

Abstract
KSHV K8 gene is activated by virally encoded transactivator RTA in delayed-early stage of viral reactivation. Three RTA-responsive elements (RREs) were identified in the promoter. Among them, RRE-II was found to be the most critical cis-acting element for RTA transactivation. In this report, the mechanism underlying RTA-mediated activation of the K8 delayed-early promoter was investigated. A DNA affinity purification study demonstrated that RRE-II was bound by cellular protein RBP-Jκ, a sequence-specific DNA binding protein and a primary target of the Notch signaling pathway. Inspection of the RRE-II sequence revealed a potential recognition sequence for RBP-Jκ (GTGAGAA) between the nucleotides -102 and –108 relative to the transcription initial site. Removal or mutation of the motif abolished RBP-Jκ binding to the K8 promoter and as a consequence, RTA failed to bind to and activate the promoter. An essential role of RBP-Jκ in the transcription of the K8 promoter was demonstrated by diminishment of the promoter activity in RBP-Jκ-null murine embryonic fibroblasts. Taken together, RTA activates K8 promoter through an indirect binding mechanism, i.e. being recruited to the K8 promoter by interacting with RBP-Jκ bound to an RBP-Jκ motif in the promoter.
Keywords: Kaposi’s sarcoma-associated herpesvirus (KSHV), Human herpesvirus-8 (HHV-8), K8 promoter, Replication and transcription activator (RTA), RBP-Jκ, Gene expression regulation
Introduction
Kaposi’s sarcoma-associated herpesvirus (KSHV), also referred to as human herpesvirus-8 (HHV-8), is a gammaherpesvirus, belonging to Rhadinovirus genus (Chang et al., 1994; Moore et al., 1996). This virus has been proven to be associated with several malignancies including Kaposi’s sarcoma (KS) (Ambroziak et al., 1995), primary effusion lymphoma (PEL) (Cesarman et al., 1995) and multicentric Castleman’s diseases (MCD) (Soulier et al., 1995).
The open reading frame K8 of KSHV encodes a bZip protein, namely K8 protein or replication-associated protein (RAP) or K-bZip (Lin et al., 1999; Zhu et al., 1999). Three functions have been reportedly associated with this protein. (i) K8/RAP was found to bind to the origin of viral lytic DNA replication (ori-Lyt) of KSHV and its binding is absolutely required for the DNA replication (Lin et al., 2003; Wang et al., 2004b). (ii) K8/RAP causes cell cycle arrest at G1 phase through induction of C/EBP α and p21 (Wu et al., 2002). (iii) K8/RAP interacts with virally encoded replication and transcription activator (RTA) and represses the transcription of viral delayed-early genes by RTA (Izumiya et al., 2003; Liao et al., 2003). The former function in ori-Lyt-dependent DNA replication appears to be required for viral lytic replication in the delayed-early stage, while the latter two functions are needed in the immediate-early stage. Therefore, K8/RAP is an important regulatory protein in both immediate-early and delayed-early stages of reactivation of KSHV.
Consistent with the functions of K8/RAP protein in the immediate-early and delayed-early stages, the K8 gene was found to be expressed in both stages during KSHV reactivation. We previously reported that transcription of the K8 gene in these two stages is controlled by two distinct promoters, yielding two transcripts, an immediate-early mRNA of 1.5 kb and a delayed-early mRNA of 1.3 kb. The transcription from the immediate-early promoter was inducible by sodium butyrate or 12-O-tetradecanoylphorbol-13-acetate (TPA), but not responsive to ORF50/RTA. The delayed-early promoter of K8 showed little response to sodium butyrate and TPA, but can be fully induced by ORF50/RTA (Wang et al., 2004a).
The current study focused on the mechanism of transactivation of the K8 delayed-early promoter by RTA. Our previous study revealed three RTA responsive elements (RREs) in the K8 delayed-early (DE) promoter. The first RRE (designated K8 RRE-I) is located between −72 and −61 and contains a functional C/EBP binding site. The second RRE (K8 RRE-II), between −133 and −95, is a novel cis-acting element for K8 gene activation by RTA. The third RRE (K8 RRE-III), between −171 and −161, shares a sequence homology with RRE-II. It was shown that RRE-I had trivial effects in RTA transactivation in BCBL-1, BJAB and BL41 cells, but contributed more to the promoter activation in embryonic kidney fibroblast 239 cells. In contrast, RRE-II was found to be a crucial cis-acting element for RTA transactivation in BCBL-1, BJAB, BL41 and 293 cells because deletion of RRE-II almost completely ablated transcriptional activity in these cells (Wang et al., 2004a). A similar observation of the roles of RRE-I and RRE-II in the activation of the K8 DE promoter in B cells and non-B cells was also reported by Seaman and Quinlivan (Seaman and Quinlivan, 2003).
RTA is the most crucial factor for the switch of KSHV between latency and lytic replication (Reviewed in Dourmishev et al., 2003). It has been shown that ectopic expression of RTA in primary effusion lymphoma cell lines harboring latent KSHV episome activates expression of downstream delayed-early and late viral lytic genes and drives viral lytic cascade to completion (Lukac et al., 1998; Sun et al., 1998). RTA activates viral lytic genes through at least two distinct mechanisms, by direct binding to DNA and by an indirect binding mode. RTA responsive elements (RREs) in PAN, K12 and ori-Lyt-associated promoters were identified, and it was demonstrated that RTA directly bound to these RREs with high affinities. RREs in PAN, K12 and ori-Lyt promoters share a common consensus motif (Chang et al., 2002; Song et al., 2002; Wang et al., 2004b). RTA also acts on RREs in the promoter of K9, but none of these RREs were found to be bound by RTA directly (Ueda et al., 2002). An RRE in the ORF57 promoter has low affinity for direct binding of RTA in an electrophoretic mobility shift assay (EMSA), but encompasses strong binding sites for cellular DNA binding protein RBP-Jκ, which mediates cooperative transactivation (Lukac et al., 2001; Liang et al., 2002). RBP-Jκ was also found to mediate RTA-driven activation of the promoters of ORF6 (SSB), K14 (vGPCR), LANA and RTA itself (Liang et al., 2002; Liang and Ganem, 2003; 2004; Lan et al., 2005). RBP-Jκ is a sequence-specific DNA binding protein and a primary target of the Notch signaling pathway. In the absence of a Notch signal, RBP-Jκ acts as a transcriptional repressor. Ligand-mediated Notch activation leads to the conversion of RBP-Jκ from a repressor to a transcriptional activtor (Reviewed in Mumm and Kopan, 2000; Hayward, 2004). RTA, which appears to mimick a Notch signal, activates some of its target promoters by binding to the repression domain of RBP-Jκ and conferring the promoters an active status (Liang et al., 2002). Besides RBP-Jκ, other cellular factors such as Oct-1, C/EBP α, and K-RBP have also been suggested to play roles in RTA transactivation in certain target promoters (Sakakibara et al., 2001; Wang et al., 2001; 2003).
In this report, we further investigated the activation of K8 gene by RTA in delayed-early phase. We found that RTA activates K8 delayed-early promoter mainly through an indirect binding mechanism, i.e. being recruited to the K8 promoter by interaction with RBP-Jκ which is bound to an RBP-Jκ site in the promoter.
