

Abstract
A thorough understanding of the structure of fusion domains of enveloped viruses in changing lipid environments helps us to formulate mechanistic models on how they might function in mediating viral entry by membrane fusion. We have expressed the N-terminal fusion domain of HIV-1 gp41 as a construct that is water-soluble in the absence of membranes, but that also binds with high affinity to lipid micelles and bilayers in their presence. We have solved the structure and studied the dynamics of this domain bound to dodecylphosphocholine micelles by homo- and heteronuclear NMR spectroscopy. The fusion peptide forms a stable hydrophobic helix from Ile4 to Ala14, but is increasingly more disordered and dynamic in a segment of intermediate polarity that stretches from Ala15 to Ser23. When bound to lipid bilayers at low concentration, the HIV fusion domain is also largely α-helical, as determined by CD and FTIR spectroscopy. However, at higher protein/lipid ratios, the domain is partially converted to form β-structures in lipid bilayers. Controlled lipid mixing occurs at concentrations that support the α-helical, but not the β-strand conformation.
INTRODUCTION
Like many other enveloped viruses, human immunodeficiency virus (HIV) enters cells by binding to receptors on the surface of susceptible cells followed by fusion of the viral envelope with the cell's plasma membrane. HIV binds to and infects cells that express the receptor CD4 and a chemokine co-receptor such as CCR5 or CXCR4. These receptors are recognized by the gp120 subunit of the surface-expressed viral spike glycoprotein gp120/gp41. Receptor and co-receptor recognition triggers a conformational change in gp120/gp41, which in turn initiates the merging of the viral and cell membranes by membrane fusion. The gp41 subunits of the gp120/gp41 trimer are thought to be primarily responsible for the fusion reaction. Indeed, a major restructuring of the gp41 subunits occurs because of receptor binding (1–3). This conformational change exposes the hydrophobic N-terminal domain of gp41 to the cell surface and makes it available for binding and insertion into the lipid bilayer of the target cell membrane (4). Since hydrophobic domains like the one observed at the N-terminus of gp41 are highly conserved within but not across different enveloped virus families and since even very minor mutations in their sequences can lead to severe defects in membrane fusion, these domains have been termed “fusion peptides.” It has been shown for a few viruses, most notably influenza virus, that their fusion peptides are the major domains besides the C-terminal transmembrane domains that interact with the lipid bilayers of the fused membranes. The other domains (ectodomains) probably interact only minimally with lipid bilayers, although some sequences of the intraviral C-terminal domain of gp41 form strongly amphipathic membrane-interactive helices that may contribute to the mechanism of gp41-mediated membrane fusion (5).
The fusion domain or fusion peptide of HIV-1 gp41 comprises ∼15 apolar residues followed by another eight moderately polar residues (Fig. 1). A determination of the structure of this domain in lipid bilayers or other membrane-mimetic environments and biophysical studies of its interaction with membranes should help us to better understand how this domain might help fusing two otherwise inert membranes. Unfortunately, the strong apolar nature of the HIV fusion peptide causes it to not only strongly associate with lipid bilayers, but also endows it with a high propensity for self-association in solution. These properties have complicated the structural analysis and biophysical interaction studies with membranes and are probably the root cause for many diverging reports on this topic in the literature. It seems quite clear that the size, the model membrane system, the hydration state, solvents used for preparing peptide-lipid complexes, and other environmental factors contribute sometimes critically to the observed results. Early CD and FTIR studies using either short peptide models or peptide-lipid complexes at low protein concentration indicated that these peptides inserted as oblique helices into lipid model membranes (6–8). Later investigations with longer peptide models favored a transition to β-sheet structures at higher protein/lipid ratios (6,9) and concluded that the ability to form β-structures correlated with the ability of these domains to fuse liposomes (10,11). Conformational mapping by isotope-edited FTIR spectroscopy indicated an α-helical structure of the first 16 residues of the gp41 fusion domain in lipid bilayers at moderate protein/lipid ratios (12). However, fusion domains in solution or in the presence of lipid bilayers at high protein concentrations aggregated into β-sheets (12). To disperse the hydrophobic fusion domains in solution, they were often added from organic solvents to lipids in organic solvent or to preformed liposomes in aqueous solution in the studies described so far. Structural studies by solid-state NMR confirmed that fusion domains bound to mixed lipid bilayers containing cholesterol at protein/lipid molar ratios between 1:20 and 1:200 and measured at −50°C formed extended β-strands (13). Subsequent solid-state NMR REDOR experiments of the same fusion domain and a variant with three C-terminally attached lysines established that the membrane-bound β-strands aggregated in parallel and anti-parallel arrangements, which were each approximately equally populated (14). The fusion domains with the trilysine tags were shown to be soluble and monomeric in the aqueous phase before they were bound to the bilayers and the protein-lipid complexes were pelleted for the solid-state NMR measurements (15). Even though physiological lipid mixtures were used in these studies, concerns remain that the peptides may have been expelled from the membrane when they were cooled to −50°C, where most lipids form phases with highly ordered acyl chains. In contrast to the work in bilayers by solid-state NMR, solution-state NMR of short and long fusion domain constructs in SDS micelles favored helical conformations even at the relatively high protein concentrations that are needed for NMR (16,17). The questions then arise whether the strong ionic detergent SDS is too harsh to represent the structure of the fusion domain in membranes and whether the structures observed in bilayers are representative of structures that are physiologically delivered to lipid bilayers in the liquid-crystalline state through aqueous environments. DPC is a much milder detergent than SDS; it shares its phosphocholine headgroup with that of the most abundant phospholipid class in cell membranes. Previous work also showed that the HIV fusion domain remained α-helical in DPC, but the samples were either not stable enough (17) or the homonuclear resonance lines were not resolved well enough for a complete structure determination by NMR (18) in DPC micelles that were successful for solving the structure of the influenza hemagglutinin fusion domain (19). Adding a highly polar tag to the C-terminus of the fusion domain dramatically increases its solubility and permits binding of the domain to micelles or lipid bilayers from solution rather than from organic solvents (20). This strategy has proven key to the success with the structure determination of wild-type and several mutant influenza fusion domains (21) and has also been employed by Jaroniec et al. (17) and in the solid-state NMR work by Yang et al. (14,15). We have made a construct consisting of the first 23 residues of the fusion domain linked to a pentalysine moiety with a flexible triple-glycine linker (Fig. 1). This protein domain is soluble in aqueous buffers and binds strongly to lipid micelles and bilayers. Its structure and dynamics were determined by solution NMR in DPC micelles and its conformational plasticity was examined by CD and FTIR spectroscopy in lipid bilayers. Our data indicate that the monomeric helical structure remains helical in bilayers at low concentrations, but converts to structures containing substantial amounts of β-sheet at higher protein concentrations in lipid bilayer model membranes. Fusion domain concentrations that form α-helices are sufficient to mediate efficient fusion (lipid mixing) of model membranes.
