

Abstract
We perform a systematic analysis of mechanisms of feedback regulation that underlie short-term adaptation in intracellular signaling systems. Upon receiving an external cue, these systems generate a transient response that quickly returns to basal levels even if the stimulus persists. Signaling pathways capable of short-term adaptation are found in systems as diverse as the high osmolarity response of yeast, gradient sensing in Dictyostelium, and the cytokine response in vertebrates. Using mathematical analysis and computational experiments, we compare different feedback architectures in terms of response amplitude and duration, ability to adapt, and response to variable stimulus levels. Our analysis reveals three important features of these systems: 1), multiple step signaling cascades improve sensitivity to low doses by an effect distinct from signal amplification; 2), some feedback architectures act as signal transducers converting stimulus strength into response duration; and 3), feedback deactivation acts as a dose-dependent switch between transient and sustained responses. Finally, we present characteristic features for each form of feedback regulation that can aid in their identification.
INTRODUCTION
Intracellular signaling pathways are an important component of the biochemical systems that allow cells to survive and proliferate in constantly changing environmental conditions. These pathways convert an external cue, such as a hormone, growth factor, or environmental stress, into an intracellular signal that generates an appropriate response to the challenge (1). Such responses can include changes in genetic expression and regulation of metabolic processes. The genetic program a cell follows is determined by both the amplitude and duration of the signal generated by the stimulus. For example, it has been demonstrated that epidermal growth factor causes transient activation of the ERK MAP kinase and leads to cell proliferation, while nerve growth factor causes sustained ERK activation and results in cell differentiation (2). It has also been suggested that, in yeast, sustained activation of the MAPK Kss1 leads to invasive growth, whereas transient Kss1 activation is required for a proper mating response (3). Moreover, abnormal or inappropriate activation of MAP kinase activity can lead to a number of diseases, including asthma and cancer. Therefore, understanding the control mechanisms that regulate the activity of signaling pathways is a fundamental problem in cell biology.
The external stimulus received by a cell, as well as the elicited response, can be temporally transient or sustained. However, the time-dependent behavior of the two does not have to coincide. For example, signaling systems termed adaptive generate a transient response in the presence of a sustained stimulus. Long-term adaptation results from induced changes in genetic expression. For example, stimulus-dependent induction of Socs1/3 is thought to inhibit Janus kinase's ability to phosphorylate Stat1/3, leading to long-term adaptation in the cytokine immune response (4). Short-term adaptation requires feedback or feed-forward regulation mediated by protein-protein interactions, phosphorylation events, or other biochemical mechanisms. In this case, the pathway not only functions as information transduction system but also plays an important role in regulating the whole cellular response. For example, we recently demonstrated that in the high osmolarity pathway of yeast, feedback phosphorylation of the osmosensor Sho1 by the MAPK Hog1 plays an important role in rapid signal attenuation after cells are exposed to high osmotic stress (5). To establish Sho1 as a target of feedback regulation, we combined experimental analysis with mathematical modeling. These investigations motivated the theoretical and computational studies presented below.
To provide a better understanding of the control mechanisms that lead to short-term adaptation, we performed a systematic analysis of various pathway architectures that regulate signaling through feedback inhibition. The models are compared in terms of their ability to produce a strong response followed by good adaptation, and the advantages and shortcomings of each signaling system are discussed. Also investigated is the ability of these systems to perform in the presence of changing conditions. Our results reveal that multilevel cascades improve the sensitivity of adapting systems by a mechanism distinct from signal amplification. We also demonstrate how feedback deactivation can be used to construct a dose-dependent switch between transient and sustained signaling and illustrate how certain pathway architectures convert stimulus strength into signal duration. The biological implications of these results are discussed. Additionally, we present characteristic features of each model system that can be used to help establish the underlying mechanism of adaptation in signaling pathways.
