

Abstract
The Saccharomyces cerevisiae RecQ-mediated genome instability (Rmi1) protein was recently identified as the third member of the slow growth suppressor 1–DNA topoisomerase III (Sgs1–Top3) complex, which is required for maintaining genomic stability. Here, we show that cells lacking RMI1 have a mitotic delay, which is partly dependent on the spindle checkpoint, and are sensitive to the microtubule depolymerizing agent benomyl. We show that rmi1 and top3 single mutants are defective in sister chromatid cohesion, and that deletion of SGS1 suppresses benomyl sensitivity and the cohesion defect in these mutant cells. Loss of RAD51 also suppresses the cohesion defect of rmi1 mutant cells. These results indicate the existence of a new pathway involving Rad51 and Sgs1-Top3-Rmi1, which leads to the establishment of sister chromatid cohesion.
Keywords: DNA replication, sister chromatid cohesion, Sgs1, Rmi1, Top3
Introduction
The RecQ family of DNA helicases has important roles in the maintenance of genomic integrity. In humans, five genes encoding the RecQ family of DNA helicases—RECQL1, BLM, WRN, RECQL4 and RECQL5—have been identified, and defects in three—BLM, WRN and RECQL4—cause Bloom syndrome, Werner syndrome and Rothmund–Thomson syndrome, respectively; all these disorders are associated with genomic instability (Ellis et al, 1995; Yu et al, 1996; Kitao et al, 1999). In the budding yeast Saccharomyces cerevisiae, only a single gene encodes the RecQ family DNA helicase slow growth suppressor 1 (SGS1), and cells lacking SGS1 show phenotypes similar to those observed in cells from patients with Bloom syndrome and Werner syndrome, including hyper-recombination (Watt et al, 1996).
A mutant allele of SGS1 was identified as a suppressor of the slow-growth phenotype of the DNA topoisomerase III (TOP3) mutant (Gangloff et al, 1994), and Sgs1 interacts physically with Top3 (Bennett et al, 2000). Sgs1 and Top3 are required for damage-induced recombination repair (Ui et al, 2001, 2005), and are thought to be involved in the resolution of recombination intermediates (Ira et al, 2003). RecQ-mediated genome instability (RMI1) was identified as the third member of the Sgs1–Top3 complex, and cells lacking SGS1, TOP3 or RMI1 show slow growth, poor sporulation, genome instability and hyper-recombination (Chang et al, 2005; Mullen et al, 2005). In addition, sgs1 and rmi1 mutant cells show the same range of synthetic lethality/sickness interactions with rrm3, slx1, slx4, mus81 and mms4 mutations (Tong et al, 2004). Analyses of the synthetic lethality between sgs1 and the above mutations showed that the Sgs1–Top3–Rmi1 complex seems to function in the repair or bypass of spontaneous S-phase damage (Fabre et al, 2002; Fricke & Brill, 2003; Schmidt & Kolodner, 2004; Torres et al, 2004).
Accurate transmission of chromosomes to daughter cells is important for the maintenance of genomic integrity. It is well known that sister chromatid cohesion—the physical association of replicated sister chromatids—has an important role in the precise segregation of chromosomes, thereby ensuring high-fidelity chromosome transmission (Koshland & Guacci, 2000; Nasmyth et al, 2000). The cohesion of sister chromatids is mediated by a cohesin complex consisting of Smc1, Smc3, Scc1 and Scc3, and its establishment is coupled with DNA replication (Uhlmann, 2004). A screen for noncohesin components involved in sister chromatid cohesion identified several proteins involved in the DNA-damage response (Warren et al, 2004). It has also been reported that cohesin is recruited to sites of double-strand breaks during G2 (Strom et al, 2004). Here, we present a new finding that Rmi1 and Top3 function in sister chromatid cohesion downstream of Sgs1 in a pathway involving Rad51.