Results
RBP-Jκ binds to K8 RRE-II and recruits RTA to the promoter
Given that the RRE-II has been shown to be the most critical cis-acting element in the K8 DE promoter for RTA-mediated transcriptional activity, we would further characterize it for the mechanism of RTA-mediated transactivation through this RRE. Since the RRE-II shows no sequence homology to the consensus DNA sequence for direct RTA binding that derived from RREs of PAN promoter (Song et al., 2002), K12 promoter (Chang et al., 2002) and ori-Lyt-associated promoter (Wang et al., 2004b), we speculated that RTA acts on the RRE-II through an indirect DNA binding mechanism, whereby RTA is recruited to the RRE-II through interaction with an as yet unidentified factor. Thus, we sought to identify the factor by employing a DNA affinity purification approach. A 132-bp DNA fragment of the K8 promoter derived from the region between −161 – −30 (PK8-wt) and its deletion mutant, in which the RRE-II sequence has been deliberately deleted (PK8-Δ2), were synthesized by PCR using PK8-DE250 plasmid DNA or its RRE-II deletion mutant (PK8-DE250Δ2) as templates (Wang et al., 2004a). One of the PCR primers was labeled with a biotin residue at its 5′ end so that the PCR products were 5′ biotinylated. The PK8-wt DNA fragment contained RRE-I and RRE-II motifs, while the PK8-Δ2 DNA carried RRE-I but no RRE-II sequence. These biotinylated DNAs were coupled to streptavidin-conjugated magnetic beads. The beads were incubated with nuclear extract derived from TPA-induced BCBL-1 cells. The bound materials were washed with 150 mM KCl-containing buffer and eluted with 300 mM, 500 mM and 1 M KCl, sequentially. The eluted materials were analyzed on Western blots with antibodies against some cellular DNA binding proteins including RBP-Jκ, C/EBP α, C/EBP β, Sp-1 and NF-κB. The result revealed that RBP-Jκ was bound to wild-type K8 promoter DNA and eluted at the 500 mM KCl step elution (Fig. 1). In contrast, RBP-Jκ was not able to bind to the mutated promoter where RRE-II sequence had been deleted (PK8-Δ2). Binding of RBP-Jκ in RRE-II of the K8 promoter raised a possibility that RTA activates K8 DE promoter by targeting RRE-II through interaction with an RBP-Jκ molecule. The Western blotting with antibodies against C/EBP α and other cellular proteins failed to detect their antigens in the eluted materials (data not shown).
Fig. 1.
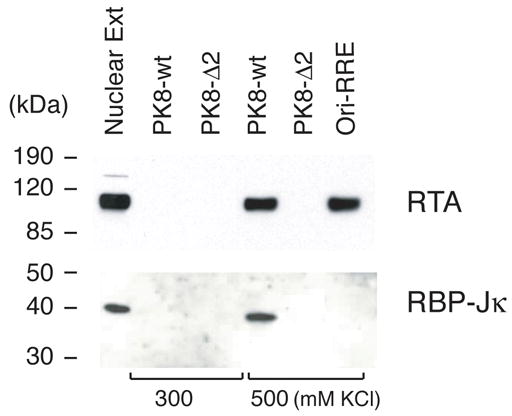
Identification of K8 promoter RRE-II binding proteins by using DNA affinity purification. TPA-treated BCBL-1 nuclear extracts were subjected to DNA affinity purification with immobilized DNA fragments of the K8 delayed-early promoter (PK8-wt), the RRE-II deletion mutant of the promoter (PK8-Δ2), and the ori-Lyt-associated promoter sequence (ori-RRE). After washing with 150 mM KCl-containing buffer, the affinity-purified proteins were eluted with 300 mM KCl followed by 500 mM KCl. 5% of the eluted materials as well as 0.5% of the input material were resolved in a SDS-PAGE and analyzed by Western blotting with various specific antibodies.
The DNA affinity purified materials were then analyzed for RTA using a specific antibody against the protein. Western blot showed that RTA bound to the wt DNA and eluted with 500 mM KCl buffer, but did not bind to the Δ2 DNA. It appears that RTA failed to bind to the promoter when RBP-Jκ was not on the DNA. Taken together, our data suggest that cellular protein RBP-Jκ binds to RRE-II DNA and recruits RTA to the RRE-II in the K8 promoter by interacting with it. As a control, ori-Lyt-associated promoter was not bound by RBP-Jκ, but RTA can bind to it directly (Fig. 1).
To our surprise, our DNA affinity study did not detect RTA in the material eluted from the Δ2 DNA with 300 and 500 mM KCl (Fig. 1). RTA was also not found in the 1M KCl eluate as well as on the DNA-conjugated beads after elution (data not shown). This result suggests that RTA apparently only binds to RRE-II of the K8 promoter, but not to RRE-I in the condition used in this study. In addition, C/EBP α was not detected in this DNA affinity study with either the wild-type or Δ2 DNA although RRE-I is present in both DNAs (data not shown).
RBP-Jκ recognition sequence is essential for RRE-II-mediated transactivation of the promoter by RTA
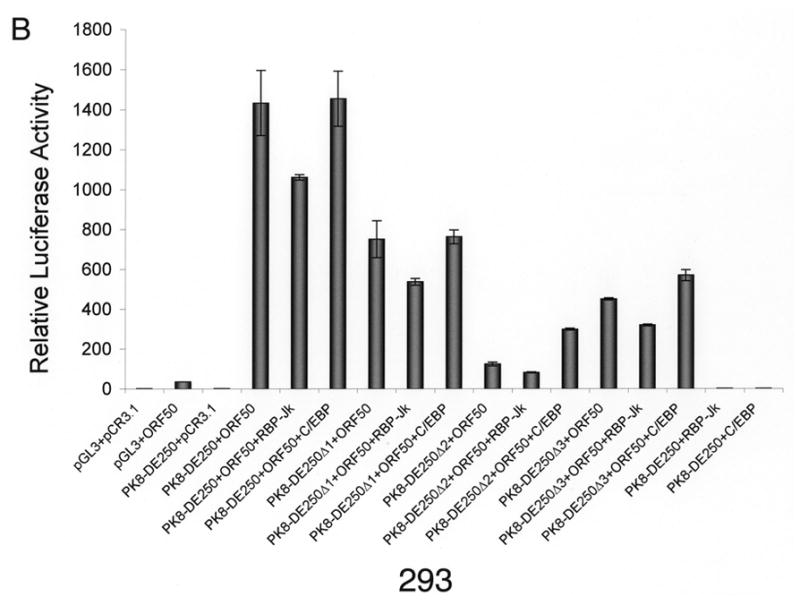
Inspection of the DNA sequence within RRE-II revealed a potential recognition sequence for RBP-Jκ (GTGAGAA) between the nucleotides -102 and -108 relative to the transcription initial site. This motif is not a prototypic RBP-Jκ consensus sequence, but Liang and Ganem (2004) recently showed that RBP-Jκ recognizes it in the K14/vGPCR promoter. To confirm the role of the RBP-Jκ recognition site in RTA-mediated transactivation of the K8 promoter, we made a mutation in the K8 promoter, which altered the RBP-Jκ recognition motif GTGAGAA to a Bam HI recognition site (Fig. 2A). This mutation was introduced into a K8 promoter-reporter construct PK8-DE250 and the effect of this RBP-Jκ motif mutation on RTA-mediated transactivation of the promoter was analyzed using a luciferase assay. Reporter constructs of wild-type K8 promoter (PK8DE-250), RBP-Jκ site mutant (PK8DE-250mJκ) as well as RRE-I, RRE-II and RRE-III deletion mutants (PK8DE-250Δ1, Δ2 and Δ3) were introduced into BL41 and 293 cells together with an RTA-expressing vector (pCR3.1-ORF50) or empty pCR3.1 vector. The result showed that (i) in agreement with previous data, deletion of RRE-II caused a dramatic reduction of RTA-mediated K8 promoter transactivation by 94% in 293 cells and over 99% in BL41 cells; (ii) mutation of RBP-Jκ recognition site brought about similar effects (95% reduction in 293 cells and 99% in BL41 cells) as seen with RRE-II deletion mutant (Fig. 2B and C). This result indicates that the central theme of RRE-II is the RBP-Jκ recognition motif which plays a critical role in the K8 promoter for RTA-mediated transactivation.
