

Abstract
The role of CD4 molecules in the autoimmune and lymphoproliferative syndrome caused by murine Fas mutations was studied using the novel systemic lupus erythematosus (SLE) model, MRL-Faslprcg/Faslprcg (MRL-lprcg) mice, in combination with the novel mutant CD4 gene producing soluble CD4 (sCD4) instead of membrane-bound CD4 (mCD4). For this purpose, various autoimmune manifestations were compared among MRL-lprcg mice homozygous (CD4slprcg), heterozygous (CD4s/mlprcg), and wild-type (CD4mlprcg) for the CD4 mutation. The mortality, glomerulonephritis, proteinuria, and lymphadenopathy were significantly ameliorated in CD4slprcg compared with CD4mlprcg and CD4s/mlprcg mice, both being comparable in these clinical characteristics. In parallel with the clinical improvement, the serum levels of immunoglobulin, anti-DNA antibodies, anti-nuclear antibodies and immune complexes, and the extent of glomerular immune deposition, were significantly lower in the former. The results indicate that mCD4 is important and can not be replaced by sCD4 in full development of SLE-like manifestations, and suggest that CD4+ T cells may aggravate the autoimmune disease by stimulating autoreactive B cells to produce autoantibodies through their helper activity in Fas mutant models. The sCD4 levels in the serum and spleen elevated with the increased accumulation of B220+CD4−CD8− (double-negative (DN)) T cells in CD4slprcg mice. This, together with the significantly milder lymphadenopathy associated with lower DN T cell contents in CD4slprcg than CD4mlprcg mice, implies that some of abnormal DN T cells may be derived from cells of the CD4 lineage.
Keywords: lupus nephritis, autoimmunity, MRL-Faslprcg mice, CD4 molecules
INTRODUCTION
MRL/MpJ-Faslpr/Faslpr (MRL-lpr) mice spontaneously develop a severe autoimmune syndrome associated with progressive lymphoproliferation and autoantibody production. Since this autoimmune syndrome has many similarities to human systemic lupus erythematosus (SLE) in the development of immune complex (IC)-associated end organ disease of the kidney and salivary glands, and the production of anti-DNA and other autoantibodies, MRL-lpr mice have been widely used as an animal model for SLE [1,2]. In this model, T cells, more specifically autoreactive CD4+ αβ+ T cells, have been shown to be essential for the full induction of the SLE-like manifestations including nephritis by neonatal thymectomy [3], immunodepletion with antibodies against Thy-1, CD3 and CD4 [4–11], and genetic deletion of CD4+ T cells by targeting class II and CD4 genes [12,13]. Faslpr (lpr) is defined as the insertion of an early retrotransposon in the second intron of Fas that interferes with normal transcription of the gene [14]. The most characteristic phenotype of lpr is lymphoproliferation due to the accumulation of B220+TCRαβ+ CD4−CD8− (double-negative (DN)) T cells. Another mutation of Fas, Faslprcg (lprcg), encodes in the Fas cytoplasmic death domain a single amino acid substitution that abolishes the ability of Fas to transduce apoptotic signal into cells despite its binding to Fas ligand (FasL) [15]. Thus, unlike lpr, lprcg can induce lymphadenopathy by interacting with Faslgld (gld) in the double heterozygous state [16]. Importantly, analysis of the Fas locus in human patients with autoimmune lymphoproliferative disease associated with peripheral lymphadenopathy, splenomegaly and autoantibody production indicated that they appeared to be heterozygous for a mutant Fas allele [17,18]. MRL-lprcg/lprcg (MRL-lprcg) mice also develop a complex autoimmune disease closely mimicking SLE with IC-mediated glomerulonephritis and production of a wide variety of autoantibodies [19] and provide a unique SLE model that enables us to test the possible influences of the functions of Fas displayed through the region outside the death domain on disease development. Recently, we happened upon mutant mice characterized by the production of soluble CD4 (sCD4) instead of membrane-bound CD4 (mCD4) due to the substitution of one thymine for 14 base pairs at the junction between exon VIII and intron VIII in the genomic DNA [20].
In this study, we introduced the unique mutant CD4 gene into the novel SLE model, MRL-lprcg mice, to investigate the effects of the presence of sCD4 and the absence of mCD4 on the development of autoimmunity.
MATERIALS AND METHODS
Mice
MRL-lprcg female mice were mated with CD4-mutant males, and their female offspring were backcrossed to MRL-lprcg males to produce N2 offspring. A single-cell suspension was prepared from a biopsied inguinal lymph node (LN) and screened for CD4 expression by flow cytometry. N2 males heterozygous for the CD4 mutation with intermediate expression of mCD4 were selected, and the mice homozygous for lprcg were further selected by development of clear LN swelling within 3 months of age and backcrossed to MRL-lprcg females to produce N3 offspring. From them, the mice expressing mCD4 were selected and intercrossed to obtain N3F1 mice. The Fl mice were separated into three groups by the extent of mCD4 expression on LN cells and expressed as follows: CD4slprcg mice which are homozygous for lprcg and do not express mCD4 but produce sCD4 in a homozygous state, CD4s/mlprcg mice which are homozygous for lprcg and produce both mCD4 and sCD4 in a heterozygous state, and CD4mlprcg mice which are homozygous for lprcg and express mCD4 in a homozygous state. These mice segregated with the expected Mendelian frequency and were expected to be 87·5% MRL genetic background on the average. Wild-type MRL (MRL-+) mice were purchased from SLC (Hamamatsu City, Japan) and used for comparison.
Lymphoid hyperplasia
Mice were deeply anaesthetized and killed by drawing blood from the heart. LN and spleens were excised and weighed wet separately. The weights of the cervical, axillary, brachial, inguinal and other enlarged subcutaneous LN were added and presented as the external LN weight, those of the mediastinal, renal, lumbar and sciatic LN were also added and presented as the internal LN weight, and the mesenteric LN weight was presented separately. Sera were individually isolated and stored at −20°C until use.