FIGURE 1.

Strategy for expression and purification of a soluble version of the HIV-1 gp41 fusion domain. See text for details.
MATERIALS AND METHODS
Expression and purification of HIV-1 gp41 fusion domain for NMR studies
The fusion protein construct used for expression of the HIV fusion domain consisted of KSI-M-P23H8-M-His6, in which KSI is the 125 amino-acid ketosteroid isomerase, M is methionine, P23H8 is the fusion domain consisting of the 23 most N-terminal residues of HIV-1 gp41 (LAVmal strain) and the eight-residue host peptide of three glycines and five lysines, and His6 is a hexahistidine-tag for purification. Methionines were introduced before and after the fusion domain as cut sites to remove KSI and the His tag. The DNA template was synthesized by the Biomolecular Research Facility at the University of Virginia. The single methionine residue in position 19 of the wild-type sequence of the fusion domain was mutated to a valine with the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) to avoid cleavage at this position. The final expression plasmid was constructed by inserting P23H8-M-His6 coding sequence at the AlwNI site of the pET31b(+)vector (Novagen, Madison, WI). The plasmid was transformed into BLR(DE3)pLysS cells for large scale expression. 15N-labeled protein was expressed in EMBL minimal media supplemented with (15NH4)2SO4 and 15N-labeled Bioexpress (Cambridge Isotope Laboratories, Andover, MA). The cells were induced with 1 mM IPTG at an OD600 of 0.3–0.5 at 37°C and grown for another 4 h.
Harvested cells were resuspended in binding buffer (5 mM imidazole, 40 mM Tris-HCl, 500 mM NaCl, pH 7.9), sonicated on ice until no longer viscous, and centrifuged at 12,000 g for 10 min at 4°C. The majority of the KSI-M-P23H8-M-His6 protein was found as inclusion bodies in the insoluble fraction, and was extracted with binding buffer containing 6 M guanidinium-HCl. The dissolved material was loaded on a nickel affinity column, and washed with six bed volumes of washing buffer (16 mM imidazole, 40 mM Tris-HCl, 500 mM NaCl, pH 7.9) containing 6 M guanidinium-HCl. The fusion protein was eluted from the column with six bed volumes of elution buffer (300 mM imidazole, 40 mM Tris-HCl, 500 mM NaCl, pH 7.9) containing 6 M guanidinium-HCl. Three-milliliter fractions were collected and the peak fractions (∼12 ml) were combined and dialyzed overnight at room temperature against three 1-liter changes of H2O in 8000 cut-off dialysis bags. The majority of the protein formed a white precipitate and was pelleted by centrifugation at 2000 g for 10 min at 4°C. To remove the KSI fusion protein and His-tag from the fusion domain, the dense white pellet was dissolved in 50% trifluoroacetic acid/H2O, and 100 mol CNBr for each mol of methionine (there are four methionines in the whole construct) was added to the solution. After stirring overnight at room temperature under nitrogen and in the dark, the reactant mixture was diluted 10-fold with water and lyophilized. The powder was dissolved in 30% CH3CN/H2O and purified by reverse-phase HPLC (Beckman Coulter, Fullerton, CA) on a model No. 218TP510 column (Grace-Vydac, Deerfield, IL) with a gradient of water/acetonitrile containing 0.1% trifluoroacetic acid. Peptide homogeneity and identity were analyzed by analytical HPLC and matrix-assisted laser desorption/ionization time-of-flight mass spectroscopy, respectively, and the purity was estimated >95%. CNBr cleavage results in a homoserine lactone instead of free carboxyl group at the C-terminus of P23H8 (see Fig. 1). The purified fusion domain was lyophilized and stored at −20°C before further use.
Liposomes
Small unilamellar vesicles were prepared by mixing four parts of POPC and one part of POPG (Avanti Polar Lipids, Alabaster, AL) in chloroform, evaporating the solvent under a stream of nitrogen, followed by evacuation for >3 h to remove residual solvent. The lipid dispersions were rehydrated and vortexed in 5 mM HEPES, 10 mM MES buffer, pH 7.4, followed by sonication with a probe sonicator for 45 min at 50% duty cycle on an ice-water bath. Large unilamellar vesicles of the same composition were prepared by extrusion (15 passes) of the vortexed and freeze-thawed (five cycles) lipid dispersions through two polycarbonate membranes (100 nm pore size) using a Liposofast extruder (Avestin, Ottawa, ON).