Results And Discussion
G2/M delay in rmi1 cells depends on the spindle checkpoint
Previous work has shown that cells lacking RMI1 show slow growth and a large-budded morphology (Chang et al, 2005; Mullen et al, 2005). To determine whether the slow growth phenotype of rmi1 mutant cells was accompanied by a specific defect in cell-cycle progression, we examined the cells by using flow cytometry. Consistent with previous reports, we found that, after release from α-factor, synchronous cultures of rmi1 cells passed through G1 and S phase, and remained blocked in G2/M phase until the end of the experiment (240 min; Fig 1A). Microscopy analysis of asynchronous rmi1 cells also showed an increase in the percentage of large-budded cells, with the nucleus located at or beyond the mother-bud neck, a morphology characteristic of the G2/M phase (Fig 1B). When we examined spindle morphology, we found that a higher proportion of rmi1 cells had intermediate-length spindles compared with wild-type cells, suggesting that rmi1 cells arrested at metaphase (supplementary Fig S1 online). In addition, a marked increase in the number of cells with aberrant spindle structures was observed in the rmi1 mutant cell population (supplementary Fig S1 online).
Figure 1.
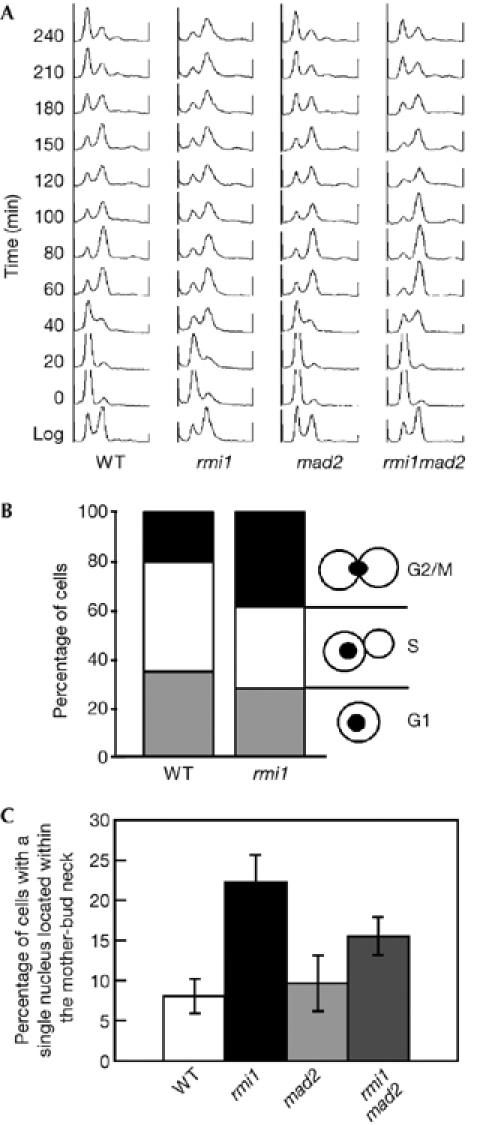
Deletion of RMI1 results in a G2/M-phase delay that is partly dependent on the mitotic spindle checkpoint. (A) Log-phase cultures of wild-type (WT; YK402a), rmi1 (YK402ar1), mad2 (YK402am2) and rmi1mad2 (YK402ar1m2) cells were arrested with α-factor, released into YPD and processed for flow cytometry. (B) Log-phase cultures of wild-type (WT; yMP10381) and rmi1 (SCRr1) cells were fixed with ethanol and stained with DAPI to visualize the DNA. Cells with no bud (G1), cells with bud (S) and large-budded cells with a single nucleus located within the mother-bud neck (G2/M) were scored. (C) Log-phase cultures of wild-type (WT; YK402a), rmi1 (YK402ar1), mad2 (YK402am2) and rmi1mad2 (YK402ar1m2) cells were fixed with ethanol and stained with DAPI to visualize the DNA. Large-budded cells with a single nucleus located within the mother-bud neck were scored. The data shown represent the average of two independent experiments. One hundred cells were counted in each experiment. Bars indicate standard deviation. DAPI, 4,6-diamidino-2-phenylindole.
