

Abstract
MRL/Mp-lpr/lpr (MRL/lpr) mice develop glomerular lesions with regular variations in their histopathological manifestations, similar to those in lupus nephritis. These lesions are mainly either cell-proliferative or wire loop-like and are associated with glomerular deposits of immunoglobulins, most frequently IgG and IgM. We previously established a nephritogenic IgG3-producing hybridoma clone, B1, from an MRL/lpr mouse, which induces only a ‘wire loop-like’ type of glomerular lesion when injected into SCID mice. Injection of SCID mice with an anti-trinitrophenyl IgM antibody-producing hybridoma clone, Sp6, following injection of the B1 clone, however, resulted in the development of a ‘cell-proliferative’ type of glomerular lesion, associated with an accumulation of both antibodies in glomeruli. This accumulation occurred even though Sp6 IgM antibodies did not react with B1 IgG3 antibodies and vice versa. A mutant clone of Sp6, T/13μE/3.1, which produces antibodies deficient in C1q binding, produced a similar effect as that of the Sp6 clone, i.e. ‘cell-proliferative’ lesions. Again the B1 antibodies did not react with T/13μE/3.1-IgM antibodies and vice versa. We therefore conclude that bystander IgM antibodies contribute to the remodelling of glomerular lesions in situ, following glomerular injury by the nephritogenic antibodies.
Keywords: lupus nephritis, nephritogenic antibody, IgM antibody, complement
INTRODUCTION
Glomerular injury is very common in systemic lupus erythematosus (SLE) and is termed lupus nephritis. It is not unusual to see various types of glomerulonephritis (GN) which differ in their histopathological manifestations and their various transitional and combined forms.
Animal models of SLE, such as the MRL/Mp-lpr/lpr (MRL/lpr) strain of mice, are important tools for clarifying the pathogenesis of lupus nephritis. MRL/lpr mice spontaneously develop a lethal GN with regular variations in histopathological manifestations. These lesions may consist of diffuse cell-proliferative, crescentic and/or wire loop-like forms, closely resembling various aspects of human lupus nephritis [1,2]. The lesions are characterized by the deposition of immune complexes associated with an increase in serum levels of autoantibodies such as rheumatoid factors, anti-DNA and anti-glycoprotein 70 antibodies [3–5] which are thought to play a major role in the histopathogenesis of lupus nephritis [6–8]. Although the autoantibodies thought to be responsible for such glomerular lesions in murine models have been studied at the monoclonal level [9–13], the mechanisms which induce regular variations in the histopathological manifestations of lupus nephritis are still controversial.
In previous studies, we found that IgG3 production plays a critical role in the development of GN in MRL/lpr mice [14]. Subsequently we developed nephritogenic IgG3-producing hybridomas from an unmanipulated MRL/lpr mouse [15,16] which, at the monoclonal level, induce at least two different types of glomerular lesions when injected into SCID mice: a wire loop-like lesion and an endocapillary proliferative lesion [16]. This may suggest that the histopathology of lupus nephritis depends on the clonality of the B cells producing nephritogenic antibodies and their combinations.
In addition to B cell clonality, several serum components might modulate glomerular lesions in lupus nephritis. The composition of the deposits in the glomerular lesions of SLE varies, but generally they contain several immunoglobulins, most frequently IgG, but also IgM and IgA, as well as components of the complement system (C3, C1q, C4) [17]. For instance, although anti-dsDNA IgM antibodies are negatively associated with lupus nephritis [18], IgM deposits are remarkable in the glomeruli of SLE patients [17]. We have observed similar deposits in the glomerular lesions of MRL/lpr mice. However, it is still unclear whether these molecules are nephritogenic by themselves or, if they are not, whether they act as accelerators in the development of lupus nephritis following the event induced by nephritogenic antibodies.
