

Abstract
The contribution of CCR6 and phagocyte recruitment to the initiation of T cell responses to a local pathogen is unclear. CD4 T cell activation to an injected soluble antigen occurred rapidly and was completely CCR6-independent. In marked contrast, the tempo of pathogen-specific CD4 T cell activation depended on whether the antigen was secreted or cell-associated. Furthermore, lymph node pathogen-specific CD4 T cell activation required CCR6 and cell migration from the site of infection. Surprisingly, adoptive transfer of wild-type blood phagocytes rescued bacteria-specific T cell activation in CCR6-deficient mice, even when these cells were unable to participate in direct antigen presentation. These data demonstrate that T cell responses to a local bacterial infection follow a distinct tempo, largely determined by bacterial protein secretion, and that CCR6-mediated blood phagocyte recruitment to the site of infection is a critical step in the initiation of pathogen-specific immune responses in skin draining lymph nodes.
Keywords: antigen presentation, bacteria
It is clear that dendritic cells (DCs) play a critical role in the initiation of naïve T cell responses to microbial pathogens (1, 2); however, few studies have directly visualized antigen presentation and pathogen-specific T cell activation simultaneously in vivo (3). We previously demonstrated two distinct “waves” of antigen presentation in the lymph node after injection of a soluble protein (4). The first wave involved presentation of lymph-borne antigen, whereas the second wave was mediated by DCs migrating from the injection site. Antigen presentation and T cell activation to a local bacterial infection are likely to be considerably more complex than injection of single soluble protein (5). It remains unclear whether migrating skin DCs (4, 6), recruited bloodborne DC precursors (7, 8), or lymph node resident DCs (9) are involved in T cell activation to local bacterial infection.
CCL20 is rapidly secreted in response to inflammatory stimuli, attracting CCR6 expressing cell populations, including immature DCs and blood monocytes (8). Although CCR6 is known to play an important role in initiating T cell activation at mucosal surfaces (2, 8), the contribution of CCR6 to T cell activation at other sites of infection has not been examined. Indeed, the kinetics of antigen presentation and T cell activation in response to local infection have not been examined in any detail and it remains unclear whether secreted or cell-associated antigens induce different responses in vivo.
Here, we demonstrate that antigen presentation and T cell activation in response to secreted versus cell-associated bacterial antigens are temporarily distinct, and that CCR6-mediated recruitment of blood phagocytes to the inflammatory site is an essential component of pathogen-specific T cell activation in the draining lymph node. Surprisingly, these recruited blood phagocytes are not required to present antigen directly to lymph node T cells and may simply shuttle antigen to the draining lymph node to increase the efficiency of antigen presentation.
Results
Detection of Bacterial Antigen Presentation in Vitro and in Vivo.
The Y-Ae antibody recognizes a peptide from Eα bound to I-Ab and can be used to examine antigen presentation directly in vivo (4). To examine Salmonella antigen presentation, Salmonella strains expressing red fluorescent protein (RFP), or EαRFP were constructed, and both fluoresced red [supporting information (SI) Fig. 8]. A significant proportion of CD11c+ splenic DCs were Y-Ae+, 24 h after in vitro incubation with purified EαRFP or Heat-killed Salmonella-EαRFP, but not with Salmonella-RFP (Fig. 1A, in vitro). Furthermore, Y-Ae+ CD11c+ cells were detected in the draining lymph node of mice, 24 h after injection of EαRFP or heat-killed Salmonella-EαRFP (Fig. 1A, in vivo). These data demonstrate that the Y-Ae antibody can detect Salmonella antigen presentation in vitro and in vivo.
Fig. 1.

Detection of bacterial antigen presentation in vitro and in vivo. (A) Spleen cells were isolated from C57BL/6 mice and incubated with soluble EαRFP, heat-killed Salmonella-RFP, or heat-killed Salmonella-EαRFP for 24 h before staining with Y-Ae and an antibody specific for CD11c (in vitro). Alternatively, C57BL/6 mice were immunized s.c. with soluble EαRFP, heat-killed Salmonella-RFP, or heat-killed Salmonella-EαRFP, and draining lymph nodes were recovered 24 h later and stained with Y-Ae, isotype control, or anti-CD11c (in vivo). Numbers show the percentage of Y-Ae+ cells within the boxed gate. Data are representative of three different experiments. (B) Cervical lymph nodes were harvested from C57BL/6 mice 14 h after s.c. injection of 2 × 105 Salmonella-RFP or Salmonella-EαRFP and frozen sections fixed and stained with Y-Ae, isotype control, and anti-B220 and analyzed by confocal microscopy. Images depict Y-Ae (red) and B220 (blue) staining in the same lymph node sections. The optical thickness of each image is 9 μm.
Detection of Bacterial Antigen-Presenting Cells During Live Infection.