Generally, mitotic delays could reflect activation of either the DNA-damage checkpoint (Weinert & Hartwell, 1988) or the mitotic spindle checkpoint (Li & Murray, 1991). Chang et al (2005) reported that the Rad53-dependent DNA-damage checkpoint is activated in rmi1 cells. Therefore, we examined whether the accumulation of rmi1 cells in G2/M phase was due to the activation of the mitotic arrest-deficient 2 (Mad2)-dependent spindle checkpoint. Flow cytometry analysis showed that the accumulation of rmi1 cells in G2/M phase was partly suppressed by deletion of MAD2 (Fig 1A). In addition, the percentage of large-budded cells with the nucleus located at or beyond the mother-bud neck, which was increased in rmi1 cells, was also partly reduced by the deletion of MAD2 (Fig 1C). The activation of the Mad2–spindle checkpoint stabilizes securin (Pds1) by inhibiting anaphase promoting complex (APC); therefore, we monitored levels of Pds1 throughout the cell cycle and found that Pds1 persisted for a long time in rmi1 cells (supplementary Fig S2 online). These results indicate that the absence of RMI1 activates the Mad2–spindle checkpoint, resulting in a delay in the progression of M phase.
Defective sister chromatid cohesion in rmi1 and top3 cells
The spindle checkpoint is required for correct chromosome segregation (Li & Murray, 1991). We were interested in whether Rmi1 was involved in some aspect of chromosome segregation as the mitotic spindle checkpoint was activated in rmi1 cells. To assess the sensitivity of rmi1 cells to perturbation of the chromosome segregation machinery, we exposed them to benomyl, a microtubule-depolymerizing drug. As shown in Fig 2A, rmi1 cells were moderately sensitive to benomyl, compared with wild-type cells.
Figure 2.
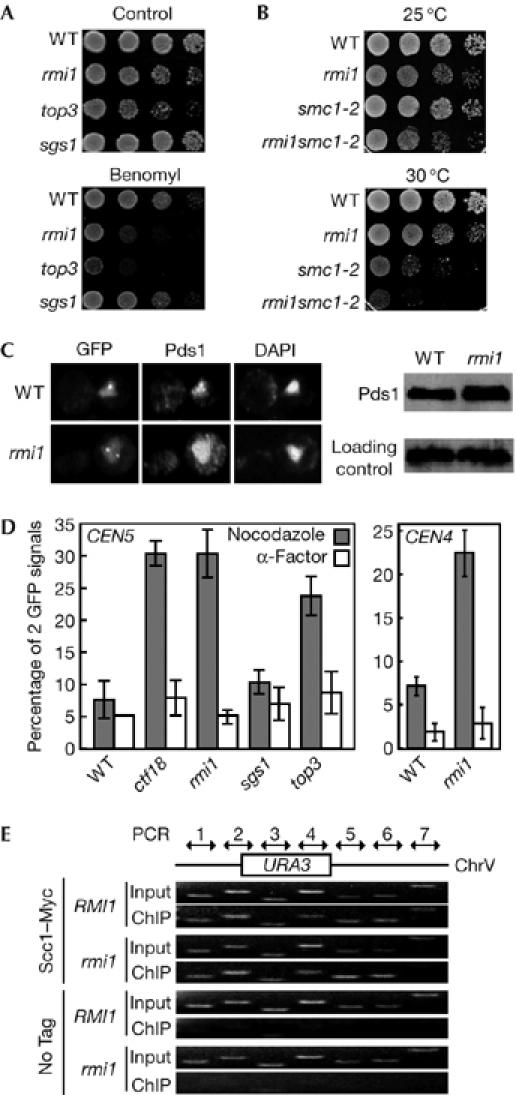
rmi1 and top3 mutants are sensitive to benomyl and defective in sister chromatid cohesion. (A) Serial tenfold dilutions of log-phase cultures of wild-type (YPH1477), rmi1 (YPH1477r1), top3 (YPH1477t3) and sgs1 (YPH1477s1) cells were spotted onto growth plates with or without 20 μg/ml benomyl, incubated at 30°C for 3 days and then photographed. (B) Serial tenfold dilutions of log phase cultures of wild-type (982-6a), rmi1 (982-6ar1), smc1-2 (1355-1a) and rmi1smc1-2 (1355-1ar1) cells were spotted onto growth plates, incubated at 25°C (top) or 30°C (bottom) for 3 days and then photographed. (C) Fluorescence micrographs of wild-type (YPH1477) and rmi1 (YPH1477r1) cells showing GFP loci on sister chromatids (GFP), Pds1 and DNA (DAPI) (left panels). Cells expressing Pds1–13Myc in wild-type (YPH1477b1) or rmi1 (YPH1477b1r1) cells were arrested in M phase (15 μg/ml nocodazole for 5 h) and analysed by western blotting (right panels). Histone H3 was used as a loading control. Pds1–13Myc and histone H3 were detected using Myc (9E10) monoclonal antibody and histone H3 antibody, respectively. (D) Wild-type (YPH1477; tet operator locus located 35 kb from CEN5), ctf18 (YPH1477c18), rmi1 (YPH1477r1), sgs1 (YPH1477s1) and top3 (YPH1477t3) cells (left panel) and wild-type (Y819; tet operator locus located 12.7 kb from CEN4) and rmi1 (Y819r1) cells (right panel) were arrested in M-phase with nocodazole or in G1-phase with α-factor and then fixed with paraformaldehyde. One hundred cells of each strain were scored for the number of cells with two GFP signal foci. The data shown represent the average of three experiments. Bars indicate standard deviation. (E) Chromatin immunoprecipitation (ChIP) analysis at the URA3 locus. Seven PCR products designed to span the URA3 locus are shown (Lam et al, 2006). Scc1–13Myc ChIP was carried out in the presence (YK402aSCC1) or absence (YK402aSCC1r1) of RMI1. Cells were arrested in M-phase (15 μg/ml nocodazole for 5 h) before proceeding with ChIP. Input is the chromatin solution used to perform ChIP. A no-tag control is shown for each experiment. ChrV, chromosome 5; DAPI, 4,6-diamidino-2-phenylindole; GFP, green fluorescent protein; WT, wild-type.