Mechanisms of adaptation
Broadly speaking, there are three strategies for achieving adaptation: integral control, feed-forward motifs, and negative feedback loops (Fig. 1). Integral control uses the time integral of the difference between the response and its prestimulus level to regulate signaling. This control mechanism has been discussed extensively in the literature in the context of bacterial chemotaxis (6–9). Systems containing feed-forward motifs, in which two stimulus-dependent pathways converge on a common signaling component, also produce adaptation when the parallel pathways have opposing effects on the common component. Feed-forward motifs have received considerable attention in the literature and underlie the regulation of genetic networks as well as signaling systems (9–12). Negative feedback loops, the focus of this article, are pervasive in signaling systems, and many models of pathway regulation based on negative feedback have been proposed (13–16). In these systems, adaptation is achieved when a signaling species initiates a feedback loop that negatively regulates its own activity either directly or indirectly by targeting an upstream pathway component. Here, regulation occurs as a result of the transduced signal (output), whereas in feed-forward architectures, regulation is mediated by upstream components (input) independently of the pathway's output. As a result, in feedback-based systems, the strength of upstream pathway inhibition is determined by the magnitude of the downstream response. For this reason, feedback is usually the method of choice for engineered systems in which control is exercised as a function of how the actual measured output compares to the desired target value. Negative regulation in signaling cascades can assume many forms including deactivation (17), desensitization (5,16), sequestration of an upstream species (18), spatial relocalization (19,20), or stimulus-dependent degradation of a pathway component (21). Both integral control and feed-forward motifs are capable of perfect adaptation in the sense that the signal returns exactly to its prestimulus level. Negative-feedback-based regulation does not, in general, produce perfect adaptation. However, in many cases it is capable of near-perfect adaptation that for biological purposes is indistinguishable from strict perfect adaptation. It is important to note that adaptation to sustained stimuli must meet two criteria: the propagated signal must be of sufficient amplitude and appropriate duration to elicit the required response, and, at the same time, return to basal or near-basal levels in the presence of a sustained stimulus. As we show in this work, these requirements are often in conflict with each other.
FIGURE 1.

Mechanisms for adaptation. (A) Integral control. (B) Feed-forward regulation. (C) Feedback inhibition through decreased activation (*) or increased deactivation (**).
METHODS
Mathematical modeling
In all the models, we assume activation and deactivation reactions follow Michaelis-Menten kinetics. This choice was made because Michaelis-Menten is the simplest form of saturable kinetics and is used extensively to describe enzymatic reactions. The rates of these reactions have the form
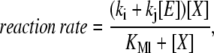 |
(1) |
where [E] is the concentration of the enzyme (e.g., kinase or phosphatase), [X] is the concentration of the substrate (e.g., unphosphorylated or phosphorylated pathway component), kj [E] is the maximum reaction velocity, KMl is the Michaelis constant, and ki represents basal enzymatic activity not associated with [E]. Note that for simplicity the Michaelis constant of the basal enzymatic activity is assumed to be the same as that of the enzyme E. Protein degradation involves enzymatic steps, and, therefore, should also follow some form of saturable kinetics. For simplicity, we assume the reaction rate for protein degradation also follows Eq. 1. The limit of first-order kinetics for protein degradation is achieved by taking KMl ≫ [X]. Receptor/ligand binding follows mass-action kinetics. We use an asterisk to denote an activated protein (e.g., [X*]), and a dash to denote an inhibited protein (e.g., [X−]). In the models that do not involve production and/or degradation, the concentrations have been normalized to the total amount of each species and normalization factors have been included in the kinetic constants. The differential equations describing our models were solved numerically using the software Mathematica (Wolfram Research, Champaign, IL). The programs used to simulate the models are available upon request. In all the experiments, the models were subject to a small basal activity and run to steady state before each experiment.
Model criteria and parameter selection
Parameter selection was performed using a uniform set of criteria for all the models. A signaling pathway must produce a response of sufficient strength and duration to be recognized by downstream machinery, while at the same time being capable of adapting to a wide range of stimulus doses. To assess the models' ability to meet these criteria, we make use of the following definitions. Let [X*] denote the time-dependent concentration of the active form of the signaling molecule. The response amplitude is defined as the maximum concentration of [X*] after stimulation. The response duration is defined as the time between half maxima of [X*]. The adaptation level is defined as the steady-state level of [X*] in the presence of a sustained stimulus. The recovery time is defined as the time for the system to return to its prestimulus state after removal of the stimulus. Based on examples from the literature, we required that the models be able to generate signals with duration between 5 min and 2 h and for adequate adaptation we require that [X*] returns within a range of 20% of its basal level. We require that the signal amplitude be at least five times the basal concentration of the signaling species, and for the models that do not include production and degradation, we require the amplitude to be a significant fraction (>10%) of the total available signaling pool. Mathematical analyses of the models allowed us to pick parameter sets by hand that met the above criteria. The cases for which this was not possible are discussed below.
