

Abstract
In this study we have tested the concept of using wild-type p53 gene for immunotherapy of cancer. Dendritic cells (DC) were transduced with a human wild-type p53 containing recombinant adenovirus (Ad-p53). About a half of DC transduced with this virus expressed p53 protein by FACS analysis 48 h after infection. Mice immunized twice with Ad-p53 DC developed substantial cytotoxic T lymphocyte (CTL) responses against tumour cells expressing wild-type and different mutant human and murine p53 genes. Very low CTL responses were observed against target cells infected with control adenovirus (Ad-c). Immunization with Ad-p53 provided complete tumour protection in 85% of mice challenged with tumour cells expressing human mutant p53 and in 72.7% of mice challenged with tumour cells with murine mutant p53. Treatment with Ad-p53-transduced DC significantly slowed the growth of established tumours. Thus, DC transduced with wild-type p53 may be a promising new tool for the immunotherapy of cancer.
Keywords: p53, tumour immunology, dendritic cells
INTRODUCTION
Many types of cancer have been found to be associated with mutations in the p53 gene. These mutations usually result in the over-expression of the mutant p53 protein in tumour cells, and only rarely in loss of expression (review in [1]). Significant over-expression may also occur in tumours without mutations in p53. Over-expression of p53 may lead to the generation of multiple epitopes that can be targets for immunotherapy. We and others have demonstrated the generation of anti-tumour immune responses using mutant p53 peptides as antigens [2–4]. However, this approach to cancer immunotherapy has serious limitations. p53 mutations occur at many different sites in the protein, making it necessary to identify the site of mutation in each patient before constructing a customized mutant peptide for therapy. Furthermore, not all mutations are contained in regions of the protein known to bind to MHC molecules. These weaknesses stimulated the search for antigenic epitopes in wild-type sequences common to the vast majority of tumour-derived p53 proteins. Recently, wild-type p53 peptide-specific cytotoxic T lymphocytes (CTL) were generated from human and murine responding lymphocytes, some of which recognized p53-over-expressing tumours in vitro [5–7]. However, since presentation of antigens is MHC class I-restricted, only certain oligopeptides can be successfully used in certain patients, because of highly polymorphic MHC class I peptide binding site. Besides that, a peptide vaccine that does bind to an individual's class I MHC may not be sufficiently presented by MHC class II, the molecules that play a crucial role in induction of CD4+ T cell immune responses. Additionally, responses directed against only a single epitope may not be as effective as polyepitope responses. One strategy to overcome these problems is not to rely on a single mutant peptide, but rather to immunize with the intact wild-type p53 to take advantage of the relative over-expression of the whole p53 molecule in most human tumours. In order to be effective this approach should generate immune response against tumour cells expressing p53 genes with mutations at different sites.
Perhaps the cell type best suited for vaccine antigen delivery is the DC. They are the most potent antigen-presenting cells (APC), very effective in stimulation of primary and secondary immune responses [8]. Murine and human DC have been reported to induce specific cytotoxic immune responses against tumour antigens and tumour cells [3,4,9,10]. DC efficiently take up soluble and cell-derived protein antigens, but several groups have recently shown that transfection of DC results in effective presentation of the recombinant protein antigen.
Adenovirus provides high-level transduction efficacy of many cell types, regardless of the mitotic status of the cell [11]. Replication-defective adenoviruses with deletions in the E1 region have been directly injected into people in several human clinical trials with no major adverse reactions (review in [12]). Recently successful transduction of DC with green fluorescent protein and model antigens has been reported [13–15].
Here, we report our findings that DC transduced with wild-type 53 are able to elicit potent anti-tumour immune responses specific for a variety of cells expressing different mutant p53 proteins, and that this may provide the basis for a simple and effective new approach to immunotherapy of cancer.
MATERIALS AND METHODS
Animals
Six-to-eight-week-old female BALB/c and CBA mice were purchased from Harlan Inc. (Indianapolis, IN) and were housed in specific pathogen-free units of the Division of Animal Care at Vanderbilt University Medical Center.
Reagents and cell lines
Tumour cells D459 were constructed by transfection of BALB/c 3T3 cells with EJ ras and a mutant human p53 expression vector. Details of this cell lines were described elsewhere [16,17]. MethA sarcoma cells were obtained from Dr L. J. Old. This is a transplantable 3-methylcholanthrene-induced sarcoma of BALB/c origin passaged as an ascitic tumour. P815 mouse mastocytoma cell lines transfected with mutant human p53 genes were also described elsewhere [18]. These two cell lines contain p53 genes with two different mutations, one in codon 135 (P815–135) and the other one in codon 173 (P815–173).
Control adenovirus (Ad-c) was prepared by deletion of E1 region from adenovirus serotype 5. Adenovirus containing human wild-type 53 (Ad-p53) was a generous gift from Introgen Therapeutics Inc. (Houston, TX).