In the present study, we examined whether non-nephritogenic bystander IgM antibodies are deposited in glomeruli in association with the nephritogenic B1 antibodies derived from an MRL/lpr mouse, the latter antibodies possessing weak DNA binding activity and lacking rheumatoid factor and gp70 binding activities [15,16]. We present evidence that bystander IgM antibodies are deposited in glomeruli and contribute to the remodelling of the glomerular lesions in situ.
MATERIALS AND METHODS
Mice
All experiments were performed using 8–12-week-old C.B-17/Inc-scid/scid(SCID) mice [19], which were kindly donated by Dr S. Ikehara (Kansai Medical University, Japan). They were bred in the Experimental Animal Institute of Tohoku University School of Medicine.
Hybridoma clones
A nephritogenic antibody-producing hybridoma clone, B1, derived from an unmanipulated MRL/lpr mouse, was used in this study. The B1 clone is a subclone of clone 7B6.8 and it induces glomerular lesions of the wire loop type throughout the progression of disease when injected into normal or SCID mice [15,16].
As a source of non-nephritogenic antibody-producing hybridoma clones, two IgM-producing hybridomas, Sp6 [20] and T/13μE/3.1, both of which are anti-trinitrophenyl (TNP) IgM-producing hybridomas, were used. T/13μE/E/3.1 is a mutant clone of Sp6 and its antibody lacks the ability to bind to C1q via its Fc portion [21].
These IgM antibodies did not bind to B1 antibodies and vice versa as determined by the ELISA method [14].
Injections of hybridomas
The B1 clone (1 × 107 cells) was injected intraperitoneally into SCID mice. After 7–10 days, either of the hybridoma clones (Sp6 or T/13μE/3.1) producing non-nephritogenic IgM antibodies (1 × 107 cells) were then injected intraperitoneally. In the mice injected with the B1 clone alone, significant changes in renal glomeruli characterized by wire loop-like lesions were found 20–25 days after the injection. After more than 26 days, the injected mice started dying, possibly of renal insufficiency and/or intraperitoneal bleeding due to vascular invasion by hybridoma cells. Thus, in this study 20–25 days after the first injection, serum samples were collected from the mice under ether anaesthesia, and kidneys, heart, lungs, liver, pancreas and salivary glands were removed for histopathological examination.
To quantify IgG3 and IgM in the sera of mice injected with the hybridomas, single radial immunodiffusion (SRID) was performed according to a method described elsewhere [22].
Histopathological examination
Tissue samples were fixed with 10% formalin in 0.01 mol/l phosphate buffer pH 7.2 and embedded in paraffin. They were stained with haematoxylin and eosin (H–E) or periodic acid-Schiff (PAS) for histological examination by light microscopy. An individual positive for glomerular lesions was defined as one having at least wire loop-like and/or proliferative glomerular lesions in 20 renal glomeruli. The details are given in Table 1.
Table 1.
Incidence of histopathological types of glomerular lesions in SCID mice injected with hybridomas

Immunohistochemical procedures were based on a method described elsewhere [23]. In brief, samples of kidneys, lungs and liver obtained at autopsy were frozen in OCT compound (Miles Inc., Elkhart, IN) and 3 μm thick cryostat sections were cut. IgG, IgM and C3 were detected by a direct method using FITC-conjugated goat anti-mouse IgG (Zymed Inc., San Francisco, CA), goat anti-mouse IgM and rabbit anti-mouse C3 antibodies (Zymed), respectively. The staining of C1q and C4 in glomeruli was performed using rabbit anti-mouse C1q and C4 antibodies [24,25], respectively, followed by biotinylated goat anti-rabbit IgG antibodies (Vector Labs Inc., Burlingame, CA) and FITC-labelled avidin (Vector). To detect Mac-2 antigens [26], kidney samples were fixed overnight at 4°C with a periodate-lysine-paraformaldehyde solution [27] prior to freezing. Immunostaining was performed using rat anti-Mac-2 antibodies (Hybritech Inc., San Diego, CA), biotinylated rabbit anti-rat IgG antibodies (Vector) and FITC-labelled avidin.