Fig. 2.

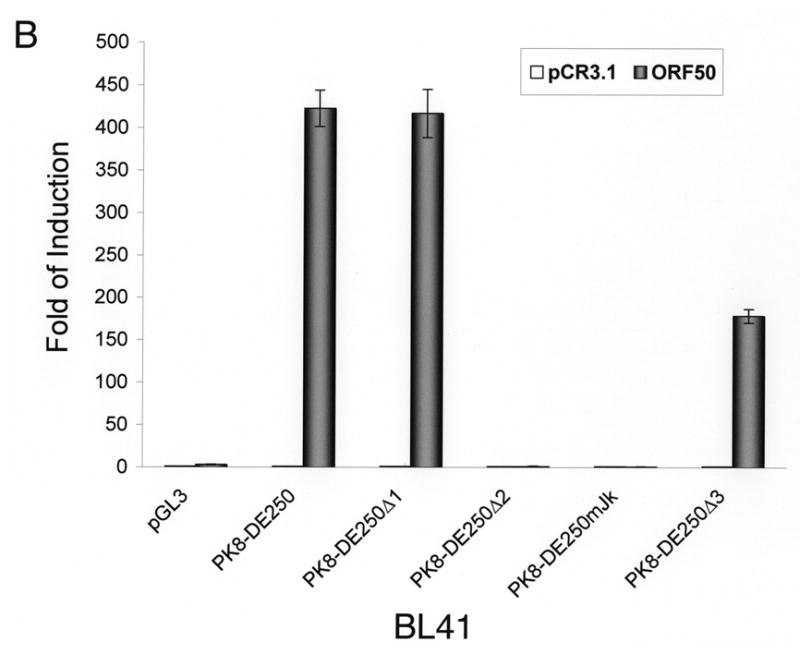
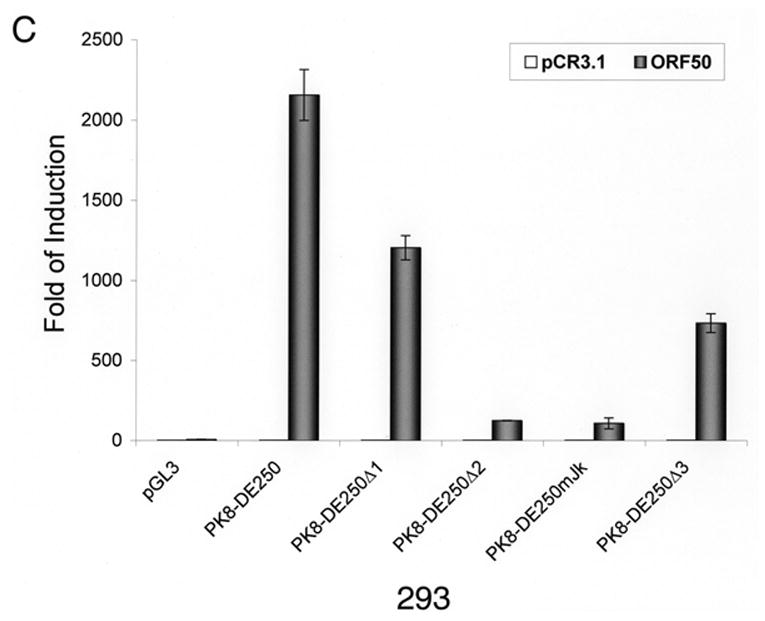
Mutational analysis of RBP-Jκ binding motif in the K8 DE promoter. The K8 DE promoter and its derivatives including deletion mutants of each RRE and substitutive mutant of RBP-Jκ are illustrated schematically in panel A. The potential RBP-Jκ motif is marked with a box and three RREs are underlined. BL41 (B) and 293 cells (C) were cotransfected with a K8 promoter-luciferase reporter or its mutants shown in panel A and pCR3.1-ORF50 or pCR3.1 empty plasmid. Luciferase activities were analyzed by the dual-luciferase reporter assay. Relative luciferase activities were measured as fold activity induced by RTA relative to the basal level of luciferase activity in cells that are cotransfected with blank plasmid pCR3.1, with the error bars representing standard deviations of the results from two independent experiments.
RRE-II/RBP-Jκ-mediated RTA transactivation contributes most to transcription of K8 delayed-early gene
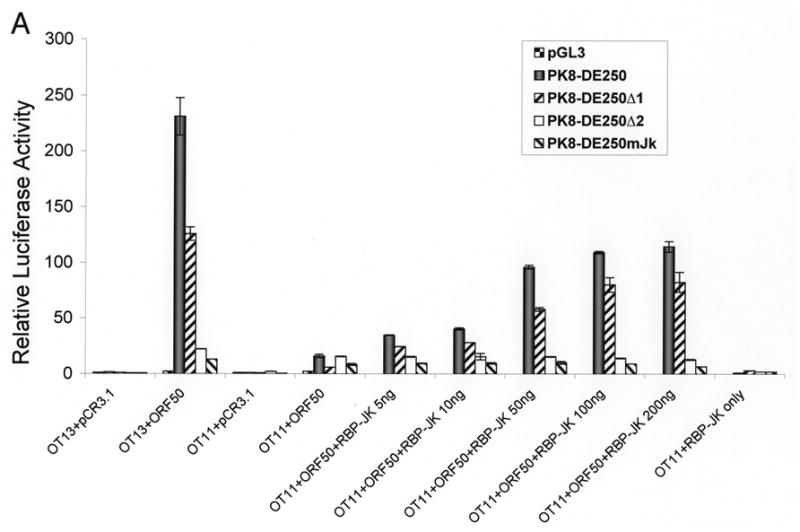
Our data demonstrated that the RBP-Jκ motif resided in RRE-II is a crucial cis-acting element for RTA activation of K8 DE promoter. However, RRE-I was also reported to contribute to the K8 promoter activation by RTA and shown to be directly bound by purified RTA in vitro (Lukac et al., 2001). Furthermore, a functional C/EBP α binding site (designated C/EBP-II) was found within the RRE-I and ectopic expression of C/EBP α was able to enhance RTA transactivation of the K8 promoter in a transient reporter gene assay as well as K8 gene expression in PEL cells (Wang et al., 2003). These data suggest that the regulation of the K8 promoter might involve multiple mechanisms, including direct DNA binding mechanism and indirect mechanisms with RBP-Jκ and C/EBP α, respectively. However, in the DNA affinity binding assay, we failed to detect the direct RTA binding and C/EBP α-mediated indirect binding of RTA to RRE-I (Fig. 1). One possible explanation for this result is that the affinities of RTA directly binding to RRE-I and C/EBP α binding to the RRE were low; the concentrations of RTA and C/EBP α in the BCBL-1 nuclear extract were lower than the binding constants (Kd’s) for RTA-RRE-I and C/EBP α-RRE-I interactions, so that RTA binding to RRE-I, either in a direct mode or through C/EBP α, was not detected in the condition used in our DNA affinity assay. To clarify this issue and assess the contribution of RRE-I and RRE-II to the activation of the K8 promoter by RTA, we examined these two RREs that allows comparison of their contributions to RTA-mediated activation of the promoter in parallel. BL-41 and 293 cells were transfected with RBP-Jκ and C/EBP α expression vectors, respectively, as well as the combination of RBP-Jκ or C/EBP α with RTA. The effects of these cellular proteins on RTA-mediated K8 promoter activation were examined using K8 promoter reporters that were cotransfected into these cells. First, ectopic expression of RBP-Jκ and C/EBP α alone (in the absence of RTA) did not activate the K8 DE promoter in either BL41 or 293 cells (Fig. 3). Second, cotransfection with RBP-Jκ and RTA brought about small increases in the transcription of the K8 promoter-reporter in both cell lines. This suggests that the physiological concentration of RBP-Jκ was saturated for RRE-II occupation and the ectopic expression of the protein did not have much effect on the promoter activity (Fig. 3). Third, the contribution of endogenous C/EBP α to the K8 promoter activity and the effects of ectopic expression of C/EBP α on the promoter were found to vary between BL41 and 293 cells.