Lipid mixing assay for membrane fusion
Membrane fusion was measured by fluorescence resonance energy transfer (22) using a Fluorolog-3 spectrofluorometer (Jobin-Yvon, Edison, NJ). Two populations of large unilamellar vesicles composed of POPC/POPG (4:1), one unlabeled and one labeled with 1% each of n-[7-nitro-2-1,3-benzoaxadiazole-4-yl]-egg-phosphatidylethanolamine and n-[lissamine rhodamine B]-egg-phosphatidylethanolamine, were mixed at a 9:1 unlabeled/labeled ratio and 200 μM total lipid in 1.2 mL HEPES/MES buffer at pH 7.4 at 25°C. After equilibration of the vesicles, 10–50 μl of a 1 mg/ml fusion domain stock solution were added to give 3–15 μM final protein concentrations. The fluorescence before protein addition and after the addition of 120 μL of 20% Triton X-100 to give 2% final Triton X-100 were taken as 0 and 100% fusion, respectively. All experiments were performed in triplicates, and representative curves are shown.
Circular dichroism
Fusion domains in 5 mM HEPES, 10 mM MES buffer, pH 7.4 were added to small unilamellar vesicles composed of POPC/POPG (4:1) or DPC (Avanti Polar Lipid, Alabaster, AL) micelles at a ratio of 1:100 protein/lipid to give 0.1 mM protein and degassed for 5 min at room temperature before measurements. CD spectra were collected at 25°C on an AVIV model 215 spectropolarimeter (AVIV, Lakewood, NJ). The signals from pure small unilamellar vesicles or DPC micelles were subtracted from the sample spectra as blanks. Helical fractions, fH, were estimated from the molar ellipticities at 222 nm, θ222, and the expression fH = −(θ222 − 895)/34,870, which is derived from more general expressions applied to the length of our protein and the measuring temperature (23,24).
Fourier transform infrared spectroscopy
The ATR-FTIR spectra were recorded on a Vector 22 Fourier transform infrared spectrometer (Bruker, Billerica, MA). Planar phospholipid bilayers supported on germanium ATR plates were prepared by Langmuir-Blodgett deposition and fusion of small unilamellar vesicles as described previously (25). The substrate-supported monolayer was 1,2-dimyristoyl-3-sn-phosphatidylcholine, and the monolayer exposed to the buffer compartment was POPC/POPG (4:1). Fusion domains at 10 μg/ml in 5 mM HEPES, 10 mM MES buffer, pH 7.4 were added to the sample cell, incubated for 5 min at room temperature, and excess unbound protein was washed away with 3 vol of the same buffer made in D2O.
Sample preparation for NMR
Peptides were dissolved into d38- DPC micelles in 20 mM d4-acetic acid pH 7 buffer (95% H2O/5% D2O), containing 5 mM dithiothreitol and 0.05% NaN3. The final 15N-labeled P23H8 samples contained 1 mM protein and 200 mM d38-DPC (Cambridge Isotope Laboratories), and the unlabeled samples contained 2 mM peptide and 400 mM d38-DPC.
NMR spectroscopy
NMR spectra were recorded at 30°C on Varian Innova 500 and 600 MHz (Palo Alto, CA) and Bruker Avance 600 MHz spectrometers (Madison, WI) equipped with triple-resonance z-gradient cryogenic probes. Resonances were assigned from TOCSY (mixing times 50 and 80 ms), NOESY (mixing times 80 and 120 ms), and 15N-HSQC-NOESY (mixing times 120 ms) spectra. 3JHNHα couplings were measured using a three-dimensional HNHA experiment (26). The {1H}-15N heteronuclear NOEs were measured as described with a 5-s saturation delay (27). All NMR spectra were processed in NMRPipe (28) and analyzed in SPARKY (29).
Structure calculation
Dihedral angles φ were calculated from experimental 3JHNHα couplings according to
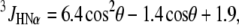 |
(1) |
where θ = |φ − 60°| (30). Structures were calculated with the program DYANA (31). The input consisted of the NOE upper distance constraints and dihedral angle constraints derived from NOESY spectra using the program HABAS and from the 3JHNHα experimental data, respectively. A total of 300 structures were calculated, and the 40 lowest energy conformers were selected for further optimization by using the AMBER force field implemented in the program OPAL (32). The resulting energy-minimized conformers were used to represent the structure of the fusion domain in DPC micelles.
Protein structure accession number
The coordinates of the structure of the HIV-1 gp41 fusion domain in DPC micelles have been deposited in the Protein DataBank under accession No. 2PJV.
RESULTS AND DISCUSSION
Multiple strategies were pursued to obtain sufficient amounts of a soluble HIV fusion domain construct for structural and biophysical studies on the interaction of these domains with lipid bilayers. Preliminary studies with synthetic peptides showed that a host peptide with a minimum of five lysines at the C-terminus were necessary to solubilize the HIV fusion domain; four lysines were sufficient in the case of the influenza HA fusion domain (20). Preliminary studies also showed that homonuclear NMR with these constructs was not sufficient to resolve all resonances needed for a structure determination. Therefore, we expressed and purified the 15N-labeled fusion domain (P23H8) as a fusion protein with KSI as outlined in Fig. 1 and described in more detail in Materials and Methods. The KSI-HIV fusion protein, which also contained a C-terminal His-tag for purification, could be expressed in high yields. Typically, we obtained 35 mg of fusion protein per liter of culture in minimal media. Initial attempts to express P23H8 either by itself or as a fusion protein with glutathione transferase failed. The fusion domain appears to be toxic to cells when expressed as a soluble construct. However, when expressed as a fusion protein with KSI, it is directed to inclusion bodies and apparently does not interfere with cell growth. The KSI and His tags were removed from P23H8 by cyanogen bromide cleavage, which cleaves after methionines and converts the methionine into a peptidyl homoserine lactone at the C-terminus of the target protein (Fig. 1). To avoid internal cleavage of the HIV fusion domain, methionine 19 was changed to a valine. This substitution did not affect CD or homonuclear NMR spectra in lipid bilayers or DPC micelles (data not shown). The final product contained the first 23 residues of the HIV-1 fusion domain, followed by three glycines providing a flexible linker, and five lysines substituting for the ectodomain of gp41. The fusion domain is soluble in various aqueous buffers, and binds to lipid micelles and bilayers with high affinity. We anticipate that the general expression and purification strategy that was developed here for the HIV fusion domain will also be useful for the preparation of other toxic hydrophobic viral fusion domains and pore-forming bacterial toxins.