RESULTS
Bystander IgM antibodies induce a cell-proliferative type of glomerular lesion
Glomerular lesions induced by the B1 clone alone were wire loop-like (14 of the 16 treated mice), but not cell-proliferative (Table 1) and appeared as severe hyaline material deposits along the glomerular capillary walls (Fig. 1a), as previously reported [16]. However, the glomerular lesions induced by the combination of the B1 and Sp6 clones were different. Eight of the 18 treated mice revealed cell-proliferative lesions, characterized by inflammatory cell infiltrates (Table 1) (Fig. 1b). Thus, the B1 plus Sp6 clones significantly induced a type of glomerular lesions different from those in the mice injected with the B1 clone alone (χ2 = 8.88; P = 0.0029). Some of the glomeruli in these mice exhibited a segmental lesion consisting of a mixture of cell-proliferative and wire loop types (Fig. 1c). Mice injected with the Sp6 clone alone did not develop any glomerular lesions (Fig. 1d). In addition, no other lesions were observed in kidneys (except for the glomeruli), heart, lungs, liver, pancreas and salivary glands from either group of mice, at least by light microscopy.
Fig. 1.
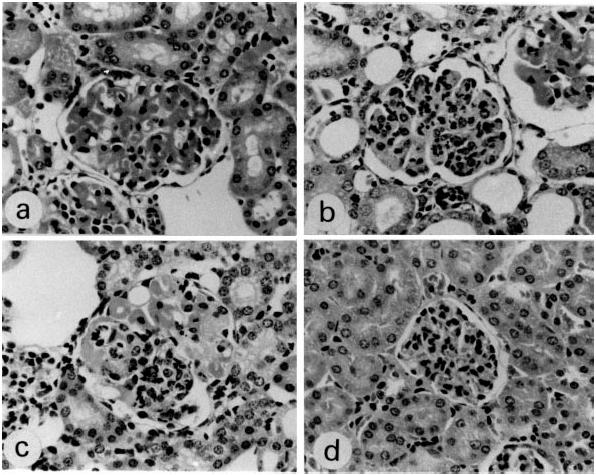
Histopathological manifestations in glomeruli of SCID mice injected with hybridomas (H–E, × 300). (a) Glomerular lesions induced by the B1 clone alone, as seen 25 days after the injection, manifesting glomerular enlargement and hyaline deposits along the capillary wall. This type of lesion is designated ‘wire loop-like’. (b) Glomerular lesions induced by B1 and Sp6 clones, characterized by extreme cell proliferation with an accumulation of macrophage-like and polymorphonuclear cells. This type of lesion is termed ‘cell-proliferative’. (c) Glomerular lesions induced by B1 and Sp6 clones, characterized by the cell-proliferative type mixed with segmental wire loop-like lesions. (d) Glomeruli in mice injected with the Sp6 clone alone were normal in size and did not develop any lesions.
Because SCID mice are severely deficient in immunoglobulin stem cells and are hypogammaglobulinaemic, any immunoglobulin deposits in glomeruli of the injected mice would have originated from the injected hybridomas. In immunohistological studies, IgG but not IgM deposits were observed in the mice injected with the B1 clone (Fig. 2a,c). In glomerular lesions induced by the combination of the B1 and Sp6 clones, deposits of both IgG and IgM were observed and these would have arisen from the B1 and Sp6 clones, respectively (Fig. 2b,d). IgG and/or IgM deposits were not detected in kidneys (except for the glomeruli), lungs and liver, including Kupffer's cells. Glomerular IgM deposits were not observed in mice injected only with the Sp6 clone (data not shown).
Fig. 2.
