

Abstract
The chemoattractant cytokine IL-16 has been reported to suppress lymphocyte activation and to inhibit HIV-1 replication in acutely infected T cells. We have cloned and expressed human IL-16 in Escherichia coli and investigated whether the recombinant protein could regulate the level of lymphocyte apoptosis from HIV-1-infected subjects. After purification and refolding, only 2–10% of the recombinant cytokine was present in a biologically active homotetrameric form. This could explain the need for high concentrations of the bacterially derived IL-16 to induce significant inhibition of HIV-1 replication. Addition of IL-16 to unstimulated peripheral blood mononuclear cell (PBMC) cultures from HIV-1-infected subjects did not modify the observed level of spontaneous lymphocyte apoptosis. In contrast, IL-16 added to PBMC cultures stimulated with anti-CD3, anti-CD95 or dexamethasone reduced significantly the percentage of lymphocytes undergoing AICD. This effect was found to correlate with the ability of the cytokine to decrease CD95 expression on activated CD4+ T cells. Comparative studies on PBMC from healthy individuals indicated that the regulation of apoptosis levels by IL-16 is a complex phenomenon and could depend on the nature of the activator used and/or the immune status of lymphocytes tested. The outcome of CD4 cross-linking on T cells by various ligands is discussed in the context of the observed beneficial activities of IL-16 and its potential role in the treatment of HIV disease.
Keywords: HIV-1 replication, apoptosis, IL-16
INTRODUCTION
In HIV infection, functional defects and deletion of antigen-reactive T cells are more frequent than can be explained by direct viral infection. Cells from infected subjects were found to undergo in vitro a spontaneous process of programmed cell death (PCD). This process also appeared to increase in magnitude upon cell activation with different stimuli [1,2]. Viral antigens including gp120 and tat have been reported to accelerate, even in non-infected lymphocytes, the AICD and are therefore believed to contribute to CD4+ T cell depletion in HIV disease [3,4]. The relationship between T cell depletion and PCD in HIV infection has been further substantiated by the in vivo findings of high apoptosis levels, occurring predominantly in bystander cells, in lymph nodes of infected subjects [5]. The increased intensity of the apoptotic phenomenon in lymph nodes was also linked to a general state of immune activation and was shown to be independent of disease progression or of the level of viral load [6]. However, in peripheral lymphocytes of HIV-infected subjects, the increased susceptibility to apoptosis was correlated with disease progression [7,8] or with high viral load [9] and could be prevented through cosignals delivered simultaneously by IL-1 and IL-2 [10]. In addition, the inhibition of AICD could similarly be achieved by costimulation of cell cultures with type-1 cytokines, interferon-gamma (IFN-γ), IL-2 or IL-12 [11,12]. Nevertheless, a correlation between the ability of a cytokine to inhibit HIV replication and to block the increased sensitivity of lymphocytes to PCD is not yet evident. This would necessitate a thorough evaluation of the capacity of HIV-suppressive cytokines to inhibit in parallel the enhanced level of lymphocyte apoptosis.
Among the mechanisms known to play an important role in the apoptosis of CD4+ lymphocytes following infection with HIV are the increased expression of the death receptor CD95 (Fas) and the increased production of CD95-ligand by peripheral blood mononuclear cells (PBMC) [13]. Cross-linking the CD4 antigen, using anti-CD4 antibodies or HIV-1 envelope glycoprotein, was similarly shown to enhance T cell apoptosis by up-regulating CD95 and CD95-ligand expression on lymphocytes and monocytes, respectively [14]. On the other hand, the natural ligand for CD4 has been identified as the chemoattractant cytokine IL-16 [15]. This molecule has been reported to be active only when present in homotetrameric form, to inhibit lymphocyte activation [16] and to suppress HIV-1 replication [17,18]. Therefore, in this study we addressed the question whether CD4 cross-linking by its natural ligand could regulate the level of spontaneous and activation-induced cell death in cultures from HIV-1-infected subjects or healthy controls. In addition, we cloned, expressed and evaluated the inhibitory activity of recombinant IL-16 on the replication of macrophage-tropic (M-tropic) and T-tropic HIV-1 isolates in human PBMC. We present data to demonstrate that the HIV-suppressing activity of IL-16 is mediated by the tetrameric form. However, the latter constitutes only up to 10% of the total protein produced in the bacterial expression system. Furthermore, we provide evidence for a protective activity of the recombinant cytokine against AICD and we show that this effect is mediated, at least in part, by down-regulation of CD95 expression on CD4+ lymphocytes from HIV-infected subjects.
SUBJECTS AND METHODS
Study subjects
A total of 34 HIV-1 infected subjects and 18 seronegative healthy controls was studied. Among the HIV group, 15 were asymptomatics (grade A according to the classification of the Centers for Disease Control) and 19 were symptomatics (grade B). At the time blood samples were drawn, only one asymptomatic and five symptomatics were not receiving anti-retroviral therapy. The rest of the subjects were on treatment with either two reverse transcriptase inhibitors or a three-drug regimen containing one protease inhibitor. The CD4 counts (mean ± s.e.m.; 662 ± 74 cells/mm3; range 300–1458 cells/mm3) and viral loads (median 1992 copies/ml; range 150–293 695 copies/ml) in the asymptomatics were comparable to those detected in symptomatic patients (570 ± 43 cells/mm3; range 394–1061 cells/mm3 and median 5306; range 200–199 970 copies/ml). None of the subjects presented with opportunistic infections during the study period. Because no significant differences (P > 0.05; Mann–Whitney U-rank test) could be noted between grade A and grade B infected subjects with regard to clinical parameters or to the effects of IL-16 described below, data on all subjects from both categories were pooled to represent a single group.
Reagents used
Murine anti-human MoAbs used either in culture or for cytofluorometric detection were CD4–FITC, CD95–PE (UB2), IgG1–FITC, IgG1–PE, CD3 (UCHT1) and purified azide-free IgM anti-human CD95 (CH11) (Immunotech, Marseille, France). Other reagents were dexamethasone (DEX), phytohaemagglutinin (PHA; Sigma, La Verpillière, France), YOPRO-1 dye (Molecular Probes Inc, Eugene, OR), polyrA and oligo dT (Pharmacia, Saclay, France), and 3H-thymidine triphosphate (Amersham, Les Ulis, France). Gel filtration standards were from BioRad (Ivry-sur-Seine, France). IL-2 was purchased from Boehringer Mannheim (Meylan, France) and macrophage inflammatory protein-1α (MIP-1α) from R&D Systems (Abingdon, UK).