It has been shown that chromosome transmission fidelity (CTF) ctf7 and ctf18 mutant cells, which have defects in sister chromatid cohesion (Skibbens et al, 1999; Hanna et al, 2001; Mayer et al, 2001), show MAD2-dependent M-phase arrest and have intermediate-length spindles. As rmi1 cells showed a similar phenotype to ctf7 and ctf18 mutants, we next tested whether Rmi1 has a role in sister chromatid cohesion.
First, we assessed the genetic interactions between RMI1 and SMC1—a gene that functions in sister chromatid cohesion, the conditional allelle (smc1-2) of which confers a synthetically lethal phenotype in the absence of CTF8 (Mayer et al, 2001). We observed conditional synthetic sickness between rmi1 and smc1–2, suggesting that RMI1 is involved in cohesion (Fig 2B).
Next, we assessed cohesion directly by using cells that were constructed as described previously (Michaelis et al 1997). When arrested in M phase with nocodazole, wild-type cells showed one green fluorescent protein (GFP) signal, which is indicative of nonseparated sister chromatids, whereas mutants defective in cohesion showed two GFP signal foci, which is indicative of separated sister chromatids (Fig 2C). We compared the efficiency of sister chromatid pairing in wild-type and rmi1 cells at two different chromosome loci: at a Tet operator locus located 35 kb from the centromere of chromosome 5 (CEN5) and another located 12.7 kb from CEN4. When the CEN5 locus was monitored in nocodazole-arrested cells, 8% of M-phase wild-type cells had two GFP signal foci, whereas approximately 30% of M-phase rmi1 cells had two GFP signals, indicating that rmi1 cells, similar to ctf18 cells, are defective in sister chromatid cohesion (Fig 2D, left panel). A similar result was obtained with the Tet operator at the CEN4 locus (Fig 2D, right panel). When cells were arrested in G1 phase, the percentage of cells with two GFP signal foci was similar between wild-type and rmi1 cells, excluding the possibility that the increase seen in M-phase-arrested rmi1 cells was due to aneuploidy (Fig 2D).
To eliminate the possibility that the cohesion defect in rmi1 cells was caused by premature destruction of the separin (Esp1) inhibitor Pds1, we monitored the levels of Pds1 in M-phase-arrested cells. We confirmed that Pds1 was present in similar amounts in rmi1 cells showing two GFP signals and in wild-type cells (Fig 2C).
To investigate whether the RMI1 mutation affects the recruitment of cohesin to the Tet operator locus, we carried out chromatin immunoprecipitation to examine the association of sister chromatid cohesion 1 (Scc1) at the uracil 3 (URA3) locus, where the Tet operator is integrated. Myc-tagged Scc1 was immunoprecipitated from extracts prepared from M-phase-arrested cells and the location of Scc1 was identified by performing PCR. We used the series of PCR primer pairs described by Lam et al (2006) to map Scc1 association over the entire URA3 region (Fig 2E). We found that Scc1-binding profiles were indistinguishable between rmi1 and wild-type cells (Fig 2E), suggesting that Rmi1 is not involved in the recruitment of cohesin. Therefore, it appears that Rmi1 functions in the establishment of sister chromatid cohesion.