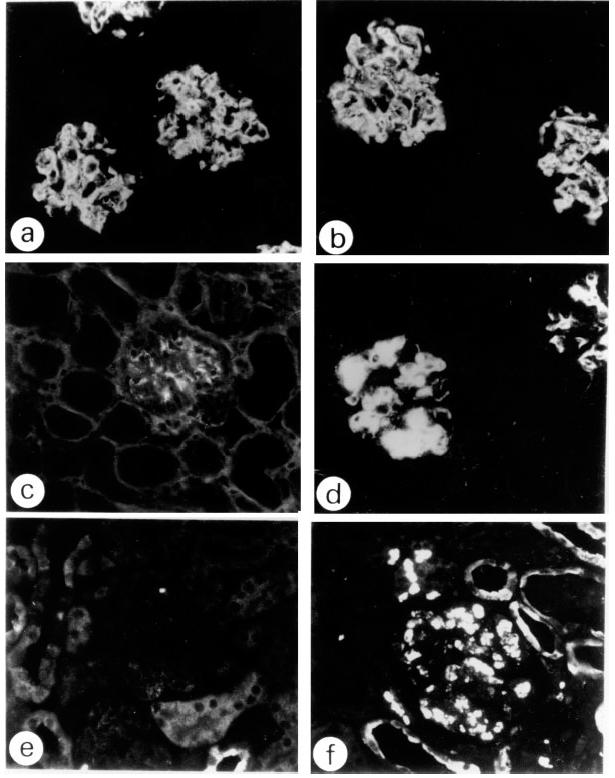
Immunofluorescent studies of glomerular lesions in injected SCID mice (× 300). (a,c,e) Glomeruli of mice injected with the B1 clone alone. (b,d,f) Glomeruli of mice injected with both the B1 and Sp6 clones. (a,b) IgG. (c,d) IgM. (e,f) Mac-2 staining. In the glomeruli of the mice injected with the combination of the B1 and Sp6 clones both IgG and IgM deposits are remarkable (b,d), and a number of Mac-2+ cells are observed (f), while in the case of the B1 clone alone, IgG was positive (a), but IgM was negative (c), and there was an absence of Mac-2+ cells (e). Glomeruli of the mice injected with the Sp6 clone alone were negative for all the staining reactions (not shown).
In glomerular lesions caused by the B1 clone alone, there were very few Mac-2+ cells (Fig. 2e), whereas when both the B1 and Sp6 clones were injected, the glomerular lesions were characterized by a remarkable accumulation of Mac-2+ cells, suggesting that macrophages are involved in the cell-proliferative type of glomerular lesions (Fig. 2f). This suggests that these cells act as scavengers of the B1 and/or Sp6 antibodies deposited in the glomeruli.
We next examined IgG3 and IgM levels in the sera of these mice. Mice injected with both the B1 and Sp6 clones and manifesting cell-proliferative lesions had increased levels of both IgG3 and IgM in their sera (Table 2). However, mice exhibiting wire loop-type lesions, despite being injected with both clones, had undetectable levels of IgM. In these cases it is likely that the injected Sp6 cells had not proliferated. These results indicate that cell-proliferative glomerular lesions result from increased serum levels of both Sp6 and B1 antibodies.
Table 2.
Serum levels of IgM and IgG3 in SCID mice injected with B1 and/or Sp6 hybridomas
A mutant IgM antibody defective in binding C1q also induces a cell-proliferative type of glomerular lesion
To determine whether the change in the type of glomerular lesion brought about the bystander Sp6-IgM is associated with the classical pathway of complement activation in situ, we analysed the histopathological changes which occurred when mice were injected with the Sp6 mutant hybridoma, T/13μE/3.1, whose IgM cannot bind to C1q [21]. The T/13μE/3.1 clone caused a cell-proliferative type of glomerular lesion in the presence of the B1 clone (Fig. 3) more frequently than was the case with the Sp6 clone (T/13μE/3.1; 19/31, Sp6; 8/18), even though the level of IgM in the sera of these mice was considerably lower than that in the sera of mice injected with the Sp6 clone (T/13μE/3.1 0.10 ± 0.01 mg/ml; Sp6 0.53 ± 0.17 mg/ml). The mice injected with only the T/13μE/3.1 clone did not develop any glomerular lesions (n = 7). In immunohistochemical studies C1q, C4 and C3 depositions were observed equally in the glomeruli of mice injected with the B1 clone alone or with both the B1 and T/13μE/3.1 clones (data not shown). These findings may indicate that the mechanism of IgM deposition in glomeruli does not involve the binding of circulating IgM antibodies to C1q already deposited in glomeruli by B1 antibodies.