Cloning and expression of human IL-16
Total RNA from PHA-activated PBMC was isolated using RNA Plus and following the manufacturer's recommendations (Bioprobe Systems, Montreuil, France). Reverse transcription was performed in 25 μl final volume containing 0.5 μg total RNA which was incubated for 5 min at 65°C before the addition of 0.25 mm mix dNTPs, 1× reverse transcriptase (RT) buffer (50 mm Tris–HC1 pH 8.3, 75 mm KCl, 3 mm MgC12, 8 mm DTT, 4 U RNase inhibitor) and 200 U Mu-MLV RT. The reaction was allowed 1 h at 37°C and 3 min at 92°C. To produce recombinant human IL-16, we amplified 2 μl of the product of reverse transcription in the presence of IL-16-specific primers flanked with BamH1 at the 5′ end and HindIII site at the 3′ end. The primers derived from the published sequence of IL-16 [19] were: sense: GAGAGGATCCATGCCCGACCTCAACTCCTCC, antisense: GAGAAAGCTTCTAGGAGTCTCCAGCAGCTGT.
The amplified product of the right size was purified, digested with BamH1 and HindIII, subcloned in pQE 30 expression vector (Qiagen GmbH, Hilden, Germany) and entirely sequenced. The obtained sequence was identical to the published one [19]. The production of IL-16 was done in Escherichia coli following the manufacturer's recommendations and was rendered endotoxin-free (< 0.125 endotoxin unit (EU)/20 μg protein) by several passages over polymyxin-B column (Pierce, Montluçon, France).
Further purification of IL-16 by medium pressure liquid chromatography was performed using a gel-filtration column (BioSilect SEC125.5, 300 × 7.8 mm; BioRad) connected to the Biologic Workstation (BioRad). The column was equilibrated with 50 mm phosphate buffer, 200 mm NaC1, pH 6.8 at a flow rate of 1 ml/min. Fractions (0.5 ml) were collected and were tested for inhibition of HIV replication as described below. Molecular weights of eluted forms of recombinant hIL-16 were deduced from a curve calculated by plotting the decimal logarithm of standard molecular weight protein markers versus their elution time expressed in seconds.
Culture conditions
PBMC were isolated from heparinized blood samples by Ficoll–Hypaque density gradient centrifugation, and cultured in RPMI 1640 (GIBCO, Courbevoie, France) supplemented with 10% fetal calf serum (FCS; Boehringer), 2 mm glutamine (GIBCO) and 1% gentamycin (Schering Plough, Levallois-Perret, France). Cells were cultured for 24 h in 96-well plates (Costar, Brumath, France) at 2.5 × 105/ml or at 5 × 105/ml in 48-well plates (Costar). Lymphocyte apoptosis was induced by stimulation with DEX (10−7 m), PHA (1 μg/ml) or anti-CD3 MoAb (0.5 μg/ml). The effect of CD95 cross-linking was also explored by incubating cells with the CD95 MoAb CH11 (3 μg/ml). When added to the cultures, MIP-1α and IL-16 were used at 0.2 and 1 μg/ml, respectively, unless otherwise stated.
Measurement of apoptosis
Apoptosis was measured by either optical or cytofluorometric analysis of cell death, as previously described [12,20]. Briefly, cells were counted in a haemocytometer in the presence of 0.5% trypan blue. Cells counted as apoptotic included those with characteristic nuclear chromatin condensation and fragmentation as well as dead cells that had lost trypan blue exclusion capacity. Cytofluorometric analysis of cell death was assessed using the impermeant DNA intercalatant YOPRO-1 dye, which was added at 10 μm final concentration to 5 × 104 cells and was allowed to stand for 5 min at room temperature in the dark. Cell analysis was performed on a Vantage cell sorter (Becton Dickinson) using an argon-ion laser tuned to 488 nm. Green cell fluorescence, gated on forward and side light scatter, was collected using a 525 ± 10 nm band-pass filter and displayed using a logarithmic amplification. Inhibition of cell death by costimulation with cytokines was calculated as follows:
The reproducibility and specificity of detection of apoptotic cells were verified randomly on samples from five different subjects by (i) assessing the apoptosis levels on the same samples in triplicate and obtaining < 5% variation among triplicate measurements, and (ii) cell sorting the populations representing apoptotic cells, following flow cytometry, and demonstrating that these cells did not show any uptake of trypan blue.
Phenotypic analysis of living cells
In some experiments, the expression of CD95 antigen on the surface of living CD4+ T cells was examined. PBMC stimulated for 24 h with DEX, PHA or anti-CD3, in the presence or absence of IL-16 (1 μg/ml) were first incubated with FITC- and PE-conjugated MoAbs (10 μg for 105 cells in a 100-μl final volume) for 30 min at 4°C in the dark. Cells were subsequently washed twice with cold PBS containing 10% bovine serum albumin (BSA). Live lymphocytes were gated using their higher forward and side light scatter intensities and were analysed for simultaneous double staining by emission of green and orange fluorescences for CD4 and CD95 expression, respectively. Percentages of CD95-expressing CD4+ T cells were determined after correcting for non-specific double staining.
HIV-1 infection in vitro
PHA blasts generated in 3 days' culture of PBMC from healthy donors were incubated for 2 h at 37°C with a dose of HIV-1 corresponding to 5000 ct/min of RT activity per 106 cells. Following two washings, cells were cultured at 5 × 105/ml in medium containing 10 U/ml IL-2 and were fed twice a week by replacing 0.5 ml of the supernatant with fresh medium. Cytokines evaluated for inhibitory effects on HIV replication were added immediately upon seeding and were present continuously in culture. Infection was performed with one of three isolates: the T-tropic strain HIV-1 IIIB, the M-tropic strain HIV-1Bal and a primary non-syncytium-inducing, M-tropic strain CHR4, which was isolated from an HIV-1+ individual and was phenotyped in our laboratory. Viral replication was evaluated by the level of RT activity in culture supernatants, as previously described [21].