Rmi1 interacts physically and functionally with Sgs1 and Top3 (Chang et al, 2005; Mullen et al, 2005). Next, we examined whether Sgs1 or Top3 was also involved, along with Rmi1, in sister chromatid cohesion. We found that top3 mutants, but not sgs1 mutants, showed higher sensitivity to benomyl compared with wild-type cells (Fig 2A). In addition, sister chromatid cohesion was defective in top3 mutants, but not in sgs1 mutants (Fig 2D).
Deletion of SGS1 or RAD51 suppresses the cohesion defect
Top3 functions downstream of Sgs1 to resolve DNA substrates created by Sgs1. Most of the defects shown by top3 mutants are suppressed by mutation of SGS1 (Gangloff et al, 1994). Recent studies indicate that the growth defect and sensitivity to DNA-damaging agents of rmi1 cells are also suppressed by SGS1 deletion, suggesting that Rmi1 functions downstream of Sgs1, in a manner similar to Top3 (Chang et al, 2005; Mullen et al, 2005). As sgs1 cells did not show benomyl sensitivity or a defect in sister chromatid cohesion (Fig 2), we examined whether these phenotypes were suppressed in rmi1 and top3 cells by deletion of SGS1. As shown in Fig 3A, deletion of SGS1 suppressed the benomyl sensitivity of rmi1 and top3 cells. The cohesion defect in the mutant cells was also considerably suppressed by deletion of SGS1 (Fig 3B). In addition, rmi1top3 double mutants showed similar levels of benomyl sensitivity and cohesion defects to those of the corresponding single mutants, suggesting that Rmi1 and Top3 act at the same stage of cohesion establishment (Fig 3C,D).
Figure 3.
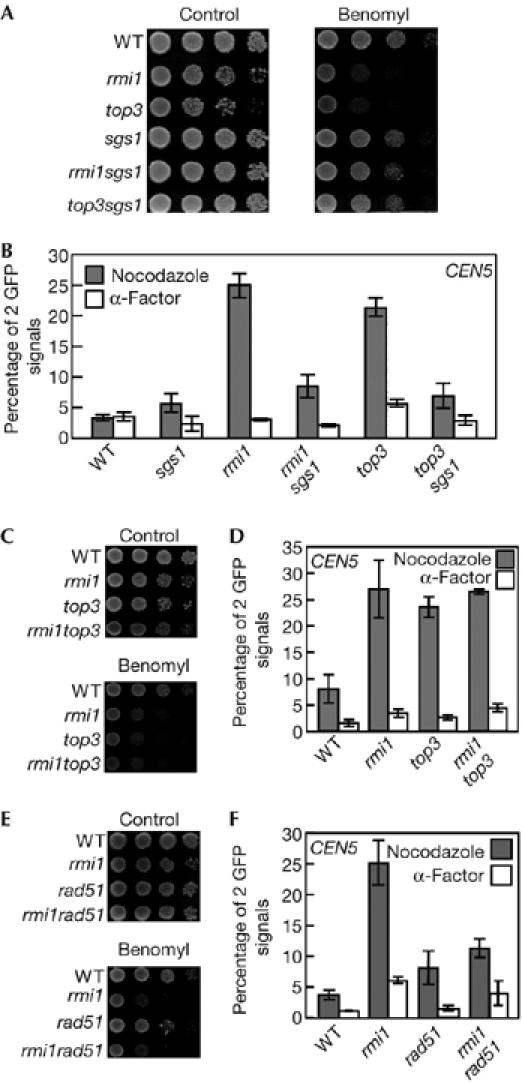
Deletion of SGS1 or RAD51 suppresses cohesion defects in rmi1 and top3 cells. (A,C,E) Serial tenfold dilutions of log phase cultures of (A) wild-type (YPH1477), rmi1 (YPH1477r1), top3 (YPH1477t3), sgs1 (YPH1477s1), rmi1sgs1 (YPH1477r1s1) and top3sgs1 (YPH1477t3s1) cells, (C) of wild-type (YPH1477), rmi1 (YPH1477r1), top3 (YPH1477t3) and rmi1top3 (YPH1477r1t3) cells, and (E) of wild-type (YPH1477), rmi1 (YPH1477r1), rad51 (YPH1477r51) and rmi1rad51 (YPH1477r1r51) cells were spotted onto growth plates containing 0, 20 (A,E) or 22 μg/ml (C) benomyl, incubated at 30°C for 3 days and then photographed. (B,D,F) The same combination of cells in (A), (C) and (E) were used. Cells were arrested in M-phase with nocodazole or in G1 phase with α-factor and then fixed with paraformaldehyde. One hundred cells of each strain were scored for number of cells with two green fluorescent protein (GFP) signal foci. The data shown represent the average of three experiments. Bars indicate standard deviation. WT, wild type.