Proteinuria
Urinary protein levels were monitored in freshly voided urine with Albustix strips (Bayer-Sankyo Co., Ltd, Tokyo, Japan) before autopsy, and scored as 0–4: 0, negative; 0·5, ±; 1, +(30 mg/dl); 2, ++ (100 mg/dl); 3, +++ (300 mg/dl); and 4, ++++ (≥ 1000 mg/dl).
Histopathological analysis
Kidney specimens were fixed in 10% formalin in PBS, embedded in paraffin, and sectioned at 3 μm. After staining with haematoxylin and eosin or periodic acid-Schiff, they were examined in a blinded fashion and scored to provide a semiquantitative measure of glomerulonephritis by the criteria described [21]: 0, normal; 0·5, a few glomeruli are slightly hypercellular; 1, many glomeruli are slightly hypercellular; 2, many glomeruli are hypercellular with focal changes, and foci of necrosis or crescent in a few glomeruli are found; and 3, diffuse glomerular changes with crescent and necrosis, and some completely destroyed or fibrotic glomeruli are found. On the other hand, fresh kidney fragments were embedded in OCT compound (Sakura, Tokyo, Japan), frozen, and sectioned at 4 μm in a cryostat. Sections were fixed in acetone, air-dried, and stained with FITC-conjugated goat anti-mouse C3 antibody (Cappel, Malvern, PA) or FITC-conjugated goat anti-mouse IgG2a or IgG3 antibody (Cappel), followed by fluorescent microscopy. Severity of IC deposition was evaluated in a blinded fashion on a 0–3 scale based on the intensity of the staining in mesangium and peripheral loop areas.
Quantification of serum immunoglobulin levels
Scrum IgM and IgG levels were determined by sandwich ELISA. Microtitre plates (Becton Dickinson, Mountain View, CA) were coated with 1 μg/ml of anti-mouse IgM MoAb (PharMingen, San Diego, CA) or 2 μg/ml of anti-mouse IgG MoAb (Zymed, San Francisco, CA) at 4°C overnight, and blocked with 3% bovine serum albumin (BSA) in PBS. Sera were added at appropriate dilutions in duplicate. After washing, biotin-conjugated goat anti-mouse immunoglobulin antibody (PharMingen) was added, followed by addition of horseradish peroxidase (HRP)-conjugated streptavidin (Zymed). After washing, the plates were incubated with o-phenylenediamine (Sigma, St Louis, MO) in 0·1 m citrate buffer and 0·05% H2O2. The reaction was stopped with 2 n H2SO4, and then the absorbance at 490 nm was determined in Microplate Reader Model 550 (BioRad, Hercules, CA). The total IgM and IgG concentrations were calculated with reference to the calibration curve generated with mouse immunoglobulin reference serum (ICN, Costa Mesa, CA).
Quantification of serum anti-double-stranded DNA antibody levels
Anti-double-stranded (ds)DNA antibodies of IgM and IgG classes and IgG isotypes were also determined by sandwich ELISA. Microtitre plates were coated with 50 μg/ml of poly-l-lysine (Sigma) in PBS. After washing, the plates were incubated with 2 μg/ml of calf thymus DNA (Sigma) in PBS at 37°C overnight. Biotin-conjugated antibody against mouse IgM, IgG and IgG isotypes (Zymed) were used as a secondary antibody. Sera were diluted appropriately and applied in duplicate for quantification. Pooled serum from 4-month-old MRL-lprcg mice was used for standardization.
Quantification of IC in serum
Circulating IC levels were assessed using the C1q solid-phase assay kit (Sankyo Junyaku, Tokyo, Japan) with some modifications. Briefly, human C1q was applied to microtitre plates at a concentration of 10 μg/ml and incubated overnight at 4°C. After blocking, the wells were incubated with 50-fold diluted and 0·1 m EDTA-treated serum samples for 1 h at room temperature. After washing, alkaline phosphatase-conjugated anti-mouse IgG antibody (Cappel) was added for 1 h at room temperature. After washing, the plate was further incubated with the mixture of 5 mm phenylphosphate disodium and 2·5 mm 4-aminoantipyrine. Finally, the colour developed with potassium peroxide was read at 490 nm. The amount of IC was standardized with soluble aggregated mouse gammaglobulin as described [22].
Quantification of anti-nuclear antibody in serum
Anti-nuclear antibody (ANA) levels were determined by an indirect immunofluorescence technique. Small liver fragments were obtained from a 6-week-old C57Bl/6 mouse, frozen in OCT compound (Sakura), sectioned at 5 μm, fixed in cold acetone, and dried. Sera were serially two-fold diluted from 1:20 to 1:640, placed on the sections, and incubated for 30 min at 37°C. After washing in PBS, the sections were incubated with FITC-conjugated goat anti-mouse immunoglobulin antibody (PharMingen) for 30 min at 37°C. Nuclear fluorescence was evaluated in a blinded manner. ANA levels were assessed semiquantitatively by the end point positive titre for detection of staining and scored from 1 to 6 which corresponds to 1:20 to 1:640 dilutions, respectively.
Quantification of sCD4 in serum and spleen
A single-cell suspension was prepared from spleen, and 5 × 107 cells were solubilized at 4°C in 1 ml of 10 mm Tris–HCl buffer pH 7·8 containing 1% NP-40, 0·15 m NaCl, 5 mm EDTA, 10 μg/ml of aprotinin and 2 mm sodium orthovanadate, and centrifuged to obtain a supernatant. Supernatants and sera were assayed for sCD4 by ELISA. A microtitre plate (Becton Dickinson) was coated with anti-CD4 MoAb (GK1.5), and incubated with 100 μl of 10-fold diluted sera or undiluted supernatants in duplicate. After washing, the plate was incubated with biotinylated anti-CD4 MoAb (RM4-4) and then with HRP-labelled streptavidin. sCD4 concentrations were photometrically determined and standardized [20].