Recombinant mouse granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-4 were obtained from R&D Systems (Minneapolis, MN).
FITC- and PE-labelled antibodies used in flow cytometry were purchased from Pharmingen (San Diego, CA): anti-CD11c (N418), anti-CD-86 (B7-2), anti-CD40 and anti-I-Ad FITC- and PE-conjugated isotype-matched IgG were used in controls. Anti-p53 antibody and matched isotype control mouse IgG2b were obtained from Dako Corp. (Carpinteria, CA). FITC-labelled anti-mouse immunoglobulin was obtained from Sigma (St Louis, MO).
Anti-CD4 (L3T4, TIB-207) and anti-CD8 (Ly-2.2, TIB-210) antibodies were used as culture supernatants of hybridomas obtained from the American Type Tissue Collection (Manassas, VA).
Cell preparation and infection with adenovirus
Bone marrow cells were prepared as described earlier [2]. Briefly, bone marrow cells were obtained from the femurs and tibias of BALB/c mice. Mononuclear cells were placed in tussie flasks at a concentration 5 × 105/ml in complete culture medium (CCM) (RPMI 1640 (Gibco BRL, Gaithersburg, MD) with 100 U/ml penicillin, 0.1 mg/ml streptomycin, 1 × 10−5m 2-mercaptoethanol (2-ME) and 10% fetal calf serum (FCS; HyClone, Logan, UT)) supplemented with rmGM-CSF at a final concentration 3 ng/ml and rmIL-4 at a final concentration of 5 ng/ml. After 3 days, half of the medium was removed after gentle swirling and replenished with an equivalent amount of fresh GM-CSF and IL-4 supplemented medium. Three to four days later, clusters of DC were dislodged. The purity of DC prepared in this fashion was > 60% at > 95% viability.
Splenic DC were prepared as described [16]. A single-cell suspension was prepared by pressing the spleens through a wire mesh. Cells were then washed and incubated overnight in CCM. Non-adherent cells were layered onto a metrizamide (Nygaard, Oslo, Norway) gradient (14.5 g plus 100 ml RPMI 1640 medium) and centrifuged for 10 min at 600 g. Cells at the interface were washed once and resuspended in CCM. DC were identified by their distinctive morphology and by labelling with N418 (CD11c) antibody and had a purity > 40% with > 95% viability.
T cells were isolated from lymph nodes using nylon wool columns as described elsewhere [2].
Spleen cells from immunized mice were centrifuged over Lympholyte M (Cedarlane, Hornby, Ontario, Canada) gradient and incubated for 60 min in tissue culture flasks in complete culture medium. Non-adherent cells were collected, washed and used as effector cells in CTL assay.
DC (106) obtained either from bone marrow or from spleen were infected with adenovirus at various multiplicities of infection (MOI) for 60 min in 1 ml of serum-free medium in 24-well plates. After that time, 1 ml of fresh medium supplemented with GM-CSF, IL-4 and 20% FCS was added. No IL-4 was added to splenic DC. Cells were incubated for another 24, 48, 72 or 120 h. After that time, cells were washed in PBS before use.
Tumour induction and immunization procedures
For immunization, bone marrow-derived DC were used. Two hundred thousand dendritic cells were injected intravenously, intraperitoneally or subcutaneously into BALB/c mice. Two hundred thousand D459 cells or 6 × 105 MethA sarcoma cells were injected subcutaneously into the shaved backs of mice. These doses of tumour cells were chosen after preliminary experiments showed that they resulted in tumour formation in 100% of the mice (data not shown).
T cell proliferation assay
DC infected with Ad-p53 or Ad-c were irradiated (2000 cGy) and added in triplicate to 5 × 104T cells obtained from BALB/c mice immunized with Ad-p53 DC or, for studies of allogeneic mixed leucocyte reaction (MLR), DC were cultured with T cells obtained from CBA mice. After a 3-day incubation in 96-well U-bottomed plates, the cultures were pulsed with 1 μCi 3H-thymidine (Amersham, Arlington Heights, IL) for 8–12 h. 3H-thymidine uptake was counted using a liquid scintillation counter.
Analysis of the p53 protein expression and expression of surface molecules
The efficiency of DC transduction was tested based on the over-expression of human p53 protein by FACS analysis. Briefly, DC after infection with Ad-p53 or Ad-c were fixed for 30 min with 2% paraformaldehyde, permeabilized for 60 min with 0.2% Tween-20 and stained with anti-p53 antibody. FITC-conjugated anti-mouse immunoglobulin was used as a secondary antibody. Non-specific binding was measured using secondary antibody alone. Cells were analysed using flow cytometer FACScalibur (Becton Dickinson, Mountain View, CA) with gates set around clusters of large cells. Expression of the surface molecules was studied on non-fixed, non-permeabilized DC using MoAbs specific for B7-2, CD40, and I-Ad and analysed by flow cytometry. Non-specific binding was measured using isotype-matched mouse immunoglobulin.