Statistical analysis
Statistical comparisons were made using the non-parametric Wilcoxon matched-pairs test or Mann–Whitney U-rank test. Probability values < 0.05 were considered significant.
RESULTS
Effect of IL-16 on HIV-1 replication in human PBMC
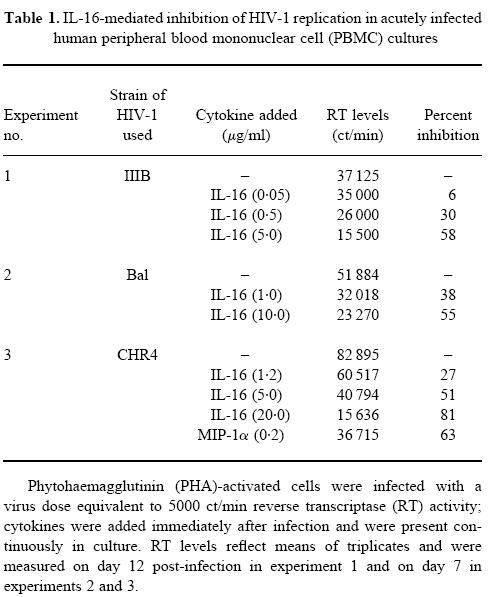
PHA-activated cells from different donors were infected either with T-tropic or M-tropic HIV-1 isolates and were maintained in culture in the absence or presence of various concentrations of IL-16. The level of RT in the supernatants was assessed between days 7 and 14 and results from three representative experiments are shown in Table 1. It was noted that a 50–60% inhibition of viral replication could be achieved with a concentration ≥ 5 μg/ml of the recombinant cytokine. This effect was evident on all viral strains tested, including the primary isolate CHR4. The replication of this latter was also found to be inhibited (63%) by the continuous presence of MIP-1α in the culture at a concentration of 0.2 μg/ml (Table 1). The maximum level of inhibition observed under our experimental conditions was 90% using the highest endotoxin-free concentration of our recombinant IL-16 (20 μg/ml). We did not attempt to use higher concentrations to achieve 100% inhibition due to the potential and probable effect of minute contaminants, which could lead either to false-negative or to false-positive results.
Table 1.
IL-16-mediated inhibition of HIV-1 replication in acutely infected human peripheral blood mononuclear cell (PBMC) cultures
The relatively high concentration of IL-16 needed to induce significant inhibition of viral replication prompted us to evaluate the percentage of the biologically active tetrameric form of IL-16 in our recombinant preparation. Thus, 25 μg of the cytokine were passed over gel filtration column and 0.5-μl fractions of different molecular sizes were collected as described in Subjects and Methods. The fractions were filtered through 0.22-μm Millipore filters and were tested at 1:20 dilution (v/v), for the inhibitory activity on HIV-1 Bal replication. Results shown in Fig. 1 demonstrate that the virus-inhibiting activity of IL-16 was retained in the fraction containing the tetrameric form (50–60 kD), but which only represented 2–10% of the total protein (in six different batch preparations). Extrapolations from these results suggest that the observed IL-16-mediated inhibition on HIV-1 replication occurs at a concentration range of 100–500 ng/ml of the active homotetramer.
Fig. 1.
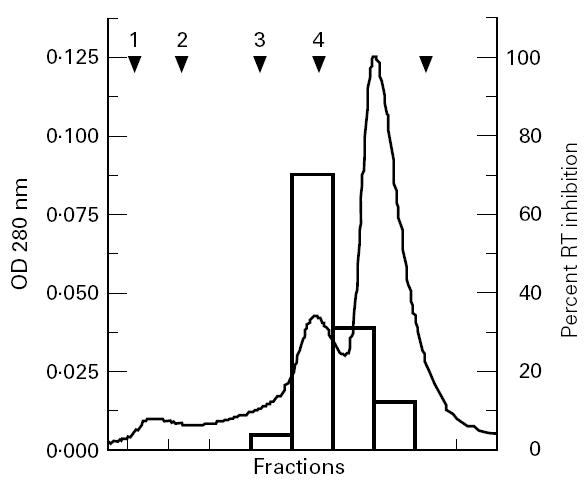
Separation of various oligomeric forms of IL-16 in the recombinant protein and evaluation of their HIV-suppressing activities. Purified IL-16 (25 μg) was loaded onto gel filtration column at a flow rate of 1 ml/min. Fractions (0.5 ml) were collected, diluted to 1:20 and tested for the inhibition of HIV-1Bal replication in acutely infected phytohaemagglutinin (PHA) blasts. The solid line represents the optical density (OD) values at 280 nm and open bars reflect the percentage inhibition of reverse transcriptase (RT) levels measured on day 14 post-infection. Arrows indicate the position at which standard molecular weight proteins were eluted from the column: 1 is thyroglobulin (670 kD), 2 is IgG (150 kD), 3 is human serum albumin (67 kD), 4 is ovalbumin (44 kD) and 5 is horse myoglobin (17 kD).