RESULTS
Because we are interested in mechanisms of short-term adaptation, we use the term response (output) to refer to the activity level (e.g., phosphorylation state of a MAP kinase) of the downstream pathway component responsible for relaying the signal that results from an external stimulus (input). We refer to the transcriptional program and other nongenetic processes initiated by this output as the cellular response. Deactivation is taken to mean a process by which an active signaling species is transformed back to its original inactive state, but is available for reactivation if the stimulus persists. An example of deactivation is dephosphorylation of kinase that becomes active upon phosphorylation. Desensitization refers to the conversion of a signaling species to a form that cannot propagate the signal even when the pathway component is activated. In general, desensitization is reversible, and when the reverse steps are relatively fast, desensitization is equivalent to deactivation. In the case of degradation, the protein is permanently removed from the signaling pool. Sequestration involves the removal of a species from the signaling pool by the formation of protein complexes or spatial relocalization of signaling components. For our purposes, it can be considered a form of desensitization. The nature of the negative feedback (deactivation, desensitization, or degradation) places different constraints on the system's ability to signal and adapt. To understand the advantages and disadvantages of these mechanisms, we analyze several simple negative feedback architectures; three based on feedback deactivation, Models I–III, and two that rely on desensitization or stimulus-dependent degradation, Models IV, A and B.
Model I: Feedback deactivation generates a dose-dependent switch
In Model I the signaling component directly activates its own negative regulator (Fig. 2 A). This mechanism is inspired by experimental evidence showing that the activity of some phosphatases can be increased upon phosphorylation by their substrate kinase (14). This feature suggests a scenario in which the stimulus leads to phosphorylation and activation of a kinase. In turn, the kinase regulates its activity level by phosphorylating and activating a phosphatase. Note that rather than increasing the rate of deactivation of a pathway component, it is possible that the negative feedback decreases the rate at which the component is activated. We investigated this scenario as well and found no significant differences between the two mechanisms.
FIGURE 2.

Model I—feedback deactivation. (A) Schematic diagram of the model. (B) The phase plane of the system showing nullclines for [K*] and [P*]. (C) Response curves for K* as function of the stimulus dose: maximum [K*] amplitude with negative feedback (shaded curve) and without (dashed curve), and the steady-state level of [K*] with negative feedback (solid curve). (D) Response duration (solid curve) and time to maximum amplitude (shaded curve) as a function of the dose. The inset shows an amplification of the low dose regime. (E) Time series for [K*] generated using the stimulus levels indicated in panel D. The model equations and parameter values used to generate this figure are given in Appendices and Table 1.
In its simplest form, Model I can be written in terms of two variables, [K*] and [P*], the concentrations of the phosphorylated forms of the kinase and phosphatase, respectively. The model equations are given in Eqs. 2 and 3. Because this model consists of only two variables, its behavior can be understood by considering the phase plane for the system (Fig. 2 B). That is, a graph whose axes are [K*] and [P*]. Two special curves on the phase plane are the nullclines defined by the conditions d[K*]/dt = 0 and d[P*]/dt = 0. These are shown in Fig. 2 B for two stimulus levels. The K* nullcline can be interpreted as the signal-response curve for the activated kinase concentration as a function of the active phosphatase concentration. Similarly, the P* nullcline can be thought of as a dose-response curve for the active phosphatase concentration as a function of the active kinase concentration. Notice that the stimulus does not affect the P* nullcline. The intersection of the nullclines represents the steady state of the system. In the absence of a stimulus, the steady state corresponds to point A in Fig. 2 B. When the stimulus is present, the K* nullcline shifts to the right and the new steady state becomes point C. With the right choice of parameters, the new steady-state value of K* is very similar to its prestimulus level. This occurs because the P* nullcline remains close to the horizontal axis (low K* concentration) as P* increases.
The second requirement for adaptation is that the stimulus generates a response of sufficient amplitude and appropriate duration to elicit the correct cellular response. Clearly if the phosphatase responds very rapidly to changes in the activation level of K, then the K* concentration will not increase significantly above basal levels and no transient behavior will be observed. If, however, the activation kinetics of the kinase are fast, the K* concentration will rise rapidly and closely approach point B shown in Fig. 2 B, which represents the quasi-steady-state value of [K*] for the prestimulus [P*] concentration. The activated phosphatase concentration then slowly increases, bringing [K*] back to near-basal levels.