Fig. 3.
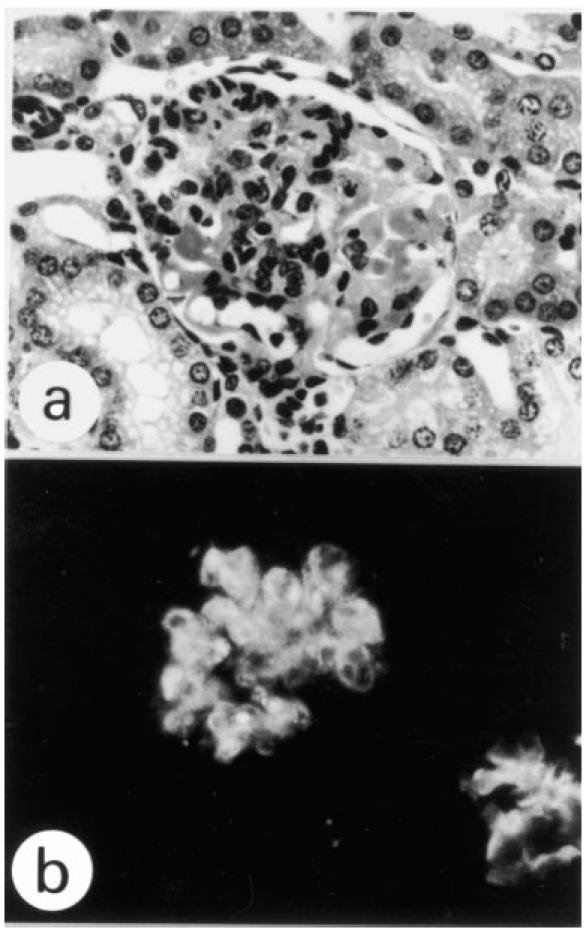
A glomerular lesion induced by the B1 and T/13μE/3.1 clones was mixed, with cell-proliferative and wire loop-like types (a) and associated with IgM deposits (b) (a, H–E; b, immunostaining, × 300).
DISCUSSION
The results of the present study using SCID mice suggest that the histopathological features of glomerular lesions clonally restricted by the type of nephritogenic antibody used could be changed by the presence of non-nephritogenic bystander IgM antibodies. That is, glomerular lesions induced by the B1 clone alone exhibited severe hyaline deposits in mesangial and subendothelial regions which were not accompanied by inflammatory cell infiltrates. However, the additional presence of non-nephritogenic IgM antibodies, which are specific for TNP and do not interact with B1 antibodies by themselves and vice versa (see Materials and Methods), resulted in modification of the lesions to the cell-proliferative form, characteristic of macrophage and polymorphonuclear cell accumulation, although there was no significant difference in proteinuria between the two groups as measured with urinary protein test tapes (Pretest; Wako Chemicals, Tokyo, Japan) in our preliminary study. These altered lesions were associated with deposits of B1-IgG3 and coincidentally with IgM antibodies in situ. Serum levels of both IgG3 and IgM correlated well with the changes in the lesions. It appears that non-nephritogenic bystander IgM antibodies have a remodelling potency in situ in the presence of particular nephritogenic antibodies, although the molecular mechanism was not clarified in this study.
