

Abstract
Nasal administration of beta cell-derived auto-antigens has been reported to suppress the development of autoimmune diabetes. We investigated the tolerogenic effects of insulin conjugated to the B subunit of cholera toxin (CTB). Nasal administration of 1 µg of CTB-insulin significantly delayed the incidence of diabetes in comparison to CTB treated mice. However, administration of 4 or 8 µg of the conjugate had no protective effect. Protection induced by CTB-insulin was transferred to naive recipients by splenic CD4+ T cells. This result favours an active cellular mechanism of regulation, which was lost using higher (4–8 µg) or lower (0·1–0·5 µg) amounts of the conjugate. When co-administered with diabetogenic T cells, splenic T cells from CTB-insulin-treated mice reduced the lymphocytic infiltration of the islets. Reverse transcription-polymerase chain reaction analysis of recipients’ pancreatic glands revealed an increase of TGF-β and IL-10 transcripts after donor mice tolerization, while levels of IFN-γ and IL-4 RNAs were unchanged. We observed a significant increase of T cell proliferation after unspecific stimulation in the spleen and pancreatic lymph nodes 24 h after CTB-insulin administration in comparison to control treatment. Higher amounts of IL-4 and IFN-γ were noticed in pancreatic lymph nodes of tolerized mice upon in vitro stimulation. Antigen-specific unresponsiveness after immunization and upon subsequent in vitro exposure to homologous antigen was obtained in nasally treated animals. Our results underlined the importance of nasal mucosa as an inducing site of tolerance and provided evidence for similar mechanisms of action to what has been described for the oral route, which favoured a CTB-insulin specific effect.
Keywords: CTB-insulin, diabetes, nasal tolerance, NOD mice
INTRODUCTION
The non-obese diabetic (NOD) mouse develops a spontaneous form of autoimmune diabetes that closely resembles human type 1 diabetes [1]. Autoreactive T cells play a major pathogenic role and lead to destruction of β cells [2]. T cell specificity has been studied in the past few years, underlining the respective contribution of specific beta cell antigens that also elicit specific antibodies. Among these antigens, insulin, glutamic acid decarboxylase (GAD) and islet-associated protein 2 (IA-2) have been proposed as the main antigen determinants that drive the immune response leading to beta cell destruction. It has been suggested that the mechanisms that are responsible for the induction and maintenance of self-tolerance are defective in NOD mice [3,4].
Mucosal sensitization with auto-antigens is an established strategy to prevent the development of autoimmune diseases by inducing peripheral immunological tolerance [5,6]. In particular, nasal antigen administration is a highly efficient route of inducing clinically relevant tolerance [7]. It is due to the nasal associated lymphoid tissue (NALT), which is a bilateral strip of non-encapsulated lymphoid tissue underlying the epithelium of the nasal tract [8]. Several investigators have reported that nasal administration of auto-antigens effectively prevents several experimental autoimmune diseases. Nasal administration of myelin basic protein (MBP) or acetylcholine receptor (AchR), respectively, prevented experimental autoimmune encephalomyelitis (EAE) [9–11] and suppressed experimental autoimmune myasthenia gravis (EAMG) [12–14]. Nasal delivery of type II collagen suppressed collagen-induced arthritis (CIA) [15,16]. In NOD mice, administrations of insulin [17,18], GAD [19], GAD65-derived peptides [20], B9-23 peptide [21] or B24-36 peptide [22] by nasal route have the properties to prevent the autoimmune diabetes. Depending on the dose of antigen administered, anergy/deletion of antigen-specific T cells or selective expansion of cells producing immunosuppressive cytokines are two major mechanisms proposed to explain mucosal tolerance induction.
The feasibility of transferring experimental data to humans is hampered by the amount of antigen needed. When adapted to humans, the experimental doses needed to induce nasal tolerance represent several grams of antigen. We demonstrated that the protective effects observed with oral insulin could be potentiated by the conjugation of insulin to the B moiety of cholera toxin (CTB) [23,24], allowing a decrease in the amount of antigen needed. Effective oral tolerance induction in NOD mice necessitated only 4 µg of the conjugate. Due to the limitations of antigen doses, CTB-insulin conjugate is a promising alternative for human trials in prediabetic subjects.
The attractive potentialities of CTB-insulin, and the necessity to define the modalities of tolerance induction in perspective of human application, prompted us to address the question of the tolerogenic properties of CTB-insulin after its nasal administration in the experimental model of NOD mice. We demonstrated here the protective capacities of CTB-insulin administered by the nasal route and understood mechanisms supporting this effect. Moreover, we outlined the critical aspects of antigen dosage conditioning the outcome of this therapy.
MATERIALS AND METHODS
Mice
NOD Thy-1,2 mice were bred under standard conditions in our animal facility. In our colony, spontaneous diabetes starts at 12 weeks in females and the incidence of diabetes reaches 81% in females and 15–20% in males by 30 weeks. Diabetes was characterized by polydipsia, weight loss, glycosuria as assessed by urine chemstrips (Bayer, Germany) and persistent hyperglycaemia (above 200 mg/dL), which was determined with blood glucose chemstrips (Lifescan, Mountain View, CA, USA). Diabetic NOD females served as donors of autoreactive T cells during co-transfer experiments.