Flow cytometry
Single-cell suspensions were prepared from swelled LN and spleens of 4–5-month-old CD4slprcg and CD4mlprcg mice. Samples of 106 cells were treated with Fc blocking antibody (PharMingen), washed and surface-stained with FITC-labelled anti-CD8 (53-5.3), PE-labelled anti-B220 (RA4-6B2) and with biotinylated anti-CD4 (GK1.5) or biotinylated anti-α TCR (H57-597) MoAb, followed by 30 min incubation with streptavidin–PE. All labelled MoAbs were purchased from PharMingen. Stained cells were analysed on a FACScan using Lysis II software (Becton Dickinson) for data analysis. Dead cells positively stained with 7-aminoactinomycin D (Sigma) were gated out.
Statistical analysis
Statistical evaluation was performed with Fisher’s exact test or Student’s t-test. P <5% was considered statistically significant.
RESULTS
sCD4 in serum and spleen cells
The mutant CD4 gene produces sCD4 instead of mCD4 molecules. Therefore, to investigate the expression of the gene, sCD4 was quantified in sera from CD4slprcg, CD4s/mlprcgand CD4mlprcg mice. As expected, sCD4 was detectable in CD4slprcg and CD4s/mlprcg but not in CD4mlprcg mice (Table 1). Noticeably, the serum sCD4 level rose with age or development of lymphadenopathy in CD4slprcg mice. The level in disease-free C57BR and C57Bl/6 mice carrying the mutant gene remained unchanged through life (data not shown) and was comparable to that in 1-month-old CD4slprcg mice which had not yet developed lymphadenopathy. This age-dependent increase was also seen in spleen cells of CD4slprcg mice. Therefore, it appeared that abnormally expanded cells were involved in sCD4 production.
Table 1.
Soluble CD4 (sCD4) levels in sea and spleen cells form CD4slprcg, CD4s/mlprcg and CD4mlprcg mice
| sCD4 levels at the age of | ||||
|---|---|---|---|---|
| Mouse strain | Sex | 1 month | 3 months | 5 months |
| Serum (U/ml) | ||||
| CD4slprcg | Male | 78·2 ± 2·0 (6)* | 137 ± 10 (10) | 239 ± 64 (11) |
| Female | 74·0 ± 2·0 (8) | 147 ± 12 (10) | 202 ± 33 (11) | |
| CD4s/mlprcg | Male | ND† | ND | 206 ± 28 (11) |
| Female | ND | ND | 152 ± 24 (11) | |
| CD4mlprcg | Male | ND | ND | 0 |
| Female | ND | ND | 0 | |
| Spleen cells (U/5 × 106 cells) | ||||
| CD4slprcg | Female | 1·04 ± 0·10 (5) | ND | 2·82 ± 0·49 (5) |
Mean ±s.e.m. for the number of mice indicated in parentheses.
Not determined.
Observation of animals
Twenty-five males and 25 females in CD4slprcg and CD4mlprcg, and 15 males and 15 females in CD4s/mlprcg were observed for survival up to 5 months of age. None of them died before 3 months old. At this age, 10 males and 10 females were autopsied in CD4slprcg and CD4mlprcg. None of 15 males and one of 15 females in CD4slprcg, two of 15 males and two of 15 females in CD4s/mlprcg, and four of 15 males and four of 15 females in CD4mlprcg died during the fourth month of birth. All dead mice in CD4s/mlprcg and CD4mlprcg, but not the sole dead one in CD4slprcg had clearly swollen LN. In addition, the subcutaneous LN were confirmed by palpation to begin to swell later in CD4slprcg than in CD4mlprcg and CD4s/mlprcg. As nephritis is the main cause of death in lpr mice, the disease appeared to progress more slowly in the absence of mCD4 or CD4+ T cells. Finally, all survivors in CD4mlprcg, and 11 male and 11 female survivors selected at random in CD4slprcg and CD4s/mlprcg, were autopsied at 5 months old.
Lymphoid hyperplasia
The severity of lymphoid hyperplasia was compared between groups by each of the external, internal and mesenteric LN, and spleen weights (Fig. 1). Generally, no significant differences were found in these parameters between CD4mlprcg and CD4s/mlprcg at both 3 and 5 months old. It was first noted that the hyperplasia of the external and internal LN was highly significantly less severe in CD4slprcg than in CD4mlprcg and CD4s/mlprcg, but there was no significant difference in splenomegaly among them. Interestingly, mesenteric LN were intermediate in terms of this anomaly. Conspicuous swelling of mesenteric LN was noticed, since they scarcely enlarged in CBA-lprcg/lprcg mice [16].
Fig. 1.
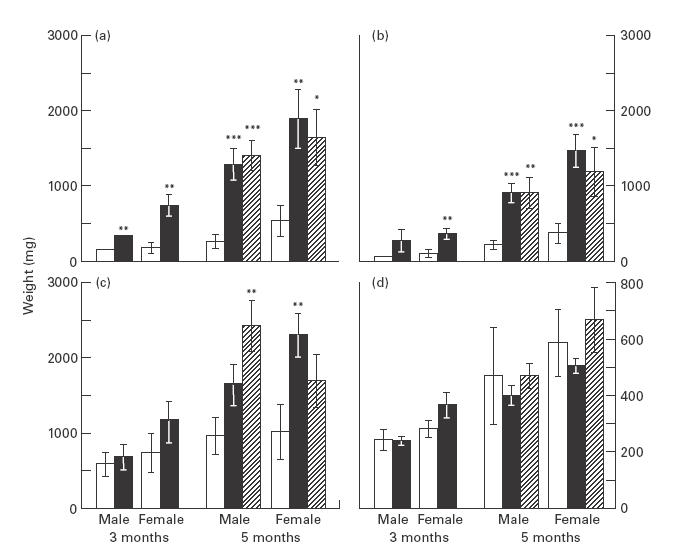
Lymphadenopathy and splenomegaly in CD4slprcg (□), CD4mlprcg (▪), and CD4s/mlprcg (hatched) groups. External lymph nodes (LN) (a), internal LN (b), mesenteric LN (c) and spleen (d) weights were determined at 3 months old in 10 males and 10 females in the first two groups, and at 5 months old in 11 males and 11 females in all groups, and the results are presented as mean ±s.e.m. Significant difference compared with sex- and age-matched CD4slprcg mice at *P < 0·05, **P <0·01, and ***P <0·001, by t-test.