Effect of IL-16 on spontaneous and activation-induced cell death
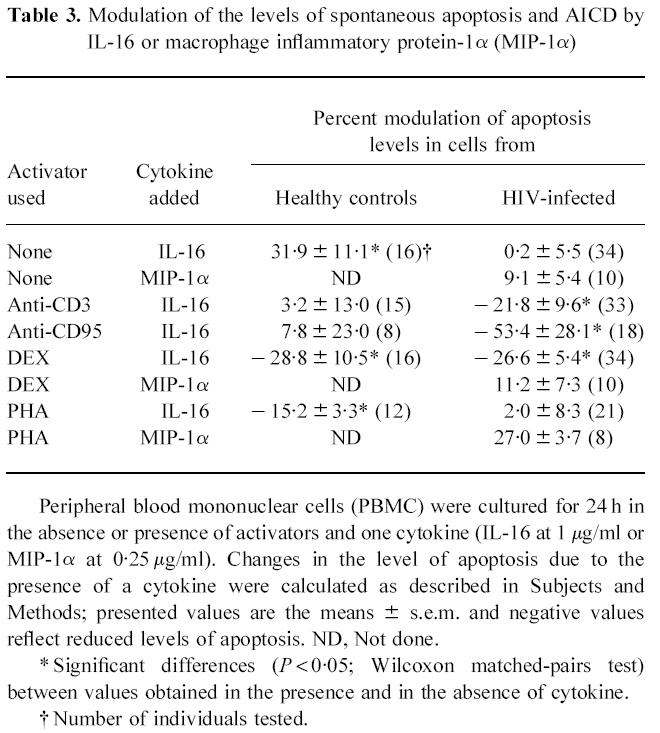
We then evaluated the ability of IL-16 to regulate the level of spontaneous lymphocyte apoptosis and AICD detectable in vitro from cells of HIV-infected subjects or healthy controls. Results shown in Table 2 represent the level of lymphocyte apoptosis detectable under different culture conditions, from the two tested groups. The percentage of spontaneous apoptosis in cells from HIV-1-infected subjects was significantly higher than, and over twice as much as that observed in cells from healthy controls. Similarly, the net percentage of AICD by anti-CD3 or by DEX was significantly elevated in cells from HIV-1-infected individuals, whereas that induced by IgM anti-CD95 or by PHA was similar and not significantly different among the two groups (Table 2). In preliminary experiments on cells from three subjects, addition of 0.01, 0.1 or 1 μg/ml of IL-16 to 24 h cultures demonstrated a dose-dependent inhibition of AICD which was maximal at the highest concentration tested. Thus, 1 μg/ml of IL-16 was then used in all further experiments to evaluate the effects on apoptosis level. As shown in Table 3, cells from healthy individuals incubated for 24 h in the presence of IL-16 showed a significant increase (32%) in the detectable level of spontaneous apoptosis. In contrast, IL-16 did not exert any detectable change in the level of spontaneous apoptosis when added to cells from HIV-1-infected subjects. Addition of the β-chemokine MIP-1α to cell cultures from the HIV group slightly increased the percentage of lymphocytes undergoing spontaneous apoptosis, although this effect did not attain statistical significance (P > 0.05, Wilcoxon matched-pairs test). On the other hand, IL-16 appeared to exert a different profile of activity on the level of AICD (Table 3). Thus, in cell cultures from healthy controls, the co-addition of IL-16 with DEX or with PHA reduced significantly by 29% and 15%, respectively, the level of AICD. No significant effect could be attributed to IL-16 in the protection against anti-CD3- or anti-CD95-induced apoptosis. Under the same culture conditions, costimulation of cells from HIV-1-infected subjects with IL-16 reduced significantly the apoptosis level induced by anti-CD3 (22%), by anti-CD95 (53%) or by DEX (27%), without modifying the level induced by PHA (2%). However, costimulation with MIP-1α at 0.25 μg/ml did not appear to exert any protective activity against DEX- or PHA-induced cell death in cultures from HIV-1-infected individuals (Table 3).
Table 2.
Levels of spontaneous apoptosis and AICD in lymphocytes from healthy controls and HIV-infected subjects
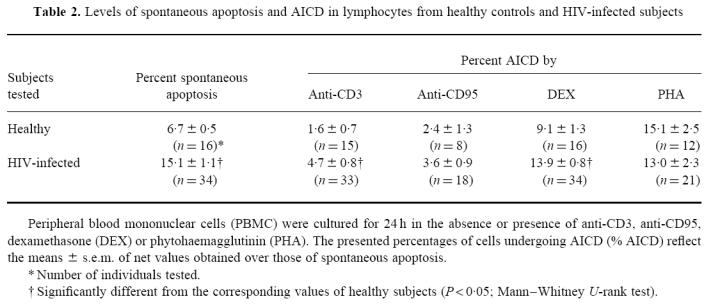
Table 3.
Modulation of the levels of spontaneous apoptosis and AICD by IL-16 or macrophage inflammatory protein-1α (MIP-1α)
Representative data, using flow cytometric analysis, of the protective activity of IL-16 against DEX-induced apoptosis in cells from one infected subject are shown in Fig. 2. Cultures maintained in medium without any stimulants for a period of 24 h showed 14.3% YOPRO-1+ cells undergoing spontaneous apoptosis (Fig. 2a). The percentages of YOPRO-1+ cells in parallel cultures stimulated either with DEX (Fig. 2b) or with DEX and IL-16 (Fig. 2c) were found to increase to 34.8% or to 24.6%, respectively. Thus, addition of IL-16 to DEX-treated cultures resulted in 50% protection against AICD.
Fig. 2.
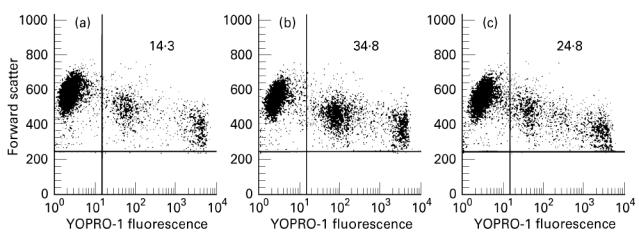
Flow cytometric analysis of the protective effect of IL-16 on dexamethasone (DEX)-induced apoptosis. Peripheral blood mononuclear cells (PBMC) from one HIV-infected subject were cultured for 24 h with (a) medium, (b) DEX and (c) DEX and IL-16. Cells undergoing apoptosis were quantified by staining with YOPRO-1 dye and the percentage of positive cells in the upper right quadrant reflects that of apoptotic cells.
Effect of IL-16 on CD95 expression
The level of AICD in CD4+ lymphocytes has been attributed to increased expression of the death receptor CD95 [13,14]. In an attempt to understand the mechanism of the protective activity of IL-16 on AICD, we analysed the level of surface expression of CD95 on CD4+ lymphocytes following stimulation of PBMC with DEX, with anti-CD3 or with PHA in the absence or presence of IL-16. Results presenting the percentages of CD4+ and CD95+ gated lymphocytes in cultures from nine HIV-1-infected subjects are shown in Fig. 3. The mean of double-positive lymphocytes in DEX-treated cultures was 21.1%, and this was found to become significantly reduced in cultures stimulated with DEX and IL-16 (Fig. 3a). Similarly, co-addition of IL-16 with anti-CD3 decreased the percentage of CD4 and CD95 double-positive cells from 36% to 28%. Although this effect did not attain statistical significance (P > 0.05), the reduction in the expression of CD95 on CD4+ lymphocytes could be observed in cultures from seven out of nine tested subjects (Fig. 3b). On the other hand, no change in the percentage of CD4+ lymphocytes expressing CD95 could be attributed to IL-16 in PHA-stimulated cultures (Fig. 3c).