CTL assay
Cell cytotoxicity was measured in a standard 6-h 51Cr-release assay. Briefly, 2 × 106 spleen cells isolated from immunized mice were restimulated for 6 days with 2 × 105 splenic DC infected either with Ad-p53 or Ad-c in 24-well plates. Effector lymphocytes were incubated in duplicate with 51Cr-labelled target cells. Supernatants were harvested with a Skatron Harvesting System (Skatron, Lier, Norway) and radioactivity was counted on a gamma counter. The percentage specific lysis was calculated as 100 × [(experimental release — spontaneous release)/maximum release — spontaneous release)].
RESULTS
In preliminary experiments the most effective dose of Ad-p53 was determined. Ad-p53 and Ad-c at doses of 50–200 MOI did not significantly affect DC viability, which remained > 95%. Higher doses of virus resulted in significant loss of viability (< 50% at doses > 500 MOI). No differences between the effect of Ad-c and Ad-p53 on cell viability were observed. This indicates that loss of viability at high MOI was not due to over-expression of p53, but rather non-specific viral toxicity. The efficiency of transduction was estimated using intracellular staining with an anti-p53 antibody. The maximum level of p53 was detected at an Ad-p53 MOI of 100 plaque-forming units (PFU)/cell. At this dose 40–45% DC were positive for p53 (Fig. 1). This dose of adenovirus was used in all subsequent experiments. A 48-h incubation with Ad-p53 resulted in the highest levels of p53 protein, which slightly decreased 24 h later, and was undetectable by day 5. Thus, these preliminary experiments demonstrated that Ad-p53 at a dose of 100 MOI was non-toxic for DC, and that Ad-p53-transduced DC expressed detectable levels of p53 protein. Infection of DC with adenovirus did not affect the ability of these cells to stimulate allogeneic T cells, and slightly increased expression of B7-2 and CD40 molecules on their surface (data not shown).
Fig. 1.
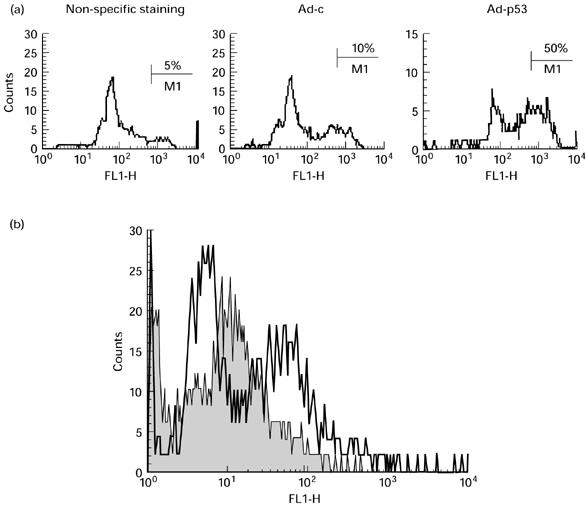
Expression of p53 protein in dendritic cells (DC) infected with human wild-type p53 containing recombinant adenovirus (Ad-p53). DC generated from bone marrow were infected with 100 MOI control adenovirus (Ad-c) or Ad-p53 for 48 h, washed, fixed, permeabilized and stained with anti-p53 antibody and analysed as described in Materials and Methods. (a) Non-specific staining: Ad-p53-infected DC stained only with secondary antibody. Ad-c and Ad-p53: DC infected with corresponding virus stained with anti-p53 antibody. Typical results of one of three experiments performed are shown. (b) Ad-p53-infected DC labelled with isotype control (shaded area) or anti-p53 antibodies and secondary anti-mouse FITC antibody.
We then asked whether immunization with Ad-p53-transduced DC would result in induction of a specific anti-p53 immune response, and how many immunizations would be required to achieve this goal. Mice were immunized with 2 × 105DC infected 48 h before with either Ad-p53 or Ad-c. Three routes of immunization were tested (s.c., i.p. and i.v.), and immune responses were assayed using five different target tumours: P815 cells, P815 cells infected with control adenovirus (P815-Ad-c), P815 cells infected with Ad-p53 (P815-Ad-p53), P815-135 cells and P815-173 cells. P815-135 and P815-173 are two cell lines transfected with mutant human p53 genes with two different point mutations in positions 135 and 173, respectively. Mice were immunized once or twice with a 2-week interval. Ten to 14 days after the last immunization, spleen cells were isolated and restimulated with Ad-p53-infected splenic DC. No CTL were detected after a single immunization using any of the tested routes of immunization. However, two immunizations resulted in significant CTL responses (Fig. 2a). The highest response was observed against Ad-p53-infected P815 cells, but significant responses were also seen using P815-135 and P815-173 as targets. It is important to note the very low CTL responses detected against P815 cells infected with the control adenovirus. In three experiments performed no differences in the level of anti-p53-specific CTL responses were found between the different routes of immunization (data not shown). To clarify the nature of the cells responsible for the tumour cells' killing, effector and target cells were incubated in the presence of anti-CD4 or anti-CD8 antibody. Anti-CD8, but not anti-CD4 antibody blocked the cytotoxicity (Fig. 2b). This indicated that observed cytotoxicity was mediated by CD8+ CTL. In another group of experiments tumour D459 cells were used as targets. As shown in Fig. 2c, significant levels of CTL response were observed in Ad-p53 DC-immunized mice. This response was also mediated by CD8+ cells (Fig. 2d).