The initiation of this morphological change seems to involve the entrapment of IgM antibodies by the glomeruli, perhaps due to an increase in the permeability of the endothelium induced by B1 antibodies, but not the binding of IgM to C1q which had already been deposited in the affected glomeruli. The findings that IgM antibodies did not react with B1 antibodies and vice versa and that neither IgG nor IgM deposits were observed in the Kupffer's cells in the livers of mice injected with both B1 and Sp6 clones may negate the possibility that these antibodies may have circulated as immune complexes or aggregates.
Generally, complement is thought to play an important role in generating cell-proliferative lesions following antibody deposition in glomeruli. In human and murine SLE, one major serological abnormality is the depression of complement levels, associated with the deposition of complement in glomeruli [2,28–30]. However, in our experimental model system using nephritogenic MoAbs, the results suggest that at least the classical pathway of complement activation does not contribute to the generation of the cell-proliferative lesions, since T/13μE/3.1 antibodies, which lack the ability to bind to C1q, could also generate such lesions. Furthermore, the classical complement pathway seemed to be almost equally activated in both the wire loop-type and cell-proliferative type of glomerular lesion in situ, as evidenced by C1q and C4 deposits.
The mechanisms of development of glomerular lesions induced by B1 antibodies remain unclear. The antigen binding specificities of B1 antibodies are unclear, except it is shown that they have weak DNA binding activity but lack rheumatoid factor and gp70 binding activities. In immunohistochemical studies, we observed that they do not react with renal glomeruli. An IgG3 rheumatoid factor-producing hybridoma clone, 6-19, derived from an MRL/lpr mouse, was also found to induce similar glomerular lesions, which histopathologically resembled those induced by the B1 clone [9]. Reininger et al. [31] replaced the native light chains of 6-19 and found that these chimeric 6-19 antibodies were still capable of inducing glomerular lesions, despite their loss of rheumatoid factor activity. Thus, it appears that the antigen specificities of both B1 and 6-19 antibodies seem not to be critical determinants of glomerular pathogenicity.
Previously we suggested that the variations in glomerular lesions result from a combination of expanded B cell clones which produce antibodies with different pathogenic potencies [13]. In this study, we demonstrated that these are induced not only by a combination of nephritogenic antibodies but also by the concomitant presence of non-nephritogenic bystander IgM antibodies. We speculate that circulating macromolecules such as anti-DNA antibody immune complexes themselves are not always primarily nephritogenic but rather may act as modifiers or accelerators in the development of lupus nephritis, as a result of their entrapment in glomerular lesions in the same manner as bystander IgM antibodies.
Acknowledgments
We wish to thank Ms Y. Takei for technical help in SRID. We also thank Drs W. Campbell and H. Schulman for reviewing the manuscript and Mr M. Arita and Ms M. Aibara for its preparation. This work was supported by grants from the Research Committees of the Ministry of Health and Welfare of Japan and a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan.