In our experience, high-resolution NMR spectra of viral fusion domains can only be obtained when they are prepared as soluble constructs that can be bound to lipid micelles or bilayers from solution. Recombinants of detergents and peptides prepared in organic solvent and subsequently hydrated do not yield highly resolved NMR spectra in our hands. An HSQC spectrum of the HIV fusion domain P23H8 in DPC micelles obtained at 30°C is shown in Fig. 2. This sample was stable and its spectrum did not change for several months. Cross-peaks for all residues of the P23 moiety except for Ala1 and Val2 are shown. The backbone and side-chain resonances were assigned from TOCSY, NOESY, and 15N-HSQC NOESY experiments recorded at 600 MHz. The complete resonance assignments are shown in Supplementary Material Table S1.
FIGURE 2.

15N-1H HSQC spectrum of the HIV-1 gp41 fusion domain bound to DPC micelles at 30°C.
A total of 63 NOE upper distance constraints were obtained from the NOESY and 15N-HSQC NOESY spectra. Medium-range NOEs were observed between residues 3 and 15, indicating that this region is likely helical when the fusion domain is bound to DPC micelles (Fig. 3). Negative (positive) differences between the observed Hα (Hβ) chemical shifts and tabulated chemical shifts for random coil (33) were present between residues 4 and 12, confirming a helical conformation of this segment (Fig. 4). Scalar 3JHNHα couplings were obtained from a three-dimensional HNHA experiment. Dihedral angles derived from these measurements are listed in Supplementary Material Table S2. Secondary structures predicted based on these J-coupling- and chemical shift-derived dihedral angles provide further evidence for a high propensity of α-helix between residues 4 and 12 (Fig. 3). There was no evidence for ordered secondary structures from Ala15 to Ser23. To confirm that this latter segment did not adopt a defined secondary structure, we measured {1H}-15N heteronuclear NOEs of the fusion domain bound to DPC micelles (Fig. 5). Relatively high values at ∼0.6 indicating a reasonably well-ordered structure were observed between Ile4 and Ala14. The heteronuclear NOEs gradually decreased from Ala15 and reached negative values between Gly20 and Ser23, confirming a highly flexible conformation at the C-terminus. Therefore, all structural and dynamical parameters (NOEs, chemical shifts, scalar couplings) consistently support a model of the DPC-bound fusion domain with a relatively well-ordered α-helix in the N-terminal portion and a disordered region in the C-terminal portion of the domain.
FIGURE 3.

Sequence, secondary structure propensities based on dihedral bond angles, and NOE connectivities of the HIV-1 gp41 fusion domain bound to DPC micelles at 30°C. In the rows showing φ- and ψ-dihedral angles, triangles indicate α-helix or 310-helix, circles indicate random coil, and φ- and ψ-angles of residues marked with stars are consistent with either α-helix or β-structure. In the row showing the χ1 torsion angle, the sizes of squares indicate whether the experimental values allow for one (large), two (medium), or all three (small) staggered rotamer positions χ1 = −60, 60, 180. Torsion angle restraints that exclude all three rotamer positions are shown as solid circles. The horizontal bars in the rows marked with d indicate distances between respective protons that have been observed by measurements of the corresponding NOEs.
FIGURE 4.

Secondary chemical shifts of HIV-1 gp41 fusion domain bound to DPC micelles at 30°C. (Upper panel) Difference of Hα shifts δobserved – δrandom coil. (Lower panel) Difference of Hβ shifts δobserved – δrandom coil.
FIGURE 5.

{1H}-15N heteronuclear NOEs of HIV-1 gp41 fusion domain bound to DPC micelles at 30°C.
Using the experimentally determined distance and dihedral angle constraints, a structure of the micelle bound fusion domain was calculated. Structural families of the N-terminal portion (residues 1–18) and the entire domain (residues 1–24) are presented in Fig. 6, a and b, respectively. The corresponding structural statistics are presented in Table 1. Consistent with the data of Fig. 5, residues 1–17 are quite well ordered (heavy atom RMSD 1.00 ± 0.23 Å) and residues 4–14 are very well ordered (heavy atom RMSD 0.79 ± 0.24 Å). Fig. 6 c shows a typical structure with the side chains inserted and Fig. 6 d shows the same structure in a surface representation. This figure shows that the surface of the helical portion of the HIV fusion domain is quite rough. Several prominent features protrude from the surface, most notably those formed by Phe8 and Phe11, but also those of Ile4 and Leu9. Interestingly, the tandem FLG repeat of the HIV fusion domain sequence is quite highly conserved throughout many strains of HIV (34). Several depressions are also seen on the surface, but they do not correspond to the exact positions of the many glycines (marked with asterisks in Fig. 6 d), which are also prominent in many fusion domain sequences including those of HIV gp41 (35). The fitting of hydrophobic knobs into hydrophobic holes on membrane-inserted helix surfaces is known to promote helix interactions in membranes (36) and thus could provide a mechanism for the gp41 fusion domain to interact with itself and/or the transmembrane domain at different stages of membrane fusion. An interaction of the fusion domain with the transmembrane domain has been postulated to be an important late step in the mechanism of influenza hemagglutinin-mediated membrane fusion (35).
