

Abstract
In transcriptional regulatory networks, the coincident binding of a combination of factors to regulate a gene implies the existence of complex mechanisms to control both the gene expression profile and specificity of the response. Unraveling this complexity is a major challenge to biologists. Here, a novel network topology-based clustering approach was applied to condition-specific genome-wide chromatin localization and expression data to characterize a dynamic transcriptional regulatory network responsive to the fatty acid oleate. A network of four (predicted) regulators of the response (Oaf1p, Pip2p, Adr1p and Oaf3p) was investigated. By analyzing trends in the network structure, we found that two groups of multi-input motifs form in response to oleate, each controlling distinct functional classes of genes. This functionality is contributed in part by Oaf1p, which is a component of both types of multi-input motifs and has two different regulatory activities depending on its binding context. The dynamic cooperation between Oaf1p and Pip2p appears to temporally synchronize the two different responses. Together, these data suggest a network mechanism involving dynamic combinatorial control for coordinating transcriptional responses.
Keywords: Oaf3p, oleate, peroxisome, regulatory network, stress response
Introduction
Dynamic transcriptional regulatory networks underlie most complex cellular responses. Understanding the structure and coordinated behavior of these networks is fundamental to systems biology. Yeast has proven to be an excellent model for understanding eukaryotic transcriptional regulatory networks. Genome-wide chromatin localization and expression data reveal a general hierarchical four-tiered network structure (Yu and Gerstein, 2006), and superimposed on this structure are compact units of recurring patterns in network architecture (Shen-Orr et al, 2002). Each of these network motifs confers specific properties to the system including temporal control, coordinate expression with other genes, speed or stability of responses, sensitivity to continuous or transient stimulus and noise suppression (Becskei and Serrano, 2000; Shen-Orr et al, 2002). Multi-input motifs involve regulation of a group of targets by multiple factors and have been attributed to a response being required for multiple growth conditions, or the involvement of the target genes in multiple metabolic pathways. Here, different stimuli activate different transcriptional regulators leading to the activation of both common and distinct classes of genes (Kashtan et al, 2004). The effect of concomitant control of a multi-input motif, that is multiple factors regulating the same targets at the same time, has not yet been investigated on a global scale. However, it has been suggested that because there is so much overlap between the targets of individual regulators, this is likely an important mechanism to confer specificity to transcriptional responses (Luscombe et al, 2004).
The transcriptional response to fatty acid exposure serves as an excellent model for studying the concomitant control of multi-input motifs. Yeast cells respond to oleic acid exposure by inducing genes responsible for fatty acid metabolism, but, as with other external stimuli, they additionally coordinate other related processes (e.g. glucose metabolism, stress response, etc.). The transcriptional response controlling fatty acid metabolism has been characterized and is outlined in Supplementary Figure 1A. For many genes related to this process, heterodimers of Oaf1p and Pip2p bind to upstream oleate response elements (OREs) and activate transcription in the presence of oleate (Gurvitz and Rottensteiner, 2006). The specificity of the response is controlled at two levels: Oaf1p is activated directly by oleate (Phelps et al, 2006), and the expression of PIP2 is activated by Oaf1p/Pip2p heterodimers, as it has an upstream ORE. A third transcriptional activator, Adr1p, which is also involved in regulating genes involved in the metabolism of other carbon sources (Schuller, 2003; Tachibana et al, 2005), directly activates PIP2 (Rottensteiner et al, 2003b) and some other ORE-containing targets involved in fatty acid metabolism (feedforward regulation). Adr1p is necessary for full ORE-mediated activation and may also be necessary for derepression of these genes in the absence of glucose (Schuller, 2003; Young et al, 2003; Rottensteiner et al, 2003b). The extent of the influence of these factors on the transcriptome, and how the response is propagated to coordinate other cellular processes have not been systematically explored.
Among the numerous coordinated cellular processes are two different stress responses (Koerkamp et al, 2002; Smith et al, 2002). One of these is an acute and immediate activation of an oxidative stress response, which is proposed to involve Yap1p and to be a response to fatty acid-induced uncoupling of the respiratory chain (Koerkamp et al, 2002). The other is a downregulation of general stress response genes, which appears to be related to metabolic reprograming in response to the environmental change and mediated, in part, by the exit of Msn2p and Msn4p from the nucleus (Koerkamp et al, 2002).
Here, complementary high-throughput experimental techniques and various data integration strategies, including a novel network topology-based clustering method, were used to characterize a dynamic transcriptional regulatory network controlling fatty acid metabolism. Genome-wide condition-specific chromatin localization data were generated and used to construct physical interaction networks in the presence and absence of oleate. For each network, targets were clustered based on their network topology and the control of each cluster was inferred from statistical analysis of its size, and expression and functional properties of its members. This approach, combined with targeted experimental validation of the network, demonstrates that in this context, Oaf3p (heretofore uncharacterized) acts as a negative transcriptional regulator implicated in multiple cellular responses, and reveals a dynamic multi-input network structure in which Oaf1p acts as both a negative regulator of the general stress response and a positive regulator of the fatty acid metabolism response. The behavior of Oaf1p in this network suggests a mechanism by which the same regulator can control and synchronize related biological processes through involvement in different multi-input network motifs.
Results
Dynamic network increases in size and connectivity in response to fatty acids
We characterized a dynamic regulatory network involving four fatty acid-responsive transcriptional regulators and their primary targets to gain insight into the effects of their combinatorial control. The network was seeded with Oaf1p, Pip2p and Adr1p, the three activators known to conditionally cooperate to activate expression of genes involved in fatty acid metabolism (Schuller, 2003), as well as Ykr064p, which is renamed Oaf3p and which has been implicated as a transcription factor (MacPherson et al, 2006; Titz et al, 2006), but has not been characterized (see below). Oaf3p was included because preliminary data suggested that it plays a role in regulating fatty acid-responsive genes: OAF3 was first identified as one of 224 genes that displayed expression profiles similar to genes implicated previously in fatty acid metabolism or peroxisome biogenesis (Smith et al, 2002) (see also Supplementary Figure 1B), and it is a predicted Zn(2)-Cys(6) transcriptional regulator (MacPherson et al, 2006; Titz et al, 2006). Oaf3p is also localized to the nucleus (Huh et al, 2003), and a BLAST search of the yeast proteome revealed that Oaf3p is most similar to Pip2p and Oaf1p, with P-values of 3.6 × 10−7 and 5.1 × 10−7, respectively (using WU-BLAST 2.0 with default parameters; released May 10, 2005; Gish, W (1996–2004) http://blast.wustl.edu) (Altschul et al, 1990).
The targets of Oaf1p, Pip2p, Oaf3p and Adr1p were determined after growth in the presence of 0.1% glucose and 5 h after a switch to medium containing the fatty acid oleate as the sole carbon source. The 5 h time point was chosen to maximize the detection of targets of each factor based on the average expression profile of peroxisome-related genes during oleate induction (Smith et al, 2002). At 5 h, upregulation of these genes is robust, but not yet maximal, suggesting that oleate-induced transcriptional regulation is active and not declining. The targets of each factor were identified by genome-wide chromatin localization analysis of myc-tagged versions of each factor (Ren et al, 2000). Each strain was analyzed by chromatin immunoprecipitation (ChIP) followed by two-color DNA microarray analysis comparing DNA in the ChIP fraction to that in a whole-cell extract (WCE) on yeast intergenic microarrays (see Materials and methods). Data for three biological replicates of each experiment were merged and normalized and differential enrichment ratios were calculated. Next, VERA and SAM analysis tools were used to identify intergenic regions significantly enriched in the IP fractions (Ideker et al, 2000, 2001). This was carried out by using an estimated error model to improve the accuracy of the differential enrichment ratio and to assign a λ value representing the likelihood of differential enrichment for each intergenic region. For each experiment, a λ value threshold was chosen to yield an estimated false discovery rate (FDR) of 0.001 (see Materials and methods). For each condition, intergenic regions enriched in the ChIP fraction that were above the threshold were selected as targets for each factor (Supplementary Table 1).
