show a preference for polar side chains. The optimal three-dimensional model of peptide A bound within the HLA-DQ2 binding groove suggests that residues L31 and G32 are found at the N-terminus of the binding groove, with the side chain of Q33 anchored within the P1 pocket and Q41 at the C-terminus (Fig. 9a,b).
Fig. 9.
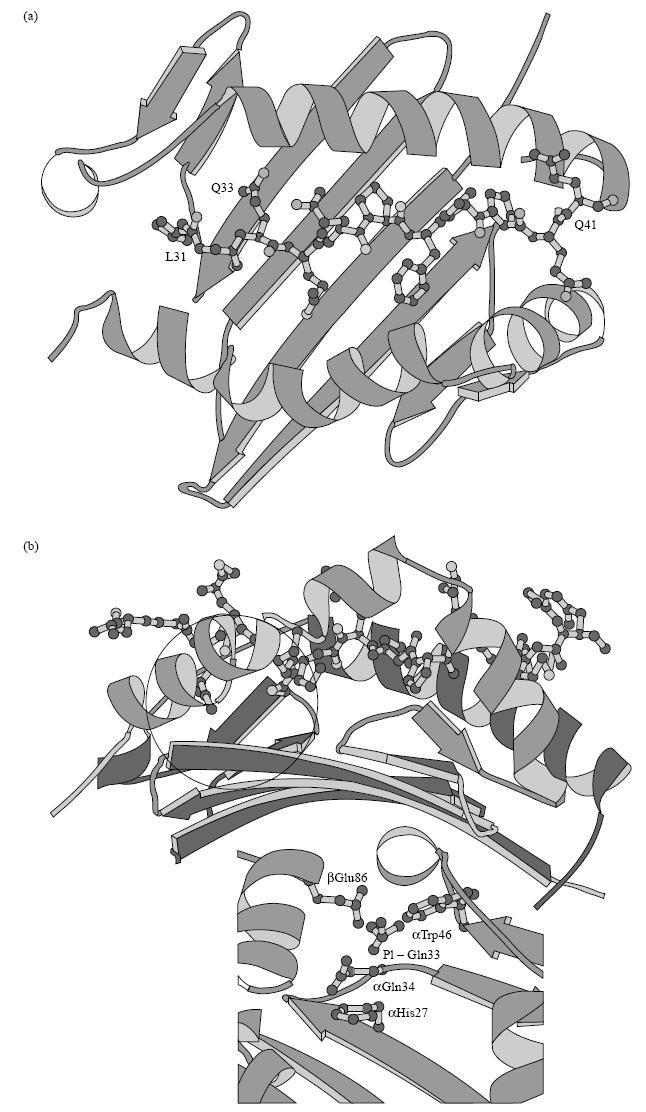
(a) Top view and (b) side view of a three-dimensional model showing peptide A bound within the HLA-DQ2 (α1*0501, β1*0201) binding cleft. Inset: HLA-DQ α- and β-chain residues important in forming the negatively charged anchoring P1 pocket. The circle in (b) outlines the anchoring side chain of the glutamine residue corresponding to amino acid 33 in peptide A within the P1 pocket.
DISCUSSION
We have demonstrated that a peptide corresponding to residues 31–49 of A-gliadin (‘peptide A’) binds to DQ2 in a cellular assay. This is important, given the strong association between CD and this class II restriction molecule and the fact that peptide A has been shown to exacerbate the coeliac lesion both in vitro [11] and in vivo [12]. Johansen et al. [14] have previously shown binding of this same peptide to affinity-purified DQ2 molecules in a biochemical competitive inhibition assay. We have found a similar level of binding (i.e. moderate/weak), confirming the reproducibility of this finding, using an alternative system which more closely resembles the in vivo situation. Further to this, Gjertsen et al. [13] have established a gluten-sensitive T cell clone from the peripheral blood of a CD patient that recognizes peptide A and is restricted by the DQ2 heterodimer. Johansen et al. have used the same T cell clone to establish a functional DQ2 peptide binding assay.
The significance of the binding of peptide B to DR3 (Fig. 6) is unclear. Gjertsen et al. [33] have reported several gluten-responsive T cell clones from the peripheral blood of treated CD patients that are DR-restricted, although the epitope involved is not known. These findings suggest that DR heterodimers can bind gluten-derived peptides and present them to T cells in the peripheral blood. Whether the same is true in the intestine will require further work, but Lundin et al. [6] did find one gluten-specific, small intestinal derived T cell clone, restricted by DR.