Antigens
CTB was purified and conjugated to recombinant human insulin (Novo-Nordisk, Baegsvaerd, Denmark) as described previously [23]. The resulting CTB-insulin conjugates were purified by gel filtration and characterized for their receptor-binding and serological reactivity by a solid phase enzyme-linked immunosorbent assay (ELISA) using immobilized GM1 ganglioside as capture system and enzyme-linked antibodies to CTB and insulin as detection systems, respectively [23]. The purified conjugates were found to contain an insulin/CTB ratio of 2·2. In all protocols, the doses of CTB-insulin given represent the amount of insulin in such conjugates and for CTB the corresponding doses according to CTB/insulin ratio.
Nasal tolerization protocols
To evaluate the effect of nasal administration of candidate antigens on the spontaneous incidence of diabetes, 4–6-week-old female mice were used. Nasal administration consisted in a single infusion of 20 µl of antigen diluted in PBS. For insulin experiments, a dose of 10 µg was chosen in order to reduce the antigenic challenge. For CTB-insulin protocols, mice were treated once with 0·1, 0·5, 1, 4 or 8 µg of CTB-insulin or equivalent amounts of unconjugated CTB. During adoptive transfer protocols, donor mice were treated once 1 week prior cell transfer. One week after treatment, 5 × 106 splenic T cells were mixed with 5 × 106 T cells from diabetic NOD females. The resulting cell mixture was injected i.v. into 8–10-week-old irradiated (750 rads) NOD males, as described previously [23].
Immunization
Ten days after the nasal administration of CTB-insulin, treated or untreated mice were immunized at the base of the tail with 50 µg of porcine insulin or ovalbumin (OVA) emulsified in an equal volume of complete Freund's adjuvant (CFA). Eight days later, a cell suspension was prepared from the spleen and para-aortic lymph nodes. T cells, 5 × 105, were cultured in 96-well plates in RPMI 10% FCS together with 5 × 105 syngenic irradiated spleen cells and 20 µg/ml of porcine insulin or OVA. Incorporation of [3H]-thymidine was measured after 72 h in culture. IL-2 content of culture supernatants was evaluated using IL-2 dependent CTLL cell line. We analysed six mice per group during two independent experiments and performed proliferation test in sextuplicate.
Cell preparation and analysis
After treatment, spleens, pancreatic lymph nodes (PLN) and cervical lymph nodes (CLN) were removed aseptically and single cell suspensions were prepared by mechanical dispersion according to standard procedures in Hanks's balanced salt solution (HBSS). T cell-enriched populations were obtained by elimination of B cells and macrophages by panning on plastic dishes coated with rabbit antimouse IgG (H + l) antibody (Rockland, Gilbertsville, PA, USA). The resulting cell populations comprised more than 90% T cells, as indicated by flow cytofluorometry using a FITC-labelled hamster antimouse CD3 antibody (clone CT-CD3, Caltag, San Francisco, CA, USA). CD4+ or CD8+ T cell-enriched populations were selected negatively using antibody coated magnetic beads (Dynal, Oslo, Norway). Controls by FACS analysis of cell preparations indicated more than 95% purity of the CD4 or CD8 T cell subsets using appropriate antibodies (anti-CD4 clone H129·19 and anti-CD8 clone 53–6·7 from Pharmingen). The percentages of CD4+, CD8+ and CD25+ T cells in the spleen and cervical lymph nodes from nasally treated mice were compared by FACS analysis (anti-CD25 clone 7D4, Pharmingen).
Histological and immunohistochemical examination
Frozen sections of pancreas (5 µm) were obtained. Infiltration of the islets was scored by two independent examinators and was expressed in percentage of each grade: 0 = no infiltration, 1 = peri-islet infiltration, 2 = intra-islet infiltration <50%, 3 = extensive islet infiltration. A mean score was calculated which represented the contribution of all degrees of insulitis.
Cell proliferation and cytokine analysis
Cells from nasally treated mice were cultured in flat-bottomed 96-well microtitre plates. Cell proliferation and cytokine concentrations in cell-free supernatants were determined after stimulation with plate-bound anti-CD3 and anti-CD28 monoclonal antibodies (MoAbs) (10 µg/ml per clone, clone 145–2C11 and clone 37·51, Pharmingen). Cell proliferation was evaluated after 72 h of culture. We used three to four mice per group for each experiment and performed three wells per condition. The cytokine analysis was performed in three independent experiments. Supernatants were collected after 48 h (IL-4, IL-10, IFN-γ) or 72 h (TGF-β) of culture and analysed for cytokine content using specific enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Abingdon, UK) following the manufacturer's instructions. The optical density of samples was determined at 492 nm, 450 nm and 650 nm for IL-4 and IL-10, TGF-β, respectively, using an automated microplate reader (Labsystems Multiskan MCC/340). Evaluation of cytokine mRNA content was also performed using unstimulated cells as described previously [24] after RNA extraction of cell pellets with a phenol–chloroform procedure and RT-PCR analysis using β2-microglobulin, IL-4, IL-10, TGF-β or IFN-γ oligonucleotide primers (Sigma-Genosys, St Quentin Fallavier, France).