Physical interaction networks representing chromatin localization data for all four factors were generated for each growth condition using Cytoscape software (Shannon et al, 2003). Networks of factor binding on oleate and low glucose are shown in Figure 1 (left panels). Each network consists of the four regulators (labeled nodes) connected by directed edges to the intergenic regions to which they bind (unlabeled nodes). After exposure to oleate, the number of targets in the network increased from 221 to 571. Specifically, the number of targets for Oaf1p, Pip2p, Adr1p and Oaf3p increased from 128, 52, 53 and 26 to 394, 212, 137 and 261, respectively. There was also an increase in network connectivity, as reflected by the >3-fold increase in connectivity score (targets with multiple edges/total number of targets) (Figure 1). These data suggest the existence of oleate-specific functions for the four factors involving multi-input motifs.
Figure 1.

Identification of overrepresented multiple input network motifs in the presence of low glucose (A) and 5 h after a switch to oleate (B). Left panels are condition-specific physical interaction networks between regulators (labeled nodes) and the intergenic regions with which they interact (unlabeled nodes) identified by large-scale genome localization analysis. Protein–DNA interactions (FDR of 0.001) for the four transcriptional regulators are shown as directed edges. Intergenic regions with same network topology (i.e. targeted by the same group of factors) are clustered and the number of targets per cluster is given beside each cluster. The network expands and there is more combinatorial control in the presence of oleate. Right panels are bar graphs showing the significance of overrepresentation for each cluster. On each graph, each cluster is labeled with up to four letters, representing the regulators targeting the cluster under that condition (A, Adr1p; O, Oaf1p; P, Pip2p; Y, Oaf3p). Overrepresented clusters have bars, the height of which reflects the significance of enrichment of that cluster in the network; note that as the P-values have been converted to significance scores (−log10(P-value)), one unit on the scale corresponds to a 10-fold difference in P-value. In all panels, significantly overrepresented topology clusters are colored in shades of red. Clusters with P-values of less than 1 × 10−25, 1 × 10−5 and 1 × 10−2 (significance scores greater than 25, 5 and 2) are colored dark red, red and pink, respectively.
Analysis of overrepresented multi-input motifs suggests multiple condition-specific combinatorial control mechanisms
The large-scale ChIP networks were analyzed for multi-input motifs appearing to represent functionally relevant trends as described below. For each condition-specific network, targets were grouped into clusters based on their network topology (Figure 1). For each cluster, the significance of the cluster size was determined by calculating the probability of having the observed size or greater (using the cumulative distribution function (CDF) with error correction) with the null hypothesis that each binding event of the four factors is independent (see Materials and methods). The results are displayed in Figure 1 (right panels) where network topology clusters are listed along the x-axis and significance of overrepresentation for each is represented by a bar.
For the glucose-specific network, only three topology clusters had significance scores greater than 2 (CDF P-values <0.01) (colored clusters) and only one (AO; bound by Adr1p and Oaf1p) had a score greater than 25 (P-value <1 × 10−25), reflecting very high confidence in cooperation of Adr1p and Oaf1p in the glucose network. In the oleate network, seven clusters had significance scores greater than 2 and three (AOY, OPY and AOPY; bound by combinations of Adr1p (A), Oaf1p (O), Pip2p (P) and Oaf3p (Y)) had scores greater than 25, suggesting more cooperation among the factors in the presence of oleate than in glucose. The conditional overrepresentation of these network motifs suggests oleate-specific regulation by at least three different multi-input motif instances (represented by AOY, OPY and AOPY clusters) with potentially different regulatory mechanisms and outputs.
The topology clusters were also analyzed using an FDR of 0.01 for the large-scale ChIP data. This analysis yielded similar results, but increased the significance scores for highly connected, high-scoring network clusters (AOY, AOPY and OPY), in part, because lowering the stringency reduces false negatives in the network. False negatives have the effect of reducing the population of highly connected clusters and erroneously increasing the occupancy of various less-connected clusters. However, the lower stringency networks are larger (801 and 570 targets for oleate and glucose networks, respectively) and presumably contain an increased number of false positives. Therefore, to maximize the accuracy of the networks and to increase statistical power of subsequent analyses, ‘combined threshold' networks were generated, for which membership in the network was determined using the high stringency threshold (FDR ∼0.001) and topology of the network was determined using the FDR threshold of ∼0.01. Intergenic regions and their topologies for these networks are listed in Supplementary Table 1. These networks were used for all subsequent analyses.
Network structure is an indicator of gene function and condition-specific expression
To facilitate the integration of gene expression data with the network, each intergenic region was translated into target genes with adjacent start sites using the genome annotations generated by MacIsaac et al (2006). Because of errors introduced by this conversion and technical limitations inherent to the genome-wide ChIP analysis (Lee et al, 2002; Hwang et al, 2005), rather than focusing on the characterization of individual targets in network clusters, we identified significantly overrepresented gene properties in each network topology cluster. This was performed using the CDF with error correction (see Materials and methods) to determine the probability of finding the observed (or greater) overlap between genes with a given property and genes in a topology cluster. The null hypothesis was that the given property is independent of membership in a topology cluster. This method of analysis not only provides a measure of confidence in the results, but also broadens the analysis to global trends and therefore is likely to reveal an insight into system level network regulatory function.
The first gene properties measured were the results of a time-course transcriptome profiling study (Koerkamp et al, 2002), which was conducted under conditions similar to those of the ChIP experiments performed here (i.e. carbon source was switched from low glucose to oleate). For this analysis, several measurements were obtained within minutes after the switch to oleate, a time when transcription appears to be changing most rapidly; thus they classified responses that might otherwise be indistinguishable. This study identified five distinct expression profile clusters of oleate-responsive genes, and integration of gene ontology (GO) and DNA motif data revealed that the expression clusters represented different biological processes including oxidative stress response, general stress response and peroxisome biology (Figure 2A).
Figure 2.

Network topology reflects target gene expression profile and function. (A) Oleate-responsive genes cluster into five groups based on their expression profiles (Koerkamp et al, 2002). The mean profiles of three gene clusters relevant to the regulatory network are shown and are annotated with biological processes associated with them. (B, C) Statistical analysis of the network distribution of the peroxisome-related (B) and the general stress response (C) expression profile clusters. Network topology clusters with an overrepresentation of peroxisome-related genes and general stress response genes have dark orange and dark blue bars, respectively. The height of each bar reflects the significance of the enrichment. In the presence of oleate, topology cluster AOY (genes targeted by Adr1p, Oaf1p and Oaf3p) is significantly enriched for the expression cluster representing general stress response genes (P-value∼5 × 10−7), whereas topology cluster OPY (genes targeted by Oaf1p, Pip2p and Oaf3p) is enriched for expression cluster representing peroxisome-related genes (P-value∼2 × 10−6). For both expression clusters, significance scores are lower and targeting is less cooperative in the glucose network. Statistical analyses of the distributions of OREs and Msn2p/Msn4p binding sites in the networks are also shown. These data are similar to those of the peroxisome-related and general stress response genes, respectively.