The results of the truncation experiments were similar to the findings of Johansen et al. [14], who in addition found that although peptide A and A-gliadin 31–47 stimulated the peptide A-specific clone of Gjertsen et al., A-gliadin 31–48 did not. Thus, although all these three peptides bind to DQ2, T cell recognition is abolished when proline is at the C-terminal end (i.e. A-gliadin 31–48). Why this should be so is not clear; presumably the conformation of the residues facing out of the class II binding groove is altered in such a way as to be unrecognizable by the T cells.
The peptide binding motif of DQ2 has been established [16, 17]. Vartdal et al. [17] have characterized the five pockets (P1, P4, P6, P7, P9) within the DQ2 binding groove important for binding. Briefly, large hydrophobic residues at relative positions P1 and P9 are preferred for binding to the large pockets at either end of the groove; P4 and P7 positions are best filled with negative side chains, the pocket at P4 also accommodating aliphatic residues, and small residues required at P6. In our computer model of the best fit for peptide A within the binding groove, Q33 is not ideal for P1, being hydrophilic, but it is polar and such residues can also interact with the pocket corresponding to this site. Alanine substitution at Q33 resulted in reduced binding, which is surprising as it is hydrophobic, although it is a small residue. Pro 36 occurs at P4, which would lead to a poor interaction, as it is not negative. Alanine substitution resulted in a marked increase in binding, as expected, since hydrophobic aliphatic residues can also make reasonable anchors at this position. Pro 39 at P7 makes a poor interaction, alanine substitution resulting in reduced binding; so of these two residues, proline, though not ideal, confers better binding characteristics at this position. Q41 at P9 is similarly not ideal, but surprisingly alanine substitution resulted in reduced binding affinity, despite being aliphatic and hydrophobic.
The interactions made by peptide A with the DQ2 binding groove in the computer model reported here, and with the binding motif described by Vartdal et al. [17], are generally poor, non-ideal matches, which explains the weak/moderate binding of peptide A observed by us and others [14]. However, only two of the five alanine substitutions made at the anchor sites give the expected changes in binding affinity, based on the described binding motif. It may be that conformational changes in the peptide backbone are induced by these substitutions, leading to the observed results. Alternatively, intracellular processing may alter the peptide in some way.
Differences between the cellular binding method described here and binding to purified class II molecules are likely to arise from intracellular processing events, though these events are currently not known. Further work on the effect of metabolic inhibitors in a cellular assay may elucidate this.
In soluble assays binding occurs best at acidic pH, correlating with the known optimum antigen/MHC class II interaction taking place in an acidic intracellular compartment, in live antigen-presenting cells [34, 35]. It was therefore unnecessary to maintain an extracellular acidic environment in our assay.
This study confirms the work of others by using a cellular assay which is closer to the disease situation. There is now considerable evidence for peptide A being involved in the molecular pathogenesis of CD, as a toxic epitope within gliadin—probably one of many [10]—presented in association with DQ2 to small intestinal T cells. Johansen et al. [14] have speculated that T cell epitopes with high binding affinity to DQ2 are more likely to be pathogenic. This is not necessarily the case, as shown by the example of peptide A. The amino acid residues pointing out of the grove and interacting with the T cell receptor are more important than the anchor residues allowing class II binding. It is these residues that allow T cell recognition and subsequent activation. Clearly there are situations where strong DQ2 binding occurs without T cell recognition. There may thus be many epitopes within gliadin that bind with perhaps varying affinity to DQ2, but which share a similar conformation of residues for T cell recognition. Further work will be required to verify or refute this hypothesis.
Acknowledgments
We are grateful to Nicola O'Reilly and Elizabeth Li for synthesizing our peptides, Drs K. Welsh and R. Vaughn (Transplantation Unit, Guy's Hospital, London, UK) for PCR-SSP genotyping, and Professor I. Hart (Imperial Cancer Research Fund, London, UK) for the K562 cell line. This study was supported by grants from the St Thomas' Research (Endowments) Committee and the National Institutes of Health, Bethseda, MD (RO1 DK47716).