FIGURE 6.

Structural models of HIV-1 gp41 fusion domain bound to DPC micelles at 30°C. (a) Forty lowest energy structures with first 18 residues aligned. (b) Same as panel a, but residues 1–24 are shown. (c) Two different views of closest-to-the-mean structure of residues 1–24 with side chains shown. (d) Same as panel c, but only first 18 residues in an electrostatic surface potential representation. Negative potentials are shown in red and positive potentials are shown in blue. Asterisks mark the positions of the Cα atoms of glycines 3, 5, 10, 13, and 16.
TABLE 1.
Structural statistics of the HIV fusion domain in DPC micelles at 30°C
| Target function (Å) | 0.25 ± 0.001 |
| Experimental NMR constraints | |
| Total constraints | 92 |
| NOE distance constraints | 63 |
| J-coupling constraints | 18 |
| Dihedral constraints | 11 |
| NMR constraint violations | |
| NOE constraint violations | |
| Sum (Å) | 1.2 ± 0.0 |
| Maximum (Å) | 0.18 ± 0.00 |
| Angle constraint violations | |
| Sum (o) | 11.4 ± 0.0 |
| Maximum (o) | 2.9 ± 0.0 |
| AMBER energy (kcal/mol) | |
| Root mean-square deviation from mean structure (Å) | |
| Backbone atoms of all residues 1–24 | 3.45 ± 1.28 |
| Heavy atoms of all residues 1–24 | 3.84 ± 1.24 |
| Backbone atoms of residues 1–17 | 0.60 ± 0.21 |
| Heavy atoms of residues 1–17 | 1.00 ± 0.23 |
| Backbone atoms of residues 4–14 | 0.20 ± 0.10 |
| Heavy atoms of residues 4–14 | 0.79 ± 0.24 |
| Ramachandran statistics analyzed using PROCHECK-NMR residues in allowed regions | 90.9+9.1% for residues 3–20; 100% for residues 4–17* |
90.9% in allowed region, 9.1% in additionally allowed region for residues 3–20; residues 4–17 are all in allowed region.
To examine whether the structure obtained in DPC micelles is likely representative of the structure in lipid bilayers, we recorded CD spectra in both environments and in solution (Fig. 7). Based on these CD spectra, the HIV fusion domain is unstructured in buffer with no or 0.2 M additional salt. The HIV fusion domain adopts a partially helical structure with characteristic CD minima at 208 and 222 nm when bound to 10 mM DPC micelles. The CD spectra virtually did not change in the peptide/lipid range 1:50–1:200, indicating the high affinity of the domain for these micelles and their complete binding under these conditions (Supplementary Material Fig. S1 A). All NMR spectra were recorded at 1:200 peptide/lipid, i.e., under saturating binding conditions. The fusion domain is ∼60% α-helical in DPC micelles based on quantitative estimates of the CD spectra in this environment. This α-helical content is consistent with the NMR structure presented in Fig. 6. When bound to 10-mM lipid bilayers composed of POPC and POPG (4:1), the negative molar ellipticity increased further. A quantitative estimate indicates that the α-helical content of the fusion domain is ∼70% in lipid bilayers. Clearly, bilayers are less dynamical than lipid micelles, which could account for the increased helical structure in bilayers. We estimate approximately four more residues to be helical in bilayers compared to the micelle structure. According to the dynamical data of Fig. 5, Gly3 at the N-terminus and Ala15 through Thr18 at the C-terminal end of the existing helix are the most likely to become ordered enough to increase the length of the helix in lipid bilayers. CD spectra of the fusion domain in 1 M NaCl and in the absence of lipid were also recorded. Under these conditions, the protein assumes some, but not as much secondary structure as in DPC micelles. Presumably the screening of the charges of the pentalysine tail used for solubilization lead to some aggregation with an associated change in secondary structure in 1 M NaCl. This experiment validates our design and reinforces why it is essential to introduce this highly charged polar tail to keep the fusion domain monodisperse for binding to lipid micelles and bilayers and for structural studies. Similar effects were observed with the fusion domains of influenza hemagglutinin, which also self-associate in 1 M NaCl (37).
FIGURE 7.

CD spectra of HIV-1 gp41 fusion domain in solution, bound to DPC micelles, and bound to lipid bilayers composed of POPC/POPG (4:1). (Solid line) 0.1 mM P23H8 in 5 mM HEPES, 10 mM MES buffer, pH 7.4. (Dash-dotted line) Same with 0.2 M NaCl added. (Dash-dot-dot line) Same with 1 M NaCl added. (Dashed line) 0.1 mM P23H8 bound to 10 mM DPC micelles. (Dotted line) 0.1 mM P23H8 bound to 10 mM POPC:POPG (4:1) bilayers. All spectra were recorded at 25°C.