To analyze the relationship between network structure and oleate-specific expression, we searched for significant overrepresentation of genes of each expression cluster in each network topology cluster. The analysis revealed significant enrichment of two of the five expression profile clusters in the networks (P-value <0.01 for at least one topology cluster). Each of these clusters (peroxisome-related genes and general stress response genes) appears to be regulated by multiple factors in an oleate-specific manner. In the presence of low glucose, genes of the peroxisome-related expression cluster were overrepresented in the cluster targeted by Pip2p with low confidence, but were significantly enriched in the cluster targeted by Oaf1p, Pip2p and Oaf3p (OPY) or all four factors (AOPY) in the presence of oleate with higher confidence (Figure 2B). In contrast, the general stress response expression cluster was most significantly enriched in topology clusters targeted by Oaf1p (and/or Adr1p) (O/AO) in the presence of glucose, but by Adr1p, Oaf1p and Oaf3p in the presence of oleate (AOY) (Figure 2C).
As mentioned above, the expression profile clusters were named based on correlations found between expression profiles and either GO annotations or the presence of transcription factor binding motifs. Therefore, network enrichments of relevant DNA binding motifs and GO Slim terms, which are a high-level view of GO terms (Hong et al, 2006), were also analyzed (see Materials and methods). OREs (Figure 2B) and Msn2p/Msn4p targets (Figure 2C) had similar profiles to the corresponding expression clusters, peroxisome-related and general stress response, respectively. In addition, the distribution of OREs in the glucose network further suggests that some of these elements can be bound by a combination of Adr1p, Oaf1p and Pip2p in the absence of oleate. The GO Slim terms analyzed also had similar network distributions to the corresponding gene expression clusters (data not shown). For this analysis, genes annotated with ‘peroxisome' (component slim term) and ‘response to stress' (process slim term) were significantly enriched in oleate network clusters OPY (P-value of 1.9 × 10−7) and AOY (P-value of 8.8 × 10−3), respectively. Together these data suggest the dynamic regulation of two biological functions in the network, the oleate-induced upregulation of peroxisomes and downregulation of general stress response, which appears to involve oleate-specific targeting of the OPY and AOY topology clusters, respectively.
Influence of network transcription factors on gene expression is context specific
To determine the transcriptional response of each topology cluster, the transcriptional activity of the oleate network was measured. First, microarray analysis was used to compare poly A+ RNA isolated from yeast grown in the presence of 0.1% glucose to that of yeast grown for an additional 5 h in the presence of oleate (see Materials and methods). Replicate experiments were merged and processed as described previously (Smith et al, 2002), and VERA and SAM analysis tools (Ideker et al, 2000) were used to generate an error model that was used to generate a λ value reflecting the likelihood of differential expression for each gene. Log10 expression ratios and λ values for each experiment are provided in Supplementary Table 2. Next, genes with significant differential expression in response to oleate were identified (see Materials and methods). A total of 79 and 137 genes significantly increased and decreased in expression in response to oleate, respectively. The abundance of each class of genes in each topology cluster in the oleate network was statistically analyzed. This was performed using CDF with error correction as described above except that the null hypothesis was that the environmental change has no effect on genes in the topology cluster (i.e. the frequency of genes up- and downregulated by oleate in the cluster is equal to the estimated FDR in the expression data) (see Materials and methods). The results are shown in Figure 3A. The significance of enrichment of genes up- and downregulated after a 5-h oleate induction is represented by red (above the x-axis) and green bars (below the x-axis), respectively. The OPY (and OP, AOPY and O) topology clusters were enriched for genes that increased in expression in response to oleate, whereas the AOY (and OY) clusters were enriched for those that decreased in response to oleate. These data are consistent with the previous analysis (Figure 2) and a similar network topology analysis of different microarray expression data (Smith et al, 2002) comparing glycerol-grown cells to those grown in oleate for 6 h (data not shown).
Figure 3.

Statistical analysis of the influence of network regulators on topology clusters in the oleate network. Discrete microarray expression data were overlaid onto the network, and topology clusters significantly enriched for up- or downregulated genes were identified. Results are displayed as in Figure 2 except that up- and downregulated gene enrichments are shown as red bars above the x-axis and green bars below the x-axis, respectively. Also, for significantly enriched clusters, the number of up- or downregulated genes is given. For the overlay of oleate induction expression data (5 h oleate induction versus low glucose (A)), genes upregulated by oleate are most significantly enriched in three topology clusters binding Oaf1p and Pip2p, whereas downregulated genes are enriched in two clusters binding Oaf1p and not Pip2p, most prominently the AOY cluster. The integration of transcription factor deletion array data (deletion strain induced for 5 h versus wild-type strain induced for 5 h; B–E) with the network suggests that Oaf1p contributes to both of these effects, whereas Pip2p and Adr1p contribute only to the gene activation observed in the presence of oleate. In addition, genes targeted by only Oaf1p are enriched for genes upregulated by oleate and genes upregulated by Adr1p. No significant effect of deleting OAF3 was observed (E).
To study the influence of each factor on the network, microarray analysis was used to determine the transcriptome profiles of each deletion strain (ΔOAF1, ΔPIP2, ΔADR1 and ΔOAF3) compared to that of wild type after a 5-h induction in oleate. The deletion experiments identified 194, 104, 175 and 76 genes with significant differential expression, respectively. Genes significantly up- and downregulated were identified and data were overlaid onto the oleate network and statistically analyzed as described above. The topology clusters with the highest significance scores (Figure 3B–D) were the same as those identified in the analysis of overrepresented clusters with data from oleate-induced wild-type cells (Figures 2 and 3A).
Clusters OPY (and AOPY and OP) were enriched for genes whose expression was reduced by deletion of OAF1, PIP2 or ADR1. These data reflect the role of these three factors in the upregulation of peroxisome-related genes in the presence of fatty acids (Gurvitz and Rottensteiner, 2006), and the role of Adr1p in directly upregulating PIP2 (Rottensteiner et al, 2003b), which is also supported by the Adr1p–PIP2 interaction identified here (Supplementary Table 2). There is also evidence of regulation of the AOY (and OY) clusters by Oaf1p and Adr1p. Adr1p appears to have a positive influence on genes of the AOY cluster, whereas Oaf1p appears to negatively regulate genes in this cluster (and in the OY cluster). This role in negative regulation appears to be independent of Pip2p because deletion of PIP2 had no significant influence on the expression of these clusters. The negative regulatory activity of Oaf1p is not likely controlled by absence of dimerization with Pip2p as it has previously been shown that overexpression of OAF1 can activate transcription from an ORE-containing promoter in a PIP2 deletion strain (Baumgartner et al, 1999). Instead, regulation of this activity appears to involve a DNA-binding context because unlike clusters positively regulated by Oaf1p, those under negative regulation (AOY and OY in Figure 3B) are not significantly enriched for OREs (Figure 2B).
Interestingly, these data suggest that a third cluster, genes targeted by only Oaf1p in the oleate network, might have biological significance. This cluster is enriched for genes that increase in expression in response to oleate (panel A) and those that are positively regulated by Adr1p (panel D). We do not know the biological function of this enrichment, but it may be due in part to the presence of genes in the O cluster that bind both Oaf1p and Pip2p that are false negatives for Pip2p binding. This is consistent with the weak enrichment of OREs in this cluster (Figure 2B).
To determine if the influences of the regulators detected here contribute to the oleate-specific expression patterns that are enriched in the network (Figure 2), overlap between the three data sets (oleate topology clusters, expression profile clusters and discrete transcription factor deletion data) was analyzed. Topology clusters AOY and AOPY/OPY were reduced to include only the relevant expression cluster genes (14 general stress cluster genes and 10 peroxisome cluster genes, respectively) and then reanalyzed for significant enrichment of genes affected by transcription factor deletions. The intersection of the AOY cluster and the general stress response expression cluster was enriched for genes that are negatively influenced by Oaf1p, whereas the intersection of the OPY cluster and the peroxisome-related expression cluster was enriched for genes that are positively influenced by Oaf1p, Pip2p and Adr1p (all P-values ⩽1 × 10−11), suggesting that the factors indeed contribute to oleate-specific expression patterns associated with these clusters.