Fig 3.
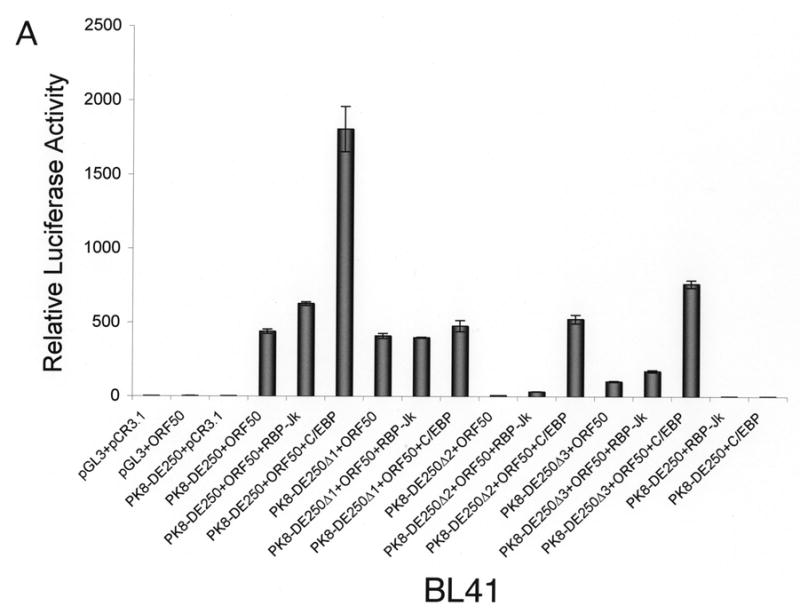
Effects of ectopic expression of RBP-Jκ and C/EBP α on transcription activity of the K8 DE promoter in BL41 and 293 cells. A K8 promoter-luciferase reporter and its RRE deletion mutants were introduced into BL41 and 293 cells with RBP-Jκ or C/EBP α expression vector alone or together with RTA expression vector (pCR3.1-ORF50) or pCR3.1 empty plasmid. Luciferase activities were analyzed by the dual-luciferase reporter assay. Relative luciferase activities were measured as fold activity relative to the basal level of luciferase activity in cells that are cotransfected with blank plasmids pGL3 and pCR3.1, with the error bars representing standard deviations of the results from two independent experiments.
Introduction of C/EBP α and RTA expression vectors to BL41 cells led to a substantial increase (four-fold) in transcription of the K8 promoter reporter in comparison to transfection with RTA alone. The effects of C/EBP α ectopic expression on RTA-mediated transactivation were greater with the RRE-II and RRE-III deletion mutants of the K8 promoter reporter, 20-fold with PK8-DE250Δ2 and 10-fold with PK8-DE250Δ3, respectively. The effect diminished in the RRE-I deletion of the reporter ((PK8-DE250Δ1), confirming that the C/EBP α effect on the promoter was through interaction with RRE-I that contains a functional C/EBP site (Wang et al., 2003). This result can be explained as follows. The physiological concentration of C/EBP α in BL41 cells is perhaps much lower than the dissociation constant (Kd) of the protein binding to RRE-I and therefore the contribution of C/EBP α and RRE-I to the promoter activation is insignificant in this type of cells. However, ectopic expression of C/EBP α elevated the level of the protein in the cells so that C/EBP α began to bind to RRE-I and recruited RTA to the RRE for transactivation.
In contrast, ectopic expression of C/EBP α in 293 cells brought about little change in transactivation of the K8 promoter by RTA. Although ectopic expression of C/EBP α is found to enhance the RTA-mediated transactivation of the promoter where RRE-II has been deleted (PK8-DE250Δ2), the C/EBP α effect was very limited (twofold). These data suggest that endogenous C/EBP α has probably reached a saturated concentration in 293 cells and is able to activate the promoter by binding to RRE-I and recruiting RTA to the promoter. It is worthy to note that although the physiological concentrations of both RBP-Jκ and C/EBP α in 293 cells are high enough for them to bind to their motifs, the contribution of RBP-Jκ to the K8 promoter activity is obviously much greater than that of C/EBP α because deletion of RRE-II or mutation of RBP-Jκ resulted in loss of 94–95% of total promoter activity in contrast to the removal of RRE-I, which only costs less than 50% of the activity in 293 cells (Fig. 2C and 3B).
Inability of transactivation of the K8 promoter in RBP-Jκ-null cells and restoration of the promoter activity by ectopic expression of RBP-Jκ
To further analyze the role of RBP-Jκ in activation of the K8 promoter by RTA, we obtained a mouse embryonic fibroblast (MEF) cell line derived from an RBP-Jκ −/− mouse (Oka et al., 1995). This RBP-Jκ-null line (OT11) and its wild-type counterpart OT13 were used to analyze activation of the K8 promoter and its deletion mutants by RTA. Reporter constructs of the K8 promoter as well as its mutant derivatives were introduced into OT11 and OT13 cells along with RTA-expression vector pCR3.1-ORF50 or empty pCR3.1 plasmid. As shown in Fig. 4, RTA was able to fully activate the K8 promoter (PK8-DE250) in wild-type OT13 cells. In contrast, cotransfection of RBP-Jk −/− MEF OT11 cells with K8 reporter construct and RTA-expression vector resulted in very low luciferase activity, indicating that RTA itself was not able to efficiently activate the K8 promoter in the absence of RBP-Jκ. Co-expression of RTA and increasing amounts of RBP-Jκ in OT11 cells resulted in progressively higher levels of K8 promoter activities in the wild-type promoter (pK8-DE250) and its RRE-I deletion mutant (pK8-DE250Δ1) (Fig. 4A). However, ectopic expression of RBP-Jκ alone did not activate the K8 promoter at all (Fig. 4A). The RBP-Jκ-dependent transactivation of the K8 promoter by RTA was greatly compromised when RRE-II-deleted (PK8-DE250Δ2) or RBP-Jκ motif-mutated reporter constructs (pK8-DE250mJκ) were used in the assay in RBP-Jκ-null cells, confirming the role of RBP-Jκ motif in RRE-II in the K8 promoter activity (Fig. 4A). The expression of RBP-Jκ in the transfected cells were monitored by Western blot with an antibody specific to RBP-Jκ (Fig. 4B). We noticed that although ectopic expression of RBP-Jκ in OT11 cells partially restored RTA-mediated transcriptional activity of the K8 promoter, the levels of the promoter activity were lower than that in wild-type OT13 cells. We think that it may be caused by a compromised RBP-Jκ activity (either in DNA binding or in interaction with RTA) associated with the attachment of a myc tag to the protein or it may just represent a difference between two cell lines (OT11 vs. OT13). Nevertheless, the inability of transactivation of the K8 promoter by RTA in RBP-Jκ-null cells and restoration of the transcriptional activity of the promoter by ectopic expression of RBP-Jκ conclusively demonstrated an essential role of RBP-Jκ in transactivation of the K8 delayed-early promoter.