In marked contrast, no Y-Ae+ cells were detected by flow cytometry in the draining lymph nodes after injection of live Salmonella-EαRFP, in the Peyer's patches after oral infection, or in the spleen after i.v. infection (data not shown). As an alternative approach, we examined Y-Ae staining by immuno-fluorescent staining of lymph node sections. Although background fluorescence near the capsule was detected in most sections (Fig. 1B, Isotype staining), Y-Ae staining was detected in the draining lymph nodes 14 h after s.c. infection with Salmonella-EαRFP (Fig. 1B, Salmonella-EαRFP), but not with Salmonella-RFP (Fig. 1B, Salmonella-RFP). At this time point, Y-Ae staining was localized immediately below B cell follicles.
Delayed Bacterial Antigen Presentation Requires Migration from the Site of Infection.
We examined the kinetics of Salmonella antigen presentation, using histological staining. There was little or no detection of Eα/I-Ab complex in the draining lymph nodes of mice infected for 4 or 8 h with Salmonella-EαRFP (Fig. 2 A and B). However, Eα/I-Ab complex was detected throughout the paracortex 24 h after infection (Fig. 2C). YAe staining was only detected in lymph nodes of mice with an intact injection site (Fig. 2D) and was ablated if the injection site was removed (Fig. 2E).
Fig. 2.

Bacterial antigen presentation requires cell migration from the site of infection. (A–C) C57BL/6 mice were infected s.c. with Salmonella-EαRFP for 4 (A), 8 (B), or 24 (C) h before cervical lymph nodes were recovered, and Eα:I-Ab complexes and B cell follicles were detected by staining with Y-Ae and anti-B220. (D and E) In a separate experiment, C57BL/6 mice were infected s.c. with Salmonella-EαRFP for 24 h with an intact injection site (D) or after injection site removal 1 h after infection (E). Cervical lymph nodes were recovered and processed as above. The images depict the location of pEα:I-Ab complexes (red) and B cell-follicles (blue). The optical thickness of each image is 9 μm.
Kinetics of Bacterial T Cell Activation Are Regulated by Bacterial Antigen Expression.
We used Eα-specific TEa T cell antigen receptor (TCR) transgenic mice to confirm these histological observations. C57BL/6 mice were adoptively transferred with 5- and 6-carboxyl-fluorescein diacetate succinimidyl ester (CFSE)-labeled TEa T cells and injected with purified EαRFP or infected with Salmonella-EαRFP. TEa T cells had undergone several rounds of cell division and increased expression of CD11a in both groups of mice 3 days later (Fig. 3A). Thus, TEa T cells proliferate similarly whether antigen is injected as a soluble protein, or expressed by live bacteria.
Fig. 3.

Bacteria-specific CD4 T cell activation kinetics are determined by bacterial antigen secretion. C57BL/6 mice were adoptively transferred with 2 × 106 CFSE-labeled, CD90.1 congeic, TEa, or SM1 T cells and infected s.c. the next day with 2 × 105 Salmonella-RFP, Salmonella-EαRFP, and Salmonella-FlagEα or immunized with 50 μg of EαRFP. (A) Three days after infection or immunization, cervical lymph nodes were harvested and stained with antibodies specific for CD4 and CD90.1 to detect TEa T cells. Plots show CD11a expression and CFSE-dye dilution after gating on CD90.1 TEa T cells from uninfected mice (Transfer Only), mice infected with Salmonella-EαRFP, or mice immunized with EαRFP. (B) Twelve hours after infection or immunization, cervical lymph nodes were harvested and stained with antibodies specific for CD4 and CD90.1 to detect TEa T cells. Plots show surface expression of CD25 and CD69 after gating on CD90.1 TEa T cells from uninfected mice (Transfer Only), mice infected with Salmonella-RFP, Salmonella-EαRFP, or Salmonella-FlagEα or immunized with EαRFP. (C) Twelve hours after infection or immunization, cervical lymph nodes were harvested and stained with antibodies specific for CD4 and CD90.1 to detect SM1 T cells. Plots show surface expression of CD25 and CD69 after gating on CD90.1 SM1 T cells from uninfected mice (Transfer Only), mice infected with Salmonella-EαRFP or Salmonella-FlagEα or immunized with EαRFP. (D) Twelve hours after infection or immunization, cervical lymph nodes were harvested and stained with antibodies specific for CD4 and CD90.1 to detect TEa T cells. Plots show surface expression of CD25 and CD69 after gating on CD90.1 SM1 T cells from uninfected mice (Transfer Only), mice infected with Salmonella-RFP or Salmonella-EαRFP or immunized with EαRFP or a mix of Salmonella-EαRFP and EαRFP. (E) Three days after infection, cervical lymph nodes were harvested and stained with antibodies specific for CD4 and CD90.1 to detect TEa T cells. Plots show CD11a expression and CFSE-dye dilution after gating on CD90.1 TEa T cells from uninfected mice (Transfer Only) or mice infected with Salmonella-EαRFP after administration of 20 μl of 100 nM BW245C or vehicle control.