Proteinuria
It is generally accepted that urinary protein levels ≥300 mg/dl are pathologically significant in mice [23]. According to this criterion, the incidence of proteinuria tended to be lower in CD4slprcg than in CD4mlprcg and CD4s/mlprcg mice at both 3 and 5 months old (Table 2). Especially, it was noted that the incidence in females was significantly lower in CD4slprcg than CD4mlprcg and CD4s/m -lprcg at 5 months old, since the autoimmune symptoms were more serious in females than in males (see below). Moreover, there were no significant differences in the incidence between CD4mlprcg and CD4s/mlprcg. The results suggested that proteinuria might progress more slowly in the absence of CD4+ T cells even when high levels of sCD4 existed. This suggestion was supported by the proteinuria score, which was significantly lower in the absence than in the presence of CD4+ T cells at 5 months old (1·19 ± 0·15 (n = 22) versus 1·80 ± 0·17 (n = 44); P < 0·05 by t-test).
Table 2.
Development of pathologically significant proteinuria in CD4slprcg, CD4mlprcg and CD4s/mlprcg mice
| No. of mice with proteinuria/no. of mice examined | ||||
|---|---|---|---|---|
| Age (months) | Sex | CD4slprcg | CD4mlprcg | CD4s/mlprcg |
| 3 | Male | 0/10 (0)* | 1/10 (0) | |
| Female | 0/10 (0) | 2/10 (20) | ||
| Total | 0/20 (0) | 3/20 (15) | ||
| 5 | Male | 1/11 (9) | 2/11 (18) | 2/11 (18) |
| Female | 0/11 (0) | 5/11 (45)† | 2/11 (18) | |
| Total | 1/22 (5) | 7/11 (32)† | 4/22 (18) | |
Incidence of proteinuria in percent (≥ 300 mg/dl) is shown in parentheses.
Significant difference compared with CD4 s-lprcg mice: P < 0·05 by Fisher’s exact test.
Renal pathology
As shown in Fig. 2a, highly advanced glomerulonephritis scored 2 or 3 did not occur in any 3- and 5-month-old CD4slprcg mice except for the sole 3-month-old male scored 2. In contrast, advanced disease was found in one of 10 males and two of 10 females in CD4mlprcg at 3 months; and in one of 11 males and five of 11 females in CD4mlprcg and in five of 11 males and four of 11 females in CD4s/mlprcg at 5 months old. In addition, the number of moderately affected mice scored 1 was also smaller in CD4slprcg than in CD4s/mlprcg and CD4mlprcg (in sum: 2/32 versus 10/32 versus 7/22). Thus, the development of renal disease was clearly suppressed in the absence of CD4+ T cells. This, together with the far lower incidence of proteinuria, was more significant, considering that more mice died of illness before 5 months of age when CD4+ T cells existed. In the severely diseased mice, the typical renal histology was characterized by lobulation and necrosis of glomerular tufts, crescent formation, and hyaline cylinders in the tubules indicative of profuse proteinuria. The less severe cases showed small areas of necrosis with slight hypercellularity and widening of mesangial regions in some glomeruli.
Fig. 2.
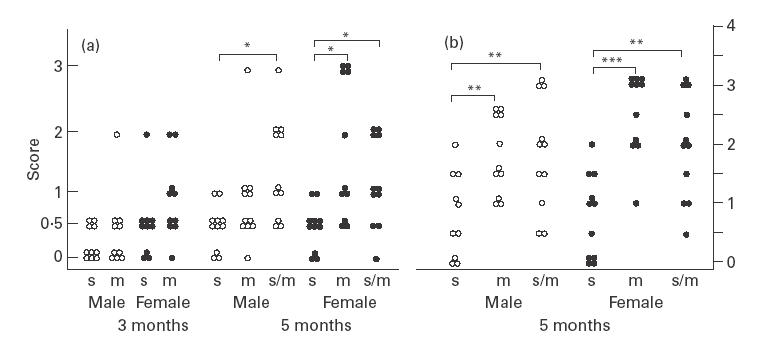
Renal histopathology (a) and glomerular immune deposits (b) in CD4slprcg (s), CD4mlprcg (m), and CD4s/mlprcg (s/m) groups. The severity of glomerulonephritis at 3 and 5 months old, and the extent of immune deposition detected with anti-C3 antibody at 5 months old were scored on a 0–3 scale as described in Materials and methods. Open and solid circles represent single male and female mice, respectively. Significant difference between the two groups at *P < 0·05, **P <0·01, and ***P <0·001, by t-test.
Immune deposits to glomeruli and circulating IC
Glomerular immune deposits were assessed at 5 months old. Immune deposits detected with anti-IgG2a antibody were smaller in quantity in CD4slprcg than in CD4mlprcg and CD4s/mlprcg mice, significantly in females but insignificantly in males, and those detected with anti-IgG3 antibody were not significantly different among three groups of males and females (data not shown). However, it is generally accepted that glomerular C3 deposition is a more reliable marker of IC-mediated nephritis. Importantly, C3 deposition was highly significantly lower in extent in CD4slprcg than in CD4mlprcg and CD4s/mlprcg mice of both sexes (Fig. 2b), in accord with the less severe glomerulonephritis in the former (Fig. 2a). Intensive to moderate granular C3 deposition along the glomerular basement membrane and in the mesangial areas was observed in severely diseased kidneys. Circulating IC levels at 3 and 5 months old were significantly lower, consistent with the significantly milder glomerular C3 deposition in CD4slprcg than in CD4mlprcg and CD4s/mlprcg mice (Table 3).
Table 3.
Circulating immune complex levels in CD4slprcg, CD4mlprcg and CD4s/mlprcg mice
| Mouse | ||||
|---|---|---|---|---|
| Age (months) | Sex | CD4slprcg | CD4mlprcg | CD4s/mlprcg |
| 3 | Male | 240 ± 40† | 598 ± 93**** | ND‡ |
| Female | 355 ± 55 | 629 ± 79** | ND | |
| 5 | Male | 41 ± 5 | 565 ± 154**** | 390 ± 95*** |
| Female | 163 ± 45 | 1264 ± 425** | 1230 ± 461* | |
Mean ±s.e.m. in μg/ml aggregated mouse gammaglobulin equivalent for 10 and 11 mice at 3 and 5 months of age, respectively.