Fig. 4.
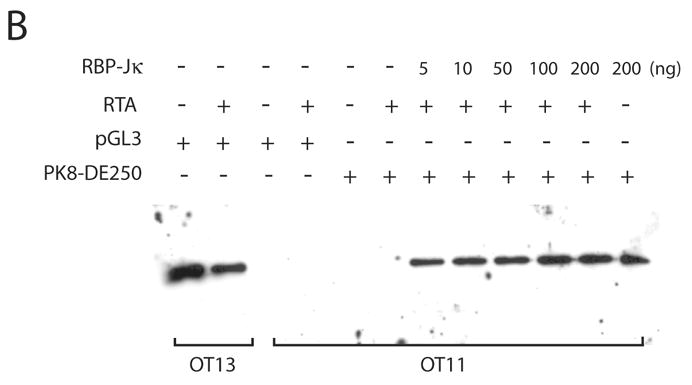
Contribution of RBP-Jκ to transactivation of the K8 DE promoter. Wild-type (OT13) and RBP-Jκ-null (OT11) mouse embryonic fibroblast cell lines were cotransfected with 100 ng of K8 promoter-luciferase reporters and 100 ng RTA expression vector (pCR3.1-ORF50 or empty pCR3.1) or 100 ng of RBP-Jκ expression vector or combination of pCR3.1-ORF50 and increasing amounts (0–200 ng) of RBP-Jκ expression vector. (A) Luciferase activities were analyzed by the dual-luciferase reporter assay. Relative luciferase activities were measured as fold activity relative to the basal level of luciferase activity in cells that are cotransfected with blank plasmids, with the error bars representing standard deviations of the results from two independent experiments. (B) Western blotting showing the levels of endogenous RBP-Jκ in OT13 cells, the absence of endogenous RBP-Jκ in OT11 cells and ectopic expression of RBP-Jκ in OT11 cells.
Finally, although it was firmly shown that RBP-Jκ is absolutely required for activation of the K8 and many other promoters by RTA, it has not been demonstrated whether RTA cannot bind to RRE-II in the K8 promoter but is recruited to the promoter through interaction with RBP-Jκ molecules. By taking advantage of RBP-Jκ-null MEF cells (OT11), we studied interaction of RTA with the K8 promoter DNA in the absence and the presence of RBP-Jκ molecules using a DNA affinity purification approach. The K8 promoter DNA (132-bp) immobilized to magnetic beads was incubated with nuclear extracts from OT11 and OT13 cells. After washing, bound proteins were eluted with 500 mM KCl and analyzed by Western blotting with anti-RBP-Jκ and anti-RTA antibodies. RTA was found to be able to bind the DNA with OT13 nuclear extract, but not able to bind with OT11 nuclear extract, confirming that RTA does not bind to K8 promoter DNA in the absence of RBP-Jκ (Fig. 5). When the nuclear extracts from OT11 cells that had been transfected with RTA- and RBP-Jκ-expression vectors are used in the assay, RTA was found to be able to bind the DNA with RBP-Jκ. RBP-Jκ binds to the K8 promoter DNA regardless the presence of RTA molecules (Fig. 5). Taken together, our results clearly demonstrated that binding of RTA to the K8 promoter through RRE-II and the activation of the promoter is dependent on the presence of RBP-Jκ. RBP-Jk plays an essential role in activate the K8 DE promoter.
Fig. 5.
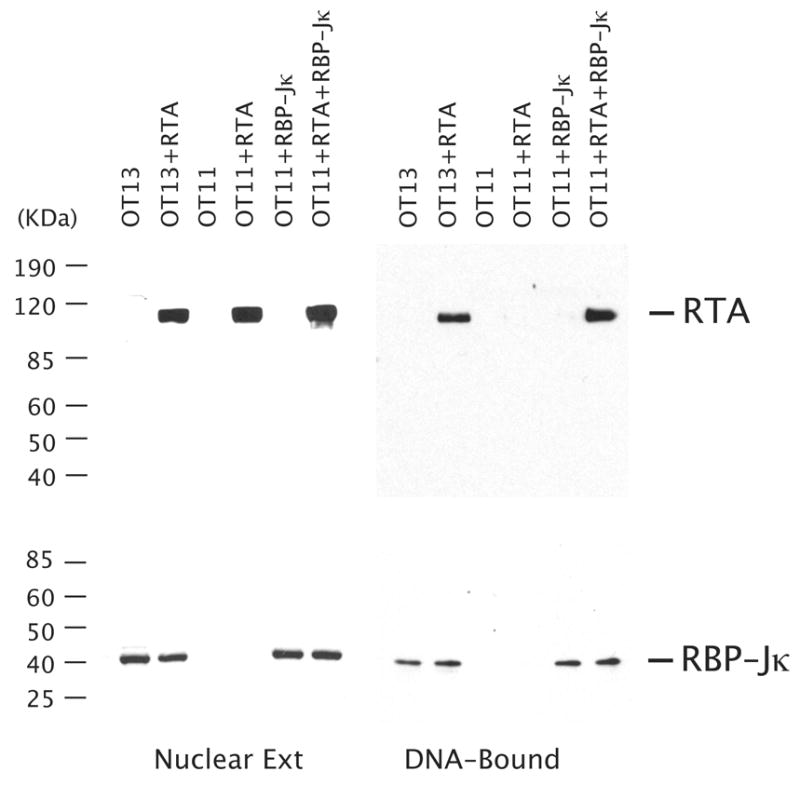
Binding of RTA to the K8 promoter is dependent on the presence of RBP-Jκ. RBP-Jκ-null MEF cells (OT11) were transfected with RTA-, RBP-Jk-expression vectors or both. The wild type MEF cells (OT13) were also transfected with RTA-expression vector (pCR3.1-ORF50) or empty vector (pCR3.1) as controls. The nuclear extracts of the transfected cells were subjected to DNA affinity purification with immobilized DNA fragments of the K8 delayed-early promoter (PK8-wt). After washing with 150 mM KCl-containing buffer, the affinity-purified proteins were eluted with 500 mM KCl. 10% of the eluted materials as well as 1% of the input material were resolved in a SDS-PAGE and analyzed by Western blotting with antibodies against RTA and RBP-Jκ.
Discussion
In this study, we attempted to reveal the mechanisms underlying activation of the K8 gene during the reactivation of KSHV. The K8 delayed-early promoter is known to be activated by the transcription activator RTA (Lukac et al., 2001; Seaman and Quinlivan, 2003; Wang et al., 2004a). In this report, we demonstrated that RTA is recruited to one of its responsive elements, namely RRE-II, in the K8 promoter through interaction with RBP-Jκ, a Notch signal transcription factor, and activates the promoter. The RBP-Jκ-mediated RTA transactivation is the most crucial regulation in the activation of the gene. The following observations are relevant to these conclusions. (i) RBP-Jκ was found to bind the K8 DE promoter at the RRE-II in a DNA affinity assay. (ii) A mutagenesis study showed that the recruitment of RTA to the promoter was dependent on the binding of RBP-Jκ on the promoter at the RRE-II site. (iii) Transient transactivation studies in RBP-Jκ-null mouse MEF (OT11) cells showed that the K8 DE promoter can not be activated by RTA in the absence of RBP-Jκ, while the promoter can be activated by nearly 250 fold in the syngeneic control cells (OT13). These data undeniably demonstrated that RBP-Jκ is essential for activation of the K8 DE promoter by RTA.