Statistical analysis
The effects of nasal treatment and cell transfers were analysed using Wilcoxon's rank-sum test and Fischer's exact test analysis. Survival analysis were also performed with Kaplan–Meier analysis and log-rank test using the SPSS software. Scores of insulitis, proliferation tests and cytokine analysis were compared using Student's t-test for unpaired samples.
RESULTS
Effects of nasal administration of CTB-insulin on the spontaneous diabetes incidence
In order to test the effect of nasal administration of CTB-insulin conjugate on the protection against spontaneous diabetes, we treated 4-week-old females with a single nasal administration with 1 or 4 µg of CTB-insulin and the equivalent amount of CTB for control animals. As shown in Fig. 1, a single nasal administration of 1 µg of CTB-insulin delayed the onset of diabetes in comparison to CTB administration with 4/9 versus 9/10 diabetic mice at 35 weeks of age (log rank test, P = 0·02). However, nasal administration of 4 µg of CTB-insulin did not reduce diabetes incidence (5/10 versus 7/10 diabetic mice at 35 weeks, P = n.s.).
Fig. 1.

Nasal tolerance with 1 µg of CTB-insulin delayed spontaneous diabetes in NOD mice; 4-week-old NOD females were treated nasally with either 1 µg of CTB-insulin (filled squares) or equivalent dose of CTB (open squares) and tested for spontaneous diabetes incidence.
Transfer of protection after nasal treatment
We investigated the capacity of splenic T cells from nasally treated mice to protect recipients against diabetes transfer. One week after the administration of 0·1, 0·5, 1, 4 or 8 µg of CTB-insulin, we co-transferred splenic T cells from these mice together with diabetogenic T cells to irradiated recipients (Fig. 2). Interestingly, only treatment with 1 µg of CTB-insulin reduced diabetes incidence in the recipients in comparison to CTB administration (1/8 versus 4/5 diabetic mice at 48 days, P = 0·03). Histological analysis of the pancreas of the seven non-diabetic recipients 50 days after the transfer revealed mild islet infiltration with predominant figures of peri-insulitis (mean ± s.d. score of insulitis of 1·3 ± 0·3). Reverse transcription-polymease chain reaction (RT-PCR) analysis of cytokine transcripts within the pancreas of the recipients showed some differences in the levels of transcripts. While TGF-β RNA was detected in 4/5 pancreatic glands from the CTB group, higher levels were observed in 5/6 mice from the CTB-insulin group. The same observation was seen for IL-10 RNAs. However, no difference in expression could be noticed for IFN-γ and IL-4 RNA expression. Results from a representative experiment are shown in Fig. 3. Interestingly, the mouse, which had the highest level of IL-4 RNA, had no IFN-γ RNA expression and the lowest insulitis score.
Fig. 2.
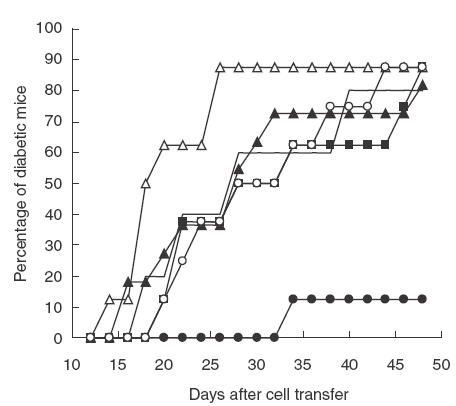
Cells from mice treated nasally with 1 µg of CTB-insulin reduced diabetes incidence in NOD mice; 8-week-old irradiated NOD males were transferred with 5 × 106 splenic T cells from diabetic mice together with 5 × 106 splenic T cells from mice treated nasally with 0·44 µg of CTB (solid line, n = 5) or CTB-insulin: 0·1 µg (open triangles, n = 8), 0·5 µg (dark triangles, n = 11), 1 µg (dark circles, n = 8), 4 µg (dark squares, n = 8) and 8 µg (open circles, n = 8).
Fig. 3.

Insulitis score and RT-PCR cytokine analysis of mRNA from pancreas of mice co-transferred with diabetogenic T cells and splenic T cells from CTB (1, 2, 3) or CTB-insulin (4, 5, 6)-treated mice. ND = not determined.
Contribution of T cell subsets to protection
To further characterize the regulatory cells that could be induced by nasal treatment with 1 µg of CTB-insulin, we analysed the respective contribution of CD4+ and CD8+ T cells subsets in the protection during adoptive transfer experiments. One week after nasal administration of 1 µg of CTB-insulin or the equivalent amount of CTB, we co-transferred splenic T cells or purified splenic CD4+ T cells or CD8+ T cells together with diabetogenic T cells to irradiated recipients. Results shown in Fig. 4 revealed marked differences between the two cell subsets. Only 3/8 mice receiving CD4+ T cells from tolerized donors became diabetic in contrast to 8/8 mice receiving CD8+ T cells 40 days after cell transfer (log rank test, P = 0·0036). We also performed flow cytometry analysis of CD4+, CD8+ and CD25+ T cells in the spleen and cervical lymph nodes 7 days after nasal administration. We did not find any significant differences between CTB and CTB-insulin treated mice in spleen and CLN, respectively, for the percentages of CD4+ (24·4 ± 11% versus 20·4 ± 8·3%, P = 0·6 and 40·9 ± 6·7% versus 33·4 ± 2·6%, P = 0·2), CD8+ (13·8 ± 6% versus 10·8 ± 3%, P = 0·5 and 33·3 ± 17% versus 21 ± 4%, P = 0·34) and CD25+ cells (10·8 ± 0·9% versus 13·6 ± 1·6%, P = 0·07 and 8·5 ± 2·2% versus 10·9 ± 2·1%, P = 0·24).