Significantly different from age- and sex-matched CD4slprcg mice at
P < 0·05
P < 0·02
P < 0·002
P < 0·005 by t-test.
Not determined.
Serum immunoglobulin and autoantibody levels
At 5 months old, both serum IgM and IgG levels were significantly lower in CD4slprcg than in CD4mlprcg and CD4s/m -lprcg males (Fig. 3). They showed a similar tendency in females, although the IgG level was significantly different only between CD4slprcg and CD4mlprcg. At 3 months old, the IgG level in males was also significantly lower in CD4slprcg than in CD4m -lprcg. It was noted that the age-dependent increase was more conspicuous in IgG than IgM. Serum anti-dsDNA antibody levels are shown in Fig. 4. IgM and IgG anti-dsDNA antibody levels displayed practically the same tendencies as serum IgM and IgG levels, respectively, at both 3 and 5 months old. The IgM antibody level was significantly lower in CD4slprcg than in CD4mlprcg and CD4s/mlprcg females at 5 months old. The IgG antibody level was significantly lower in CD4slprcg than in CD4mlprcg mice of both sexes at 3 and 5 months, and than CD4s/mlprcg males at 5 months old. Moreover, the IgG anti-dsDNA antibody level was reflected in the levels of anti-dsDNA antibody of IgG1, IgG3 and IgG2a isotypes exactly in males and approximately in females at 3 and 5 months old. The IgG2b antibody level was significantly lower only in 5-month-old CD4slprcg males compared with the CD4mlprcg and CD4s/mlprcg counterparts. Of note, the age-dependent increase was far larger in CD4slprcg than in CD4mlprcg mice and the autoantibody production at 5 months old in CD4slprcg was suppressed near to MRL-+ levels in all classes and subclasses. The ANA levels determined at 5 months old were also lower in CD4slprcg than in CD4mlprcg and CD4s/mlprcg mice, although the difference reached a significant level only in males, as observed in most of the above-mentioned analyses (data not shown).
Fig. 3.
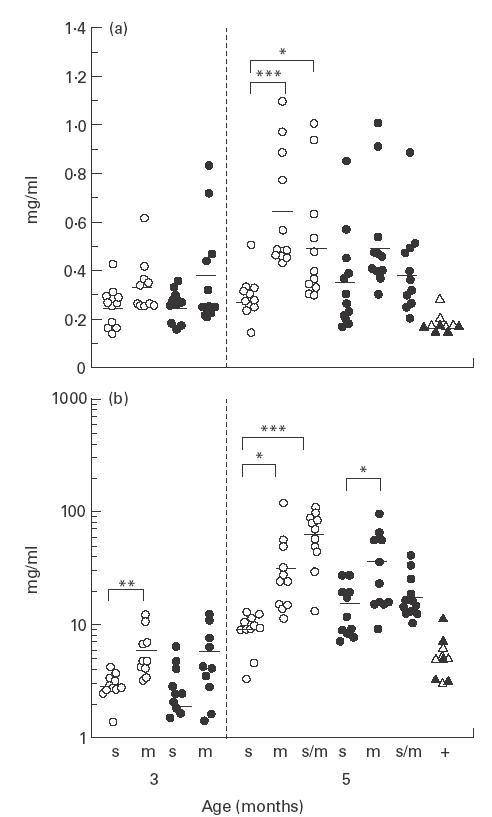
Serum IgM (a) and IgG (b) levels in CD4slprcg (s), CD4mlprcg (m), and CD4s/mlprcg (s/m) groups. Total IgM and IgG in the serum were determined at 3 months old in 10 males and 10 females in the first two groups, and at 5 months old in 11 males and 11 females in all groups. The levels in MRL-+ (+) mice aged 5–10 months are included for comparison. Open and solid symbols represent single male and female mice, respectively, and the horizontal bars show the mean. Significant difference between the two groups at *P < 0·05 and ***P < 0·001, by t-test.
Fig. 4.
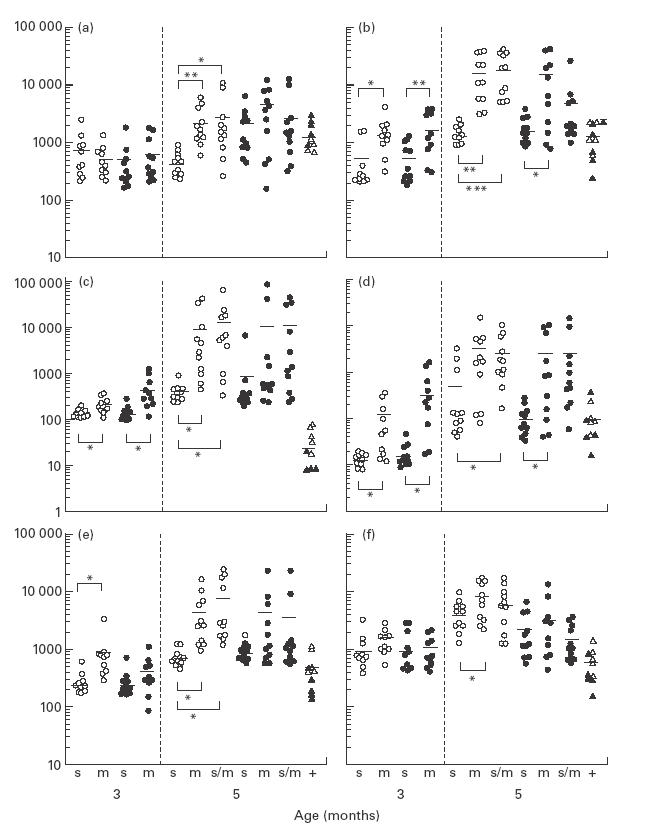
Serum levels of IgM (a), IgG (b), IgG1 (c), IgG2a (d), IgG2b (e), and IgG3 (f) anti-dsDNA antibody in CD4slprcg (s), CD4mlprcg (m), and CD4s/mlprcg (s/m) groups. These levels were determined at 3 months old in 10 males and 10 females in the first two groups, and at 5 months old in 11 males and 11 females in all groups. The levels in MRL-+ (+) mice aged 5–10 months are included for comparison. Open and solid symbols represent single male and female mice, respectively, and the horizontal bars show the mean. Significant difference between two groups at *P < 0·05, **P <0·01, and ***P <0·001, by t-test.