Fig. 3.
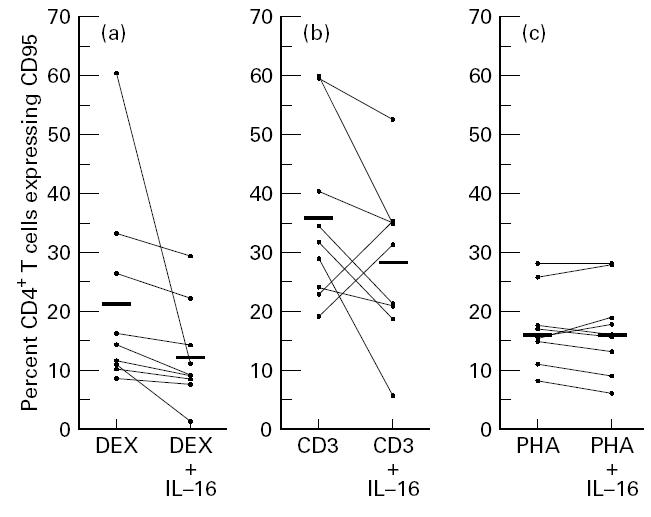
Effect of IL-16 on CD95 expression in CD4+ lymphocytes. Peripheral mononuclear cells from HIV-infected subjects were cultured for 24 h with (a) dexamethasone (DEX), (b) anti-CD3 antibodies and (c) phytohaemagglutinin (PHA), in the absence or presence of IL-16 (1 μg/ml). Cells were then double-stained with fluorescein-conjugated anti-CD4 and PE-conjugated anti-CD95. Plotted values represent the percentage of CD4+ lymphocytes expressing CD95. Bars indicate the arithmetic mean values.
DISCUSSION
Important advances have recently been made in the recognition of processes involved in HIV-1 entry into cells and in the identification of cytokines with virus-suppressing activities [22–25]. Several α and β chemokine receptors were found to serve as cofactors used by different HIV-1 isolates to infect different cell populations [22,23]. The respective ligands of these receptors were in turn reported to block viral entry [24,25]. On the other hand, IL-16, the lymphocyte chemoattractant that binds to the major HIV-1 receptor CD4, has also been identified as inhibitor of HIV replication in acutely infected PBMC cultures [17,26]. Our results, using M- and T-tropic virus isolates, confirm these earlier findings and the requirement to use 5 μg/ml of the recombinant cytokine produced in the E. coli expression system, to achieve 50% inhibition of viral replication. The need for such a high concentration of recombinant IL-16 was attributed to different folding characteristics of the bacterially derived protein [17]. Our data support these findings and show that the protein derived from expression in E. coli is mostly in monomeric or dimeric forms which lack HIV-suppressing activity. Analysis of different batch preparations of IL-16 revealed that the tetrameric form responsible for the virus-inhibiting effect did not exceed 10% of the total protein. Although these results do not clarify the reasons for the improper folding of the bacterially derived IL-16, they clearly suggest that when present in properly folded active form, tetrameric IL-16 exerts potent HIV-suppressing activity at nm concentrations. Similar virus-inhibiting concentrations of the cytokine have been recently reported in Jurkat cells transfected with IL-16 cDNA [18]. The mechanism of HIV suppression by IL-16 has been shown not to involve steric inhibition of viral binding to CD4, but to be mediated through repression of HIV long-terminal repeat promoter activity [27]. Interference with the transcription of early HIV regulatory genes has also been suggested as a complementary or alternative mechanism [18]. All these observations have been made on CD4+ lymphoid cells, but whether similar IL-16-mediated effects operate on reservoir cells, including macrophages and dendritic cells, is not yet known and needs to be addressed in future studies. Another issue which still awaits clarification is related to the question of whether the 130 amino acid recombinant IL-16 represents a major form of the naturally secreted cytokine [28]. This would be relevant for a proper understanding of the physiological role of natural IL-16 in the inhibition of HIV replication in vivo. However, clarification of this issue will not modify the potential value of recombinant IL-16 in the therapeutic approaches against HIV infection.
Cross-linking CD4 molecules by HIV-envelope protein gp160 or by anti-CD4 antibodies has been shown to induce lymphocyte apoptosis in PBMC cultures from normal individuals. This effect was found to be related to an up-regulation of CD95 expression on lymphocytes through the induced secretion of tumour necrosis factor-alpha (TNF-α) and IFN-γ [29,30]. We have also observed a small yet significant increase in spontaneous apoptosis of lymphocytes from healthy donors following incubation with IL-16. However, this was not the case when cells from HIV-infected subjects were treated with the cytokine, and no modification of the already elevated level of spontaneous apoptosis could be detected. Although we have not analysed the mechanisms of this phenomenon, the difference in the effects of IL-16 could be explained by a difference in the responsiveness of CD4 lymphocytes from patients to the chemoattractant cytokine. This may well be attributed to an altered state of cell activation, to an altered level of CD4 expression and/or to an altered efficiency in signal transduction. Alternatively, the explanation may be an indirect one related to defective responses of accessory cells from patients to IL-16. This is supported by two independent findings: (i) an abnormal function of monocytes in HIV-1 infected subjects [31], and (ii) the need for the presence of accessory cells in the PBMC populations in order to induce lymphocyte apoptosis through CD4 cross-linking [29].