Fig. 4.

CD4+ T cells from CTB-insulin nasally treated mice reduced diabetes incidence in adoptively transferred mice; 8-week-old NOD males were transferred with 5 × 106 splenic T cells from diabetic mice together with 5 × 106 splenic T cells from CTB-treated mice (open triangles), 5 × 106 splenic T cells from CTB-insulin-treated mice (open squares), 5 × 106 CD4+ T cells from CTB-insulin-treated mice (dark squares) or 5 × 106 CD8+ T cells from CTB-insulin-treated mice (dark circles).
Analysis of T cell responses
In order to understand the mechanisms of protection induced by nasal administration of CTB-insulin, we analysed T cell immune responses both after an unspecific T cell stimulation and after insulin immunization. Proliferation of anti-CD3 anti-CD28 stimulated T cells isolated 4 or 24 h after nasal administration of CTB-insulin from spleens, pancreatic (PLN) and cervical (CLN) lymph nodes are presented in Fig. 5. In the spleen of tolerized animals, CTB-insulin was associated with an increase of the proliferative rate in comparison to CTB both 4 h (51 642 ± 7444 cpm versus 36 234 ± 462 cpm, P = 0·025) and 24 h (217 295 ± 8594 cpm versus 196 237 ± 2507 cpm, P = 0·012) after CTB-insulin administration. More importantly, CTB-insulin induced a strong proliferative response at 24 h in the PLN (121 385 ± 37 913 cpm versus 3931 ± 2606 cpm, P = 0008) while CTB gave no response. This difference was not observed in the CLN. These changes were associated with modification in cytokine content from culture supernatants (Table 1). We observed a significant increase of IL-4 (28 ± 23 versus 8 ± 10 pg/5·105 cells, P = 0·03) and IFN-γ (33 483 ± 16 078 versus 333 ± 57 pg/5·105 cells, P = 0·02) secretion in the PLN 24 h after nasal CTB-insulin treatment in comparison to CTB administration.
Fig. 5.

Nasal administration of CTB-insulin increased the proliferation of T cells in the PLN; 4 h or 24 h after nasal administration of CTB (white) or CTB-insulin (black), we measured T cell proliferation on plate-bound anti-CD3 anti-CD28 antibodies in the spleen, pancreatic (PLN) and cervical (CLN) lymph nodes. *P < 0·05, **P < 0·01
Table 1.
Cytokine secretion of T cells from the spleen, the PLN and the CLN isolated 4 h or 24 h after CTB or CTB-insulin treatment. Results were obtained from nine mice per experimental group during three independent experiments. For IFN-γ, only one representative experiment is presented
| Spleen | PLN | CLN | |||||||
|---|---|---|---|---|---|---|---|---|---|
| CTB-INS | CTB | P | CTB-INS | CTB | P | CTB-INS | CTB | P | |
| IL-4(pg/5·105 cells) | |||||||||
| 4 h | 233 ± 166 | 227 ± 72 | n.s. | 48 ± 73 | 17 ± 22 | n.s. | 26 ± 19 | 23 ± 23 | n.s. |
| 24 h | 145 ± 59 | 134 ± 37 | n.s. | 28 ± 23 | 8 ± 10 | 0·03 | 15 ± 12 | 47 ± 58 | n.s. |
| IL-10 (pg/5·105 cells) | |||||||||
| 4 h | 1046 ± 376 | 807 ± 306 | n.s. | 40 ± 29 | 54 ± 72 | n.s. | 59 ± 17 | 58 ± 15 | n.s. |
| 24 h | 923 ± 339 | 986 ± 287 | n.s. | 43 ± 22 | 36 ± 26 | n.s. | 73 ± 42 | 127 ± 156 | n.s. |
| IFN-γ (pg/5·105 cells) | |||||||||
| 4 h | 17218 ± 922 | 17585 ± 3138 | n.s. | 279 ± 352 | 0 | n.s. | 13810 ± 5725 | 14650 ± 677 | n.s. |
| 24 h | 109950 ± 1125 | 109316 ± 1133 | n.s. | 33483 ± 16 078 | 333 ± 57 | 0·02 | 110600 ± 0 | 107216 ± 5860 | n.s. |
| TGF-β (pg/5·105 cells) | |||||||||
| 4 h | 1417 ± 90 | 1392 ± 230 | n.s. | 834 ± 188 | 932 ± 183 | n.s. | 1159 ± 117 | 1102 ± 145 | n.s. |
| 24 h | 1444 ± 191 | 1393 ± 219 | n.s. | 985 ± 114 | 853 ± 165 | n.s. | 1189 ± 151 | 1140 ± 128 | n.s. |
To address the question of the specificity of T cell responses, we evaluated the influence of nasal treatment on the response to immunization with insulin or ovalbumin (OVA) at the base of the tail. As shown in Fig. 6, T cell response to insulin immunization (8848 versus 3557 cpm, P = 0·002, mean proliferation index = 4·93) was completely abrogated by CTB-insulin treatment (3657 versus 2624 cpm, P = n.s., mean proliferation index = 1·66 and 3657 versus 8848 cpm, P < 0·001). However, response to OVA immunization (25 483 versus 6074 cpm, P < 0·00001, mean proliferation index = 4·27) was maintained after nasal CTB-insulin administration (14278 versus 4644 cpm, P < 0·0001, mean proliferation index = 4·20), suggesting an insulin-specific effect. Furthermore, this proliferation was correlated with the amount of IL-2 from culture supernatants of splenic T cells or lymph nodes. As shown in Fig. 7, immunization with OVA in CTB-insulin tolerized mice led to an increase in IL-2 secretion after an in vivo challenge with OVA but not with insulin. In contrast, immunization with insulin in CTB-insulin tolerized mice did not produce such an IL-2 increase.