Cell surface markers of LN and spleen cells
As expected, CD4+ cells were not detected at all in CD4slprcg mice. Accumulation of B220+ DN T cells in lymphoid organs is the most characteristic phenotype of lpr and lprcg mice [19,24]. The proportion of these abnormal T cells was reduced clearly in LN but not in spleen in CD4slprcg compared with CD4mlprcg mice (Table 4), in accord with the significantly smaller degree of LN hyperplasia in the former and the same degree of splenomegaly in both mice (Fig. 1). Of note, the proportions of B220− DN T cells in both LN and spleen were far larger in CD4slprcg than in CD4mlprcg mice (Table 2). These cells in LN were also increased in proportion in the CD4-mutant C57BR mice compared with wild-type counterparts [20]. As expected from the far higher content of CD8+ T cells in LN and spleen in the original mutants, the proportion of B220−CD8+ cells was also significantly larger in CD4slprcg than in CD4mlprcg mice.
Table 4.
Flow cytometric analysis of lymph node (LN) and spleen cells from 4–5-month-old CD4slprcg and CD4mlprcg mice
| LN cells | Spleen cells | |||
|---|---|---|---|---|
| Cell surface markers | CD4slprcg | CD4mlprcg | CD4slprcg | CD4mlprcg |
| B220+CD4−CD8−TCRαβ+ | 55·9 ± 9·4* | 82·6 ± 0·2 | 47·8 ± 4·6 | 46·4 ± 1·2 |
| B220−CD4−CD8−TCRαβ+ | 19·6 ± 6·9 | 0·9 ± 0·15 | 21·1 ± 3·8 | 7·1 ± 1·35 |
| B220+CD4+ | 0 | 11·7 ± 1·8 | 0 | 3·3 ± 1·0 |
| B220−CD4+ | 0 | 5·5 ± 0·5 | 0 | 14·3 ± 1·6 |
| B220+CD8+ | 0·77 ± 0·21 | 0·05 ± 0 | 0·49 ± 0·09 | 0·14 ± 0·02 |
| B220−CD8+ | 12·9 ± 6·0 | 2·1 ± 0·5 | 14·7 ± 2·7 | 6·6 ± 0·9 |
| B220+TCRαβ− | 9·1 ± 3·5 | 6·3 ± 0·8 | 2·4 ± 0·6 | 6·1 ± 0·4 |
Mean ±s.e.m. for three mice in percent of total cells.
DISCUSSION
Autoreactive CD4+αβ+ T cells have been shown to play a crucial role in the maximal induction of SLE-like syndrome in MRL-lpr mice [7,8,12,13]. Thus, we investigated the role of the CD4 molecule in induction of lpr disease by using the novel CD4 mutation characterized by production of sCD4 instead of mCD4 [20] and the novel Fas mutation, lprcg[19]. For this purpose, CD4slprcg, CD4mlprcgand CD4s/mlprcg mice were constructed and compared with respect to various autoimmune indices. It was first noted that the mortality and clinical manifestations including glomerulonephritis and proteinuria, a sensitive marker of renal injury, were clearly improved in CD4slprcg compared with CD4mlprcg mice (Fig. 1, Table 2). Thus, the lpr disease progressed in a milder form in the lack of mCD4 or CD4+ T cells despite the presence of sCD4, indicating that mCD4 is necessary for the full development of the disease and that sCD4 can not replace mCD4 in this respect. On the other hand, CD4s/mlprcg mice were comparable to CD4mlprcg in the severity of these clinical symptoms and to CD4slprcg in serum sCD4 (Table 1), indicating that sCD4 has no influence on the course of lpr disease when CD4+ T cells exist, in accord with the report that high levels of sCD4 produced by the transgenic technique did not influence immune functions [25]. In parallel with the clinical improvement, the glomerular deposition of IC was suppressed to a significant degree, and the circulating IC, immunoglobulin, anti-DNA antibody and ANA levels in the blood were markedly reduced in CD4slprcg mice (Table 3; Figs 3 and 4). Such salutary effects on autoimmunity of the lack of CD4+ T cells have been observed in MRL-lpr mice depleted of these cells by knockout (KO) of the class II or CD4 gene [12,13]. In addition, the development of crescentic glomerulonephritis after injection of anti-mouse glomerular basement membrane globulin was almost completely inhibited in CD4KO mice [26]. Therefore, the present results provide additional evidence for the crucial role of CD4+ T cells in the development of IC-associated renal disease. In accord with this, autoimmune manifestations were improved by anti-CD4 antibody treatment [6–10].
The fundamental abnormality in MRL-lpr and MRL-lprcg mice is the mutation of Fas. However, many lines of evidence have provided evidence that other background genes contribute to autoimmune manifestations in them. In this study, the CD4 mutant gene on chromosome 6 was introduced into MRL-lprcg from C57BR mice by backcrosses. Therefore, it was possible that the genes on this chromosome were unequally distributed among CD4slprcg, CD4mlprcg and CD4s/mlprcg mice and affected autoimmune manifestations. However, this possibility is unlikely, since the several background genes reported to modify autoimmune disease so far are not on chromosome 6 [27–29].