Accumulating evidence indicates that RTA activates its target genes through at least two modes of action: direct DNA binding and indirect mechanisms. RTA was shown to bind to the RREs in the PAN, K2, K12, ori-Lyt promoters (Chang et al., 2002; Deng et al., 2002; Song et al., 2002; Wang et al., 2004b), but more RREs in other promoters are not directly bound by RTA (Liang et al., 2002; Liang and Ganem, 2004; Sakakibara et al., 2001; Ueda et al., 2002; Wang et al., 2003). In latter cases, cellular factors were found to be required for RTA transactivation function. The cellular factors that are reportedly involved in RTA-mediated transactivation include Oct-1, C/EBP α, K-RBP and RBP-Jκ (Liang et al., 2002; Sakakibara et al., 2001; Ueda et al., 2002; Wang et al., 2001; Wang et al., 2003). Among them, RBP-Jκ has been reported to be involved in RTA-mediated transcription of ORF57 (Mta), ORF6 (SSB), K14 (vGPCR), LANA and ORF50/RTA itself (auto-activation) (Liang et al., 2002; Liang and Ganem, 2004; Lan et al., 2005). In addition, RTA also induces cellular CD21 and CD23a gene expression through interaction with RBP-Jκ molecule on RBP-Jκ binding sites in the first intron of CD21 and in the CD23a core promoter, respectively (Chang et al., 2005). In this report, the K8 delayed-early promoter was found to be activated by RTA through interaction with RBP-Jκ. This finding, together with previous reports regarding involvement of RBP-Jκ in RTA-mediated activation of many other viral lytic promoters, suggest that interaction with RBP-Jκ is a common mechanism by which RTA activates lytic genes during reactivation. A central role of RBP-Jκ in KSHV reactivation has been demonstrated by the inability of KSHV reactivation in the RBP-Jκ-null murine embryonic fibroblast cell line. The results showed that the reactivation of lytic gene expression, viral DNA replication, and the release of progeny viruses were dramatically inhibited in the absence of RBP-Jκ (Liang and Ganem, 2003). The RBP-Jκ-RTA interaction is critical for lytic viral replication.
The RRE-I-mediated activation of K8 promoter by RTA has been investigated previously. This element was shown to be a direct binding site for purified RTA (Lukac et al., 2001). The element also contains a functional binding site for C/EBP α and a cooperative transactivation of the K8 promoter by RTA and C/EBP α has been demonstrated (Wang et al., 2003). Although we did not detect direct RTA binding or C/EBP α binding to the promoter DNA in the DNA affinity assay (despite that RRE-I was present in the biotinylated promoter DNAs, i.e. wt and Δ2), it does not necessarily suggest any discrepancy between our present data and the results previously published. (i) We were able to demonstrate the contribution of RRE-I to the activation of the promoter by RTA in our transient transactivation assay. Deletion of RRE-I from the K8 promoter resulted in a reduction in RTA transactivation by nearly 50% in 293 cells, which is consistent with previous publication (Lukac et al., 2001; Seaman and Quinlivan, 2003; Wang et al., 2004a). However, the contribution of RRE-I to the promoter in B cells (BCBL-1, BJAB and BL41 cells) appeared to be less significant. (ii) One explanation for the failure to detect the association of C/EBP α with the DNA probe of the K8 promoter might be the concentration of C/EBP α in the nuclear extract as well as in B cells is probably much lower than the dissociation constant (Kd) of the protein to RRE-I DNA. This notion is supported by the fact that C/EBP α-mediated RTA activation through RRE-I was dramatically increased with ectopic expression of C/EBP α in B cells (Fig. 3A). Thus, although the functional consequence of the weak RTA binding to RRE-I can be detected by luciferase reporter assay, the binding was too weak to be detected in our in vitro experimental condition with the nuclear extract from TPA-induced BCBL-1 cells. (iii) It is conceivable that binding affinity of RTA to RRE-I DNA either by a direct DNA binding or through interaction with a C/EBP α molecule is much lower than that of RTA to RRE-II through interaction with RBP-Jκ, as RRE-II displays a greater effect than RRE-I on the gene activation by RTA based on the results of the transient promoter activation assay. (iv) Finally, our observation may suggest a synergy in RTA binding to all RREs. It is possible that association of RTA with RRE-I could be facilitated if RTA has bound to RRE-II through RBP-Jκ. Deletion of RRE-II may not only abolish the association of RTA/RBP-Jκ to this element, but also weaken the binding of RTA to RRE-I. Although RRE-II is the most crucial cis-acting element, binding of RTA to RRE-I (as well as RRE-III) may synergistically enhance transcription of the K8 promoter. Transcriptional synergy among cis-acting elements and transactivitors has been demonstrated in EBV ZTA transactivation system (Chi et al., 1995). Therefore, our finding on the major role of RRE-II in K8 promoter activation and the involvement of RBP-Jκ in the recruitment of RTA to the promoter present no discrepancy, but an addition to our knowledge about RTA activation of the K8 delayed-early promoter.
Based on the data and observations from the current and previous studies, a model for K8 DE promoter activation by RTA can be established as follows. (i) RTA activates the K8 DE promoter mainly through RRE-II by interaction with RBP-Jκ protein. (ii) RTA also binds weakly to RRE-I either by direct DNA binding or through interaction with C/EBP α. However, the RTA bound to RRE-I may be synergized with the RTA/RBP-Jκ on RRE-II or vice versa to provide the maximal transcription efficiency of the K8 promoter. (iii) Although RRE-II is the most crucial cis-acting element for RTA-mediated transactivation of the K8 promoter, the contribution of RRE-I may vary due to differential availability of certain cellular factors (such as C/EBP α) in different types of cell, providing opportunities that allow K8 gene being efficiently regulated in different cell environments. Considering that KSHV naturally persists in B cells and endothelial cells, the differential regulation of a viral gene such as K8 in different cell types would be of biological significance.
Materials and Methods
Cell culture
BCBL-1 (Renne et al., 1996) cells were obtained from the NIH AIDS Research and Reference Reagent Program and grown in RPMI 1640 medium supplemented with 10% fetal bovine serum. BL-41 (an EBV-negative, KSHV-negative Burkitt’s lymphoma cell line) cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum. Human embryonic kidney fibroblast 293 cells were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum. Mouse RBP-Jκ−/− (OT11) and WT (OT13) embryonic fibroblast cell lines (Oka et al., 1995) were kindly provided by T. Honjo (Kyoto University, Kyoto, Japan) and were grown in high-glucose DMEM supplemented with 10% fetal bovine serum and 100 U/ml of mouse interferon gamma (PBL Biomedical Laboratories, Piscataway, NJ).
Reporter plasmids and expression vectors
Reporter plasmids (PK8-DE250, PK8-DE250Δ1, PK8-DE250Δ2 and PK8-DE250Δ3) were constructed and described previously (Wang et al., 2004a). PK8-DE250mJκ was generated using a PCR-based site-directed mutagenesis system, namely QuickChange Multi (Stratagene). In brief, a pair of overlapping oligonucleotides K8-RRE2 quick-1 (5′-TCT ACT TAA AAT AGC TCA TTT GGA TCC GAA TCT GGT TGA TTG TGA CTA-3′) and K8-RRE2 quick-2 (TAG TCA CAA TCA ACC AGA TTC GGA TCC AAA TGA GCT ATT TTA AGT AGA-3′), that contain desired mutations toward opposite directions, were used in a high-fidelity PCR with PK8-DE250 as a template. After PCR, the parental DNA template was removed by complete digestion with restriction enzyme Dpn I, which did not degrade PCR-sythesized DNA. The PCR product was used to transform E. coli competent cells. The mutation was verified by DNA sequencing.