Interestingly, for a sufficiently large stimulus, this mechanism loses its ability to adapt and the resulting response becomes sustained. This occurs when the K* nullcline shifts far enough to the right so that the level of active phosphatase saturates and is insufficient to counteract the stimulus-induced activation of the kinase. If the activation kinetics of the kinase K are ultrasensitive (23) with respect to the stimulus, then the transition between a transient response and a sustained one occurs in a switchlike manner (see Appendices). A transition from transient to sustained signaling was proposed to underlie a cell fate decision in yeast (3). In this system, transient MAPK activation promotes a mating response, whereas sustained activation leads to filamentous growth. We note that this design also provides a mechanism for ensuring the pathway is not activated by a spurious low-level stimulus. Below the threshold, the response is transient and rapidly returns to near-basal levels. These two properties of feedback deactivation are considered further in the Discussion.
Fig. 2 C depicts the typical dose-dependence of the response amplitude (shaded curve) and steady-state level (solid curve) for this architecture. Fig. 2 D shows a similar plot for the response duration (solid curve) and time for the response to reach its maximum value (shaded curve). For very low stimuli doses, the response amplitude (Fig. 2 C, shaded curve) increases with the dose and can be estimated from the K* signal-response curve for the case in which the phosphatase is absent (Fig. 2 C, dashed line). In this regime, the duration of the response is roughly dose-independent (Fig. 2 D, inset, solid curve). At higher doses, when the stimulus level is sufficient to activate almost the entire pool of K, the amplitude becomes dose-independent while the response duration starts to increase (Fig. 2 D, solid curve). In this regime, the system is converting dose information into response duration (see Discussion). Fig. 2 E shows time series generated by this model using the stimulus levels labeled in Fig. 2 D.
For this model, the recovery time is intrinsically linked to the adaptation time. When the stimulus disappears, K* quickly return to basal levels but, because of its slow kinetics, it takes a considerable time for P* to subside. In the best-case scenario, recovery can happen within the same timescale as adaptation. However, the constraints set by the requirements of good adaptation and strong signaling often necessitate that the rate of P* deactivation be significantly slower than that of its activation resulting in long recovery times (sometimes by orders of magnitude). Additionally, the large strength of the negative feedback needed to produce adaptation in this model means that signaling is strongly inhibited during most of the recovery phase, thereby generating a refractory period in which the system is not able to respond to a new challenge.
Model II: Direct deactivation of a positive regulator fails to produce good adaptation
A second strategy for producing adaptation through negative feedback is for the signaling component to deactivate an upstream element. The simplest architecture, albeit the least biologically realistic, is one in which the signaling molecule K is directly responsible for deactivating the pathway component KK located directly upstream (Fig. 3 A). As we show, this model does not produce good adaptation, but its study highlights the specific benefits of more complex systems as well as the limitations of a feedback mechanism in which the same molecule that transmits the signal also directly inhibits pathway activity. A potentially more biological realistic scenario is one in which the feedback effect of K is to decrease the rate at which KK is activated. Similar to Model I, this scenario does not produce qualitatively different results from the case of feedback deactivation.
FIGURE 3.

Model II—direct feedback deactivation. (A) Schematic diagram of the model. (B) The phase plane of the system showing nullclines for [K*] and [KK*]. (C) Response curves for K* as function of the stimulus dose: maximum [K*] amplitude (shaded curve) and the steady-state level of K* (solid curve) (D) Time series for [K*] generated using the stimulus levels indicated in panel C. The model equations and parameter values used to generate this figure are given in Appendices and Table 1.