Less than 1% of TEa T cells were CD25+CD69+ in the lymph nodes of naïve mice (Fig. 3B, Transfer Only). Twelve hours after injection of soluble EαRFP, a large proportion of TEa cells were activated to increase surface expression of CD25 and CD69 (Fig. 3B, Soluble EαRFP). In contrast, TEa T cells in infected mice did not increase expression of CD25 or CD69 (Fig. 3B, Salmonella EαRFP and Salmonella RFP). Indeed, activated TEa T cells were detected as early as 4 h after administration of soluble EαRFP, with peak expression occurring at 12 h (Fig. 4A). In contrast, TEa T cells did not increase expression of CD69 at any point during the first 24 h after Salmonella-EαRFP infection (Fig. 4A). These data are in broad agreement with the Y-Ae histological staining in infected mice and together suggest that rapid antigen presentation occurs after administration of soluble EαRFP but does not occur in response to local bacterial infection.
Fig. 4.

Early kinetics of T cell activation after infection or immunization. C57BL/6 mice were adoptively transferred with 2 × 106 CFSE-labeled, CD90.1 congeic, TEa or SM1 T cells and infected s.c. the following day with 2 × 105 Salmonella-RFP or Salmonella-EαRFP or immunized with 50 μg of EαRFP. At various time points after infection or immunization, cervical lymph nodes were harvested and stained with antibodies specific for CD4 and CD90.1 to identify TEa or SM1 T cells. Each point shows the mean percentage of CD69+ TEa (A) or SM1 (B) T cells in infected or immunized mice. Each time point shows the mean ± SD of three mice per group and is representative of two individual experiments.
Bacteria Do Not Inhibit Antigen Presentation in Response to Soluble Antigen Administration.
It has been reported that Salmonella can suppress T cell activation in the draining lymph node (10). We examined whether bacterial infection can inhibit early activation of TEa T cells in response to soluble antigen administration. Injection of EαRFP caused rapid TEa activation in the draining lymph node, whereas infection with Salmonella-EαRFP did not (Fig. 3D). However, TEa activation after coinjection of EαRFP and Salmonella was indistinguishable from that observed in mice administered EαRFP alone (Fig. 3D).
Migration from the Site of Infection Is Required for Bacteria-Specific T Cell Activation.
We also examined the importance of migration from the injection site by using the synthetic prostaglandin analog BW245C, which inhibits cell migration (9, 11). Injection of BW245C severely reduced the proliferation of TEa T cells in the draining lymph node, 3 days after infection with Salmonella-EαRFP (Fig. 3E). Thus, bacteria-specific CD4 T cell proliferation in the draining lymph node requires migration from the site of infection.
The Tempo of T Cell Activation Is Determined by Bacterial Antigen Location.
It seemed plausible that the delay in TEa T cell activation after Salmonella-EαRFP infection could be attributed to the cell-associated nature of EαRFP, and a secreted bacterial protein might be presented more rapidly. Flagellin is one of the most abundantly secreted Salmonella proteins (12, 13) and the target of SM1 TCR transgenic T cells (14). In contrast to TEa T cells, SM1 T cells displayed increased CD69 expression, 12 h after infection with Salmonella-EαRFP (Fig. 3C). Indeed, activation of SM1 T cells was detected as early as 4 h (Fig. 4B), very similar to the kinetics observed after soluble protein injection (Fig. 4A). We tested the hypothesis that antigen secretion determines the tempo of antigen presentation by infecting mice with a Salmonella strain expressing the Eα peptide embedded in flagellin (Salmonella-FlagEα). In contrast to our analysis of Salmonella-EαRFP, infection with Salmonella-FlagEα induced early CD25 and CD69 expression by both TEa and SM1 T cells (Fig. 3 B and C). Thus, the early kinetics of bacterial-specific T cell activation is determined by whether antigens are secreted or cell-associated.
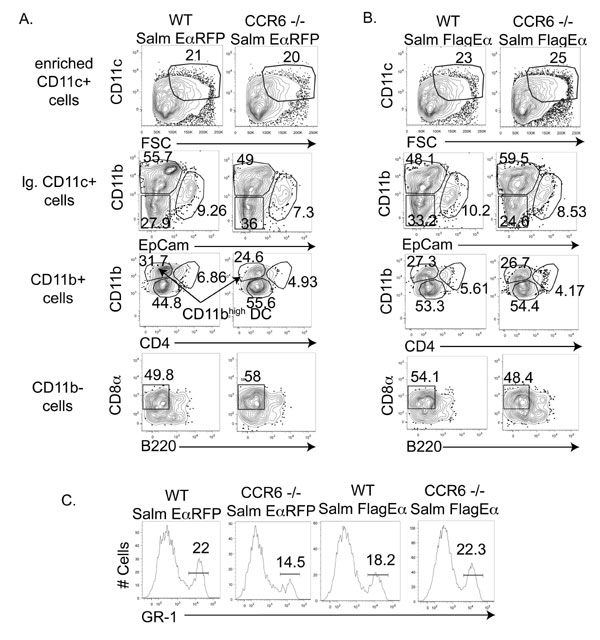
CCR6-Deficiency Does Not Alter Lymph Node DC Populations Under Steady State or Inflammatory Conditions.
We detected no obvious differences in DC populations in the lymph node of WT or CCR6-deficient mice (SI Fig. 9). Furthermore, we examined DC populations in the draining lymph node of WT and CCR6-deficient mice infected with either Salmonella-EαRFP or Salmonella-FlagEα and detected no differences (SI Fig. 10).