Previous studies indicated that the HIV fusion domain may also assume β-structures in lipid bilayers (10,11,13–15). To further characterize the secondary structure of this domain in lipid bilayers, we recorded FTIR spectra of the domain bound to planar supported bilayers composed of POPC and POPG (4:1) as a function of protein concentration. Representative FTIR spectra in the amide I region are shown in Fig. 8. A major broad band centered at ∼1650 cm∼1 dominates at all protein concentrations ranging from 10 to 40 μg/ml added protein. This band is commonly assigned to α-helical and random coil structures, which are not easily distinguished from each other in FTIR spectroscopy (25). However, a second band centered at 1630 cm−1 increases with increasing protein concentration. This band indicates increasing amounts of β-structures and the small shoulder at ∼1685 cm−1 hints at antiparallel β-sheets. (Parallel β-sheets do not exhibit this minor band, which in antiparallel β-sheets is usually ∼10% of the intensity of the main band at 1630 cm−1 (25)). Unfortunately, it is not straightforward to determine the exact protein/lipid ratios at the membrane in these single bilayer experiments for two reasons. First, the experiments are conducted under much more dilute solution conditions than the NMR and CD experiments so that we cannot assume quantitative binding of all protein to the membrane surface. For the same reason, a significant amount of the injected protein may adsorb nonspecifically to other surfaces than the membrane in the sample cell (opposite surface of the holding cell, access tubing, etc.). Nevertheless, we can make a very crude order-of-magnitude estimate. The exposed membrane area in the holding cell corresponds to ∼6 μM lipid and 10 μg/ml is equivalent to a 3.4-μM injected protein solution. If we assume that ∼10% of the injected protein actually binds to the membrane surface, we begin with a protein/lipid ratio of ∼1:20 and increase it to 4:20 as we go to the highest concentration of 40 μg/ml added fusion domain. These are significantly higher protein/lipid ratios than used in the CD and NMR experiments of this work. Clearly, increasing the protein concentration at the membrane surface gradually converts the HIV fusion domain to antiparallel β-structure by a membrane-assisted self-association process. Self-association of fusion domains likely occurred in numerous previous studies where β-structures were reported for HIV fusion domains that were either not initially dispersed by a polar tail, but mixed with lipids in organic solvents, or extruded from lipid bilayers below their gel-to-liquid-crystalline phase transition.
FIGURE 8.

ATR-FTIR spectra of HIV-1 gp41 fusion domain bound to planar-supported lipid bilayers composed of POPC/POPG (4:1) at different protein concentrations. (Solid line) Ten micrograms per milliliter P23H8 added to bilayer. (Dashed line) Twenty micrograms per milliliter P23H8 added to bilayer. (Dotted line) Thirty micrograms per milliliter P23H8 added to bilayer. (Dash-dotted line) Forty micrograms per milliliter P23H8 added to bilayer. All spectra were recorded at room temperature. The spectrum of the pure bilayer recorded before the first protein addition was subtracted from each spectrum.
A structure of the gp41 fusion domain that has been previously determined by solution NMR in SDS micelles is even more helical than ours (17). The N-terminal portions of the two structures are very similar, but the helix extends further up to Gly20 in the Jaroniec et al. (17) structure. Apparently, the relatively harsh ionic detergent SDS supports the helical conformation. Before the present work, it was not clear whether this structure was nonphysiologically induced by SDS. The fact that we observe a similar structure in a much more membranelike environment suggests that the helical structure is indeed physiological. This is supported by our CD and FTIR results, which also show helical conformations at low protein concentrations in fluid lipid model membranes. There appear to be two major differences between the structures in SDS and DPC (Supplementary Material Fig. S2). The helix is more extended toward the C-terminus in SDS and there is an apparent break in the helix around Gly10 in the DPC structure. Although we do not wish to overinterpret details of these differences (some could be due to dynamical differences and the number of collected NMR restraints in the two environments), the primary chemical shift data clearly indicate differences that for some residues are quite significant and for others more minor (Supplementary Material Fig. S3). Interestingly, the chemical shift trends seen in the titrations with DPC that were performed by Jaroniec et al. (17), but that did not lead to samples that were stable enough for a structure determination in this environment, extrapolate very nicely to the final chemical shifts that we observed in DPC (Supplementary Material Fig. S3). Therefore, Jaroniec et al. (17) appear to have recorded similar structural changes upon a partial transfer into DPC, although they were not able to obtain a structure under these conditions. An apparent detail of the differences between the two structures is the break of the helix at residue 10 in DPC, but not in SDS. However, closer inspection of the complete families of structures calculated in SDS and DPC shows that some DPC structures have a continuous helix and some SDS structures also have a break at this residue (17). The differences lie in the weights of populations, rather than actual structural differences of this region, in the two environments. The longer helix observed in the SDS structure could be induced by the anionic and much smaller headgroup of SDS compared to that of DPC or by the longer fusion domain construct that was used by these authors. Interestingly, our fusion domain construct is also 10–15% more helical in SDS than in DPC (Supplementary Material Fig. S1 B). Given the conformational plasticity of the HIV fusion domain in different environments as documented in Figs. 6 and 7 and the Supplementary Material figures, it is clear that details in sample preparation and environment are important factors for determining the functional structure of the HIV fusion domain in membranes.
To examine whether our fusion domain construct is able to fuse lipid bilayer model membranes and which conformation supports membrane fusion, we performed lipid mixing assays using a fluorescence resonance energy transfer method. Fusion domains alone usually give rise to leaky fusion, which makes fluorescent contents mixing assays not very useful for this purpose. However, lipid mixing has been observed for many active fusion domains (38), including that from influenza hemagglutinin (20). Fig. 9 shows lipid mixing and fusion between liposomes containing fluorescent probes that are quenched owing to resonance energy transfer and unlabeled liposomes as catalyzed by the gp41 fusion domain. Our soluble fusion domain construct P23H8 promoted efficient fusion between these liposomes. Control experiments using only the solubilizing host peptide H7 did not induce any lipid mixing or fusion. The rates and extents of fusion increased in the concentration range from 3 to 6 μM P23H6 (Fig. 9). At 7 μM, overall fusion still increased compared to 5 μM, but discontinuities were often observed (Supplementary Material Fig. S4). When the concentration was further increased, the dequenching decreased and the traces became increasingly more noisy (Supplementary Material Fig. S4). As was shown in Fig. 8, the α-helical form of the fusion domain is preferred at low concentrations, but the β-sheet form prevails when fusion domains are bound to membranes at high concentrations. The protein/lipid ratios in the experiment of Fig. 9 were 1:40 or lower, which is in the range where the α-helical form predominates. Significant amounts of β-sheet develop at protein/lipid ratios 1:20 and higher, which corresponds to the 10 and 15 μM traces shown in Supplementary Material Fig. S4. We conclude, therefore, that the α-helical form promotes membrane fusion whereas the β-sheet form is unable to induce a controlled fusion reaction. The α-helical structure determined by solution NMR in DPC micelles therefore likely represents the physiologically active conformation of the HIV-1 gp41 fusion domain in lipid bilayers.