The effect of IL-16 on the inhibition of AICD may reflect a major difference in the outcome of signals transduced following CD4 ligation with different ligands. Cross-linking CD4 molecules with gp120 in the presence of anti-T cell receptor stimulation has been reported to increase AICD through a mechanism involving CD95/CD95 ligand interaction [32]. Similar results have also been obtained when anti-CD4 antibodies were used as the ligand [30,33]. Our findings on the ability of IL-16 to down-regulate CD95 expression on activated CD4 cells may well explain the protective activity of this cytokine against AICD. However, other alternative or additional mechanisms can not be ruled out, including inhibition by IL-16 of the secretion of cytokines which enhance apoptosis [34]. Such a mechanism would clearly differentiate the activity of IL-16 on AICD from that of other CD4 ligands which are known to induce secretion of cytokines implicated in enhancing PCD [30,35]. Alternatively, the difference in the activity of IL-16 from that of other CD4 ligands may well be linked to the recognition of different epitopes on the CD4 molecule [15]. Signals transduced following the binding of certain anti-CD4 antibodies or gp120 to their respective epitopes have been shown to result in the inhibition of binding activities of nuclear factors that are known to up-regulate gene enhancer activity of IL-2 [36]. In addition, different CD4 epitopes have been reported to induce distinct signalling properties, and those recognized by anti-CD4 or gp120 were found inefficient in activating the transcription factor NF-AT, needed for IL-2 gene expression [37]. The latter cytokine, however, is known to possess apoptosis-inhibiting effects on cells from HIV-infected subjects [38]. Thus, it will be of high interest to examine in future studies whether signalling via the IL-16 epitope on CD4 could result in up-regulating IL-2 enhancer activity. On the other hand, the ability of all three CD4 ligands, IL-16, gp120 and anti-CD4, to inhibit HIV replication [39,40] argues that such an effect would have to be mediated by mechanisms that are different from those implicated in regulating apoptosis. This is also substantiated by our findings that MIP-1α exerts HIV-suppressing but lacks anti-apoptotic activities.
The protective effect of IL-16 against AICD appears to be dependent on the nature of the activator used and on the immune status of lymphocytes tested. In this respect, IL-16 protected lymphocytes from HIV-infected subjects against PCD induced by anti-CD3, by IgM anti-CD95 or by DEX, whereas no such effect could be noted on PHA-induced apoptosis. These findings could be explained by the inability of IL-16 to down-regulate CD95 expression in PHA-stimulated cells (Fig. 3). Alternatively, CD95/CD95 ligand interaction may not be involved in the process of AICD following PHA stimulation of cells from HIV-infected subjects. This would be in agreement with previous findings demonstrating that AICD can be mediated by multiple and/or different mechanisms [41,42]. In addition, our findings of a protective activity of IL-16 on the PHA-induced apoptosis of lymphocytes from healthy controls suggest that distinct mechanisms, which could be triggered by the same activator, may operate in health and in disease [43].
Finally, the state of chronic immune activation in HIV infection has been linked to the gradual and continuous process of immune deficiency resulting from T cell depletion [44]. Although the evidence for a direct correlation between the level of viral load and cell apoptosis is still questionable [45], results on the use of highly active anti-retroviral therapy suggest a direct relationship between sustained suppression of viral replication and enhanced lymphocyte count or immune recovery [46]. Moreover, effective anti-retroviral treatment has been recently demonstrated to result in down-regulation of CD95-ligand expression on T cells, suggesting that significant reduction in virus replication could lead to a decrease in the PCD of uninfected cells [47]. Thus, therapeutics capable of inhibiting viral replication and simultaneously blocking AICD may have stronger and more rapid effects in the treatment of HIV disease. Our results on the efficacy of IL-16 to down-regulate both processes related to the infection give a high potential and priority for the clinical development of this cytokine as adjunct therapy to anti-retrovirals.
Acknowledgments
We are grateful to Dr J. P. Kusnierz for his assistance in the flow cytometry analyses, to M. Loyens and C. Godin for the purification of IL-16, to J. Dewulf for carrying out the infection assays, and to C. Vincent for typing the manuscript. This work was supported by a research grant (no. 97088) from the Agence National pour la Recherche contre le SIDA in France.
References
- 1.Meyaard L, Otto SA, Jonker RR, Mijnster MJ, Keet RPM, Miedema F. Programmed death of T cells in HIV-1 infection. Sci. 1992;257:217–9. doi: 10.1126/science.1352911. [DOI] [PubMed] [Google Scholar]
- 2.Groux H, Torpier G, Monté D, Mouton Y, Capron A, Ameisen JC. Activation-induced death by apoptosis in CD4+ T cells from human immunodeficiency virus-infected asymptomatic individuals. J Exp Med. 1992;175:331–40. doi: 10.1084/jem.175.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banda NK, Bernier J, Kurahara DK, Kurrle R, Haigwood N, Sekaly RP, Finkel TH. Crosslinking CD4 by human immunodeficiency virus gp120 primes T cells for activation-induced apoptosis. J Exp Med. 1992;176:1099–106. doi: 10.1084/jem.176.4.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Westendorp MO, Frank R, Ochsenbauer C, Stricker K, Dhein J, Walczak H, Debatin KM, Krammer PH. Sensitization of T cells to CD95-mediated apoptosis by HIV-1 tat and gp120. Nature. 1995;375:497–500. doi: 10.1038/375497a0. [DOI] [PubMed] [Google Scholar]