CCR6 Is Required for Bacterial-Specific T Cell Responses in the Draining Lymph Node and Is Rescued by Transfer of Blood Phagocytes.
We examined whether CCR6 was required for the activation of T cells after administration of soluble protein or bacterial infection. Three days after soluble EαRFP administration, TEa T cells had completed a similar number of cell divisions in both WT and CCR6-deficient recipients (Fig. 5A), indicating that CCR6 is not required for T cell proliferation to soluble protein. In marked contrast, CCR6 was required for optimal CD4 T cell responses to bacterial infection. In WT recipients, TEa T cells had undergone several rounds of cell division in the draining lymph node after Salmonella-EαRFP infection, but this proliferation was significantly diminished in CCR6-deficient recipients (Fig. 5A). If bacteria were heat-killed before injection, TEa T cells proliferated similarly in response to Salmonella-EαRFP in WT and CCR6 recipients (Fig. 5B). Thus, CCR6 is required for optimal T cell activation to a cell associated bacterial antigen expressed by live bacteria.
Fig. 5.

Induction of CD4 T cell responses to live bacteria requires CCR6. Wild-type or CCR6-deficient mice were adoptively transferred with 2 × 106 CFSE-labeled TEa T cells, and immunized s.c. with 50 μg of EαRFP or infected s.c. with 2 × 105 Salmonella-EαRFP (A) or immunized with 2 × 105 Heat-killed Salmonella-EαRFP or infected with 2 × 105 live Salmonella-EαRFP (B). Three days after infection or immunization, cervical lymph nodes were harvested and stained with antibodies specific for CD4 and CD90.1 to detect TEa T cells. Plots show CD4 and CD90.1 expression on total lymph node cells or CD11a expression and CFSE-dye dilution after gating on CD4+CD90.1+ TEa T cells from uninfected mice (Transfer Only), mice infected with Salmonella-EαRFP, or mice immunized with EαRFP or heat-killed Salmonella-EαRFP.
Next, we examined whether reduced bacteria-specific TEa responses in CCR6-deficient recipients was affected by the transfer of CCR6-sufficient blood cells. White blood cells were harvested from the blood of WT mice and adoptively transferred into CCR6-deficient mice, 10 min before infection with Salmonella-EαRFP. The transfer of CCR6-sufficient blood cells was sufficient to rescue T cell responses in CCR6-deficient mice (Fig. 6A). To determine which blood leukocyte population was responsible for enhancing T cell responses, we FACS-sorted CD11b+Gr1+, CD11b+Gr1−, and CD11b−Gr1+ blood cells and adoptively transferred these into CCR6-deficient mice before infection. Surprisingly, the transfer of either blood subpopulation partially rescued TEa expansion and CFSE-dye dilution (Fig. 6B), whereas the transfer of blood CD4 T cells did not (data not shown). CD11b+Gr1+ and CD11b+Gr1− blood cells expressed both CCR6 and CCR2 (SI Fig. 11), but none of these populations expressed high levels of CD11c (data not shown).
Fig. 6.

Adoptive transfer of WT blood phagocytes restores T cell expansion and cell division in infected CCR6-deficient mice. Wild-type or CCR6-deficient mice were adoptively transferred with 2 × 106 CFSE-labeled TEa T cells, and immunized s.c. with 50 μg of EαRFP or infected s.c. with 2 × 105 Salmonella-EαRFP. Three days after infection or immunization, cervical lymph nodes were harvested and stained with antibodies specific for CD4 and CD90.1 to detect TEa T cells. Plots show CD11a expression and CFSE-dye dilution after gating on CD90.1 TEa T cells from uninfected mice (Transfer Only), mice infected with Salmonella-EαRFP, or mice immunized with EαRFP. Some groups of CCR6-deficient mice were injected i.v. with, 1 × 106 mononuclear blood leukocytes (A) or purified Gr1+CD11b−, Gr1+CD11b+, or Gr1−CD11b− blood monocytes from WT mice 10 min before infection (B). Expansion plots show detection of TEa T cells 3 days after infection or immunization of WT and CCR6-deficient recipients, some which were injected with sorted cells 10 min before infection. Numbers show the percentage of TEa T cells within each boxed gate. Lower plots show CD11a staining and CFSE dye dilution of gated TEa T cells from the respective TEa expansion plots. Each plot is representative to two to three mice per group and two individual experiments.
Blood Phagocytes Are Recruited to the Site of Infection but Do Not Participate Directly in Antigen Presentation.
Mice were infected with Salmonella, and 12 or 24 h later, the site of infection was removed and processed to examine the cell infiltrate. A population of autofluorescent cells was detected after tissue processing of the injection site from infected and uninfected mice (Fig. 7A). However, at 12 h after infection, a large recruitment of CD11b+Gr1+ and CD11b+Gr1− cells was detected at the site of infection and increased over the next 12 h (Fig. 7A). It seemed possible that this infiltrate captured bacterial antigen and migrated to the lymph node to activate T cell directly, or shuttled bacterial antigen to the lymph node for another cell to activate T cells (9). To discriminate between these possibilities, we adoptively transferred class-II-deficient FACS-sorted blood cells into CCR6-deficient mice. The transfer of class-II deficient blood cells resulted in a similar rescue of TEa cell division in CCR6-deficient mice (Fig. 7B), indicating that recruited blood phagocytes are not required to participate directly in antigen presentation.