Fig. 6.

Nasal administration of CTB-insulin abolished the proliferative response to insulin immunization and not OVA immunization; 4-week-old NOD female were untreated (control) or nasally treated with 1 µg of CTB-insulin (nasal treatment). Ten days later, the mice were immunized with 50 µg of porcine insulin (a) or 50 µg of OVA (b). The basal proliferative response of splenic T cells (in white) was compared with the response to 20 µg/ml insulin (a) or OVA (b) (in black) 8 days after the immunization. **P < 0·0001.
Fig. 7.

Nasal administration of CTB-insulin abolished IL-2 response to insulin immunization and not OVA immunization; 4-week-old NOD females were nasally treated with 1 µg of CTB-insulin and immunized 10 days later with 50 µg of porcine insulin (a) or 50 µg of OVA (b). IL-2 secretion was evaluated in culture supernatants of draining lymph nodes with insulin (in white) or OVA (in black).
DISCUSSION
In the present study, we investigated the tolerogenic properties of CTB-insulin administrated by nasal route on the autoimmune diabetes in NOD mice. We demonstrated that nasal administration of 1 µg of this conjugate delayed the incidence of spontaneous diabetes and prevented diabetes transfer. CTB-insulin administration resulted in an active cellular mechanism of protection that reduced the capacity of diabetogenic T cells to transfer the disease and abrogated systemic T cell response to insulin.
Tolerance induction using mucosal delivery of auto-antigens has been obtained successfully in different experimental models of autoimmune diseases, depending on the dose of antigen, route of administration and timing of treatment with regard to disease onset. In this study, we found that CTB-insulin administration via the nasal route is highly effective in protecting NOD mice against autoimmune diabetes. Furthermore, a lower dose of the conjugate was needed to induce tolerance by the nasal route than by the oral route [23]. This finding has already been reported in collagen-induced arthritis (CIA) [7] and encephalomyelitis (EAE) [6]. The better efficacy of the nasal to the oral delivery could be that nasally administered antigens have direct access to the antigen-presenting cells from the nasal-associated lymphoid tissue (NALT) [8,25,26]. This is in contrast to orally administered antigens, which are exposed to gastric and intestinal proteases before reaching the gut associated lymphoid tissue (GALT) in the small intestine [6]. CTB-insulin-mediated protection occurred in a narrow therapeutic window as doses equal to or above 4 µg did not decrease both spontaneous and accelerated forms of diabetes as well as doses below or equal to 0·5 µg. However, we did not observe a dose-dependent acceleration of diabetes transfer in non-protected animals.
Using adoptive cotransfer experiments, we demonstrated that CTB-insulin induced an active mechanism of protection, which involved regulatory CD4+ T cells. It has been reported previously that high amounts of insulin administered by aerosol induced regulatory T cells from the CD8 phenotype [18]. Our results with CTB-insulin excluded the role of γδ T cells that were described when insulin was administered to the respiratory tract using an aerosol [18] or nasally [22]. Contamination of the gut by antigen swallowing as an explanation for the involvement of CD4 cells is unlikely, as doses administered nasally were four times lower to those necessary to obtain a protection using the oral route [27]. Consistent with our findings, splenic CD4+ but not CD8+ T cells from GAD65-derived peptides treated mice inhibited the adoptive transfer of diabetes [20]. In this model, the intranasal administration of GAD65 peptides induced a diversion of the Th1 response towards a Th2 profile.
RT-PCR analysis of the pancreatic glands from protected animals revealed a heterogeneous pattern with an increase in the expression levels of TGF-β and IL-10 RNAs but no differences in IFN-γ and IL-4 levels. These patterns differed from that obtained after oral CTB-insulin administration [27], where the amounts of IL-4 transcripts were increased in CTB-insulin treated animals. However, our RT-PCR analysis was performed 50 days after cell transfer and we cannot exclude differences at earlier stages of analysis.