REFERENCES
- 1.Murphy ED, Roths JB. Autoimmunity and lymphoproliferation. In: Rose NR, Bigazzi PE, Werner NL, editors. Induction by mutant gene lpr, and acceleration by a male-associated factor in strain BXSB mice.Genetic control of autoimmune disease. New York: Elsevier North Holland Inc; 1978. pp. 207–21. [Google Scholar]
- 2.Andrews BS, Eisenberg RA, Theofilopoulos AN, et al. Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. J Exp Med. 1978;148:1198–215. doi: 10.1084/jem.148.5.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ebling F, Hahan BH. Restricted subpopulations of DNA antibodies in kidneys of mice with systemic lupus. Arthritis Rheum. 1980;32:392–403. doi: 10.1002/art.1780230402. [DOI] [PubMed] [Google Scholar]
- 4.Nardella FA, Teller DC, Izui S, Mannik M. Self-associating IgG-rheumatoid factors in MRL/l autoimmune mice. Arthritis Rheum. 1984;27:1165–73. doi: 10.1002/art.1780271013. [DOI] [PubMed] [Google Scholar]
- 5.Izui S, Elder JH, McConahey PJ, Dixon FJ. Association of circulating retroviral gp70–anti-gp70 immune complexes with murine systemic lupus erythematosus. J Exp Med. 1979;149:1099–116. doi: 10.1084/jem.149.5.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koffler D, Schur PH, Kunkel HG. Immunological studies concerning the nephritis of systemic lupus erythematosus. J Exp Med. 1967;126:607–23. doi: 10.1084/jem.126.4.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Winfield JB, Faiferman I, Koffler D. Avidity of anti-DNA antibodies in serum and IgG glomerular elutes from patients with systemic lupus erythematosus. J Clin Invest. 1977;59:90–6. doi: 10.1172/JCI108626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Madaio MP, Shlomchik MJ. Emerging concepts regarding B cells and autoantibodies in murine lupus nephritis. B cells have multiple roles: all autoantibodies are not equal. J Am Sor Nephrol. 1996;7:387–96. doi: 10.1681/ASN.V73387. [DOI] [PubMed] [Google Scholar]
- 9.Gyotoku Y, Abdelmoula M, Spertini F, Izui S, Lambert PH. Cryoglobulinemia induced by monoclonal immunoglobulin G rheumatoid factors derived from autoimmune MRL/MpJ-lpr/lpr mice. J Immunol. 1987;138:3785–92. [PubMed] [Google Scholar]
- 10.Vlahakos DV, Foster MH, Adams S, Katz M, Ucci AA, Barrett KJ, Datta SK, Madaio MP. Anti-DNA antibodies from immune deposits at distinct glomerular and vascular sites. Kidney Int. 1992;41:1690–700. doi: 10.1038/ki.1992.242. [DOI] [PubMed] [Google Scholar]
- 11.Ehrenstein MR, Katz DR, Griffiths MH, Papadaki L, Winkler TH, Kalden JR, Isenberg DA. Human IgG anti-DNA antibodies deposit in kidneys and induce proteinuria in SCID mice. Kidney Int. 1995;48:705–11. doi: 10.1038/ki.1995.341. [DOI] [PubMed] [Google Scholar]
- 12.Ray SK, Putterman C, Diamond B. Pathogenic autoantibodies are routinely generated during the response to foreign antigen: a paradigm for autoimmune disease. Proc Natl Acad Sci USA. 1996;93:2019–24. doi: 10.1073/pnas.93.5.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ravirajan CT, Rahman MA, Papadaki L, et al. Genetic, structural and functional properties of an IgG DNA-binding monoclonal antibody from a lupus patient with nephritis. Eur J Immunol. 1998;28:339–50. doi: 10.1002/(SICI)1521-4141(199801)28:01<339::AID-IMMU339>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 14.Takahashi S, Nose M, Sasaki J, Yamamoto T, Kyogoku M. IgG3 production in MRL/lpr mice is responsible for development of lupus nephritis. J Immunol. 1991;147:515–20. [PubMed] [Google Scholar]
- 15.Takahashi S, Itoh J, Nose M, Ono M, Yamamoto T, Kyogoku M. Cloning and cDNA sequence analysis of nephritogenic monoclonal antibodies derived from an MRL/lpr lupus mouse. Mol Immunol. 1993;30:177–80. doi: 10.1016/0161-5890(93)90089-t. [DOI] [PubMed] [Google Scholar]