Discussion
We have visualized antigen presentation and T cell activation during the early stages of a local bacterial infection. Antigen presentation could be visualized by histological staining but not by flow cytometry, even after enriching for antibody-binding cells (16). It seems likely that antigen presentation fell below the limit of detection for flow cytometry, but was detected in histological staining because of the use of a more sensitive biotinyl-tyramide amplification system. Low levels of presentation during live infection may result from active inhibition of antigen processing by bacteria (17, 18), or increased competition from other bacterial antigens. Either way, we were able to examine the kinetics of presentation in histological sections and confirm that these kinetics parallel bacteria-specific T cell activation in vivo.
Our study demonstrates that the kinetics of antigen presentation and T cell activation during bacterial infection are distinct from soluble antigen administration (4). After bacterial infection, Y-Ae+ cells were first detected at 12–14 h, and peak staining occurred at 24 h. TEa T cells were activated to express CD69 and CD25 within hours of soluble antigen administration, but were CD69−25− during the first 24 h of bacterial infection. These data suggest a greater dependence on cell migration when dealing with a cell-associated bacterial antigen. Indeed, removing the site of infection completely ablated bacterial antigen presentation, and blocking antigen transport with BC245C inhibited bacteria-specific TEa T cell proliferation.
It is possible that antigen dose is responsible for the differential kinetics of T cell activation in response to soluble antigen versus bacteria. However, our data demonstrate that an identical dose of Salmonella engineered to express a soluble or cell-associated antigen can induce a T cell response with substantially different kinetics (Fig. 4). Furthermore, despite the differing tempo of early activation, soluble EαRFP and Salmonella-EαRFP induced very similar TEa CFSE dye dilution profiles at day 3, suggesting that T cells were eventually stimulated to an equivalent degree. We also completed experiments titrating our bacterial challenge dose. Unfortunately, increasing the challenge dose 10-fold caused rapid death of infected mice, whereas decreasing the Salmonella dose 10-fold reduced our ability to reliably detect TEa T cell activation (data not shown). Thus, although we think it unlikely that antigen dose is responsible for these differences in early T cell activation kinetics, we cannot completely rule out this possibility.
Our data demonstrate greater dependence on antigen transport from the site, but this only applies to cell-associated bacterial proteins. A naturally secreted bacterial antigen and Eα peptide imbedded within a secreted bacterial antigen activated T cells with kinetics similar to soluble EαRFP. Thus, the kinetics of T cell activation in response to bacterial antigens can be delineated based on immediate antigen access to lymph fluid. It is tempting to speculate that such temporal differences may alter the nature of T cell responses to secreted versus cell-associated bacterial antigens, and this will be the focus of future study in our laboratory. An alternative possibility is that TLR5 ligation is responsible for these early temporal differences in T cell activation because EαFlag can bind TLR5, whereas EαRFP cannot. Indeed, the TLR11 ligand, profilin, can enhance the efficiency of antigen presentation and T cell activation to proflin epitopes (19). Although we cannot completely exclude this possibility, the rapid activation of TEa T cells in response to soluble EαRFP suggest that the soluble nature of the target antigen is the major determining factor for rapid T cell activation.
A number of reports have described the inhibition of antigen presentation and T cell activation after Salmonella infection (10, 20–23). Our data demonstrate the detection of antigen presentation and T cell activation in the draining lymph node, suggesting that significant inhibition of antigen presentation does not occur in vivo. However, it is possible that our visualization techniques are sensitive enough to detect antigen presentation and T cell activation even in the face of considerable bacterial suppression, and therefore, we do not completely rule out a role for active suppression of immune activation during Salmonella infection.
CCR6 was not required for expansion and proliferation of T cells responding to soluble antigen. Thus, neither waves of lymph node antigen presentation is CCR6-dependent, and is more likely to depended on CCR7 (24). The absence of a requirement for CCR6 after soluble antigen administration is not wholly surprising given that expression of CCL20 is induced by inflammatory stimuli (25). In contrast, presentation of a bacterial cell-associated protein was heavily dependent on CCR6. A requirement for CCR6 during skin bacterial infection is somewhat unexpected and suggests a wider role for this chemokine receptor in microbial immunity than has been previously appreciated.
Reduced bacterial-specific T cell responses in CCR6-deficient recipients were rescued by adoptive transfer of blood phagocytes, and Gr1+CD11b+ were also rapidly recruited to the infection site. These data suggest that CCL20 is induced at the site of infection and mediates CCR6-dependent recruitment of cells from the blood, although other chemokine receptors may also be involved. Although infiltration of phagocytes was required for T cell proliferation to bacterial antigen, MHC class-II expression by these cells was not required. A previous report described the role of infected Gr-1+ neutrophils as a substrate for the cross-priming of CD8+ T cells to nonsecreted Listeria antigens (26). Together, these data suggest that the influx of phagocytes to the site of infection may play an important role in amplifying T cell responses, especially to nonsecreted bacterial antigens.