- 5.Finkel TH, Tudor-Williams G, Banda NK, et al. Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV- and SIV-infected lymph nodes. Nature Med. 1995;1:129–34. doi: 10.1038/nm0295-129. [DOI] [PubMed] [Google Scholar]
- 6.Muro-Cacho CA, Pantaleo G, Fauci AS. Analysis of apoptosis in lymph nodes of HIV-infected persons: Intensity of apoptosis correlates with the general state of activation of the lymphoid tissue and not with stage of disease or viral burden. J Immunol. 1995;154:5555–66. [PubMed] [Google Scholar]
- 7.Pandolfi F, Pierdominici M, Oliva A, et al. Apoptosis-related mortality in vitro of mononuclear cells from patients with HIV infection correlates with disease severity and progression. J Acquir Immune Defic Syndr Hum Retrovirol. 1995;9:450–8. [PubMed] [Google Scholar]
- 8.Gougeon ML, Lecoeur H, Dulioust A, Enouf MG, Crouvoisier M, Goujard C, Debord T, Montagnier L. Programmed cell death in peripheral lymphocytes from HIV-infected persons: Increased susceptibility to apoptosis of CD4 and CD8 T cells correlates with lymphocyte activation and with disease progression. J Immunol. 1996;156:3509–20. [PubMed] [Google Scholar]
- 9.Carbonari M, Cibati M, Pesce AM, Sbarigia D, Grossi P, D'Offizi G, Luzi G, Fiorilli M. Frequency of provirus-bearing CD4+ cells in HIV type 1 infection correlates with extent of in vitro apoptosis of CD8+ but not CD4+ cells. AIDS Res Hum Retrovir. 1995;11:789–94. doi: 10.1089/aid.1995.11.789. [DOI] [PubMed] [Google Scholar]
- 10.Gougeon ML, Garcia S, Heeney J, et al. Programmed cell death in AIDS-related HIV and SIV infections. AIDS Res Hum Retrovir. 1993;9:553–63. doi: 10.1089/aid.1993.9.553. [DOI] [PubMed] [Google Scholar]
- 11.Clerici M, Sarin A, Coffman RL, et al. Type 1/type 2 cytokine modulation of T cell programmed cell death as a model for human immunodeficiency virus pathogenesis. Proc Natl Acad Sci USA. 1994;91:11811–5. doi: 10.1073/pnas.91.25.11811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Estaquier J, Idziorek T, Zou W, Emilie D, Farber CM, Bourez JM, Ameisen JC. T helper type 1/T helper type 2 cytokines and T cell death: preventive effect of interleukin 12 on activation-induced and CD95 (Fas/APO-1) -mediated apopotosis of CD4+ T cells from human immunodeficiency virus-infected persons. J Exp Med. 1995;182:1759–67. doi: 10.1084/jem.182.6.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sloand EM, Young NS, Kumar P, Weichold PF, Sato T, Maciejewski JP. Role of Fas ligand and receptor in the mechanism of T cell depletion in acquired immunodeficiency syndrome: effect on CD4+ lymphocyte depletion and human immunodeficiency virus replication. Blood. 1997;89:1357–63. [PubMed] [Google Scholar]
- 14.Oyaizu N, Adachi Y, Hashimoto F, McCloskey TW, Hosaka N, Kayagaki N, Yagita H, Pahwa S. Monocytes express Fas ligand upon CD4 cross-linking and induce CD4+ T cells apoptosis: a possible mechanism of bystander cell death in HIV infection. J Immunol. 1997;158:2456–63. [PubMed] [Google Scholar]
- 15.Center DM, Kornfeld H, Cruikshank WW. Interleukin-16 and its function as a CD4 ligand. Immunol Today. 1996;17:476–81. doi: 10.1016/0167-5699(96)10052-i. [DOI] [PubMed] [Google Scholar]
- 16.Cruikshank WW, Lim K, Theodore AC, Cook J, Fine G, Weller PF, Center DM. IL-16 inhibition of CD3-dependent lymphocyte activation and proliferation. J Immunol. 1996;157:5240–8. [PubMed] [Google Scholar]
- 17.Baier M, Werner A, Bannert N, Metzner K, Kurth R. HIV suppression by interleukin-16. Nature. 1995;378:563. doi: 10.1038/378563a0. [DOI] [PubMed] [Google Scholar]
- 18.Zhou P, Goldstein S, Devadas K, Tewari D, Notkins AL. Human CD4+ cells transfected with IL-16 cDNA are resistant to HIV-1 infection: inhibition of mRNA expression. Nature Med. 1997;3:659–64. doi: 10.1038/nm0697-659. [DOI] [PubMed] [Google Scholar]
- 19.Cruikshank WW, Center DM, Nisar N, Wu M, Natke B, Theodore AC, Kornfeld H. Molecular and functional analysis of a lymphocyte chemoattractant factor: association of biologic function with CD4 expression. Proc Natl Acad Sci USA. 1994;91:5109–13. doi: 10.1073/pnas.91.11.5109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Idziorek T, Estaquier J, De Bels F, Ameisen JC. YOPRO-1 allows cytofluorometric analysis of programmed cell death (apoptosis) without interfering with living cell viability. J Immunol Methods. 1995;185:249–58. doi: 10.1016/0022-1759(95)00172-7. [DOI] [PubMed] [Google Scholar]
- 21.Rey MA, Spire B, Dormont D, Barré-Sinoussi F, Montagnier L, Chermann JC. Characterization of the RNA dependent DNA polymerase of a new human T-lymphotropic retrovirus (lymphodenopathy associated virus) Biochem Biophys Res Commun. 1984;121:126–33. doi: 10.1016/0006-291x(84)90696-x. [DOI] [PubMed] [Google Scholar]
- 22.Choe H, Farzan M, Sun Y, et al. The β-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell. 1996;85:1135–48. doi: 10.1016/s0092-8674(00)81313-6. [DOI] [PubMed] [Google Scholar]
- 23.Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272:872–7. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- 24.Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P. Identification of RANTES, MIP-1α and MIP-1β as the major HIV-suppressive factors produced by CD8+ T cells. Science. 1995;270:1811–5. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- 25.Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, Springer TA. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature. 1996;382:829–33. doi: 10.1038/382829a0. [DOI] [PubMed] [Google Scholar]