Negative regulatory activity of Oaf1p is independent of Pip2p
The data suggest that the negative regulatory activity of Oaf1p is independent of Pip2p. While the Pip2p-dependant role of Oaf1p is well characterized, the Pip2-independent role is not. Therefore, we measured the effects of transcription factor deletions on representatives of the AOY cluster in the oleate network. DIP5 and ATO3, which are both negatively regulated by Oaf1p and not regulated by Pip2p (Supplementary Table 2), were used as reporters. The levels of Dip5p and Ato3p GFP chimeras were determined after exposure of wild-type and isogenic mutant strains (ΔOAF1, ΔPIP2, ΔADR1 or ΔOAF3) to oleate (Figure 4). Deletion of OAF1 resulted in increased levels of both chimeras, supporting the conclusion that Oaf1p negatively influences the expression of these genes. In contrast, deletion of PIP2 had no detectable effect on either protein, supporting the conclusion that Oaf1p does not cooperate with Pip2p in this context. Interestingly, deletion of OAF3 resulted in increased levels of Dip5p-GFP (1.4-fold), suggesting that it is a negative regulator of DIP5. Consistent with this, the deletion of OAF3 resulted in 1.5-fold increased expression of DIP5 by microarray analysis (Supplementary Table 2), but the λ value reflecting the likelihood of differential expression was 29, which fell below our threshold of 36.23. These data suggest that the influence of OAF3 deletion on network gene expression, as detected by microarrays, is modest (see also Figure 3E).
Figure 4.

Protein levels reflect the Pip2p-independent role of Oaf1p as a negative regulator. Wild-type and deletion strains each with a GFP-tagged version of an AOY cluster gene (DIP5 or ATO3) were analyzed by FACS in the presence of glycerol and 20 h (Dip5-GFP strains) or 5 h (Ato3-GFP strains) after the addition of oleate to the medium. Deletions of OAF1 but not PIP2 resulted in lower levels of fluorescence relative to wild-type levels. Deletion of ADR1 or OAF3 resulted in decreased or increased Dip5-GFP levels, respectively.
Oaf3p functions as a weak negative regulator
To investigate further the role of Oaf3p, we analyzed the OAF3 deletion array data by combining wild-type time-course expression, deletion strain expression and large-scale ChIP data sets. We first identified a set of 65 genes that were differentially expressed in the oleate time-course expression study (Smith et al, 2002), and significantly upregulated at 5 h on oleate in the OAF3 deletion strain as compared to wild type. Next, we determined the frequency of Oaf3p binding (on oleate) to the 53 of these genes that were represented on the intergenic microarray, and compared this frequency to the average frequency of Oaf3p binding among all oleate-responsive genes (see Materials and methods). Using this approach, we determined that Oaf3p binding is enriched among genes transcriptionally responsive to oleate and negatively regulated by OAF3 on oleate (P-value=0.0023). These data support a role for Oaf3p as a negative regulator of expression of its target genes in response to oleate.
We therefore looked for further validation of Oaf3p influence on network targets by looking at the protein levels of its targets under various conditions. The effect of deleting OAF3 on the levels of Cta1p (encoded by an AOPY group gene) was analyzed by immunoblotting (Figure 5A). A wild-type strain with a TAP-tagged version of Cta1p was compared to isogenic strains deleted for either OAF1 or OAF3. In the wild-type strain, levels of Cta1-TAP were not detected in cells grown in raffinose-containing media (non-inducing condition), but levels increased after growth in the presence of oleate or antimycin, a second peroxisome-inducing condition (Epstein et al, 2001). Deletion of OAF1 resulted in little or no induction as expected from the previously published data (Gurvitz et al, 2001), but deletion of OAF3 resulted in oleate expression levels that were higher than those in the wild-type strain.
Figure 5.

Oaf3p is a weak negative regulator. (A) Immunoblot analysis of Cta1-TAP in wild-type cells and in OAF1 or OAF3 deletion strains. Cta1-TAP levels increase under conditions of peroxisome induction (in the presence of oleate or antimycin). This induction is not detectable in the absence of OAF1, but is more robust in the absence of OAF3, suggesting that expression of CTA1 (gene with AOPY oleate network topology) is activated by OAF1 but repressed by OAF3 in the presence of oleate. Equal protein was loaded in each lane as indicated by the levels of Gsp1p (a protein whose corresponding gene is not in the network). (B) FACS analysis of various Oaf3p targets in GFP-tagged strains overexpressing OAF3 (from copper-inducible promoter in plasmid pYEX-OAF3; red curve) and in wild-type strain (transformed with empty plasmid, pYEX-BX; blue curve) after 20 h induction in the presence of oleate and copper. In many cases, OAF3 overexpression resulted in reduced protein levels of its target genes (oleate network topologies shown in brackets). (C) Clear zone assays measuring efficiency of fatty acid metabolism in wild-type and OAF3 overexpression strains. An equal number of cells of each strain were spotted onto turbid fatty acid plates and grown for 3 days. Spots of wild-type cells have larger clear zones than spots of the overexpression strain, suggesting that overexpression of OAF3 inhibits fatty acid metabolism.
The protein levels of other Oaf3p targets were also analyzed in an OAF3 overexpression strain. Various GFP-tagged strains were transformed with pYEX-OAF3 (a plasmid with OAF3 under the control of the copper-inducible promoter of CUP1), induced for 20 h in medium containing copper and oleate, and analyzed by fluorescence-activated cell sorting (FACS). The results are shown in Figure 5B. In most cases tested, overexpression of OAF3 (red lines) resulted in decreased protein levels of the corresponding target gene (panels 1–4; 1.5- to 2.4-fold decrease), but in some cases, no detectable effect on protein levels was evident (e.g. Faa1-GFP). This is not unexpected, as not all members of the topology groups responded identically to other perturbations.
As overexpression of OAF3 had a negative effect on the transcription of POT1, a gene involved in fatty acid metabolism, an overexpression strain was also analyzed for its ability to metabolize the fatty acid myristic acid (Figure 5C). An equal number of cells of a wild-type strain transformed with either the empty plasmid or the overexpression plasmid was induced with copper and then grown on turbid fatty acid medium. Overexpression of OAF3 resulted in a reduction in the size of the halo around the cell patches, indicating a reduced ability to metabolize fatty acids (Smith et al, 2006). Taken together, these data suggest a role for Oaf3p as a negative regulator of transcription in the presence of oleate.
Analysis of dynamic network activities reflects temporal coordination of responses
To explore the potential for temporal coordination of the network, representative GFP-tagged strains were analyzed by FACS after growth in the absence of oleate (YPBG) and after 5 and 20 h inductions in the presence of oleate (YPBO) (Figure 6). Consistent with the gene expression data (Figure 3A), levels of proteins corresponding to the AOY cluster decreased in the presence of oleate, whereas those corresponding to the OPY and AOPY clusters increased in response to oleate. Interestingly, although the decreases in expression were clear after a 5-h induction, increases in expression were prominent after 20 h in oleate. These data appear to reflect the fact that the transcriptional response of general stress response genes precedes that of the peroxisome-related genes as identified previously by microarray analysis (Koerkamp et al, 2002) (Figure 2A). This suggests temporal coordination of the two biological processes and provides insight into the function of the network as described in Discussion.
Figure 6.