These data allow us to propose the following model. After local bacterial infection, CCL20 is rapidly produced at the site, attracting blood phagocytes in a CCR6-dependent manner. These phagocytes engulf bacteria and migrate to the lymph node where antigen is transferred to resident DCs that can then mediate T cell activation.
The involvement of a CCR6-dependent, blood-derived phagocyte is somewhat unexpected, because tissue and lymph node contain numerous DCs that can presumably access bacterial antigens directly. It seems possible that this phagocyte population either increases the efficiency of DCs antigen acquisition at the site of infection or simply increases the amount of antigen being delivered to the lymph node. A similar antigen “shuttling” model has been proposed for dermal DCs during CD8 T cell activation (9), and may be a more common process in lymph node T cell activation than previously appreciated.
Materials and Methods
Mice.
C57BL/6 mice were purchased from NCI (Fredrick, MD). SM1 and TEa TCR transgenic mice expressing the Thy1.1 allele have been reported in refs. 14, 27, and 28. CCR6-deficient mice (29) were provided by L. Lefrancois (University of Connecticut Health Center, Farmington, CT). All mice were cared for in accordance with University of Minnesota and National Institutes of Health guidelines.
Plasmids and Template Plasmid Construction.
Plasmids, pKD46, pKD13, and pCP20, were obtained from the E. coli Genetic Stock Center (Yale University, New Haven, CT) (30). DNA from the pKD13 plasmid was cut with BamHI restriction enzyme and sense and antisense Eα peptide oligo flanked by BamHI residues were ligated into the BamHI site. Additional sense and antisense oligos were constructed consisting of three copies of the FLAG epitope flanked by AccI residues. The pKD13Eα plasmid was cut with AccI and FLAGx3 annealed oligos inserted.
Generation of Bacterial Strains.
Epitope-tagging of Salmonella genes has been described (31). A modification of this method was used to introduce internally tagged EαFLAGx3 into the hypervariable region of fliC (Salmonella-FlagEα). Primer pairs were designed which would anneal to the pKD13EαFLAGx3 template plasmid over constant regions and contain 45-nt extensions homologous to the middle variable region of the fliC gene (starting at 575–620 nt) in the forward primer and to a downstream region (862–907 nt of fliC). PCR products were purified and introduced into SL1344 carrying the pKD46 plasmid and recombinants selected by using kanamycin. The kanamycin resistance gene was then flipped out of the inserted EαFLAGx3 sequence by introducing pCP20. The internally tagged fliC gene had an 82-aa deletion in the variable region of the gene and an 81-aa insert consisting of EαFLAGx3 and a residual scar sequence encoding a new 27-residue peptide. Two other recombinant Salmonella strains were generated, expressing EαRFP (Salmonella-EαRFP) or RFP alone (Salmonella-RFP). Salmonella-EαRFP was generated by electroporation of plasmid pTrc-EαRFP carrying a fusion protein of RFP and Eα52−67 (4), into the virulent strain SL12023.
Salmonella Strains, Infection, and Immunization.
Bacteria were grown in LB medium, supplemented with 100 μg/ml Ampicillin (Sigma–Aldrich, St. Louis, MO). At mid-log phase, expression of recombinant protein was induced by IPTG (1 mM) for 7 h, after which bacteria were washed and resuspended in PBS. Typically, 2 × 105 bacteria were injected intradermally in a volume of 25 μl in the tip of the ear, using a Hamilton (Reno, NV) syringe fitted with a 30.5-gauge needle. Soluble EαRFP was produced as described in ref. 4, and injected into the ear in a similar manner.
T Cell Adoptive Transfer.
Spleen and lymph node cells were harvested from TEa or SM1 TCR transgenic mice and 1 × 106 TCR transgenic cells adoptively transferred into recipient mice via the lateral tail vein. The frequency of TCR transgenic T cells was determined by staining an aliquot of cells with antibodies specific for CD4, CD90.1, and Vα2 (TEa), or Vβ2 (SM1). In most experiments, TEa transgenic cells were labeled with CFSE as described in ref. 32.
DC Purification.
Cervical lymph nodes were pooled and digested with collagenase D and EDTA as described in ref. 33. DCs were enriched by CD11c-labeled magnetic bead selection from total lymph node cells (Miltenyi Biotech, Auburn, CA) and stained sequentially with specific antibodies.
Flow Cytometry.