Sgs1 functions downstream of Rad51 under certain conditions. Next, we asked whether deletion of RAD51 also suppressed the cohesion defect in rmi1 cells. As shown in Fig 3F, the cohesion defect in rmi1 cells was suppressed by the disruption of RAD51, to a similar level as that seen in rad51 cells. These results indicate that activation of a pathway involving Rad51 and Sgs1 results in sister chromatid cohesion by the functions of Top3 and Rmi1, and that dysfunction of Top3 or Rmi1 causes defects in cohesion. However, disruption of RAD51 did not suppress the benomyl sensitivity of rmi1 cells (Fig 3E), indicating that the cohesion defect does not necessarily correlate with benomyl sensitivity.
What then is the pathway involving Rad51 and Sgs1, which causes sister chromatid cohesion by the functions of Top3 and Rmi1? An attractive model involving Rad51, Sgs1 and Top3 was proposed by Liberi et al (2005). In this model, when the DNA replication fork encounters DNA lesions, a hemicatenane with double Holliday junctions is formed through the action of Rad51, and dissolved by Sgs1 and Top3, resulting in restoration of the replication fork (Liberi et al, 2005). Interpreting our results in the context of this model, Rmi1 and also Sgs1 and Top3 would be involved in the dissolution of double Holliday junctions. Indeed, the human homologue of Rmi1, BLAP75, stimulates dissolution of double Holliday junctions by BLM and Top3α (Raynard et al, 2006; Wu et al, 2006). Thus, Rmi1 together with Top3 might have a crucial role in coupling dissolution and cohesion.
The suppression of the cohesion defect of rmi1 cells by disruption of SGS1 or RAD51 can be explained by the following model. Sgs1 and Rad51 generate recombination intermediates that require Top3 and Rmi1 for processing to establish cohesion. In the presence of Sgs1, the access of other factors to recombination intermediates is inhibited. When Sgs1 is absent, a structure-specific nuclease resolves the Holliday junctions, eliminating the requirement for Top3 and Rmi1 in the establishment of sister chromatid cohesion. In the absence of Rad51, hemicatenanes with double Holliday junctions are not formed and the lesions are bypassed or repaired by systems that do not perturb the original sister chromatid cohesion.
In conclusion, we provide the first evidence, to our knowledge, indicating that Rmi1 and Top3 are involved in sister chromatid cohesion. Functional studies that explain the molecular mechanism linking Rmi1 and Top3 with cohesion, and confirm the validity of the above model are the focus of current and future efforts in our laboratory.
Methods
Yeast strains and medium. The yeast strains used are listed in supplementary Table S1 online. We thank P. Hieter and V. Guacci for their generous gift of strains. The medium used for growth was described previously (Rose et al, 1990).
Flow cytometry, DNA staining and immunofluorescence. Flow cytometry, DNA staining and immunofluorescence were carried out as described previously (Branzei et al, 2002).
Spot assay. Log-phase cells grown in YPD medium were collected, washed once with distilled water, counted and diluted appropriately. Tenfold serial dilutions of cells (105, 104, 103 and 102 cells) were spotted onto YPD with or without benomyl. The plates were incubated at 30°C for 2–4 days and then photographed.