Antigen-driven immune responses are probably reorientated by CTB. Like cholera toxin (CT), CTB may increase the amount of nasally administrated antigen that crossed the mucosal surface [28]. Alternatively, CTB can enhance the ability of dendritic cells present in the NALT to process antigen [29,30]. CTB, like CT, may also induce dendritic cell maturation and migration through the expression of chemokine receptors [31]. As suggested elsewhere, this effect on the antigen-presenting cells allows the activation of CD4 cells and induces a Th2 response [32,33]. Several examples of CD4 T cell responses following nasal administration of antigens or peptides have been observed in different experimental models. Tolerance to EAMG by nasal administration of acetylcholine receptor (AchR) also involved antigen-specific CD4+ Th3 cells producing TGF-β [14,34]. These distinct mechanisms involved, depending on the model, may reflect the diversity of the protective immune responses that can be induced by nasal delivery of antigens.
To elucidate the modifications induced directly in the nasally treated mice, we investigated T cell activation and cytokine production following nasal administration of CTB-insulin in different compartments between nasal mucosa and pancreatic islets.
Our results indicated that nasal CTB-insulin enhanced the proliferation rate of T cells from spleen and pancreatic lymph nodes on plate-bound CD3–CD28 antibodies. This observation was not associated with any change in lymphocyte subpopulations. Interestingly, this effect was observed in the spleen 4 h after antigen administration but needed 24 h to occur in the pancreatic lymph nodes. In contrast to spleen and cervical lymph nodes, CTB administration did not induce a proliferative response in the pancreatic lymph nodes, which outlined the specificity of the response to CTB-insulin in this lymphoid organ. Antibody stimulated cells from CTB-insulin treated mice produced more IL-4 and more IFN-γ in the pancreatic lymph nodes while no significant changes were noticed in the other lymphoid organs for IL-4, IL-10, TGF-β and IFN-γ. This observation is reminiscent to what has been recently reported in a model of MHC class I deficient islet grafts, where both IL-4 and IFN-γ were increased in a non-destructive peri-insular infiltrate [35]. Furthermore, Kweon et al. [36] demonstrated the lack of orally induced systemic unresponsiveness in IFN-γ knock-out mice. These findings indicate clearly a central role for IFN-γ in the induction and maintenance of mucosally induced tolerance. It is possible that transient activation of Th1-like cells may precede the induction of regulatory elements. This phenomenon has also been observed in other studies, where transient secretion of IFN-γ preceded the development of tolerance [37].
Using two unrelated antigens, we also demonstrated that nasal antigen treatment was able to abrogate T cell response to systemic immunization specifically to the antigen conjugated to CTB, i.e. insulin. This finding is essential in the perspectives of human application in order to target specifically the type of immune responses.
Our study showed that the nasal route of antigen administration is an attractive strategy for the prevention of autoimmune diabetes. Conjugation of insulin to CTB induced active mechanisms similar to that observed with the oral route. It appears that dose, frequency of intranasal antigen administration and timing of antigen delivery with regard to disease onset are critical determinants of tolerance induction and subsequent suppression of T cell-mediated autoimmune disease [38]. In addition, previous studies using orally administered antigens outlined the risk of activation of immunity instead of tolerance induction [39], and the possibility to worsen an ongoing autoimmune disease [40]. In spite of these limitations, clinical trials using inhaled insulin to prevent diabetes are already being conducted in Finland (DIPP project) and Australia (INIT trial), despite the controversial explanations of the mechanisms that trigger its effect. The importance of dose-dependent effects and the relatively narrow window of therapeutic efficacy have to be taken into consideration with regard to prevention trials in high-risk individuals using nasal insulin.
Acknowledgments
We are indebted to Cecil Czerkinsky for active and valuable support, Annie Durand and Anne Stefanutti for excellent technical assistance and Diane Le Quynh for managing the NOD colony. This work was supported in part by a grant from ALFEDIAM-Novo-Nordisk.
REFERENCES
- 1.Bach JF. Insulin-dependent diabetes mellitus as an autoimmune disease. Endocr Rev. 1994;15:516–42. doi: 10.1210/edrv-15-4-516. [DOI] [PubMed] [Google Scholar]
- 2.Cahill GF, Jr, McDevitt HO. Insulin-dependent diabetes mellitus: the initial lesion. N Engl J Med. 1981;304:1454–65. doi: 10.1056/NEJM198106113042403. [DOI] [PubMed] [Google Scholar]