FIGURE 9.

Lipid mixing between labeled and unlabeled liposomes composed of POPC/POPG (4:1) mediated by the HIV-1 gp41 fusion domain. Five (a) or three (b) μM P23H8 was added to 200 μM liposomes at time 0 s. Five μM of the control host peptide H7 (GCGKKKK) induces no lipid mixing (c).
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
This work was supported by grant No. R37 AI30557 from the National Institutes of Health.
Abbreviations used: ATR, attenuated total reflection; CD, circular dichroism; DPC, dodecylphosphocholine; FTIR, Fourier transform infrared; gp41, glycoprotein of molecular weight 41,000; gp120, glycoprotein of molecular weight 120,000; HA, hemagglutinin; HEPES, n-[hydroxyethyl]piperizine-n′-[2-ethanesulfonic acid]; HIV, human immunodeficiency virus; HSQC, heteronuclear single quantum coherence; KSI, ketosteroid isomerase; NMR, nuclear magnetic resonance; NOE, nuclear Overhauser effect; NOESY, nuclear Overhauser effect spectroscopy; POPC, 1-palmitoyl-2-oleoyl-3-sn-phosphatidylcholine; POPG, 1-palmitoyl-2-oleoyl-3-sn-phosphatidylglycerol; SDS, sodium dodecylsulfate; TOCSY, total correlated spectroscopy.
Editor: Mark Girvin.
References
- 1.Weissenhorn, W., A. Dessen, S. C. Harrison, J. J. Skehel, and D. C. Wiley. 1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 387:426–430. [DOI] [PubMed] [Google Scholar]
- 2.Kwong, P. D., R. Wyatt, J. Robinson, R. W. Sweet, J. Sodroski, and W. A. Hendrickson. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 393:648–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eckert, D. M., and P. S. Kim. 2001. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 70:777–810. [DOI] [PubMed] [Google Scholar]
- 4.Gallaher, W. R. 1987. Detection of a fusion peptide sequence in the transmembrane protein of human immunodeficiency virus. Cell. 50:327–328. [DOI] [PubMed] [Google Scholar]
- 5.Kliger, Y., and Y. Shai. 1997. A leucine zipper-like sequence from the cytoplasmic tail of the HIV-1 envelope glycoprotein binds and perturbs lipid bilayers. Biochemistry. 36:5157–5169. [DOI] [PubMed] [Google Scholar]
- 6.Rafalski, M., J. D. Lear, and W. F. DeGrado. 1990. Phospholipid interactions of synthetic peptides representing the N-terminus of HIV gp41. Biochemistry. 29:7917–7922. [DOI] [PubMed] [Google Scholar]
- 7.Martin, I., H. Schaal, A. Scheid, and J. M. Ruysschaert. 1996. Lipid membrane fusion induced by the human immunodeficiency virus type 1 gp41 N-terminal extremity is determined by its orientation in the lipid bilayer. J. Virol. 70:298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mobley, P. W., A. J. Waring, M. A. Sherman, and L. M. Gordon. 1999. Membrane interactions of the synthetic N-terminal peptide of HIV-1 gp41 and its structural analogs. Biochim. Biophys. Acta. 1418:1–18. [DOI] [PubMed] [Google Scholar]
- 9.Saez-Cirion, A., and J. L. Nieva. 2002. Conformational transitions of membrane-bound HIV-1 fusion peptide. Biochim. Biophys. Acta. 1564:57–65. [DOI] [PubMed] [Google Scholar]
- 10.Nieva, J. L., S. Nir, A. Muga, F. M. Goni, and J. Wilschut. 1994. Interaction of the HIV-1 fusion peptide with phospholipid vesicles: different structural requirements for fusion and leakage. Biochemistry. 33:3201–3209. [DOI] [PubMed] [Google Scholar]
- 11.Peisajovich, S. G., R. F. Epand, M. Pritsker, Y. Shai, and R. M. Epand. 2000. The polar region consecutive to the HIV fusion peptide participates in membrane fusion. Biochemistry. 39:1826–1833. [DOI] [PubMed] [Google Scholar]
- 12.Gordon, L. M., P. W. Mobley, R. Pilpa, M. A. Sherman, and A. J. Waring. 2002. Conformational mapping of the N-terminal peptide of HIV-1 gp41 in membrane environments using 13C-enhanced Fourier transform infrared spectroscopy. Biochim. Biophys. Acta. 1559:96–120. [DOI] [PubMed] [Google Scholar]
- 13.Yang, J., C. M. Gabrys, and D. P. Weliky. 2001. Solid-state nuclear magnetic resonance evidence for an extended β strand conformation of the membrane-bound HIV-1 fusion peptide. Biochemistry. 40:8126–8137. [DOI] [PubMed] [Google Scholar]
- 14.Yang, J., and D. P. Weliky. 2003. Solid-state nuclear magnetic resonance evidence for parallel and antiparallel strand arrangements in the membrane-associated HIV-1 fusion peptide. Biochemistry. 42:11879–11890. [DOI] [PubMed] [Google Scholar]
- 15.Yang, J., M. Prorok, F. J. Castellino, and D. P. Weliky. 2004. Oligomeric beta-structure of the membrane-bound HIV-1 fusion peptide formed from soluble monomers. Biophys. J. 87:1951–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang, D. K., S. F. Cheng, and W. J. Chien. 1997. The amino-terminal fusion domain peptide of human immunodeficiency virus type 1 gp41 inserts into the sodium dodecyl sulfate micelle primarily as a helix with a conserved glycine at the micelle-water interface. J. Virol. 71:6593–6602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaroniec, C. P., J. D. Kaufman, S. J. Stahl, M. Viard, R. Blumenthal, P. T. Wingfield, and A. Bax. 2005. Structure and dynamics of micelle-associated human immunodeficiency virus gp41 fusion domain. Biochemistry. 44:16167–16180. [DOI] [PubMed] [Google Scholar]