With regard to the role of CD4+ T cells in lymphoid hyperplasia in lpr mice, contradictory results have been reported depending on the methods of their depletion. Anti-CD4 treatment caused a dramatic reduction in splenomegaly and lymphadenopathy by decreasing the accumulation of DN T cells [7,8,10], whereas targeted disruption of the class II gene exhibited no effects on lymphadenopathy [12], and that of the CD4 gene did not affect LN hyperplasia but markedly enhanced splenomegaly and increased the proportion of DN T cells in LN and spleen [13]. In contrast, the loss of CD4+ T cells was associated with significantly reduced hyperplasia and DN T cell content of LN but had no influence on splenomegaly in CD4slprcg mice (Fig. 1; Table 4). The differences in profile of lymphoproliferation and generation of DN T cells between CD4slprcgand CD4KO MRL-lpr mice can not be explained by the functional differences between lprcg and lpr, since both mutant genes were completely the same in the ability to induce lymphoproliferation and abnormal DN T cells on the C3H/HeJ background [30]. Whether or not sCD4 causes this difference remains to be addressed. Several lines of evidence have shown that some DN T cells traverse a CD4 developmental pathway [13,31,32]. Consistent with this, sCD4 in the serum and spleen increased with the expansion of DN T cells (Table 1) and lymphadenopathy was clearly reduced in CD4slprcg mice (Fig. 1).
Serum IgM and IgG levels were comparable between the CD4-mutant and wild-type mice [20]. Although the ability to produce antibody against a T-independent antigen was also similar in both mice, antibody production against T-dependent antigen was greatly impaired in the former. In this study, IgM and IgG levels, and autoantibody levels of IgM and IgG classes and of IgG subclasses, were generally lower in CD4slprcg than in CD4mlprcg mice (Figs 3 and 4). These results indicate that autoreactive CD4+ T cells escaping the Fas-mediated deletion may stimulate the production of immunoglobulin and antibody against T-dependent autoantigens by autoreactive B cells. However, CD4slprcg mice still produced autoantibodies, suggesting that some autoreactive B cells may be T cell-independent and/or that B220− DN T cells may carry residual helper activity [20,33]. DTH reaction in CD4-mutant mice was markedly suppressed compared with CD4KO and normal mice, both being comparable in this respect ([24], unpublished data). This defect was ascribed to the lower interferon-gamma (IFN-γ) production caused by sCD4 (unpublished data). IFN-γ plays a key role in the development of glomerulonephritis in MRL-lpr mice [23]. Serum IFN-γ levels in most CD4slprcg mice were in the same range as those in MRL-+ mice (data not shown). Thus, the suppression of IFN-γ production by sCD4 associated with lower levels of nephritogenic IgG3 autoantibodies may be one of the mechanisms for reduction of renal disease in CD4slprcg mice [34].
In conclusion, our novel SLE mouse model combined with the unique CD4 mutant gene provided additional evidence for the requirement of CD4+ T cells for the full development of autoimmune renal disease as confirmed in a variety of spontaneous and induced models.
Acknowledgments
This study was supported by a Grant-in-Aid from the Ministry of Education, Science, Culture, and Sports, Japan (104700055). We thank T. Matsuzawa for expert secretarial assistance.
REFERENCES
- 1.Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Adv Immunol. 1985;37:269–390. doi: 10.1016/s0065-2776(08)60342-9. [DOI] [PubMed] [Google Scholar]
- 2.Theofilopoulos AN, Koffer R, Singer PA, et al. Molecular genetics of murine lupus model. Adv Immunol. 1989;46:61–109. doi: 10.1016/s0065-2776(08)60651-3. [DOI] [PubMed] [Google Scholar]
- 3.Steinbereg AD, Roths JB, Murphy ED, et al. Effects of thymectomy or androgen administration upon the autoimmune disease of MRL/Mp-lpr/lpr mice. J Immunol. 1980;125:871–3. [PubMed] [Google Scholar]
- 4.Seaman WE, Wofsy D, Greenspan JS, et al. Treatment of autoimmune MRL-lpr mice with monoclonal antibody to Thy-1.2: a single injection has sustained effects on lymphoproliferation and renal disease. J Immunol. 1983;130::1713–8. [PubMed] [Google Scholar]
- 5.Wofsy D, Ledbetter JA, Hendler PL, et al. Treatment of murine lupus with monoclonal anti-T cell antibody. J Immunol. 1985;134::852–7. [PubMed] [Google Scholar]
- 6.Merino R, Iwamoto M, Fossati L, et al. Polyclonal B cell activation arises from different mechanisms in lupus-prone (NZB x NZW) F1 and MRL/MpJ-lpr/lpr mice. J Immunol. 1993;151:6509–16. [PubMed] [Google Scholar]
- 7.Jabs DA, Burek CL, Hu Q, et al. Anti-CD4 monoclonal antibody therapy suppresses autoimmune disease in MRL/Mp-lpr/lpr mice. Cell Immunol. 1992;141:496–507. doi: 10.1016/0008-8749(92)90166-m. [DOI] [PubMed] [Google Scholar]
- 8.Jabs DA, Kuppers RC, Saboori AM, et al. Effects of early and late treatment with anti-CD4 monoclonal antibody on autoimmune disease in MRL/Mp-lpr/lpr mice. Cell Immunol. 1993;154:66–76. doi: 10.1006/cimm.1994.1057. [DOI] [PubMed] [Google Scholar]
- 9.O’Sullivan FX, Ray CJ, Takeda Y, et al. Long-term anti-CD4 treatment of MRL/lpr mice ameliorates immunopathology and lymphoproliferation but fails to suppress rheumatoid factor production. Clin Immunol Immunopathol. 1991;61:421–7. doi: 10.1016/s0090-1229(05)80013-3. [DOI] [PubMed] [Google Scholar]