Cohesion assay. Strains containing Tet repressor-GFP/Tet operator repeats were arrested in M phase or G1 phase by incubation with 15 μg/ml nocodazole and 5 μg/ml α-factor, respectively, for 5 h at 30°C. Cells were then fixed by incubation with an equal volume of 4% paraformaldehyde for 30 min, washed once with SK (1 M sorbitol, 0.05 M K2PO4) and resuspended in 50 μl SK for cohesion assessment.
Pds1 assay. Immunostaining and western blotting of Pds1 were carried out as described previously (Mayer et al, 2004).
Chromatin immunoprecipitation. Log-phase cells expressing Myc-tagged Scc1 were arrested in M phase by incubation with 15 μg/ml nocodazole for 5 h. ChIP was carried out using the Myc antibody as described previously (Ogiwara et al, 2007).
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
supplementary information Fig S1
supplementary information Fig S2
supplementary information Table S1
Acknowledgments
This work was supported by grants for Scientific Research and for Scientific Research on Priority Areas from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- Bennett RJ, Noirot-Gros MF, Wang JC (2000) Interaction between yeast sgs1 helicase and DNA topoisomerase III. J Biol Chem 275: 26898–26905 [DOI] [PubMed] [Google Scholar]
- Branzei D, Seki M, Onoda F, Yagi H, Kawabe Y, Enomoto T (2002) Characterization of the slow-growth phenotype of S. cerevisiae Whip/Mgs1 Sgs1 double deletion mutants. DNA Repair 1: 671–682 [DOI] [PubMed] [Google Scholar]
- Chang M, Bellaoui M, Zhang C, Desai R, Morozov P, Delgado-Cruzata L, Rothstein R, Freyer GA, Boone C, Brown GW (2005) RMI1/NCE4, a suppressor of genome instability, encodes a member of the RecQ helicase/Topo III complex. EMBO J 24: 2024–2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, Ciocci S, Proytcheva M, German J (1995) The Bloom's syndrome gene product is homologous to RecQ heliases. Cell 83: 655–666 [DOI] [PubMed] [Google Scholar]
- Fabre F, Chan A, Heyer WD, Gangloff S (2002) Alternative pathways involving Sgs1/Top3, Mus81/Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc Natl Acad Sci USA 99: 16887–16892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke WM, Brill SJ (2003) Slx1–Slx4 is a second structure-specific endonuclease functionally redundant with Sgs1–Top3. Genes Dev 17: 1768–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff S, McDonald JP, Bendixen C, Arthur L, Rothstein R (1994) The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: a potential eukaryotic reverse gyrase. Mol Cell Biol 14: 8391–8398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna JS, Kroll ES, Lundblad V, Spencer FA (2001) Saccharomyces cerevisiae CTF18 and CTF4 are required for sister chromatid cohesion. Mol Cell Biol 21: 3144–3158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ira G, Malkova A, Liberi G, Foiani M, Haber JE (2003) Srs2 and Sgs1–Top3 suppress crossovers during double-strand break repair in yeast. Cell 115: 401–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitao S, Shimamoto A, Goto M, Miller RW, Smithson WA, Lindor NM, Furuichi Y (1999) Mutations in RECQL4 cause a subset of cases of Rothmund–Thomson syndrome. Nat Genet 22: 82–84 [DOI] [PubMed] [Google Scholar]
- Koshland DE, Guacci V (2000) Sister chromatid cohesion: the beginning of a long and beautiful relationship. Curr Opin Cell Biol 12: 297–301 [DOI] [PubMed] [Google Scholar]
- Lam WW, Peterson EA, Yeung MT, Lavoie BD (2006) Condensin is required for chromosome arm cohesion during mitosis. Genes Dev 20: 2973–2984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Murray AW (1991) Feedback control of mitosis in budding yeast. Cell 66: 519–531 [DOI] [PubMed] [Google Scholar]
- Liberi G, Maffioletti G, Lucca C, Chiolo I, Baryshnikova A, Cotta-Ramusino C, Lopes M, Pellicioli A, Haber JE, Foiani M (2005) Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev 19: 339–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML, Gygi SP, Aebersold R, Hieter P (2001) Identification of RFC (Ctf18p, Ctf8p, Dcc1p): an alternative RFC complex required for sister chromatid cohesion in S. cerevisiae. Mol Cell 7: 959–970 [DOI] [PubMed] [Google Scholar]