References
- 1.Sollid L, Markussen G, Ek J, Gjerde H, Vartdal F, Thorsby E. Evidence for a primary association of celiac disease to a particular HLA-DQ α/β heterodimer. J Exp Med. 1989;169:345–50. doi: 10.1084/jem.169.1.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lundin KEA, Sollid LM, Qvigstad E, et al. T Lymphocyte recognition of a celiac disease-associated cis- or trans-encoded HLA-DQ α/β heterodimer. J Immunol. 1990;145:136–9. [PubMed] [Google Scholar]
- 3.Sollid LM, Thorsby E. HLA susceptibility genes in celiac disease: genetic mapping and role in pathogenesis. Gastroenterology. 1993;105:910–22. doi: 10.1016/0016-5085(93)90912-v. [DOI] [PubMed] [Google Scholar]
- 4.Tighe MR, Hall M, Ashkenazi A, Siegler E, Lanchbury J, Ciclitira PJ. Coeliac disease among Ashkenazi jews from Israel: a study of the HLA class II alleles and their association with disease susceptibility. Hum Immunol. 1993;38:270–5. doi: 10.1016/0198-8859(93)90554-e. [DOI] [PubMed] [Google Scholar]
- 5.Ek J, Albrechtsen D, Solheim BG, Thorsby E. Strong association between the HLA-Dw3 and coeliac disease. Scand J Gastroenterol. 1978;13:229–33. doi: 10.3109/00365527809181753. [DOI] [PubMed] [Google Scholar]
- 6.Lundin K, Scott H, Hansen T. Gliadin specific HLA-DQ (A1*0501, B1*0201) restricted T cells isolated from the small intestinal mucosa of coeliac patients. J Exp Med. 1993;178:187–96. doi: 10.1084/jem.178.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trier JS. Celiac sprue. New Engl J Med. 1991;325:1709–19. doi: 10.1056/NEJM199112123252406. [DOI] [PubMed] [Google Scholar]
- 8.Marsh MN. Gluten, major histocompatibility complex, and the small intestine. Gastroenterology. 1992;102:330–54. [PubMed] [Google Scholar]
- 9.MacDonald TT, Spencer J. Evidence that activated mucosal T cells play a role in the pathogenesis of enteropathy in human small intestine. J Exp Med. 1988;167:1341–9. doi: 10.1084/jem.167.4.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lundin KEA, Sollid LM, Anthonsen D, et al. Heterogeneous reactivity patterns of HLA-DQ restricted, small intestinal T-cell clones from patients with celiac disease. Gastroenterology. 1997;112:752–9. doi: 10.1053/gast.1997.v112.pm9041236. [DOI] [PubMed] [Google Scholar]
- 11.Shidrawi RG, Day P, Przemioslo RT, Ellis HJ, Nelufer JM, Ciclitira PJ. In vitro toxicity of gluten peptides in coeliac disease assessed by organ culture. Scand J Gastroenterol. 1995;30:758–62. doi: 10.3109/00365529509096324. [DOI] [PubMed] [Google Scholar]
- 12.Sturgess RP, Day P, Ellis HJ, et al. Wheat peptide challenge in coeliac disease. Lancet. 1994;343:758–61. doi: 10.1016/s0140-6736(94)91837-6. [DOI] [PubMed] [Google Scholar]
- 13.Gjertsen HA, Lundin KEA, Sollid LM, Eriksen JA, Thorsby E. T cells recognise a peptide derived from α-gliadin presented by the celiac disease-associated HLA-DQ (α1*0501, β1*0201) heterodimer. Hum Immunol. 1994;39:243–52. doi: 10.1016/0198-8859(94)90267-4. [DOI] [PubMed] [Google Scholar]
- 14.Johansen BH, Gjertsen HA, Vartdal H, et al. Binding of peptides from the N-terminal region of α-gliadin to the celiac disease-associated HLA-DQ2 molecule assessed in biochemical and T cell assays. Clin Immunol Immunopathol. 1996;79:288–93. doi: 10.1006/clin.1996.0081. [DOI] [PubMed] [Google Scholar]
- 15.Johansen BH, Vartdal F, Eriksen JA, Thorsby E, Sollid LM. Identification of a putative motif for binding of peptides to HLA-DQ2. Int Immunol. 1996;8:177–82. doi: 10.1093/intimm/8.2.177. [DOI] [PubMed] [Google Scholar]
- 16.van de Wal Y, Kooy YMC, Drijfhout JW, Amons R, Koning F. Peptide binding characteristics of the coeliac disease-associated DQ (α1*0501, β1*0201) molecule. Immunogenetics. 1996;44:246–53. doi: 10.1007/BF02602553. [DOI] [PubMed] [Google Scholar]
- 17.Vartdal F, Johansen BH, Friede T, et al. The peptide binding motif of the disease associated HLA-DQ (α1*0501, β1*0201) molecule. Eur J Immunol. 1996;26:2764–72. doi: 10.1002/eji.1830261132. [DOI] [PubMed] [Google Scholar]