The simplest version of this model is very similar to the one studied in the previous section with the difference being that, in this case, the species K both propagates the signal and deactivates KK*. Therefore, the analysis of this model will follow closely the one carried out in the previous section. Fig. 3 B shows the phase plane for this model, where again the signaling species K* is on the vertical axis and the horizontal axis is the activated kinase KK*. Analysis of the nullclines seems to indicate that adaptation could be possible if the K* nullcline (labeled as d[K*]/dt = 0 in Fig. 3 B) is sufficiently switchlike as a function of [KK*]. However, this system cannot fulfill the requirement of generating a significant response. To understand this shortcoming, it is useful to consider the two limiting cases in which the kinetics of K are fast or slow as compared to those of KK. When the KK kinetics are fast, the stimulus causes the KK* level to rapidly rise from its basal level (A in Fig. 3 B) to its quasi-steady-state level (B) located on the post-stimulus [KK*] nullcline. Next, K becomes activated causing the KK* level to decrease along the [KK*]-nullcline until it reaches the new steady-state C. This scenario produces a monotonic increase in K* and no transient signaling. In the second limiting case in which the kinetics of K are fast compared to those of KK, the system evolves along the [K*] nullcline toward point C. Again, the increase in K* is monotonic in time. While these two limiting cases seem to indicate that this system is not capable of both adapting and producing a significant response, they do not address the intermediate regime where the kinetics of K and KK are comparable. In fact, in this intermediate regime, the system is capable of showing some degree of transient signaling. With the right choice of parameters, the system can overshoot the equilibrium point C producing a transient increase in the [K*] level. However, this strategy has two serious caveats. First, in the presence of the stimulus, the steady-state K* concentration (C in Fig. 3 B) has to be significantly higher than the basal level, thereby precluding significant adaptation. Second, this setup is very prone to oscillations. Fig. 3 C shows the dependence of the response amplitude and steady-state level on the stimulus strength. The time-dependent response for various doses is shown in Fig. 3 D. These figures clearly show how adaptation quickly disappears and the response turns into a small transient overshoot. In this model, response duration is not a relevant quantity because the response does not return to below its half-maximum. The principal advantage of this system is its very fast recovery time after the stimulus has been removed. This is due to the fact that K* is responsible for both propagating the signal and deactivating KK*, making activation and recovery times similar.
The difference in the behavior of Models I and II may appear puzzling, especially because both rely on feedback deactivation. The major difference between the models is that in Model I feedback occurs through an intermediate step, whereas in Model II, the kinase deactivates its upstream activator directly. This difference is crucial because adaptation requires a sustained feedback that responds on a timescale slower than that of activation. Model I can produce a transient response because the activation of the signaling species K occurs faster than the feedback timescale determined by the kinetics of the phosphatase. However, in Model II, activation of the signaling species K and the onset of the feedback are the same process and therefore occur on the same timescale. Based on these observations, we find that for systems in which adaptation occurs as a result of deactivating a pathway component, intermediate steps that separate the activation timescale from that of feedback deactivation are a necessary feature. Such intermediate steps allow for a strong negative feedback capable of returning the pathway output to near-basal levels, while at the same time providing a time delay that enables a large transient response. A strong feedback is incompatible with fast kinetics because it prevents the development of a transient response. To further explore this observation, in the next section we analyze more complex variants of Model II that include intermediate steps.
Model III: Intermediate steps enhance adaptation
To better understand the dynamics of feedback deactivation we considered two extensions of Model II. The first case (Model III A, Fig. 4 A) represents a scenario in which the signaling species K inhibits an upstream activator KK through activation of a negative regulator. This can be thought of as a terminal kinase K activating a phosphatase P, which in turns dephosphorylates the kinase KK directly upstream of K. In the second scenario (Model III B, Fig. 4 C), the signaling species K deactivates a pathway component two levels upstream. Even though the direct interaction assumed in this model is unlikely to appear in real signaling systems, the analysis of this case reveals the specific effects of targeting a component further upstream.
FIGURE 4.

Intermediate pathway components. (A) Model III A is similar to Model I except that pathway deactivation occurs upstream of K. (B) Time series of K* generated by Model III A for various dose levels. (C) Model III B is similar to Model II except feedback deactivation occurs at an upstream pathway component. (D) Response curves for K* as function of the stimulus dose for Model III B: maximum [K*] amplitude (shaded curve) and the steady-state level of K* (solid curve). (E) Time series for [K*] generated using the stimulus levels indicated in panel D. The inset shows time series of the upstream species KK* and KKK* illustrating the delay effect discussed in the text. The model equations and parameter values used to generate this figure are given in Appendices and Table 1.
Model III A is almost equivalent to Model I, except that the negative regulator P now acts on an upstream component rather than the terminal signaling species K. Therefore, it is not surprising that the model is capable of near-perfect adaptation and possesses dynamics and dose-response relationships very similar to Model I (not shown). Fig. 4 B depicts the response produced by the system in the presence of various stimulus levels. As with Model I we can see that good adaptation is possible over a range of doses, but at some point the system loses the ability to adapt and a persistent response ensues. A difference between this model and Model I is that at low stimulus levels there is region of concentrations for which the response duration decreases with increasing dose. The decrease occurs in systems in which the feedback targets elements upstream of the signaling species. A key feature of this model is the relatively slow kinetics of the intermediate species P. This feature decouples the activation and feedback timescales, allowing a sufficiently strong feedback for adaptation to a wide range of doses while at the same time producing a strong transient response.