RTA expression vector (PCR3.1-ORF50) was constructed and described previously (Wang et al., 2004a). RBP-Jκ expression vector (pA3M-RBP-Jκ-myc) was provided by Erle Robertson (University of Pennsylvania) and C/EBP α expression vector (pSG5-Flag-C/EBP α) by Gary Hayward (Johns Hopkins University).
DNA affinity purification and assay
Biotinylated DNA fragments of the K8 DE promoter (PK8-wt and PK8-Δ2) were synthesized by PCR using PK8-DE250 plasmid DNA or its RRE-II deletion mutant (PK8-DE250Δ2) as templates and two oligonucleotides as primers. The oligonucleotides were K8-8 (5′-CAG TTT GGT GCA AAG TGG AGT TAA CC-3′) and K8-DE-Biotin (5′-biotin-GTC GAC AAC GGA GGA AAT ACC A-3′). The resultant biotinylated PCR fragments were coupled to streptavidin-conjugated magnetic beads (Dynal, Oslo, Norway) and then incubated with nuclear extracts prepared from tetradecanoyl phorbol acetate (TPA)-induced BCBL-1 cells or transfected mouse embryonic fibroblast OT11 and OT13 cells for 2 hours at 25°C. The bound materials were washed four times in D150 buffer (20 mM HEPES [pH 7.9], 20% glycerol, 0.2 mM EDTA, 150 mM KCl, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 0.05% NP-40) and then progressively eluted with D300 (same as above, except 300 mM KCl), D500 (500 mM KCl), and D1000 (1 M KCl). The affinity-purified materials were assayed by Western blot analysis with various antibodies.
Antibodies and Western Blotting
Rabbit polyclonal antibody to ORF50/RTA was provided by David Lukac (UMDNJ). Polyclonal antibody against RBP-Jκ was provided by Ke Lan and Erle Robertson (University of Pennsylvania). Polyclonal antibodies against C/EBP α, C/EBP β and NF-κB (p65) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Polyclonal anti-Sp1 antibody was purchased from UPSTATE (Lake Placid, NY).
Cell extracts or DNA affinity eluates were resolved on 4–20% SDS-PAGE and transferred to Hybond-ECL nitrocellulose membrane (Amersham). The membranes were blocked in 5% dry milk in PBST buffer and then incubated with diluted primary antibodies at room temperature for 2 hours or 4°C overnight. Anti-rabbit immunoglobulin G antibody conjugated to horseradish peroxidase (Amersham) was used as the secondary antibody. The ECL chemiluminescence system (Amersham) was used for detection.
DNA transfection and luciferase assays
To transfect B cells, 10 μg luciferase reporter construct, 1 μg pRL-TK plasmid and 3 μg pCR3.1-ORF50 (or pCR3.1 vector) were mixed with 107 BL-41 or BJAB cells in OPTI-MEM medium (GIBCO-BRL) and electroporated (250V, 960 μF) with a Genepulser II (Bio-Rad, Hercules, CA). Electroporated cells were then transferred to RPMI 1640 medium supplemented with 10% serum and grown for 48 hours. The pRL-TK plasmid was included as an internal control, which constitutively expresses Renilla luciferase.
To transfect human embryonic kidney 293 and mouse embryonic fibroblast cells, subconfluent cells grown in 24-well plates were transfected with 100 ng of pGL-3 reporter, 10 ng pRL-TK reporter and 100 ng pCR3.1-ORF50 using Qiagen Effectene transfection kit. Cells were harvested at 48 hours post-transfection for luciferase assay.
The dual luciferase reporter assay system (Promega) was used to examine the responsiveness of the promoters to RTA. Transfected cells were washed once with 1xPBS and suspended in 400 μl of 1 x passive lysis buffer. Cells were frozen-thawed once and centrifuged in mirocentrifuge for 1 min. Supernatants were assayed for firefly luciferase and Renilla luciferase activities using a TD-20/20 luminometer with dual auto injector (Turner Designs). The luciferase assays were carried out according to the manufacture’s instruction (Promega).
Acknowledgments
We thank Dr. Tasuku Honjo (Kyoto University) for RBP-Jκ (−/−) mouse embryonic fibroblast cell line. We thank Drs. Ke Lan and Erle Robertson (University of Pennsylvania) for RBP-Jκ expression vector (pA3M-RBP-Jκ-myc) and polyclonal antibody to RBP-Jκ, Dr. Gary Hayward (Johns Hopkins University) for C/EBP α expression vector (pSG5-Flag-C/EBP α) and Dr. David Lukac for RTA polyclonal antibody. We thank all members of Yuan Lab for constructive discussion, suggestion and critical reading of the manuscript. This work was supported by research grants from the National Institutes of Health (R01AI52789 and R01CA86839).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ambroziak JA, Blackbourn DJ, Herndier BG, Glogau RG, Gullett JH, McDonald AR, Lennette ET, Levy JA. Herpes-like sequences in HIV-infected and uninfected Kaposi’s sarcoma patients. Science. 1995;268:582–583. doi: 10.1126/science.7725108. [DOI] [PubMed] [Google Scholar]
- Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med. 1995;332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- Chang H, Gwack Y, Kingston D, Souvlis J, Liang X, Means RE, Cesarman E, Hutt-Fletcher L, Jung JU. Activation of CD21 and CD23 gene expression by Kaposi’s sarcoma-associated herpesvirus RTA. J Virol. 2005;79:4651–4663. doi: 10.1128/JVI.79.8.4651-4663.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang P, Shedd JD, Gradoville L, Cho MS, Chen LW, Chang J, Miller G. Open reading frame 50 protein of Kaposi’s sarcoma-associated herpesvirus directly activates the viral PAN and K12 genes by binding to related response elements. J Virol. 2002;76:3168–3178. doi: 10.1128/JVI.76.7.3168-3178.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994;266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- Chi T, Lieberman P, Ellwood K, Carey M. A general mechanism for transcriptional synergy by eukaryotic activators. Nature. 1995;377:254–257. doi: 10.1038/377254a0. [DOI] [PubMed] [Google Scholar]
- Deng H, Song MJ, Chu JT, Sun R. Transcriptional Regulation of the Interleukin-6 Gene of Human Herpesvirus 8 (Kaposi’s Sarcoma-Associated Herpesvirus) J Virol. 2002;76:8252–8264. doi: 10.1128/JVI.76.16.8252-8264.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dourmishev LA, Dourmishev AL, Palmeri D, Schwartz RA, Lukac DM. Molecular genetics of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus-8) epidemiology and pathogenesis. Microbiol Mol Biol Rev. 2003;67:175–212. doi: 10.1128/MMBR.67.2.175-212.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward SD. Viral interactions with the Notch pathway. Semin Cancer Biol. 2004;14:387–396. doi: 10.1016/j.semcancer.2004.04.018. [DOI] [PubMed] [Google Scholar]