Fig. 2.
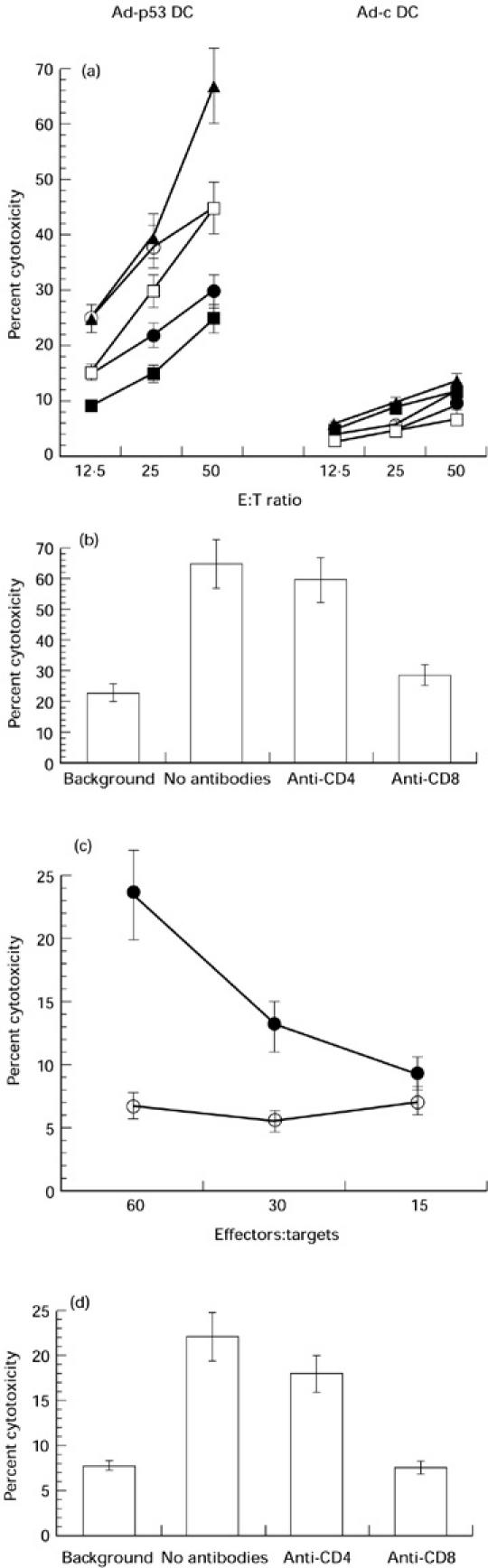
Human wild-type p53 containing recombinant adenovirus (Ad-p53)-transduced dendritic cells (DC) induce anti-p53 immune responses. (a) Cytotoxic T lymphocyte (CTL) response. Mice were immunized twice with DC infected with either control adenovirus (Ad-c) (Ad-c DC) or with Ad-p53 (Ad-p53 DC) (i.v. injections). Ten days after the last immunization, spleen cells from these mice were restimulated with Ad-p53 DC and a CTL assay was performed as described in Materials and Methods. P815-Ad and P815-Ad-p53 targets were prepared by overnight incubation of P815 cells with adenovirus at MOI 100 pfu/ml. Mean ± s.e.m. of cytotoxicity from four experiments is shown. ▪, P815; •, P815-Ad; ▴, P815-Ad-p53; ○, P815-135; □, P815-173. (b) CTL response is mediated by CD8+ cells. Spleen cells from mice immunized with Ad-p53 DC were restimulated with Ad-p53 DC as described above. After 6 days effector cells were cultured with either non-treated P815 cells (background) or with P815 cells infected with Ad-p53 (no antibodies, anti-CD4, anti-CD8) at a ratio of 50:1. Anti-CD4 or anti-CD8 antibodies (10% v/v) were added at the beginning of 6 h incubation. Mean ± s.e.m. of cytotoxicity from two experiments is shown. (c) CTL response against D459 cells. Spleen cells were obtained and restimulated as described above. D459 cells were incubated for 3 days with 250 U/ml rmIFN-γ, washed and used as targets. Mean ± s.e.m. of cytotoxicity from two experiments is shown. •, Ad-p53 DC for immunization and restimulation; ○, Ad-c DC for immunization and restimulation. (d) CTL response is blocked by anti-CD8 antibody. CTL assay was performed as described in the legend to Fig. 2c. Effector:target cell ratio of 60:1 is shown. Background: Ad-c DC were used for immunization and restimulation. No antibodies, anti-CD4, anti-CD8: Ad-p53 DC were used for immunization and restimulation. Antibodies were used at a concentration of 10% (v/v). Mean ± s.e.m. of cytotoxicity from two experiments is shown.