Model III B (Fig. 4 C) resembles Model II in that the signaling species is directly responsible for feedback deactivation causing both timescales to be intrinsically linked. Fig. 4 D shows the dose dependence of the response amplitude and steady-state level when the intermediate species KK was adjusted to react slower than the upstream component KKK. Fig. 4 E shows responses produced at different doses as well as the [KK*] and [KKK*] responses (inset). The steady-state level after adaptation is roughly similar to that of Model II. However, the presence of a slower intermediate step dramatically increases the amplitude of the response, especially at lower doses (compare with Fig. 2 C). The slow deactivation kinetics of KK produces a delay between feedback-deactivation of KKK* and its downstream effect on K*. It is important to notice that the negative feedback starts acting as soon as K* levels rise and quickly deactivates the upstream element KKK, causing different components of the pathway to adapt on different timescales. This effect is a purely transient phenomenon, and as such cannot overcome the poor adaptation (a steady-state property) observed with this model. The steady-state level of K* has to be sufficient to deactivate KKK*. Such a strong negative feedback prevents transient signaling just as in Model II. It is interesting to contrast Models III, A and B. In Model III A, the slow intermediate species delays the effects of the negative feedback while in Model III B what is delayed is the time at which the effect of the feedback reaches downstream components. This has interesting implications for cross-talk between pathways (see Discussion). As expected, adding additional levels to the pathway makes it easier to generate oscillatory responses that may be undesirable in actual signaling networks. As a matter of fact, the combination of negative feedback loops and delays is a classic recipe for oscillations (24,25). In all our examples, the strength or slow timescale of the feedback mechanisms precluded any significant oscillations. However, low amplitude ringing (very overdamped oscillations) was observed in some cases.
As one would expect, Model III A's response duration and recovery time are very similar to Model I. On the other hand, Model III B inherits Model II's fast recovery time. The only slow component in this model is the intermediate element KK, which by the time adaptation is reached, has already come back to near-basal levels. Therefore, this mechanism provides a very fast adapting system, albeit one that produces good signaling and adaptation only at low dose levels.
Model IV: Feedback degradation and desensitization provide an effective strategy for dose-independent adaptation
The reason adaptation is lost at high dose levels in the case of feedback deactivation (Models I–III) is because the species acted upon by the feedback immediately becomes available for reactivation. Additionally, the dual role played by species K, both as a signaling molecule and negative regulator, requires it to be a very effective deactivator to counteract a persistent stimulus, but not so strong that it prevents transient signaling altogether. An alternative approach is to use desensitization or degradation rather than deactivation as the feedback mechanism. In this scenario, the desensitized (or degraded) component is removed (transiently or permanently) from the signaling pool, thereby relaxing the need for a strong sustained feedback. This mechanism plays a role in Raf-1 regulation (16) and the yeast pheromone response (21), among others. Desensitization and degradation based systems display behavior markedly different from feedback deactivation. To illustrate these differences, we focus on the two models depicted in Figs. 5 and 6.
FIGURE 5.

Model IV A—feedback degradation. (A) Schematic diagram of the model. (B) The [K*] versus [R*] dose-response curve. The vertical dotted line indicates the threshold for K* activation. (C) Response curves for R* and R as function of the stimulus dose: maximum [R*] amplitude with negative feedback (shaded curve) and without (dashed curve), steady-state level of R* with negative feedback (solid curve). The dotted shaded line indicates the R* threshold for K activation. (D) Dose response curves for K*: maximum amplitude with feedback (shaded curve) and without (dashed curve) and the steady-state level with feedback (solid curve). The inset shows the response duration (solid curve) and time to maximum amplitude (shaded line). No signal is generated at very low doses due to the activation threshold (see text). (E) Time series for [K*] and [R*] (inset, dotted line indicates the activation threshold) generated using the stimulus levels indicated in panel D. The model equations and parameter values used to generate this figure are given in Appendices and Table 1.
FIGURE 6.