- 18.Johansen BH, Buus S, Vartdal S, et al. Binding of peptides to HLA-DQ molecules: peptide binding properties of the disease-associated HLA-DQ (α1*0501, β1*0201) molecule. Int Immunol. 1994;6:453–61. doi: 10.1093/intimm/6.3.453. [DOI] [PubMed] [Google Scholar]
- 19.Kagnoff MR, Raleigh KA, Hubert JJ, Bernadin JF, Kasarda DD. Possible role of a human adenovirus in the pathogenesis of coeliac disease. J Exp Med. 1984;160:1544–7. doi: 10.1084/jem.160.5.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tatham AS, Marsh MN, Wieser H, Shewry PR. Conformational studies of peptides corresponding to the coeliac-activating regions of wheat alpha gliadin. Biochem J. 1990;270:313–8. doi: 10.1042/bj2700313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walls E, Crawford D. Lymphocytes—a practical approach. In: Klaus G, editor. 1987. pp. 149–62. Oxford: IRL Press, [Google Scholar]
- 22.Long EO, Rosen-Bronson S, Karp D, Malnati M, Sekaly R, Jaraquemada D. Efficient cDNA expression vectors for stable and transient expression of HLA-DR in transfected fibroblast and lymphoid cells. Hum Immunol. 1991;31:229–35. doi: 10.1016/0198-8859(91)90092-n. [DOI] [PubMed] [Google Scholar]
- 23.Jacobson S, Sekaly R, Jacobson C, McFarland H, Long E. HLA-class II restricted presentation of cytoplasmic measles-virus antigen to cytoxic T-cells. J Virol. 1989;63:1756–60. doi: 10.1128/jvi.63.4.1756-1762.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Busch R, Strang G, Howland K, Rothbard J. Degenerate binding of immunogenic peptides to HLA-DR proteins on B cell surfaces. Int Immunol. 1990;2:443–51. doi: 10.1093/intimm/2.5.443. [DOI] [PubMed] [Google Scholar]
- 25.Busch R, Rothbard J. Detection of peptide-MHC class II complexes on the surface of intact cells. J Immunol Methods. 1990;134:1–22. doi: 10.1016/0022-1759(90)90107-7. [DOI] [PubMed] [Google Scholar]
- 26.Thorpe CJ, Travers PJ. Prediction of an HLA B44 binding motif by the alignment of known epitopes and molecular modelling of the antigen binding cleft. Immunogenetics. 1994;40:303–5. doi: 10.1007/BF00189977. [DOI] [PubMed] [Google Scholar]
- 27.Jurcavic S, Travers P, Hills A, Agrewala J, Moreno C, Ivanyi J. Distinct conformation of an HLA-permissive peptide when bound to HLA-DR1 or DRB5*0101 molecules. Int Immunol. 1996;8:1807–14. doi: 10.1093/intimm/8.11.1807. [DOI] [PubMed] [Google Scholar]
- 28.Brown JH, Jardetsky TS, Gorga JC, et al. Three-dimensional structure of class II histocompatibility antigen HLA-DR1. Nature. 1993;329:506–18. doi: 10.1038/364033a0. [DOI] [PubMed] [Google Scholar]
- 29.Stern LJ, Brown JH, Jardetsky TS, et al. Crystal structure of the human class II MHC protein HLA-DR1 complexed with an influenza virus peptide. Nature. 1994;368:215–21. doi: 10.1038/368215a0. [DOI] [PubMed] [Google Scholar]
- 30.Sutcliffe M, Haneef I, Carney D, Blundell T. Knowledge based modelling of homologous proteins. Part 1: three dimensional frameworks derived from the simultaneous superposition of multiple structures. Prot Eng. 1987;1:377–84. doi: 10.1093/protein/1.5.377. [DOI] [PubMed] [Google Scholar]
- 31.Sutcliffe M, Hayes F, Blundell T. Knowledge based modelling of homologous proteins. Part II: rules for the conformations of substituted side chains. Prot Eng. 1987;1:385–92. doi: 10.1093/protein/1.5.385. [DOI] [PubMed] [Google Scholar]
- 32.Horn G, Bugawan T, Long C, Manos M, Erlich H. Sequence analysis of HLA class II genes from insulin dependent diabetes individuals. Hum Immunol. 1988;21:249–254. doi: 10.1016/0198-8859(88)90034-1. [DOI] [PubMed] [Google Scholar]
- 33.Gjertsen HA, Sollid LM, Ek J, Thorsby E, Lundin KEA. T cells from the peripheral blood of coeliac disease patients recognize gluten antigens when presented by HLA-DR, -DQ, or -DP molecules. Scand J Immunol. 1994;39:567–74. doi: 10.1111/j.1365-3083.1994.tb03414.x. [DOI] [PubMed] [Google Scholar]
- 34.Jensen PE. Regulation of antigen presentation by acidic pH. J Exp Med. 1990;171:1779–84. doi: 10.1084/jem.171.5.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mouritsen S, Hansen AS, Petersen BL, Buus S. pH dependence of the interaction between immunogenic peptides and MHC class II molecules. J Immunol. 1992;148:1438–44. [PubMed] [Google Scholar]