- 18.Morris, K. F., X. Gao, and T. C. Wong. 2004. The interactions of the HIV gp41 fusion peptides with zwitterionic membrane mimics determined by NMR spectroscopy. Biochim. Biophys. Acta. 1667:67–81. [DOI] [PubMed] [Google Scholar]
- 19.Han, X., J. H. Bushweller, D. S. Cafiso, and L. K. Tamm. 2001. Membrane structure and fusion-triggering conformational change of the fusion domain from influenza hemagglutinin. Nat. Struct. Biol. 8:715–720. [DOI] [PubMed] [Google Scholar]
- 20.Han, X., and L. K. Tamm. 2000. A host-guest system to study structure-function relationships of membrane fusion peptides. Proc. Natl. Acad. Sci. USA. 97:13097–13102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lai, A. L., H. Park, J. M. White, and L. K. Tamm. 2006. Fusion peptide of influenza hemagglutinin requires a fixed angle boomerang structure for activity. J. Biol. Chem. 281:5760–5770. [DOI] [PubMed] [Google Scholar]
- 22.Struck, D. K., D. Hoekstra, and R. E. Pagano. 1981. Use of resonance energy transfer to monitor membrane fusion. Biochemistry. 20:4093–4099. [DOI] [PubMed] [Google Scholar]
- 23.Rohl, C. A., and R. L. Baldwin. 1997. Comparison of NH exchange and circular dichroism as techniques for measuring the parameters of helix-coil transitions in peptides. Biochemistry. 36:8435–8442. [DOI] [PubMed] [Google Scholar]
- 24.Luo, P., and R. L. Baldwin. 1997. Mechanism of helix induction by trifluoroethanol: a framework for extrapolating the helix-forming properties of peptides from trifluoroethanol/water mixtures back to water. Biochemistry. 36:8413–8421. [DOI] [PubMed] [Google Scholar]
- 25.Tamm, L. K., and S. A. Tatulian. 1997. Infrared spectroscopy of proteins and peptides in lipid bilayers. Q. Rev. Biophys. 30:365–429. [DOI] [PubMed] [Google Scholar]
- 26.Vuister, G. W., and A. Bax. 1993. Quantitative J correlation: a new approach for measuring homonuclear three-bond J(HNHalpha) couplings in 15N-enriched proteins. J. Am. Chem. Soc. 115:7772–7777. [Google Scholar]
- 27.Farrow, N. A., R. Muhandiram, A. U. Singer, S. M. Pascal, C. M. Kay, G. Gish, S. E. Shoelson, T. Pawson, J. D. Forman-Kay, and L. E. Kay. 1994. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 33:5984–6003. [DOI] [PubMed] [Google Scholar]
- 28.Delaglio, F., S. Grzesiek, G. W. Vuister, G. Zhu, J. Pfeifer, and A. Bax. 1995. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 6:277–293. [DOI] [PubMed] [Google Scholar]
- 29.Goddard, T. D., and J. M. Kneller. SPARKY 3, University of California, San Francisco, CA.
- 30.Pardi, A., M. Billeter, and K. Wuthrich. 1984. Calibration of the angular dependence of the amide proton-Cα-proton coupling constants, 3JHNα, in a globular protein. Use of 3JHNα for identification of helical secondary structure. J. Mol. Biol. 180:741–751. [DOI] [PubMed] [Google Scholar]
- 31.Guntert, P., C. Mumenthaler, and K. Wuthrich. 1997. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273:283–298. [DOI] [PubMed] [Google Scholar]
- 32.Luginbuhl, P., P. Güntert, M. Billeter, and K. Wüthrich. 1996. The new program OPAL for molecular dynamics simulations and energy refinements of biological macromolecules. J. Biomol. NMR. 8:136–146. [DOI] [PubMed] [Google Scholar]
- 33.Wishart, D. S., B. D. Sykes, and F. M. Richards. 1992. The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry. 31:1647–1651. [DOI] [PubMed] [Google Scholar]
- 34.Tamm, L. K., and X. Han. 2000. Viral fusion peptides: a tool set to disrupt and connect biological membranes. Biosci. Rep. 20:501–518. [DOI] [PubMed] [Google Scholar]
- 35.Lai, A. L., Y. Li, and L. K. Tamm. 2005. Interplay of proteins and lipids in virus entry by membrane fusion. In Protein-Lipid Interactions: From Membrane Domains to Cellular Networks. L. K. Tamm, editor. WILEY-VCH Verlag, Weinheim, Germany.
- 36.MacKenzie, K. R., J. H. Prestegard, and D. M. Engelman. 1997. A transmembrane helix dimer: structure and implications. Science. 276:131–133. [DOI] [PubMed] [Google Scholar]
- 37.Han, X., and L. K. Tamm. 2000. pH-dependent self-association of influenza hemagglutinin fusion peptides in lipid bilayers. J. Mol. Biol. 304:953–965. [DOI] [PubMed] [Google Scholar]
- 38.Durell, S. R., I. Martin, J. M. Ruysschaert, Y. Shai, and R. Blumenthal. 1997. What studies of fusion peptides tell us about viral envelope glycoprotein-mediated membrane fusion (review). Mol. Membr. Biol. 14:97–112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.