The experiments described so far were performed using constructs and tumour cell lines expressing human p53. Since there is a high homology between human and murine p53, we asked whether immunization with Ad-p53 DC would also result in CTL responses against over-expressed mutant murine p53. For these studies we used the murine MethA sarcoma tumour, a carcinogen-induced tumour bearing different point mutations in each allele of its endogenous p53 genes. Tumour cells were preincubated for 3 days with 50 U/ml recombinant murine interferon-gamma (IFN-γ) and then used as a target in CTL assay. Low but clearly significant CTL responses specific for MethA were detected in mice immunized with Ad-p53 DC (Fig. 3). We also tested whether Ad-p53 DC were able to stimulate T cell proliferation in this system. T cells were obtained from immune mice (two immunizations with Ad-p53 DC) and were cultured with either uninfected DC (background level), or DC infected with Ad-c or with Ad-p53. DC infected with Ad-p53, but not those infected with Ad-c, were able to stimulate T cell proliferation significantly higher than the control level of proliferation of T cells incubated with syngeneic DC (Fig. 4).
Fig. 3.
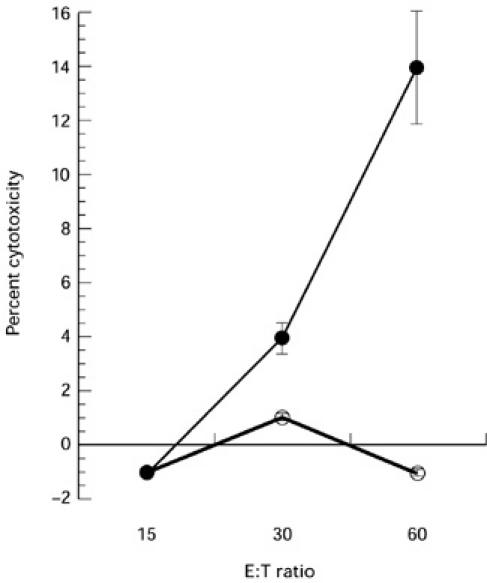
Cytotoxic T lymphocyte (CTL) responses against MethA sarcoma cells. Mice were immunized and spleen cells were restimulated either with control adenovirus (Ad-c) dendritic cells (DC) (Ad-c) (○) or with human wild-type p53 containing recombinant adenovirus (Ad-p53) DC (Ad-p53) (•) as described in the legend to Fig. 2a. Target: MethA sarcoma cells were preincubated with 50 U/ml IFN-γ for 3 days before the assay. Two experiments with the same results were performed.
Fig. 4.
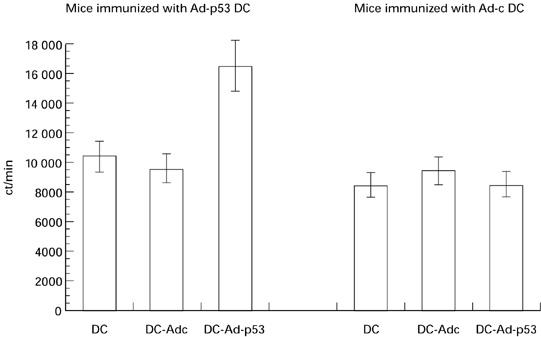
T cell proliferation in immunized mice. Mice were immunized either with human wild-type p53 containing recombinant adenovirus (Ad-p53) dendritic cells (DC) or with control adenovirus (Ad-c) DC as described in the legend to Fig. 2. T cells were isolated and cultured in triplicates at a ratio of 80:1 with either control untreated DC, Ad-c DC or Ad-p53 DC. 3H-thymidine uptake was measured on day 3 as described in Materials and Methods. Mean ± s.e.m. of thymidine incorporation from two experiments is shown.
Next, we asked whether the observed immune response would provide tumour protection. Mice were immunized twice intravenously with Ad-p53- and Ad-c-infected DC. Ten days after the second immunization they were challenged with either D459 tumour, bearing a mutant human p53 gene, or with MethA sarcoma cells, expressing mutant murine p53. Doses of tumour cells were selected which resulted in tumour formation in 100% of non-immune control mice. After immunization with Ad-p53 DC, 17 out 20 (85%) immunized mice were completely protected against D459 tumour and eight out of 11 mice (72.7%) were protected against MethA sarcoma (Fig. 5).