- 3.Kishimoto H, Sprent J. A defect in central tolerance in NOD mice. Nat Immunol. 2001;2:1025–31. doi: 10.1038/ni726. [DOI] [PubMed] [Google Scholar]
- 4.Ridgway WM, Fasso M, Lanctot A, Garvey C, Fathman CG. Breaking self-tolerance in nonobese diabetic mice. J Exp Med. 1996;183:1657–62. doi: 10.1084/jem.183.4.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiao BG, Link H. Mucosal tolerance: a two-edged sword to prevent and treat autoimmune diseases. Clin Immunol Immunopathol. 1997;85:119–28. doi: 10.1006/clin.1997.4432. [DOI] [PubMed] [Google Scholar]
- 6.Bai XF, Link H. Nasal tolerance induction as a potential means of immunotherapy for autoimmune diseases: implications for clinical medicine. Clin Exp Allergy. 2000;30:1688–96. doi: 10.1046/j.1365-2222.2000.00972.x. [DOI] [PubMed] [Google Scholar]
- 7.Higuchi K, Kweon MN, Fujihashi K, McGhee JR, Kiyono H. Comparison of nasal and oral tolerance for the prevention of collagen induced murine arthritis. J Rheumatol. 2000;27:1038–44. [PubMed] [Google Scholar]
- 8.Kuper CF, Koornstra PJ, Hameleers DM, et al. The role of nasopharyngeal lymphoid tissue. Immunol Today. 1992;13:219–24. doi: 10.1016/0167-5699(92)90158-4. [DOI] [PubMed] [Google Scholar]
- 9.Metzler B, Wraith DC. Inhibition of experimental autoimmune encephalomyelitis by inhalation but not oral administration of the encephalitogenic peptide: influence of MHC binding affinity. Int Immunol. 1993;5:1159–65. doi: 10.1093/intimm/5.9.1159. [DOI] [PubMed] [Google Scholar]
- 10.Li HL, Liu JQ, Bai XF, van der Meide PH, Link H. Dose-dependent mechanisms relate to nasal tolerance induction and protection against experimental autoimmune encephalomyelitis in Lewis rats. Immunology. 1998;94:431–7. doi: 10.1046/j.1365-2567.1998.00526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.al-Sabbagh A, Nelson PA, Akselband Y, Sobel RA, Weiner HL. Antigen-driven peripheral immune tolerance. suppression of experimental autoimmmune encephalomyelitis and collagen-induced arthritis by aerosol administration of myelin basic protein or type II collagen. Cell Immunol. 1996;171:111–9. doi: 10.1006/cimm.1996.0180. [DOI] [PubMed] [Google Scholar]
- 12.Im S, Barchan D, Fuchs S, Souroujon MC. Mechanism of nasal tolerance induced by a recombinant fragment of acetylcholine receptor for treatment of experimental myasthenia gravis. J Neuroimmunol. 2000;111:161–8. doi: 10.1016/s0165-5728(00)00395-7. [DOI] [PubMed] [Google Scholar]
- 13.Barchan D, Souroujon MC, Im SH, Antozzi C, Fuchs S. Antigen-specific modulation of experimental myasthenia gravis: nasal tolerization with recombinant fragments of the human acetylcholine receptor alpha-subunit. Proc Natl Acad Sci USA. 1999;96:8086–91. doi: 10.1073/pnas.96.14.8086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi FD, Li H, Wang H, et al. Mechanisms of nasal tolerance induction in experimental autoimmune myasthenia gravis: identification of regulatory cells. J Immunol. 1999;162:5757–63. [PubMed] [Google Scholar]
- 15.Garcia G, Komagata Y, Slavin AJ, Maron R, Weiner HL. Suppression of collagen-induced arthritis by oral or nasal administration of type II collagen. J Autoimmun. 1999;13:315–24. doi: 10.1006/jaut.1999.0320. [DOI] [PubMed] [Google Scholar]
- 16.Chu CQ, Londei M. Differential activities of immunogenic collagen type II peptides in the induction of nasal tolerance to collagen-induced arthritis. J Autoimmun. 1999;12:35–42. doi: 10.1006/jaut.1998.0255. [DOI] [PubMed] [Google Scholar]
- 17.Maron R, Melican NS, Weiner HL. Regulatory Th2-type T cell lines against insulin and GAD peptides derived from orally- and nasally-treated NOD mice suppress diabetes. J Autoimmun. 1999;12:251–8. doi: 10.1006/jaut.1999.0278. [DOI] [PubMed] [Google Scholar]
- 18.Harrison LC, Dempsey-Collier M, Kramer DR, Takahashi K. Aerosol insulin induces regulatory CD8 gamma delta T cells that prevent murine insulin-dependent diabetes. J Exp Med. 1996;184:2167–74. doi: 10.1084/jem.184.6.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramiya VK, Shang XZ, Wasserfall CH, Maclaren NK. Effect of oral and intravenous insulin and glutamic acid decarboxylase in NOD mice. Autoimmunity. 1997;26:139–51. doi: 10.3109/08916939708994736. [DOI] [PubMed] [Google Scholar]
- 20.Tian J, Atkinson MA, Clare-Salzler M, et al. Nasal administration of glutamate decarboxylase (GAD65) peptides induces Th2 responses and prevents murine insulin-dependent diabetes. J Exp Med. 1996;183:1561–7. doi: 10.1084/jem.183.4.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daniel D, Wegmann DR. Protection of nonobese diabetic mice from diabetes by intranasal or subcutaneous administration of insulin peptide B-(9–23) Proc Natl Acad Sci USA. 1996;93:956–60. doi: 10.1073/pnas.93.2.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanninen A, Harrison LC. Gamma delta T cells as mediators of mucosal tolerance: the autoimmune diabetes model. Immunol Rev. 2000;173:109–19. doi: 10.1034/j.1600-065x.2000.917303.x. [DOI] [PubMed] [Google Scholar]