Cervical lymph node cells or purified DCs were washed in HBSS containing 5% FCS and 0.05% Sodium azide before incubation on ice for 30 min in the presence of Fc block (spent 24G2 culture supernatant, 2% rat serum, 2% mouse serum and 0.01% sodium azide) and primary antibodies. FITC-, phycoerytherin (PE)-, CyChrome-, PE-Cy5-, allophycocyanin-, Pacific blue-, Pacific orange-, PE-Cy7-, Alexa 750-, or biotin-conjugated antibodies specific for CD4, CD8, CD11a, CD11b, CD11c, CD25, CD45.1, CD69, CD90.1, B220, Gr-1, EpCam, and MHC-II were purchased from eBioscience (San Diego, CA) or BD Biosciences (San Jose, CA). After staining, cells were washed and fixed in 2% formaldehyde, and data were collected by using a FACSCanto or LSR II (BD Biosciences). All FACS data were analyzed by using FlowJo software (TreeStar, Ashland, OR).
Prostaglandin D2 Analog BW245C Treatment.
In some experiments, 20 μl of prostaglandin D2 analog, BW245C (100 nM; Cayman Chemical, Ann Arbor, MI) or DMSO (vehicle control) was injected intradermally into the ear. After 20 min, mice were inoculated with Salmonella on the BW245C-treated region.
Adoptive Transfer of Blood Phagocytes.
Blood from C57BL/6 mice was diluted 1:1 with PBS, layered over LSM separation medium (Fisher, Pittsburgh, PA), and centrifuged for 30 min, and cells were aspirated from the interface layer. Recovered cells were washed with PBS and residual red blood cells were lysed by using 0.83% NH4Cl. Blood cells were stained on ice, using various antibodies and cell populations recovered by cell sorting, using a FACSAria (BD Biosciences). After sorting, 2 × 104 cells were injected intravenously into recipient CCR6-deficient mice 10 min before ear infection.
Immunohistochemistry.
Cervical lymph nodes were harvested from C57BL/6 mice at various times after ear injection or infection and snap frozen in OCT compound (Tissue-Tek, Hatfield, PA). Tissue sections (9 μm) were cut by using a cryostat, dehydrated in acetone, and stored at −80°C. Slide sections were rehydrated in PBS and endogenous peroxidase, Fc receptor and the available biotin binding sites were blocked as described in ref. 15. Y-Ae staining was detected in sections by sequential addition of biotin-labeled Y-Ae antibody, streptavidin-HRP, biotinyl tyramide amplification, and detection of signal, using streptavidin-Cy5 (Zymed, Carlsbad, CA). B cell follicles were identified by FITC-labeled anti-B220 (BD Biosciences), and DAPI (Molecular Probes, Carlsbad, CA) was used to stain nuclei. Slides were mounted by using Immuno-mount (Thermo-Electron, Pittsburgh, PA) to preserve fluorescence and images acquired by using a Zeiss 510 (Zeiss, Thornwood, NY) or a Bio-Rad (Hercules, CA) MRC-1000 confocal microscope equipped with a krypton/argon laser (Bio-Rad). Separate green and far-red (Cy5) images were collected for each section analyzed. Final image processing was performed by using Photoshop (Adobe, San Jose, CA) or Imaris software (Bitplane, Zurich, Switzerland).
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants AI056172 and AI055743 (to S.J.M.) and AI066018 (to M.K.J.).
Abbreviations
- CFSE
5- and 6-carboxyl-fluorescein diacetate succinimidyl ester
- DC
dendritic cell
- PE
phycoerytherin
- RFP
red fluorescent protein
- TCR
T cell antigen receptor.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0701363104/DC1.
References
- 1.Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, et al. Immunity. 2002;17:211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Salazar-Gonzalez RM, Niess JH, Zammit DJ, Ravindran R, Srinivasan A, Maxwell JR, Stoklasek T, Yadav R, Williams IR, Gu X, et al. Immunity. 2006;24:623–632. doi: 10.1016/j.immuni.2006.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Porgador A, Yewdell JW, Deng Y, Bennink JR, Germain RN. Immunity. 1997;6:715–726. doi: 10.1016/s1074-7613(00)80447-1. [DOI] [PubMed] [Google Scholar]
- 4.Itano AA, McSorley SJ, Reinhardt RL, Ehst BD, Ingulli E, Rudensky AY, Jenkins MK. Immunity. 2003;19:47–57. doi: 10.1016/s1074-7613(03)00175-4. [DOI] [PubMed] [Google Scholar]