Sequential changes in protein levels reflect coordination of responses by the network. FACS analysis of various GFP-tagged strains corresponding to genes under combinatorial control in the oleate network (topologies listed on the left). For each strain, fluorescence intensities of cells grown in glycerol (blue curves) were compared to those induced in oleate for 5 or 20 h (red curves). Protein levels corresponding to genes of the AOY topology are reduced in the presence of oleate, whereas those corresponding to genes with AOPY and OPY topologies increase after oleate induction. The negative effect of oleate on AOY gene expression is apparent after short and long induction periods, whereas the upregulation of gene targeted by Oaf1p and Pip2p is observed only after long induction periods.
Discussion
A dynamic transcriptional regulatory network generated from genome localization and transcriptome profiling data was characterized using a novel topology-based clustering approach. The analysis used simple, widely available tools that can be applied to the characterization of other regulatory networks. Results from this analysis were integrated with literature data to infer a dynamic model of network function (Figure 7). This was carried out by first generating a model for the 5 h time point. For this condition, oleate-specific interactions (edges) between regulators and targets were inferred from ChIP data (Figure 1; Supplementary Tables 1 and 2), negative (red) and positive (green) influences were inferred from these data along with expression and protein abundance data of wild-type, deletion and overexpression strains (Figures 3, 4 and 5 and Supplementary Table 2). The states of these network connections at other time points (before and after 5 h) are predictions made from these data along with time-course microarray data of the factors (Supplementary Figure 1B; Smith et al, 2002) and network targets (Figure 2A; Koerkamp et al, 2002; Smith et al, 2002) and from protein levels of the target genes at different time points (Figure 6). Other interactions were added from data in the literature as indicated. The network is described in detail below.
Figure 7.

Inferred dynamic model of transcriptional regulatory network. The model, based on data presented here and data from the literature, shows active paths after the switch from low glucose to oleate at four time points (immediate, early, 5 h and late). Information flow (directed edges) is shown between stimuli (diamond nodes), genes (linear nodes with arrows), proteins (circular nodes) and network topology clusters (box nodes). Activities of nodes and influences of edges are colored red or green to represent down- or upregulation, respectively. Regions of the network are highlighted in color and labeled (inverted text with black background) according to the biological response they represent. It should be noted that recent data suggest that YDR215C, the ORF immediately upstream of ADR1, is unlikely to encode a protein (Fisk et al, 2006). Accordingly, intergenic region iYDR214W is annotated as a regulator of ADR1 (Macisaac et al, 2006) and ADR1 is in the AOY topology cluster in the oleate network (Supplementary Table 2). However, owing to the preliminary nature of the evidence, these interactions do not appear in the model.
Immediately after the carbon source switch (immediate response; top panel), the presence of oleate is recognized directly by constitutively present Oaf1p (Phelps et al, 2006), which binds upstream of genes of the AOY, general stress response cluster. Genes of this cluster are known to be acutely and transiently activated when respiration is turned off (Lai et al, 2005), and may be involved in reprogramming cellular metabolism in response to changes in respiratory state (Lai et al, 2005). Here, these genes are transiently repressed by Oaf1p (and likely other factors). This Oaf1p-mediated repression appears to coincide with an efflux of Msn2p and Msn4p from the nucleus, which also contributes to maintaining these genes in an off state before subsequent metabolic reprogramming (see below) (Koerkamp et al, 2002).
The carbon source switch signals the rapid and transient increase in expression levels of ADR1 (immediate response; top panel; Supplementary Figure 1B). As ADR1 transcripts accumulate and are translated, Adr1p levels begin to rise, which positively and directly influences the expression of PIP2 (Supplementary Table 2), genes in the AOY, and AOPY clusters and likely other pathways controlling utilization of other carbon sources (Schuller, 2003; Gurvitz and Rottensteiner, 2006) (early response; second panel).
The third panel shows the response approximately 5 h after the carbon source switch. Here, the activation of PIP2 by Adr1p leads to accumulation of Pip2p, which (as a heterodimer with Oaf1p) transmits the oleate signal to genes directly involved in fatty acid metabolism and positively influences genes of the OPY and AOPY clusters. As the levels of ADR1 mRNA are declining at this time (Supplementary Figure 1B), the direct influence of Adr1p on its targets is shown as a dashed line representing weak influence in this panel. At this time, Oaf1p/Pip2p dimers also feedback positively on the expression of PIP2. This self-regulation, in combination with the Adr1p-mediated activation of PIP2 and peroxisome-related genes of the AOPY cluster, constitutes coupled feedback and feedforward circuitry to activate peroxisome-related genes.
The dual influence of Oaf1p on OPY and AOY clusters appears to mediate temporally synchronized regulation: the negative influence of Oaf1p on general stress response genes immediately follows the carbon source switch because levels of Pip2p (and thus Oaf1p/Pip2p heterodimers) in the initial condition are low. Time-delayed accumulation of Pip2p resulting from feedforward and feedback regulation (Mangan and Alon, 2003; Maeda and Sano, 2006) subsequently leads to dramatic upregulation of peroxisome-related genes by Oaf1p/Pip2p heterodimers. The late response (lower panel) shows that the influence on the peroxisome-related cluster remains strong as the negative influence on the general stress response subsides. This difference in duration reflects steady-state gene expression data (Koerkamp et al, 2002) (Figure 2A), and is likely caused by the rising Pip2p level, which increasingly draws more Oaf1p molecules from AOY to OPY and AOPY targets through heterodimerization.
As discussed earlier, Oaf3p appears to be a weak negative regulator in the network (Figure 5). Its influences in the 5 h and late response panels reflect OAF3 expression levels, which are reduced immediately after a shift to oleate and gradually increase over time (Supplementary Figure 1B). The specific function of Oaf3p in the network is not yet known. We and others have shown that transcriptional repression can ensure a more homogeneous expression level of a target gene across a cell population, in the context of network structure such as negative feedback regulation (Becskei and Serrano, 2000; Orrell and Bolouri, 2004; Ghosh et al, 2005; Ramsey et al, 2006). Kinetic modeling of the response, as performed to understand negative regulation in the galactose utilization pathway (Ramsey et al, 2006), will help to elucidate its role. Modeling can be facilitated by refining the qualitative network model by investigating other potential influences such as other transcriptional regulators not yet considered, as well as mRNA and protein degradation, which have been implicated previously in regulating carbon source utilization (Carlson, 1999).
The topology-based clustering and analysis methods outlined here identified a transcriptional regulatory network involving the participation of a bi-functional regulator in multiple multi-input network motifs to control and synchronize related biological processes. The data suggest that, beyond providing specificity in promoter binding, heterodimerization of transcription factors can contribute temporal control of tightly coordinated biological responses.
Materials and methods
Strains and growth conditions
Haploid deletion strains are isogenic to either BY4742 or BY4739, and are from the commercially available deletion set (Invitrogen, Carlsbad, CA). Haploid strains with myc-tagged genes were made by genomically tagging target genes with the sequence encoding 13 copies of the c-myc epitope from pFA6-13MYC (Longtine et al, 1998) by homologous recombination into BY4742 using a previously described PCR-based procedure (Aitchison et al, 1995). Strains with no apparent growth defects and appropriately sized fusion proteins were used. Haploid strains tagged with Aequorea victoria (S65T) GFP or TAP tag are isogenic to BY4741 and are from the commercially available GFP-clone collection (Invitrogen, Carlsbad, CA) or TAP fusion collection (Open Biosystems, Hunstville, AL), respectively. Strains containing both gene deletions and gene tags were made by mating, sporulation and tetrad dissection. For all experiments, control strains were isogenic to test strains. Unless otherwise stated, strains were grown in YPD (1% yeast extract, 2% peptone, 2% glucose); SCIM (1.7 g yeast nitrogen base without amino acids and ammonium sulfate (YNB−aa−as)/l, 0.5% yeast extract, 0.5% peptone, 0.79 g complete supplement mixture/l, 5 g ammonium sulfate/l) containing either 0.1% glucose or 0.5% Tween 40 (w/v) and 0.15% (w/v) oleate, or both; or YPB (0.3% yeast extract, 0.5% potassium phosphate (pH 6.0), 0.5% peptone) with either 3% glycerol (YPBG) or 0.5% Tween 40 (w/v) and 0.15% (w/v) oleate (YPBO).