Fig. 5.
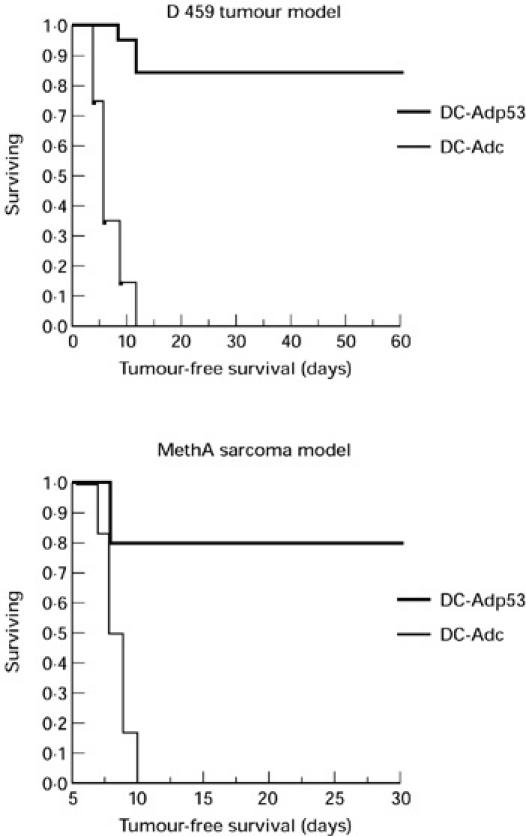
Immunization with human wild-type p53 containing recombinant adenovirus (Ad-p53) protects from tumour challenge. Mice were immunized as described in the legend to Fig. 2a. Ten days after the second immunization, mice were challenged with 2 × 105 D459 cells or with 6 × 105 MethA sarcoma cells. In experiments with D459 cells, each group included 20 mice, in experiments with MethA sarcoma they included 11 mice. Differences between groups were statistically significant (P < 0.05).
We then investigated the effect of treatment of established poor immunogenic tumours with repeated injections with Ad-p53-infected DC. D459 (2 × 105) were inoculated subcutaneously. When tumours became palpable, treatment with Ad-p53 DC was initiated. Mice (n = 10 per group) were immunized three times and tumour growth was observed for 7 weeks. Treatment with Ad-p53-infected DC significantly slowed tumour growth (Fig. 6). Mice in this group were killed due to the bulky tumour more than 2 weeks later than the mice in the control group.
Fig. 6.
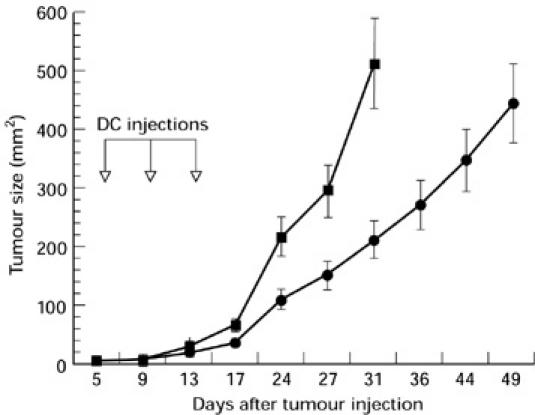
Treatment with human wild-type p53 containing recombinant adenovirus (Ad-p53) dendritic cells (DC) slowed the growth of established tumours. D459 cells (2 × 105) were inoculated subcutaneously into the shaved backs of mice. Treatment with 2 × 105 control adenovirus (Ad-c) (▪) or Ad-p53 DC (•) was initiated when tumour became palpable (day 5). DC were injected on days 5, 9 and 13. Mice in the control group were killed on day 31 due to bulky tumours, mice that received treatment with Ad-p53 DC were killed on day 49. Ten mice per group were treated. Mean ± s.e.m. is shown.
DISCUSSION
p53 is an attractive target for the immunotherapy of cancer because the frequent mutations of this gene in many tumour types often result in significant over-expression of the protein in tumour cells. Over-expression of p53 is often observed in tumour cells even without mutation in the gene [1]. p53 protein is usually undetectable in normal cells. p53 is a nucleoprotein, and peptide fragments of this protein can be presented on the cell surface associated with MHC class I or class II. Several peptides derived from wild-type p53 that bind the HLA A2 molecule have been described [5,19]. These peptides were successfully used for stimulation of anti-p53-specific CTL responses in control donors and in patients with cancer. Adoptive transfer of wild-type p53-specific CTL caused eradication of a p53+ tumour without damage of normal tissues in an animal model [20]. These data indicate that immune and anti-tumour responses can be induced against ‘self’ epitopes in the wild-type p53 protein without clinically evident autoimmunity. However, use of wild-type p53 peptides in the immunotherapy of cancer has some limitations. Patients would need to be selected for certain HLA types, and peptides selected to match only the MHC class I motif ignore the clear importance of MHC class II presentation, which plays an important role in the induction of immune response through activation of CD4+ lymphocytes. This may decrease the efficacy of these approaches to immunotherapy.