- 5.Srinivasan A, McSorley SJ. Curr Opin Immunol. 2004;16:494–498. doi: 10.1016/j.coi.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 6.Kissenpfennig A, Henri S, Dubois B, Laplace-Builhe C, Perrin P, Romani N, Tripp CH, Douillard P, Leserman L, Kaiserlian D, et al. Immunity. 2005;22:643–654. doi: 10.1016/j.immuni.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 7.Geissmann F, Jung S, Littman DR. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 8.Le Borgne M, Etchart N, Goubier A, Lira SA, Sirard JC, van Rooijen N, Caux C, Ait-Yahia S, Vicari A, Kaiserlian D, Dubois B. Immunity. 2006;24:191–201. doi: 10.1016/j.immuni.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 9.Allan RS, Waithman J, Bedoui S, Jones CM, Villadangos JA, Zhan Y, Lew AM, Shortman K, Heath WR, Carbone FR. Immunity. 2006;25:153–162. doi: 10.1016/j.immuni.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 10.Rotta G, Edwards EW, Sangaletti S, Bennett C, Ronzoni S, Colombo MP, Steinman RM, Randolph GJ, Rescigno M. J Exp Med. 2003;198:1253–1263. doi: 10.1084/jem.20030335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Angeli V, Faveeuw C, Roye O, Fontaine J, Teissier E, Capron A, Wolowczuk I, Capron M, Trottein F. J Exp Med. 2001;193:1135–1147. doi: 10.1084/jem.193.10.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Macnab RM. Annu Rev Genet. 1992;26:131–158. doi: 10.1146/annurev.ge.26.120192.001023. [DOI] [PubMed] [Google Scholar]
- 13.Salazar-Gonzalez RM, McSorley SJ. Immunol Lett. 2005;101:117–122. doi: 10.1016/j.imlet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 14.McSorley SJ, Asch S, Costalonga M, Rieinhardt RL, Jenkins MK. Immunity. 2002;16:365–377. doi: 10.1016/s1074-7613(02)00289-3. [DOI] [PubMed] [Google Scholar]
- 15.Reinhardt RL, Khoruts A, Merica R, Zell T, Jenkins MK. Nature. 2001;410:101–105. doi: 10.1038/35065111. [DOI] [PubMed] [Google Scholar]
- 16.Hataye J, Moon JJ, Khoruts A, Reilly C, Jenkins MK. Science. 2006;312:114–116. doi: 10.1126/science.1124228. [DOI] [PubMed] [Google Scholar]
- 17.Wick MJ. Curr Opin Immunol. 2002;14:437–443. doi: 10.1016/s0952-7915(02)00364-3. [DOI] [PubMed] [Google Scholar]
- 18.Bueno SM, Tobar JA, Iruretagoyena MI, Kalergis AM. Crit Rev Immunol. 2005;25:389–403. doi: 10.1615/critrevimmunol.v25.i5.40. [DOI] [PubMed] [Google Scholar]
- 19.Yarovinsky F, Kanzler H, Hieny S, Coffman RL, Sher A. Immunity. 2006;25:655–664. doi: 10.1016/j.immuni.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 20.van der Velden AW, Velasquez M, Starnbach MN. J Immunol. 2003;171:6742–6749. doi: 10.4049/jimmunol.171.12.6742. [DOI] [PubMed] [Google Scholar]
- 21.Tobar JA, Gonzalez PA, Kalergis AM. J Immunol. 2004;173:4058–4065. doi: 10.4049/jimmunol.173.6.4058. [DOI] [PubMed] [Google Scholar]
- 22.Qimron U, Madar N, Mittrucker HW, Zilka A, Yosef I, Bloushtain N, Kaufmann SH, Rosenshine I, Apte RN, Porgador A. Cell Microbiol. 2004;6:1057–1070. doi: 10.1111/j.1462-5822.2004.00418.x. [DOI] [PubMed] [Google Scholar]
- 23.van der Velden AW, Copass MK, Starnbach MN. Proc Natl Acad Sci USA. 2005;102:17769–17774. doi: 10.1073/pnas.0504382102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ohl L, Mohaupt M, Czeloth N, Hintzen G, Kiafard Z, Zwirner J, Blankenstein T, Henning G, Forster R. Immunity. 2004;21:279–288. doi: 10.1016/j.immuni.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 25.Williams IR. Ann N Y Acad Sci. 2006;1072:52–61. doi: 10.1196/annals.1326.036. [DOI] [PubMed] [Google Scholar]
- 26.Tvinnereim AR, Hamilton SE, Harty JT. J Immunol. 2004;173:1994–2002. doi: 10.4049/jimmunol.173.3.1994. [DOI] [PubMed] [Google Scholar]
- 27.Srinivasan A, Foley J, Ravindran R, McSorley SJ. J Immunol. 2004;173:4091–4099. doi: 10.4049/jimmunol.173.6.4091. [DOI] [PubMed] [Google Scholar]
- 28.Grubin CE, Kovats S, deRoos P, Rudensky AY. Immunity. 1997;7:197–208. doi: 10.1016/s1074-7613(00)80523-3. [DOI] [PubMed] [Google Scholar]
- 29.Cook DN, Prosser DM, Forster R, Zhang J, Kuklin NA, Abbondanzo SJ, Niu XD, Chen SC, Manfra DJ, Wiekowski MT, et al. Immunity. 2000;12:495–503. doi: 10.1016/s1074-7613(00)80201-0. [DOI] [PubMed] [Google Scholar]
- 30.Datsenko KA, Wanner BL. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Uzzau S, Figueroa-Bossi N, Rubino S, Bossi L. Proc Natl Acad Sci USA. 2001;98:15264–15269. doi: 10.1073/pnas.261348198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parish CR. Immunol Cell Biol. 1999;77:499–508. doi: 10.1046/j.1440-1711.1999.00877.x. [DOI] [PubMed] [Google Scholar]
- 33.Vremec D, Zorbas M, Scollay R, Saunders DJ, Ardavin CF, Wu L, Shortman K. J Exp Med. 1992;176:47–58. doi: 10.1084/jem.176.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}