Large-scale ChIP analysis
For each transcriptional regulator, genome-wide chromatin localization experiments were performed to comprehensively identify DNA-binding locations by ChIP followed by microarray analysis as developed previously (Ren et al, 2000; Lee et al, 2002). YPD-grown cultures of strains containing myc-tagged regulators were grown in SCIM medium containing 0.1% glucose for 16 h to a density of ∼8 × 106 cells/ml, and then transferred to SCIM containing 0.1% glucose or 0.15% oleate and 0.5% Tween 40, and grown for an additional 1.75 or 5 h, respectively. Proteins were crosslinked to their cognate DNA binding sites (and each other) with formaldehyde. Cells were disrupted and chromatin sheared into fragments by glass bead lysis followed by sonication. myc-tagged factors were collected by ChIP with magnetic beads. Crosslinking was reversed in fractions of the ChIP and WCE, linkers were annealed to the DNA ends and DNA was amplified and labeled by PCR in the presence of Cy5-dUTP and Cy3-dUTP, respectively. DNA in ChIP and WCE samples was compared by hybridizing both samples together to yeast intergenic DNA microarrays.
For each experiment (i.e. ChIP of one factor under one growth condition), there were three biological replicates, each hybridized to different microarrays. As each array contains four replicates of each intergenic region, the total number of replicates for each condition is 12. Data were processed by a previously published method (Ideker et al, 2001) using the SBEAMS microarray database software (Marzolf et al, 2006). Processing included normalizing scan intensities, subtracting background, merging data from replicate spots and generating an error model. A likelihood statistic, λ, was computed for each intergenic region to determine whether its abundance was significantly enriched in either the ChIP fraction or the WCE fraction. Two threshold λ values were chosen that yielded an approximate FDR of 0.01 and 0.001, corresponding to 64 and 6 false positives per 6438 intergenic regions, respectively. This was determined by utilizing the fact that differential enrichment for ChIP microarray data is one directional (i.e. DNA targets tend to be enriched in the ChIP fraction only) as opposed to expression array data, for which genes can go up or down in expression. For each experiment, the λ value was identified, above which there were 64 or 6 targets enriched in the WCE fraction instead of the ChIP fraction (false positives). Intergenic regions with λ values above this threshold (minus the known false positives enriched in the WCE fraction) were chosen as target intergenic regions. Genes with start sites adjacent to these target intergenic regions were chosen as target genes.
The chromatin localization data have been submitted to Gene Expression Omnibus database under accession number GSE5863.
Statistical analysis of overrepresented network motifs
For each condition, physical interaction data for each of the four factors were combined and graphically displayed using Cytoscape network visualization software version 2.2 (Shannon et al, 2003). Targets were grouped based on their network topology. For every topology cluster, the CDF (equation (1)) was used to calculate a P-value equal to P(X⩾x), the probability of the cluster size being equal to or greater than the observed size by chance (using R 2.3.0), with the null hypothesis that the four factors have independent sets of targets. x is the observed cluster size, n is the number of trials (the number of intergenic regions on the array in this case) and pcluster is the probability that a given target intergenic region will have this particular network topology assuming the null hypothesis. The calculation of pcluster is shown in equation (2), in which n represents an estimate of the population size and is the number of intergenics on the array and z is the number of targets of a given transcription factor. The F subscript denotes the subset of the four transcription factors analyzed that target the cluster, whereas f represents those that do not. An example calculation shows the probability of an intergenic region being in cluster AO (i.e. targeted by Oaf1p and Adr1p but not by Pip2p or Oaf3p) (equation (3)). To reduce error due to multiple statistical comparisons of a single data set, the α-level was adjusted to 15 representing each of the clusters analyzed (Bonferroni correction). In the graphs in Figure 1, bars reflecting statistical significance are shown only if the cluster contained more than two members.
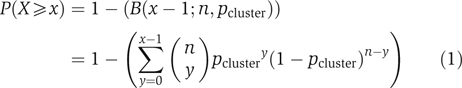 |
Gene and intergenic region attributes
Time-course expression profiles and expression profile clusters for genes in response to oleate exposure were obtained from previously published microarray analyses (Koerkamp et al, 2002; Smith et al, 2002). GO Slim terms were downloaded from the Saccharomyces Genome Database website (Hong et al, 2006). For binding motif enrichment studies, Fuzznuc, the nucleic acid pattern search algorithm component of EMBOSS software (Rice et al, 2000), was used to identify intergenic regions containing one or more ORE(s) conforming to the consensus CGGN3TN(A/G)N8−12CCG as defined previously (Rottensteiner et al, 2003a). Msn2p/Msn4p targets were defined as intergenic regions that interacted with one or both of the factors in genome localization data (Harbison et al, 2004) as determined by Macisaac et al (2006) (P<0.005 and conserved in three species). The experimental conditions of this data set include various stress-inducing environments.
Statistical analysis of enrichment of gene attributes in network clusters
To facilitate the integration of gene attribute and gene expression data with the network, each intergenic region in the combined threshold networks (Supplementary Table 1) was translated into target genes with adjacent start sites using the genome annotations generated by MacIsaac et al (2006). Topologies of gene targets are given in Supplementary Table 2. Genes assigned to more than one network topology, due to the fact that multiple intergenics regions can be assigned to one gene, have one entry with the multiple topologies merged together.
For each test, the CDF formula and Microsoft Excel software were used to calculate a P-value, equal to the probability that by chance, the enrichment of the attribute in the topology cluster is greater than or equal to that observed, with the null hypothesis that annotation with the gene attribute and membership in the topology cluster are independent. The equation used is similar to equation (1) except that x is the observed number of genes with the attribute in the topology cluster, n is the total number of genes in the topology cluster and pcluster is replaced by p, the probability of a gene with the attribute being in the cluster assuming the null hypothesis. To reduce error due to multiple statistical comparisons of a single data set, the α-level was adjusted to 15 representing each of the clusters analyzed (Bonferroni correction). In all graphs, bars reflecting statistical significance are shown only if the cluster contained more than two members with the attribute. For analysis of binding motifs, intergenic region networks were used instead of gene networks.
Generation and analysis of expression microarray data
For the comparison of mRNA levels in each of four deletion strains (ΔOAF1, ΔPIP2, ΔADR1 or ΔOAF3) to those in wild-type cells and for the wild-type versus wild-type control experiment, all strains were grown in YPD overnight and then transferred to SCIM with 0.1% glucose and without oleate and Tween 40, and grown for 16 h to ∼8 × 106 cells/ml, oleate and Tween 40 were added to the medium and cells were grown for an additional 5 h and then harvested. For the comparison of glucose- to oleate-grown cells, the experiment was carried out the same way except that the reference culture was harvested before the 5-h oleate induction. For all experiments, poly A+ RNA was extracted and cDNA was synthesized with incorporated Cy3 or Cy5 fluorescent dyes, and equimolar amounts of each label were mixed and hybridized to yeast ORF oligonucleotide microarrays as described previously (Smith et al, 2002). There were two biological replicates for each experiment, and for each replicate both label orientations were analyzed on arrays containing four replicate spots of each gene, resulting in a total of 16 replicate spots per gene. Spotfinding was performed using Analyzer DG software (Molecularware, Irvine, CA) and data analysis was carried out as described previously (Smith et al, 2002) except that the λ likelihood value threshold of 36.23 was chosen which resulted in an FDR of 0.01 as determined from a wild type versus wild-type control experiment. Genes having λ values above the threshold were annotated as up- or downregulated as a result of the deletion, and genes with λ values below the threshold were annotated as unchanged. The statistical analysis of the representation of genes up- and downregulated in each network topology cluster was performed as described above for other gene attributes except the null hypothesis was that the environmental change has no effect on genes in the topology cluster. For example, for the analysis of upregulated genes, P-value (the probability of a gene being upregulated and in the cluster assuming the null hypothesis) is 0.005, or half of the estimated FDR of differential expression. In cases where the actual rate of upregulated genes was lower than the estimated FDR, the actual rate was used.