- Izumiya Y, Lin SF, Ellison T, Chen LY, Izumiya C, Luciw P, Kung HJ. Kaposi’s sarcoma-associated herpesvirus K-bZIP is a coregulator of K-Rta: physical association and promoter-dependent transcriptional repression. J Virol. 2003;77:1441–1451. doi: 10.1128/JVI.77.2.1441-1451.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan K, Kuppers DA, Verma SC, Sharma N, Murakami M, Robertson ES. Induction of Kaposi’s Sarcoma-Associated Herpesvirus Latency-Associated Nuclear Antigen by the Lytic Transactivator RTA: a Novel Mechanism for Establishment of Latency. J Virol. 2005;79:7453–7465. doi: 10.1128/JVI.79.12.7453-7465.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Chang J, Lynch SJ, Lukac DM, Ganem D. The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jkappa (CSL), the target of the Notch signaling pathway. Genes Dev. 2002;16:1977–1989. doi: 10.1101/gad.996502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Ganem D. Lytic but not latent infection by Kaposi’s sarcoma-associated herpesvirus requires host CSL protein, the mediator of Notch signaling. Proc Natl Acad Sci USA. 2003;100:8490–8495. doi: 10.1073/pnas.1432843100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Ganem D. RBP-J (CSL) is essential for activation of the K14/vGPCR promoter of Kaposi’s sarcoma-associated herpesvirus by the lytic switch protein RTA. J Virol. 2004;78:6818–6826. doi: 10.1128/JVI.78.13.6818-6826.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao W, Tang Y, Lin SF, Kung HJ, Giam CZ. K-bZIP of Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 (KSHV/HHV-8) binds KSHV/HHV-8 Rta and represses Rta-mediated transactivation. J Virol. 2003;77:3809–3815. doi: 10.1128/JVI.77.6.3809-3815.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CL, Li H, Wang Y, Zhu FX, Kudchodkar S, Yuan Y. Kaposi’s sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: Identification of the ori-Lyt and association of K8 bZip protein with the origin. J Virol. 2003;77:5578–5588. doi: 10.1128/JVI.77.10.5578-5588.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SF, Robinson DR, Miller G, Kung HJ. Kaposi’s Sarcoma-Associated Herpesvirus Encodes a bZIP Protein with Homology to BZLF1 of Epstein-Barr Virus. J Virol. 1999;73:1909–1917. doi: 10.1128/jvi.73.3.1909-1917.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukac DM, Garibyan L, Kirshner JR, Palmeri D, Ganem D. DNA binding by Kaposi’s sarcoma-associated herpesvirus lytic switch protein is necessary for transcriptional activation of two viral delayed early promoters. J Virol. 2001;75:6786–6799. doi: 10.1128/JVI.75.15.6786-6799.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukac DM, Renne R, Kirshner JR, Ganem D. Reactivation of Kaposi’s sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology. 1998;252:304–312. doi: 10.1006/viro.1998.9486. [DOI] [PubMed] [Google Scholar]
- Moore PS, Gao SJ, Dominguez G, Cesarman E, Lungu O, Knowles DM, Garber R, Pellett PE, McGeoch DJ, Chang Y. Primary characterization of a herpesvirus agent associated with Kaposi’s sarcoma. J Virol. 1996;70:549–558. doi: 10.1128/jvi.70.1.549-558.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumm JS, Kopan R. Notch signaling: from the outside in. Dev Biol. 2000;228:151–165. doi: 10.1006/dbio.2000.9960. [DOI] [PubMed] [Google Scholar]
- Oka C, Nakano T, Wakeham A, de la Pompa JL, Mori C, Sakai T, Okazaki S, Kawaichi M, Shiota K, Mak TW, Honjo T. Disruption of the mouse RBP-Jkappa gene results in early embryonic death. Development. 1995;121:3291–3301. doi: 10.1242/dev.121.10.3291. [DOI] [PubMed] [Google Scholar]
- Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, Ganem D. Lytic growth of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat Med. 1996;2:342–346. doi: 10.1038/nm0396-342. [DOI] [PubMed] [Google Scholar]
- Sakakibara S, Ueda K, Chen J, Okuno T, Yamanishi K. Octamer-binding sequence is a key element for the autoregulation of Kaposi’s sarcoma-associated herpesvirus ORF50/Lyta gene expression. J Virol. 2001;75:6894–6900. doi: 10.1128/JVI.75.15.6894-6900.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman WT, Quinlivan EB. Lytic switch protein (ORF50) response element in the Kaposi’s sarcoma-associated herpesvirus K8 promoter is located within but does not require a palindromic structure. Virology. 2003;310:72–84. doi: 10.1016/s0042-6822(03)00095-3. [DOI] [PubMed] [Google Scholar]
- Song MJ, Li X, Brown HJ, Sun R. Characterization of Interactions between RTA and the Promoter of Polyadenylated Nuclear RNA in Kaposi’s Sarcoma-Associated Herpesvirus/Human Herpesvirus 8. J Virol. 2002;76:5000–5013. doi: 10.1128/JVI.76.10.5000-5013.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d’Agay MF, Clauvel JP, Raphael M, Degos L, Sigaux F. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood. 1995;86:1276–1280. [PubMed] [Google Scholar]
- Sun R, Lin SF, Gradoville L, Zhu FX, Yuan Y, Miller G. A viral gene that activates lytic cycle expression of Kaposi’s sarcoma associated herpesvirus. Proc Natl Acad Sci USA. 1998;95:10866–10871. doi: 10.1073/pnas.95.18.10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda K, Ishikawa K, Nishimura K, Sakakibara S, Do E, Yamanishi K. Kaposi’s Sarcoma-Associated Herpesvirus (Human Herpesvirus 8) Replication and Transcription Factor Activates the K9 (vIRF) Gene through Two Distinct cis Elements by a Non-DNA-Binding Mechanism. J Virol. 2002;76:12044–12054. doi: 10.1128/JVI.76.23.12044-12054.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Liu S, Wu MH, Geng Y, Wood C. Identification of a cellular protein that interacts and synergizes with the RTA (ORF50) protein of Kaposi’s sarcoma-associated herpesvirus in transcriptional activation. J Virol. 2001;75:11961–11973. doi: 10.1128/JVI.75.24.11961-11973.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SE, Wu FY, Fujimuro M, Zong J, Hayward SD, Hayward GS. Role of CCAAT/Enhancer-Binding Protein Alpha (C/EBP) in Activation of the Kaposi’s Sarcoma-Associated Herpesvirus (KSHV) Lytic-Cycle Replication-Associated Protein (RAP) Promoter in Cooperation with the KSHV Replication and Transcription Activator (RTA) and RAP. J Virol. 2003;77:600–623. doi: 10.1128/JVI.77.1.600-623.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Chong OT, Yuan Y. Differential regulation of K8 gene expression in immediate-early and delayed-early stages of Kaposi’s sarcoma-associated herpesvirus. Virology. 2004a;325:149–163. doi: 10.1016/j.virol.2004.04.030. [DOI] [PubMed] [Google Scholar]
- Wang Y, Li H, Chan MY, Zhu FX, Lukac DM, Yuan Y. Kaposi’s sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: cis-acting requirements for replication and ori-Lyt-associated RNA transcription. J Virol. 2004b;78:8615–8629. doi: 10.1128/JVI.78.16.8615-8629.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F, Tang Q, Chen H, ApRhys C, Farrell C, Chen J, Fujimuro M, Lane M, Hayward GS. Lytic replication-associated protein (RAP) encoded by Kaposi’s sarcoma-associated herpesvirus causes p21CIP-1-mediated G1 cell cycle arrest through CCAAT/enhancer-binding protein-alpha. Proc Natl Acad Sci USA. 2002;99:10683–10688. doi: 10.1073/pnas.162352299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu FX, Cusano T, Yuan Y. Identification of the immediate early transcripts of Kaposi’s sarcoma-associated herpesvirus. J Virol. 1999;73:5556–5567. doi: 10.1128/jvi.73.7.5556-5567.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]