- 23.Bergerot I, Ploix C, Petersen J, et al. A cholera toxoid–insulin conjugate as an oral vaccine against spontaneous autoimmune diabetes. Proc Natl Acad Sci USA. 1997;94:4610–4. doi: 10.1073/pnas.94.9.4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ploix C, Bergerot I, Fabien N, Perche S, Moulin V, Thivolet C. Protection against autoimmune diabetes with oral insulin is associated with the presence of IL-4 type 2 T-cells in the pancreas and pancreatic lymph nodes. Diabetes. 1998;47:39–44. doi: 10.2337/diab.47.1.39. [DOI] [PubMed] [Google Scholar]
- 25.Metzler B, Wraith DC. Inhibition of T-cell responsiveness by nasal peptide administration: influence of the thymus and differential recovery of T-cell-dependent functions. Immunology. 1999;97:257–63. doi: 10.1046/j.1365-2567.1999.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Metzler B, Anderton SM, Manickasingham SP, Wraith DC. Kinetics of peptide uptake and tissue distribution following a single intranasal dose of peptide. Immunol Invest. 2000;29:61–70. doi: 10.3109/08820130009105145. [DOI] [PubMed] [Google Scholar]
- 27.Ploix C, Bergerot I, Durand A, Czerkinsky C, Holmgren J, Thivolet C. Oral administration of cholera toxin B-insulin conjugates protects NOD mice from autoimmune diabetes by inducing CD4+ regulatory T-cells. Diabetes. 1999;48:2150–6. doi: 10.2337/diabetes.48.11.2150. [DOI] [PubMed] [Google Scholar]
- 28.Sun JB, Holmgren J, Czerkinsky C. Cholera toxin B subunit: an efficient transmucosal carrier–delivery system for induction of peripheral immunological tolerance. Proc Natl Acad Sci USA. 1994;91:10795–9. doi: 10.1073/pnas.91.23.10795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Porgador A, Staats HF, Itoh Y, Kelsall BL. Intranasal immunization with cytotoxic T-lymphocyte epitope peptide and mucosal adjuvant cholera toxin: selective augmentation of peptide-presenting dendritic cells in nasal mucosa-associated lymphoid tissue. Infect Immun. 1998;66:5876–81. doi: 10.1128/iai.66.12.5876-5881.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yingzi C, Weaver CT, Elson CO. The mucosal adjuvanticity of cholera toxin involves enhancement of costimulatory activity by selective up-regulation of B7.2 expression. J Immunol. 1997;159:5301–8. [PubMed] [Google Scholar]
- 31.Gagliardi MC, Sallusto F, Marinaro M, Langenkamp A, Lanzavecchia A, De Magistris MT. Cholera toxin induces maturation of human dendritic cells and licences them for Th2 priming. Eur J Immunol. 2000;30:2394–403. doi: 10.1002/1521-4141(2000)30:8<2394::AID-IMMU2394>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 32.Braun MC, He J, Wu CY, Kelsall BL. Cholera toxin suppresses interleukin (IL)-12 production and IL-12 receptor beta1 and beta2 chain expression. J Exp Med. 1999;189:541–52. doi: 10.1084/jem.189.3.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marinaro M, Staats HF, Hiroi T, et al. Mucosal adjuvant effect of cholera toxin in mice results from induction of T helper 2 (Th2) cells and IL-4. J Immunol. 1995;155:4621–9. [PubMed] [Google Scholar]
- 34.Xiao BG, Zhang GX, Shi FD, Ma CG, Link H. Decrease of LFA-1 is associated with upregulation of TGF-beta in CD4 (+) T cell clones derived from rats nasally tolerized against experimental autoimmune myasthenia gravis. Clin Immunol Immunopathol. 1998;89:196–204. doi: 10.1006/clin.1998.4537. [DOI] [PubMed] [Google Scholar]
- 35.Prange S, Zucker P, Jevnikar AM, Singh B. Transplanted MHC class I-deficient nonobese diabetic mouse islets are protected from autoimmune injury in diabetic nonobese recipients. Transplantation. 2001;71:982–5. doi: 10.1097/00007890-200104150-00025. [DOI] [PubMed] [Google Scholar]
- 36.Kweon MN, Fujihashi K, VanCott JL, et al. Lack of orally induced systemic unresponsiveness in IFN-gamma knockout mice. J Immunol. 1998;160:1687–93. [PubMed] [Google Scholar]
- 37.Hoyne GF, Askonas BA, Hetzel C, Thomas WR, Lamb JR. Regulation of house dust mite responses by intranasally administered peptide: transient activation of CD4+ T cells precedes the development of tolerance in vivo. Int Immunol. 1996;8:335–42. doi: 10.1093/intimm/8.3.335. [DOI] [PubMed] [Google Scholar]
- 38.Jiang HR, Taylor N, Duncan L, Dick AD, Forrester JV. Total dose and frequency of administration critically affect success of nasal mucosal tolerance induction. Br J Ophthalmol. 2001;85:739–44. doi: 10.1136/bjo.85.6.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hanninen A, Braakhuis A, Heath WR, Harrison LC. Mucosal antigen primes diabetogenic cytotoxic T-lymphocytes regardless of dose or delivery route. Diabetes. 2001;50:771–5. doi: 10.2337/diabetes.50.4.771. [DOI] [PubMed] [Google Scholar]
- 40.Bai XF, Li HL, Shi FD, et al. Complexities of applying nasal tolerance induction as a therapy for ongoing relapsing experimental autoimmune encephalomyelitis (EAE) in DA rats. Clin Exp Immunol. 1998;111:205–10. doi: 10.1046/j.1365-2249.1998.00467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]