A different approach has been used in this study. DC were modified to express the full open reading frame of the wild-type p53 gene. This would enable them to efficiently present multiple epitopes associated with different MHC class I and II molecules. Some of these epitopes might provide effective signals for the stimulation of specific immune responses. As was shown previously in model experiments, each of the minimal epitopes combined to a single fusion protein can be presented separately on the cell surface and be recognized by specific CTL [21]. We used adenovirus for the delivery of p53 genes into DC. As has been shown previously, adenovirus is very effective for mediating gene transfer into DC [13–15]. Despite these obvious advantages, several unanswered questions remained in the development of this method as a cancer therapeutic. It was unclear whether expression of p53 protein in DC would be sufficient for the induction of effective immune responses; whether generation of immune responses would be observed which cross-reacted with most mutant p53 proteins; whether repeated immunizations with DC infected with adenovirus would generate strong anti-adenoviral CTL responses that could potentially prevent induction of anti-p53 immune response or have acute toxicities; and finally, whether immunization with Ad-53 DC would result in the induction of significant autoimmune response. In this study we have tried to answer some of these questions.
Taking into account the non-specific background staining, p53 protein was detected in approx. 40–45% of DC infected with Ad-p53 at an MOI of 100. Infection with neither Ad-c nor with Ad-p53 resulted in significant changes in DC function or expression of the surface molecules, in accordance with previously reported data [13,15]. Over-expression of p53 did not result in decreased DC viability, probably due to the fact that DC used in this study were relatively mature non-dividing cells. Single immunizations with Ad-p53 DC were not able to generate detectable CTL response under our conditions. However, two immunizations were sufficient for the induction of immune responses against several different targets. Our data demonstrated that immunization with Ad-p53 DC generated CTL responses against tumour cells with human wild-type p53 and different mutant p53 genes, as well as tumours expressing murine mutant p53. Thus, transduction of DC with wild-type p53 may generate multiple epitopes matching different portions of the protein. It is also possible that all these mutant p53 proteins had immunogenic epitopes derived from unmutated portions of the protein and recognized by CTL. It is important to note that anti-adenoviral CTL responses were very low. This also confirms previously reported data about the low levels of anti-adenovirus antibody responses after immunization with adenovirus-infected DC [13]. The reason for these low anti-adenoviral responses is not clear, though it possibly relates to endogenous transcription of the recombinant gene product, and the passive nature of adenoviral protein product transfer. It would interesting to see whether this response would increase after multiple injections of Ad-infected DC (we used a maximum of three). In our study, immune responses were observed after immunization with Ad-p53 DC sufficient to provide a high level of protection against challenge with tumour cells expressing both human and murine mutant p53. Immunization also significantly slowed the growth of established tumours.
Thus, we have shown here for the first time that DC transduced with wild-type p53 were able to induce specific immune responses against tumour cells expressing different mutant p53 genes, including those of different species, and that this immune response was sufficient for tumour protection and had significant therapeutic effects against established tumours. Although some important questions still need to be answered, these data suggest that this approach to immunotherapy of cancer may be promising.
Acknowledgments
This work was supported by grant CA 61242-04 to D.P.C. and by ACS grant with the Kirby Foundation RPG-99-032-01-CCE to D.I.G.