- Mayer ML et al. (2004) Identification of protein complexes required for efficient sister chromatid cohesion. Mol Biol Cell 15: 1736–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis C, Ciosk R, Nasmyth K (1997) Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell 91: 35–45 [DOI] [PubMed] [Google Scholar]
- Mullen JR, Nallaseth FS, Lan YQ, Slagle CE, Brill SJ (2005) Yeast Rmi1/Nce4 controls genome stability as a subunit of the Sgs1–Top3 complex. Mol Cell Biol 25: 4476–4487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasmyth K, Peters JM, Uhlmann F (2000) Splitting the chromosome: cutting the ties that bind sister chromatids. Science 288: 1379–1385 [DOI] [PubMed] [Google Scholar]
- Ogiwara H, Ui A, Enomoto T, Seki M (2007) Role of Elg1 protein in double strand break repair. Nucleic Acids Res 35: 353–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynard S, Bussen W, Sung P (2006) A double Holliday junction dissolvasome comprising BLM, topoisomerase IIIα, and BLAP75. J Biol Chem 281: 13861–13864 [DOI] [PubMed] [Google Scholar]
- Rose MD, Winston F, Hieter P (1990) Methods in Yeast Genetics. Cold Spring Harbor, NY, USA: Cold Spring Harbor Laboratory Press [Google Scholar]
- Schmidt KH, Kolodner RD (2004) Requirement of Rrm3 helicase for repair of spontaneous DNA lesions in cells lacking Srs2 or Sgs1 helicase. Mol Cell Biol 24: 3213–3226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibbens RV, Corson LB, Koshland D, Hieter P (1999) Ctf7p is essential for sister chromatid cohesion and links mitotic chromosome structure to the DNA replication machinery. Genes Dev 13: 307–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom L, Lindroos HB, Shirahige K, Sjogren C (2004) Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol Cell 16: 1003–1015 [DOI] [PubMed] [Google Scholar]
- Tong AH et al. (2004) Global mapping of the yeast genetic interaction network. Science 303: 808–813 [DOI] [PubMed] [Google Scholar]
- Torres JZ, Schnakenberg SL, Zakian VA (2004) Saccharomyces cerevisiae Rrm3p DNA helicase promotes genome integrity by preventing replication fork stalling: viability of rrm3 cells requires the intra-S phase checkpoint and fork restart activities. Mol Cell Biol 24: 3198–3212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlmann F (2004) The mechanism of sister chromatid cohesion. Exp Cell Res 296: 80–85 [DOI] [PubMed] [Google Scholar]
- Ui A, Satoh Y, Onoda F, Miyajima A, Seki M, Enomoto T (2001) The N-terminal region of Sgs1, which interacts with Top3, is required for complementation of MMS sensitivity and suppression of hyper-recombination in sgs1 disruptants. Mol Genet Genomics 265: 837–850 [DOI] [PubMed] [Google Scholar]
- Ui A, Seki M, Ogiwara H, Onodera R, Fukushige S, Onoda F, Enomoto T (2005) The ability of Sgs1 to interact with DNA topoisomerase III is essential for damage-induced recombination. DNA Repair 4: 191–201 [DOI] [PubMed] [Google Scholar]
- Warren CD, Eckley DM, Lee MS, Hanna JS, Hughes A, Peyser B, Jie C, Irizarry R, Spencer FA (2004) S-phase checkpoint genes safeguard high-fidelity sister chromatid cohesion. Mol Biol Cell 14: 1724–1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt PM, Hickson ID, Borts RH, Louis EJ (1996) SGS1, a homologue of the Bloom's and Werner's syndrome genes, is required for maintenance of genome stability in Saccharomyces cerevisiae. Genetics 144: 935–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert TA, Hartwell LH (1988) The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science 241: 317–322 [DOI] [PubMed] [Google Scholar]
- Wu L, Bachrati CZ, Ou J, Xu C, Yin J, Chang M, Wang W, Li L, Brown GW, Hickson ID (2006) BLAP75/RMI1 promotes the BLM-dependent dissolution of homologous recombination intermediates. Proc Natl Acad Sci USA 103: 4068–4073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CE et al. (1996) Positional cloning of the Werner's syndrome gene. Science 272: 258–262 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary information Fig S1
supplementary information Fig S2
supplementary information Table S1