The gene expression data have been submitted to Gene Expression Omnibus database under accession number GSE5862.
Analysis of transcriptional regulation of oleate-responsive genes by OAF3
First, we determined the significance of enrichment of genes negatively regulated by OAF3 in a subset of genes transcriptionally responsive to oleate. Time-course microarray data measuring the transcriptional response to oleate were taken from Smith et al (2002). The λ values for all microarray expression ratios for this and the OAF3 deletion experiment were normalized by dividing by the mean un-normalized λ value for the wild-type versus wild-type control experiment described above. P-values were computed from the normalized λ values using the right-tailed CDF of the χ2 distribution with one degree of freedom (Ideker et al, 2000). For each gene, a vector of P-values was obtained from all the ratios of the time-course experiment. From this vector, a P-value threshold of 0.0012 was determined to correspond to an FDR of 0.005 (Benjamini, 1995). Using this P-value threshold, 3871 genes were determined to be significantly differentially regulated relative to the glycerol condition, in at least one replicate-combined wild-type time-course measurement. These genes were then analyzed in the ΔOAF3 versus wild-type experiment. A total of 2240 genes (of the 3871) were found to have a positive expression log-ratio ΔOAF3/wild type. From the vector of P-values for these genes, an FDR of 0.1 was found to correspond to a P-value threshold of 0.0057. A total of 65 genes were found to be significantly differentially expressed in ΔOAF3 relative to wild type, based on this significance threshold. The binding of Oaf3p to the 53 (out of 65) OAF3-repressed genes represented on the ChIP array was then analyzed. The enrichment of binding of Oaf3p (in oleate) to the 5′ flanking intergenic regions of the 53 genes was computed, relative to the background set of all 3387 oleate-responsive genes on the ChIP array (resulting in a more conservative enrichment estimate than if all intergenic regions represented on the ChIP array were used as the background set). Using the CDF of the hypergeometric distribution, the enrichment P-value was found to be 0.0023.
OAF3 overexpression experiments
The plasmid pYEX-OAF3 was made by amplifying the open reading frame of OAF3 from genomic DNA by PCR with primers containing flanking BamH1 and Kpn1 sites, and ligating into the corresponding sites of pYEX-BX (Clontech Laboratories, Mountain View, CA). Strains analyzed by FACS were grown overnight in CM−ura−leu (0.77 g CSM minus uracil and leucine (BIO 101 (Carlsbad, CA))/l, 1.7 g YNB−aa−as/l, 5 g ammonium sulfate/l) containing 3% glycerol and 0.5 mM CuSO4, transferred to CM−ura−leu containing 0.5% Tween 40, 0.15% oleate and 0.5 mM CuSO4 and grown for 23 h. Clear zone assays were performed as described previously (Smith et al, 2006) except that BY4742 cells containing either pYEX-OAF3 or pYEX-BX were grown in CM−ura containing 2% glucose overnight and induced for 2 h by adding 0.5 mM CuSO4 to the medium. Cells were washed with water and ∼10 000 cells of each strain were spotted onto solid minimal myristate medium (1.7 g YNB−aa−as/l, 5 g ammonium sulfate/l, 0.5% potassium phosphate buffer, pH 6.0, 0.77 g CSM minus uracil/l, 2% agar, 0.5% Tween 40, 0.125% myristic acid) and grown for 3 days.
FACS analysis
Fluorescence intensity of individual cells was measured using an FACS Caliber flow cytometer (BD Biosciences, San Jose, CA). Data analysis was performed using WinMDI 2.8 (available from http://FACS.scripps.edu/), a forward scatter gate of >52 units, event normalization and smoothing of 25 units.
Supplementary Material
Supplementary Figure 1
Supplementary Table 1
Supplementary Table 2
Acknowledgments
This publication was made possible by NIH grants P50 GM076547, RR022220 and GM067228. The contents of the article are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. We thank Alex Ratushny, John Boyle and Gregory Carter for helpful discussion of the manuscript.
The gene expression and chromatin localization data from this study have been submitted to Gene Expression Omnibus database under accession numbers GSE5862 and GSE5863, respectively
References
- Aitchison JD, Rout MP, Marelli M, Blobel G, Wozniak RW (1995) Two novel related yeast nucleoporins Nup170p and Nup157p: complementation with the vertebrate homologue Nup155p and functional interactions with the yeast nuclear pore-membrane protein Pom152p. J Cell Biol 131: 1133–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215: 403–410 [DOI] [PubMed] [Google Scholar]
- Baumgartner U, Hamilton B, Piskacek M, Ruis H, Rottensteiner H (1999) Functional analysis of the Zn(2)Cys(6) transcription factors Oaf1p and Pip2p. Different roles in fatty acid induction of beta-oxidation in Saccharomyces cerevisiae. J Biol Chem 274: 22208–22216 [DOI] [PubMed] [Google Scholar]
- Becskei A, Serrano L (2000) Engineering stability in gene networks by autoregulation. Nature 405: 590–593 [DOI] [PubMed] [Google Scholar]
- Benjamini YHY (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57: 11 [Google Scholar]
- Carlson M (1999) Glucose repression in yeast. Curr Opin Microbiol 2: 202–207 [DOI] [PubMed] [Google Scholar]
- Epstein CB, Waddle JA, Hale WT, Dave V, Thornton J, Macatee TL, Garner HR, Butow RA (2001) Genome-wide responses to mitochondrial dysfunction. Mol Biol Cell 12: 297–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisk DG, Ball CA, Dolinski K, Engel SR, Hong EL, Issel-Tarver L, Schwartz K, Sethuraman A, Botstein D, Cherry JM (2006) Saccharomyces cerevisiae S288C genome annotation: a working hypothesis. Yeast 23: 857–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh B, Karmakar R, Bose I (2005) Noise characteristics of feed forward loops. Phys Biol 2: 36–45 [DOI] [PubMed] [Google Scholar]
- Gurvitz A, Hiltunen JK, Erdmann R, Hamilton B, Hartig A, Ruis H, Rottensteiner H (2001) Saccharomyces cerevisiae Adr1p governs fatty acid beta-oxidation and peroxisome proliferation by regulating POX1 and PEX11. J Biol Chem 276: 31825–31830 [DOI] [PubMed] [Google Scholar]
- Gurvitz A, Rottensteiner H (2006) The biochemistry of oleate induction: transcriptional upregulation and peroxisome proliferation. Biochim Biophys Acta 1763: 1392–1402 [DOI] [PubMed] [Google Scholar]
- Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne JB, Reynolds DB, Yoo J, Jennings EG, Zeitlinger J, Pokholok DK, Kellis M, Rolfe PA, Takusagawa KT, Lander ES, Gifford DK, Fraenkel E, Young RA (2004) Transcriptional regulatory code of a eukaryotic genome. Nature 431: 99–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong EL, Balakrishnan R, Christie KR, Costanzo MC, Dwight SS, Engel SR, Fisk DG, Hirschman JE, Livstone MS, Nash R, Park J, Oughtred R, Skrzypek M, Starr B, Theesfeld CL, Andrada R, Binkley G, Dong Q, Lane C, Hitz B, Miyasato S, Schroeder M, Sethuraman A, Weng S, Dolinski K, Botstein D, Cherry JM. Saccharomyces Genome Database, http://www.yeastgenome.org/(7/19/2006) [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O'Shea EK (2003) Global analysis of protein localization in budding yeast. Nature 425: 686–691 [DOI] [PubMed] [Google Scholar]