REFERENCES
- 1.Harris CC. Structure and function of the p53 tumor suppressor gene: clues for rational cancer therapeutic strategies. J Natl Cancer Inst. 1996;88:1442–55. doi: 10.1093/jnci/88.20.1442. [DOI] [PubMed] [Google Scholar]
- 2.Gabrilovich DI, Nadaf S, Corak J, Berzofsky JA, Carbone DP. Dendritic cells in anti-tumor immune responses. II. Dendritic cells grown from bone marrow precursors, but not mature DC from tumor-bearing mice are effective antigen carriers in the therapy of established tumors. Cellular Immunol. 1996;170:111–20. doi: 10.1006/cimm.1996.0140. [DOI] [PubMed] [Google Scholar]
- 3.Mayordomo JI, Loftus DJ, Sakamoto H, et al. Therapy of murine tumors with p53 wild-type and mutant sequence peptide-based vaccines. J Exp Med. 1996;183:1357–65. doi: 10.1084/jem.183.4.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zitvogel L, Mayordomo JI, Tjandrawan T, et al. Therapy of murine tumors with tumor peptide-pulsed dendritic cells: dependence on T cells, B7 costimulation, and T hepler cell 1-associated cytokines. J Exp Med. 1996;183:87–97. doi: 10.1084/jem.183.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Theobald M, Biggs J, Dittmer D, et al. Targeting p53 as a general tumor antigen. Proc Natl Acad Sci USA. 1995;92:11993–7. doi: 10.1073/pnas.92.26.11993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ropke M, Hald J, Guldberg P, et al. Spontaneous human squamous cell carcinomas are killed by a human cytotoxic T lymphocyte clone recognizing a wild-type p53-derived peptide. Proc Natl Acad Sci USA. 1996;93:14704–7. doi: 10.1073/pnas.93.25.14704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nijman HW, Van der Burg SH, Vierboom MP, et al. p53, a potential target for tumor-directed T cells. Immunol Letters. 1994;40:171–8. doi: 10.1016/0165-2478(94)90189-9. [DOI] [PubMed] [Google Scholar]
- 8.Steinman RM. The dendritic cell system and its role in immunogenecity. Annu Rev Immunol. 1991;9:271–96. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- 9.Grabbe S, Bruvers S, Lindgren AM. Tumor antigen presentation by epidermal antigen presenting cells in the mouse: modulation by granulocyte-macrophage colony stimulating factor, tumor necrosis factor a and ultraviolet radiation. J Leukocyte Biol. 1992;52:209–17. doi: 10.1002/jlb.52.2.209. [DOI] [PubMed] [Google Scholar]
- 10.Cohen PJ, Cohen PA, Rosenberg SA, et al. Murine epidermal Langerhans cells and splenic dendritic cells present tumor-associated antigens to primed T cells. Eur J Immunol. 1994;24:315–9. doi: 10.1002/eji.1830240206. [DOI] [PubMed] [Google Scholar]
- 11.Becker TC, Noel RJ, Coats WS, et al. Use of recombinant adenovirus for metabolic engineering of mammalian cells. Meth Cell Biol. 1994;43:161–76. doi: 10.1016/s0091-679x(08)60603-2. [DOI] [PubMed] [Google Scholar]
- 12.Roth JA, Cristiano RJ. Gene therapy for cancer: what have we done and where are we going? J Natl Cancer Inst. 1997;89:21–39. doi: 10.1093/jnci/89.1.21. [DOI] [PubMed] [Google Scholar]
- 13.Brossart P, Goldrath AW, Butz EA, et al. Virus-mediated delivery of antigenic epitopes into dendritic cells as a means to induce CTL. J Immunol. 1997;158:3270–6. [PubMed] [Google Scholar]
- 14.Dietz AB, Vuk-Pavlovic S. High efficiency adenovirus-mediated gene transfer to human dendritic cells. Blood. 1998;91:392–8. [PubMed] [Google Scholar]
- 15.Wan Y, Bramson J, Carter R. Dendritic cells transduced with an adenoviral vector encoding a model tumor-associated antigen for tumor vaccination. Human Gene Ther. 1997;8:1355–63. doi: 10.1089/hum.1997.8.11-1355. [DOI] [PubMed] [Google Scholar]
- 16.Gabrilovich D, Ciernik F, Carbone DP. Dendritic cells in anti-tumor immune responses. I. Defective antigen presentation in tumor-bearing hosts. Cell Immunol. 1996;170:101–10. doi: 10.1006/cimm.1996.0139. [DOI] [PubMed] [Google Scholar]
- 17.Yanuck M, Carbone DP, Pendleton D, et al. Mutant p53 tumor suppressor protein is a target for peptide-induced CD8+ cytotoxic T-cells. Cancer Res. 1993;53:3257–61. [PubMed] [Google Scholar]
- 18.Ciernik IF, Berzofsky J, Carbone D. Mutant oncopeptide immunization induces CTL specifically lysing tumor cells endogenously expressing the corresponding intact mutant p53. Hybridoma. 1995;14:139–42. doi: 10.1089/hyb.1995.14.139. [DOI] [PubMed] [Google Scholar]
- 19.Melief CJM, Offringa R, Toes REM, Kast WM. Peptide-based cancer vaccines. Curr Opin Immunol. 1996;8:651–7. doi: 10.1016/s0952-7915(96)80081-1. [DOI] [PubMed] [Google Scholar]
- 20.Vierboom MPM, Nijman HW, Offringa R, et al. Tumor eradication by wild-type p53-specific cytotoxic T lymphocytes. J Exp Med. 1997;186:695–704. doi: 10.1084/jem.186.5.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomson SA, Khanna R, Gardner J, et al. Minimal epitopes expressed in a recombinant polyepitope protein are processed and presented to CD8+ cytotoxic T cells: implications for vaccine design. Proc Natl Acad Sci USA. 1995;92:5845–9. doi: 10.1073/pnas.92.13.5845. [DOI] [PMC free article] [PubMed] [Google Scholar]