- 16.Itoh J, Nose M, Takahashi S, Ono M, Terasaki S, Kondoh E, Kyogoku M. Induction of different types of glomerulonephritis by monoclonal antibodies derived from an MRL/lpr lupus mouse. Am J Pathol. 1993;43:1436–43. [PMC free article] [PubMed] [Google Scholar]
- 17.Churg J, Bernstein J, Glassock RJ. classification and atlas of glomerular diseases. 2. Tokyo: Igaku Shoin; 1995. Renal disease. [Google Scholar]
- 18.Witte T, Hartung K, Sachse C, et al. IgM anti-dsDNA antibodies in systemic lupus erythematosus: negative association with nephritis. SLE Study Group. Rheumatol Int. 1998;18:85–91. doi: 10.1007/s002960050063. [DOI] [PubMed] [Google Scholar]
- 19.Bosma GC, Custer RP, Bosma MJ. A severe combined immunodeficiency mutation in the mouse. Nature. 1983;301:527–30. doi: 10.1038/301527a0. [DOI] [PubMed] [Google Scholar]
- 20.Nose M, Wigzell H. Biological significance of carbohydrate chains on monoclonal antibodies. Proc Natl Acad Sci USA. 1983;80:6632–6. doi: 10.1073/pnas.80.21.6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shulman MJ, Pennell N, Collins C, Hozumi N. Activation of complement by immunoglobulin M is impaired by the substitution serine-406 → asparagine in the immunoglobulin μ heavy chain. Proc Natl Acad Sci USA. 1986;83:7678–82. doi: 10.1073/pnas.83.20.7678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garvey JS, Kremer NK, Sussdorf DH. Methods in immunology. 3. Tokyo: Benjamin Inc.; 1977. [Google Scholar]
- 23.Nose M, Kato M, Okada N, Kyougoku M, Okada H. Tissue distribution of HRF20, a novel factor preventing the membrane attack of homologous complement, and its predominant expression on endothelial cells in vivo. Immunology. 1990;70:145–9. [PMC free article] [PubMed] [Google Scholar]
- 24.Seino J, Fukuoka Y, Okuda T, Tachibana T. Isolation, molecular properties and allotype of mouse C1q. Tohoku J Exp Med. 1984;142:351–61. doi: 10.1620/tjem.142.351. [DOI] [PubMed] [Google Scholar]
- 25.Fukuoka Y, Seino J, Okuda T, Tachibana T. Purification of the fourth, second and fifth components of mouse complement. Immunology. 1984;51:493–501. [PMC free article] [PubMed] [Google Scholar]
- 26.Ho MK, Springer TA. Mac-2, a novel 32, 000 Mr mouse macrophage subpopulation-specific antigen defined by monoclonal antibodies. J Immunol. 1982;128:1221–8. [PubMed] [Google Scholar]
- 27.McLean IW, Nakane PK. Periodate-lysine-paraformaldehyde fixative: a new fixative for immunoelectron microscopy. J Histochem Cytochem. 1974;22:1077–83. doi: 10.1177/22.12.1077. [DOI] [PubMed] [Google Scholar]
- 28.Perrin LH, Lambert PH, Miescher PA. Complement breakdown products in plasma from patients with systemic lupus erythematosus and patients with membranoproliferative or other glomerulonephritis. J Clin Invest. 1975;56:165–76. doi: 10.1172/JCI108065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trinder PK, Maeurer MJ, Schorlermmer HU, Loos M. Autoreactivity to mouse C1q in a murine model of SLE. Rheumatol Int. 1995;15:117–20. doi: 10.1007/BF00302128. [DOI] [PubMed] [Google Scholar]
- 30.Williams PG, Peters DK, Fallows J. Studies of serum complement in the hypocomplementemic nephritides. Clin Exp Immunol. 1974;18:391–405. [PMC free article] [PubMed] [Google Scholar]
- 31.Reininger L, Berney T, Shibata T, Spertini F, Merino R, Izui S. Cryoglobulinemia induced by a murine IgG3 rheumatoid factor: skin vasculitis and glomerulonephritis arise from distinct pathogenic mechanisms. Proc Natl Acad Sci USA. 1990;87:10083–42. doi: 10.1073/pnas.87.24.10038. [DOI] [PMC free article] [PubMed] [Google Scholar]