- Hwang D, Smith JJ, Leslie DM, Weston AD, Rust AG, Ramsey S, de Atauri P, Siegel AF, Bolouri H, Aitchison JD, Hood L (2005) A data integration methodology for systems biology: experimental verification. Proc Natl Acad Sci USA 102: 17302–17307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideker T, Thorsson V, Ranish JA, Christmas R, Buhler J, Eng JK, Bumgarner R, Goodlett DR, Aebersold R, Hood L (2001) Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science 292: 929–934 [DOI] [PubMed] [Google Scholar]
- Ideker T, Thorsson V, Siegel AF, Hood LE (2000) Testing for differentially-expressed genes by maximum-likelihood analysis of microarray data. J Comput Biol 7: 805–817 [DOI] [PubMed] [Google Scholar]
- Kashtan N, Itzkovitz S, Milo R, Alon U (2004) Topological generalizations of network motifs. Phys Rev 70: 031909. [DOI] [PubMed] [Google Scholar]
- Koerkamp MG, Rep M, Bussemaker HJ, Hardy GP, Mul A, Piekarska K, Szigyarto CA, De Mattos JM, Tabak HF (2002) Dissection of transient oxidative stress response in Saccharomyces cerevisiae by using DNA microarrays. Mol Biol Cell 13: 2783–2794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai LC, Kosorukoff AL, Burke PV, Kwast KE (2005) Dynamical remodeling of the transcriptome during short-term anaerobiosis in Saccharomyces cerevisiae: differential response and role of Msn2 and/or Msn4 and other factors in galactose and glucose media. Mol Cell Biol 25: 4075–4091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, Hannett NM, Harbison CT, Thompson CM, Simon I, Zeitlinger J, Jennings EG, Murray HL, Gordon DB, Ren B, Wyrick JJ, Tagne JB, Volkert TL, Fraenkel E, Gifford DK, Young RA (2002) Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298: 799–804 [DOI] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14: 953–961 [DOI] [PubMed] [Google Scholar]
- Luscombe NM, Babu MM, Yu H, Snyder M, Teichmann SA, Gerstein M (2004) Genomic analysis of regulatory network dynamics reveals large topological changes. Nature 431: 308–312 [DOI] [PubMed] [Google Scholar]
- Macisaac KD, Gordon DB, Nekludova L, Odom DT, Schreiber J, Gifford DK, Young RA, Fraenkel E (2006) A hypothesis-based approach for identifying the binding specificity of regulatory proteins from chromatin immunoprecipitation data. Bioinformatics 22: 423–429 [DOI] [PubMed] [Google Scholar]
- MacPherson S, Larochelle M, Turcotte B (2006) A fungal family of transcriptional regulators: the zinc cluster proteins. Microbiol Mol Biol Rev 70: 583–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda YT, Sano M (2006) Regulatory dynamics of synthetic gene networks with positive feedback. J Mol Biol 359: 1107–1124 [DOI] [PubMed] [Google Scholar]
- Mangan S, Alon U (2003) Structure and function of the feed-forward loop network motif. Proc Natl Acad Sci USA 100: 11980–11985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzolf B, Deutsch EW, Moss P, Campbell D, Johnson MH, Galitski T (2006) SBEAMS-microarray: database software supporting genomic expression analyses for systems biology. BMC Bioinformatics 7: 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrell D, Bolouri H (2004) Control of internal and external noise in genetic regulatory networks. J Theor Biol 230: 301–312 [DOI] [PubMed] [Google Scholar]
- Phelps C, Gburcik V, Suslova E, Dudek P, Forafonov F, Bot N, MacLean M, Fagan RJ, Picard D (2006) Fungi and animals may share a common ancestor to nuclear receptors. Proc Natl Acad Sci USA 103: 7077–7081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey SA, Smith JJ, Orrell D, Marelli M, Petersen TW, de Atauri P, Bolouri H, Aitchison JD (2006) Dual feedback loops in the GAL regulon suppress cellular heterogeneity in yeast. Nat Genet 38: 1082–1087 [DOI] [PubMed] [Google Scholar]
- Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, Zeitlinger J, Schreiber J, Hannett N, Kanin E, Volkert TL, Wilson CJ, Bell SP, Young RA (2000) Genome-wide location and function of DNA binding proteins. Science 290: 2306–2309 [DOI] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A (2000) EMBOSS: the European molecular biology open software suite. Trends Genet 16: 276–277 [DOI] [PubMed] [Google Scholar]
- Rottensteiner H, Hartig A, Hamilton B, Ruis H, Erdmann R, Gurvitz A (2003a) Saccharomyces cerevisiae Pip2p-Oaf1p regulates PEX25 transcription through an adenine-less ORE. Eur J Biochem 270: 2013–2022 [DOI] [PubMed] [Google Scholar]
- Rottensteiner H, Wabnegger L, Erdmann R, Hamilton B, Ruis H, Hartig A, Gurvitz A (2003b) Saccharomyces cerevisiae PIP2 mediating oleic acid induction and peroxisome proliferation is regulated by Adr1p and Pip2p-Oaf1p. J Biol Chem 278: 27605–27611 [DOI] [PubMed] [Google Scholar]
- Schuller HJ (2003) Transcriptional control of nonfermentative metabolism in the yeast Saccharomyces cerevisiae. Curr Genet 43: 139–160 [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen-Orr SS, Milo R, Mangan S, Alon U (2002) Network motifs in the transcriptional regulation network of Escherichia coli. Nat Genet 31: 64–68 [DOI] [PubMed] [Google Scholar]
- Smith JJ, Marelli M, Christmas RH, Vizeacoumar FJ, Dilworth DJ, Ideker T, Galitski T, Dimitrov K, Rachubinski RA, Aitchison JD (2002) Transcriptome profiling to identify genes involved in peroxisome assembly and function. J Cell Biol 158: 259–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JJ, Sydorskyy Y, Marelli M, Hwang D, Bolouri H, Rachubinski RA, Aitchison JD (2006) Expression and functional profiling reveal distinct gene classes involved in fatty acid metabolism. Mol Syst Biol 2: 2006.0009 10.1038/msb4100051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana C, Yoo JY, Tagne JB, Kacherovsky N, Lee TI, Young ET (2005) Combined global localization analysis and transcriptome data identify genes that are directly coregulated by Adr1 and Cat8. Mol Cell Biol 25: 2138–2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titz B, Thomas S, Rajagopala SV, Chiba T, Ito T, Uetz P (2006) Transcriptional activators in yeast. Nucleic Acids Res 34: 955–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young ET, Dombek KM, Tachibana C, Ideker T (2003) Multiple pathways are co-regulated by the protein kinase Snf1 and the transcription factors Adr1 and Cat8. J Biol Chem 278: 26146–26158 [DOI] [PubMed] [Google Scholar]
- Yu H, Gerstein M (2006) Genomic analysis of the hierarchical structure of regulatory networks. Proc Natl Acad Sci USA 103: 14724–14731 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Table 1
Supplementary Table 2