- 10.Santro TJ, Portanova JP, Kotzin BL. The contribution of L3T4+ T cells to lymphoproliferation and autoantibody production in MRL-lpr/lpr mice. J Exp Med. 1988;167:1713–8. doi: 10.1084/jem.167.5.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henrickson M, Giannini EH, Hirsch R. Reduction of mortality and lymphadenopathy in MRL-lpr/lpr mice treated with nonmitogenic anti-CD3 monoclonal antibody. Arthritis Rheum. 1994;37:587–94. doi: 10.1002/art.1780370422. [DOI] [PubMed] [Google Scholar]
- 12.Jevinkar AM, Grusby MJ, Glimcher LH. Prevention of nephritis in major histocompatibility complex class II-deficient MRL-lpr mice. J Exp Med. 1994;179:1137–43. doi: 10.1084/jem.179.4.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koh DR, Ho A, Rahemtulla A, et al. Murine lupus in MRL/lpr mice lacking CD4 or CD8 T cells. Eur J Immunol. 1995;25:2558–62. doi: 10.1002/eji.1830250923. [DOI] [PubMed] [Google Scholar]
- 14.Watanabe-Fukunaga R, Brannan CI, Copeland NG, et al. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314–7. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- 15.Nagata S, Suda T. Fas and Fas ligand: lpr and gld mutations. Immunol Today. 1995;16(3):9–43. doi: 10.1016/0167-5699(95)80069-7. [DOI] [PubMed] [Google Scholar]
- 16.Matsuzawa A, Moriyama T, Kaneko T, et al. A new allele of the lpr locus, lprcg, that complements the gld gene in induction of lymphadenopathy in the mouse. J Exp Med. 1990;171:519–31. doi: 10.1084/jem.171.2.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fisher GH, Rosenberg FJ, Straus SE, et al. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative disease. Cell. 1995;81:935–46. doi: 10.1016/0092-8674(95)90013-6. [DOI] [PubMed] [Google Scholar]
- 18.Rieux-Laucat F, Le-Deist F, Hivroz C, et al. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995;268:1347–51. doi: 10.1126/science.7539157. [DOI] [PubMed] [Google Scholar]
- 19.Kimura M, Matsuzawa A. Autoimmunity in mice bearing lprcg: a novel mutant gene. Int Rew Immunol. 1994;11:193–210. doi: 10.3109/08830189409061727. [DOI] [PubMed] [Google Scholar]
- 20.Nagase H, Wang CR, Yoshimoto T, et al. Novel mutant mice secreting soluble CD4 without expression of membrane CD4. Eur J Immunol. 1998;28:403–12. doi: 10.1002/(SICI)1521-4141(199802)28:02<403::AID-IMMU403>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 21.Naparstek Y, Ben-Yehuda A, Madaio MP, et al. Binding of anti-DNA antibodies and inhibition of glomerulonephritis in MRL-lpr/lpr mice by heparin. Arthritis Rheum. 1990;33:1554–9. doi: 10.1002/art.1780331013. [DOI] [PubMed] [Google Scholar]
- 22.Kimura M, Ogata Y, Shimada K, et al. Nephritogenicity of the lprcg gene on the MRL background. Immunology. 1992;76:498–504. [PMC free article] [PubMed] [Google Scholar]
- 23.Haas C, Ryffel B, Le Hir M. IFN-γ is essential for the development of autoimmune glomerulonephritis in MRL/lpr mice. J Immunol. 1997;158:5484–91. [PubMed] [Google Scholar]
- 24.Cohen PL, Eisenberg RA. lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Adv Immuol. 1991;9:243–69. doi: 10.1146/annurev.iy.09.040191.001331. [DOI] [PubMed] [Google Scholar]
- 25.Weber S, Traunecker A, Karjalainen K. Constitutive expression of high levels of soluble mouse CD4 in transgenic mice does not interfere with their immune functions. Eur J Immunol. 1993;23:511–6. doi: 10.1002/eji.1830230232. [DOI] [PubMed] [Google Scholar]
- 26.Tipping PG, Huang XR, Qi M, et al. Crescentic glomerulonephritis in CD4- and CD8-deficient mice. Requirement for CD4 but not CD8 cells. Am J Pathol. 1998;152:1541–8. [PMC free article] [PubMed] [Google Scholar]
- 27.Watson ML, Rao JK, Gilkson GS, et al. Genetic analysis of MRL-lpr mice: relationship of the Fas apoptosis gene to disease manifestations and renal disease-modifying loci. J Exp Med. 1992;176:1645–56. doi: 10.1084/jem.176.6.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y, Nose M, Kamoto T, et al. Host modifier genes affect mouse autoimmunity induced by the lpr gene. Am J Pathol. 1997;151:1791–8. [PMC free article] [PubMed] [Google Scholar]
- 29.Gu L, Weinreb A, Wang X-P, et al. Genetic determinants of autoimmune disease and coronary vasculitis in the MRL-lpr/lpr mouse model of systemic lupus erythematosus. J Immunol. 1998;161:6999–7006. [PubMed] [Google Scholar]
- 30.Yasuda T, Zhang Y, Nagase H, et al. Immunological characterization of C3H mice congenic for Faslprcg, C3H/HeJ-Faslprcg/Faslprcg. Lab Anim. 2000;34:46–55. doi: 10.1258/002367700780578019. [DOI] [PubMed] [Google Scholar]
- 31.Laouar Y, Ezine S. In vivo CD4+ lymph node T cells from lpr mice generate CD4−CD8−B220+TCR-β low cells. J Immunol. 1994;153:3948–55. [PubMed] [Google Scholar]
- 32.Ohteki T, lwamoto M, Izui S, et al. Reduced development of CD4−8−B220+ T cells but normal autoantibody production in lpr/lpr mice lacking major histocompatibility complex class I molecules. Eur J Immunol. 1995;25:37–41. doi: 10.1002/eji.1830250108. [DOI] [PubMed] [Google Scholar]
- 33.Rehemtulla A, Kundig TM, Narendran A, et al. Class II major histocompatibility complex-restricted T cell function in CD4-deficient mice. Eur J Immunol. 1994;24:2213–8. doi: 10.1002/eji.1830240942. [DOI] [PubMed] [Google Scholar]
- 34.Reininger L, Berney T, Shibata T, et al. Cryoglobulinemia induced by a murine IgG3 rheumatoid factor: skin vasculitis and glomerulonephritis arise from distinct pathogenic mechanisms. Proc Natl Acad Sci USA. 1990;87:10038–42. doi: 10.1073/pnas.87.24.10038. [DOI] [PMC free article] [PubMed] [Google Scholar]