- 26.Mackewicz CE, Levy JA, Cruikshank WW, Kornfeld H, Center DM. Role of IL-16 in HIV replication. Nature. 1996;383:488–9. doi: 10.1038/383488a0. [DOI] [PubMed] [Google Scholar]
- 27.Maciaszek JW, Parada NA, Cruikshank WW, Center DM, Kornfeld H, Viglianti GA. IL-16 represses HIV-1 promoter activity. J Immunol. 1997;158:5–8. [PubMed] [Google Scholar]
- 28.Baier M, Bannert N, Werner A, Lang K, Kurth R. Molecular cloning, sequence, expression, and processing of the interleukin 16 precursor. Proc Natl Acad Sci USA. 1997;94:5273–7. doi: 10.1073/pnas.94.10.5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oyaizu N, McCloskey TW, Coronesi M, Chirmule N, Kalyanaraman VS, Pahwa S. Accelerated apoptosis in peripheral blood mononuclear cells (PBMCs) from human immunodeficiency virus type-1 infected patients and in CD4 cross-linking PBMCs from normal individuals. Blood. 1993;82:3392–400. [PubMed] [Google Scholar]
- 30.Oyaizu N, McCloskey TW, Than S, Hu R, Kalyanaraman VS, Pahwa S. Cross-linking of CD4 molecules upregulates Fas antigen expression in lymphocytes by inducing interferon-γ and tumor necrosis factor-α secretion. Blood. 1994;84:2622–31. [PubMed] [Google Scholar]
- 31.Yoo J, Chen H, Kraus T, Hirsch D, Polyak S, George I, Sperber K. Altered cytokine production and accessory cell function after HIV-1 infection. J Immunol. 1996;157:1313–20. [PubMed] [Google Scholar]
- 32.Accornero P, Radrizzani M, Delia D, Gerosa F, Kurrle R, Colombo MP. Differential susceptibility to HIV-GP120-sensitized apoptosis in CD4+ T cell clones with different T-helper phenotypes: role of CD95/CD95L interactions. Blood. 1997;89:558–69. [PubMed] [Google Scholar]
- 33.Newell MK, Haughn LJ, Maroun CR, Julius MH. Death of mature T cells by separate ligation of CD4 and the T cell receptor of antigen. Nature. 1990;347:286–9. doi: 10.1038/347286a0. [DOI] [PubMed] [Google Scholar]
- 34.Oyaizu N, McCloskey TW, Than S, Pahwa S. Inhibition of CD4 cross-linking-induced lymphocytes apoptosis by vesnarinone as a novel immunomodulating agent: vesnarinone inhibits Fas expression and apoptosis by blocking cytokine secretion. Blood. 1996;87:2361–8. [PubMed] [Google Scholar]
- 35.Clouse KA, Casentino LM, Weih KA, Pyle SW, Robbins PB, Hochstein HD, Natarajan V, Farrar WL. The HIV-1 gp120 envelope protein has the intrinsic capacity to stimulate monokine secretion. J Immunol. 1991;147:2892–901. [PubMed] [Google Scholar]
- 36.Jabado N, Le Deist F, Fischer A, Hivroz C. Interaction of HIV, gp120 and anti-CD4 antibodies with the CD4 molecule on human CD4+ T cells inhibits the binding activity of NF-AT, NF-χB and AP-1, three nuclear factors regulating interleukin-2 gene enhancer activity. Eur J Immunol. 1994;24:2646–52. doi: 10.1002/eji.1830241112. [DOI] [PubMed] [Google Scholar]
- 37.Baldari CT, Milia E, Di Somma MM, Baldoni F, Valitutti S, Telford JL. Distinct signaling properties identify functionally different CD4 epitopes. Eur J Immunol. 1995;25:1843–50. doi: 10.1002/eji.1830250708. [DOI] [PubMed] [Google Scholar]
- 38.Cordiali Fei P, Solmone M, Viora M, Vanacore P, Pugliese O, Giglio A, Caprilli E, Ameglio E. Apoptosis in HIV-infection: protective role of IL-2. J Biol Regul Homeost Agents. 1994;8:60–64. [PubMed] [Google Scholar]
- 39.Benkirane MD, Corbeau P, Housse TV, Deveaux C. An antibody that binds the immunoglobulin CDR3-like region of the CD4 molecule inhibits provirus transcription in HIV-infected T cells. EMBO J. 1993;12:4909–12. doi: 10.1002/j.1460-2075.1993.tb06185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tremblay M, Meloche S, Gratton S, Wainberg MA, Sekaly RP. Association of P56-lck with the cytoplasmic domain of CD4 modulates HIV-1 expression. EMBO J. 1994;13:774–83. doi: 10.1002/j.1460-2075.1994.tb06320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Katsikis PD, Garcia-Ojeda ME, Torres-Roca JF, Greenwald DR, Herzenberg LA, Herzenberg LA. HIV type 1 tat protein enhances activation-but not Fas (CD95)-induced peripheral blood T cell apoptosis in healthy individuals. Int Immunol. 1997;9:835–41. doi: 10.1093/intimm/9.6.835. [DOI] [PubMed] [Google Scholar]
- 42.Moreno MB, Memon SA, Zacharchuk CM. Apoptosis signaling pathways in normal T cells: differential activity of Bcl-2 and IL-1beta-converting enzyme family protease inhibitors on glucocorticoid- and Fas-mediated cytotoxicity. J Immunol. 1996;157:3845–9. [PubMed] [Google Scholar]
- 43.Kovacs B, Vassilopoulos D, Vogelgesang SA, Tsokos GC. Defective CD3-mediated cell death in activated T cells from patients with systemic lupus erythematosus: role of decreased intracellular TNF-α. Clin Immunol Immunopathol. 1996;81:293–302. doi: 10.1006/clin.1996.0192. [DOI] [PubMed] [Google Scholar]
- 44.Gougeon ML, Lecoeur H, Boudet F, et al. Lack of chronic immune activation in HIV-infected chimpanzees correlates with the resistance of T cells to Fas/Apo-1 (CD95)-induced apoptosis and preservation of a T helper phenotype. J Immunol. 1997;158:2964–76. [PubMed] [Google Scholar]
- 45.Rothen M, Gratzel S, Hirsch HH, Moroni C. Apoptosis in HIV-infected individuals is an early marker occurring independently of high viremia. AIDS Res Hum Retrovir. 1997;13:771–9. doi: 10.1089/aid.1997.13.771. [DOI] [PubMed] [Google Scholar]
- 46.Autran B, Carcelain G, Li TS, et al. Positive effects of combined antiretroviral therapy on CD4+ T cell homeostasis and function in advanced HIV disease. Science. 1997;277:112–6. doi: 10.1126/science.277.5322.112. [DOI] [PubMed] [Google Scholar]
- 47.Böhler T, Herr I, Debatin KM, Geiss M, Haas J. Downregulation of increased CD95 (APO-1/Fas) ligand in T cells from human immunodeficiency virus-type 1-infected children after antiretroviral therapy. Blood. 1997;90:886–7. [PubMed] [Google Scholar]