Model IV B—feedback desensitization. (A) Schematic diagram of the model. (B) Dose response curves for K*: maximum amplitude with negative feedback (shaded curve) and without (dashed curve) and the steady-state level with negative feedback (solid curve). (Inset) Signal duration (solid curve) and time to maximum amplitude (shaded curve). (C) Time series for [K*] using the stimulus levels indicated in panel B. The model equations and parameter values used to generate this figure are given in Appendices and Table 1.
In Model IV A (Fig. 5 A), feedback regulation targets an upstream component of the pathway for degradation, thereby permanently removing it from the signaling pool. A possible scenario is one in which a kinase K feedback-phosphorylates the receptor R and phosphorylation targets the upstream receptor for ubiquitination and degradation. Because this mechanism relies on protein degradation, it is necessary to include protein synthesis in the model to maintain a finite concentration of R. Model IV B (Fig. 6 A) involves a feedback mechanism in which the active form R* of the upstream signaling component is transformed to a desensitized form R− that cannot signal. This transformation is reversible with the desensitized component eventually reentering the signaling pool. As the rate at which desensitization is reversed becomes very small, we recover Model IV A. In theory there are two scenarios for how the desensitized element is reintroduced into the signaling pool. In the first case, removing desensitization causes the protein to reenter the active state. In the second scenario, the desensitized component must pass back through the inactive state before it can become active again. Recently, this mechanism has been suggested to describe a branch of the osmotic response in yeast (5). Under normal conditions, the Sho1 osmosensor exists as an oligomer (R) that, when exposed to osmotic stress, initiates a signaling cascade that results in the phosphorylation of the kinase Hog1. Phopsho-Hog1 then feedback-phosphorylates Sho1, causing the oligomer to dissociate and signaling to stop. The Sho1 monomers (R−) must then be dephosphorylated before reformation of the signaling-competent oligomers (R).
Models IV, A and B, work in a similar fashion producing good adaptation regardless of the stimulus strength. The mechanism of adaptation in these systems can be understood in terms of the dose-response curves of the components R and K as shown in Fig. 5, B and C, for Model IV A. As can be seen, the activation curve of K* as a function of the concentration of R* (Fig. 5 B) shows a sharp threshold below which virtually no activation occurs. Fig. 5 C shows the dose-response curves for R (shaded dashed line) and R* (solid line) when feedback is present, and for R* (dark dashed line) when the feedback loop is absent. When exposed to a sufficient stimulus level, the maximum amplitude of R* (Fig. 5 C, shaded line) transiently rises over the threshold value (dotted line, Fig. 5, B and C) for activating K and triggering feedback degradation. The systems adapt because the negative feedback is sufficient to maintain the steady-state level of R* below the K* activation threshold. This architecture results not only in the adaptation of the signaling species K*, but also of the active receptor R* (Fig. 5 E, inset). An ultrasensitive K* response curve is not required to achieve adaptation. However, for a system with a graded K* response curve to show good adaptation, the feedback must considerably reduce the level of active R*, by degrading or desensitizing virtually all of R. Fig. 5 D shows the response amplitude (shaded curve) and steady-state level (solid curve) for K* as a function of the stimulus strength. For comparison also shown in the figure is the maximum response of K* in the absence of feedback (dashed curve). Shown in the inset is the response duration (solid curve) and time to reach the maximum K* amplitude (shaded curve). Fig. 6 B shows the respective curves for Model IV B. Fig. 5 E, and 6 C, show typical time series for Models IV, A and B, respectively. In both cases, adaptation is very good; and unlike the case of feedback deactivation, it is not lost as the stimulus level increases. The response duration is mostly dose-independent, except at low stimulus levels where it decreases with dose. Fig. 5 E illustrates a very characteristic pattern in which the response duration initially becomes shorter, with increasing stimulus levels eventually becoming dose-independent. The reason for this is that, at high stimulus levels, K* has saturated—in which case, the rate of degradation or desensitization becomes signal-independent. As a result, in the high dose regime, these models generate a strong transient response, with amplitude and duration independent of the stimulus strength. The implications of this behavior are considered in the Discussion. This behavior is in contrast to feedback deactivation in which case the response length is dose-independent at low stimulus levels and increases as stimulus strength grows. The time series for Model IV A (Fig. 5 E) illustrate two interesting phenomena associated with the sharp K* response curve. The complex decay observed at different doses is caused by the interplay between R* degradation and K* deactivation kinetics. At high doses, elevated K* levels cause the rapid degradation of R* (see Fig. 5 E, inset) without producing a significant drop in K*. As R* activation decays beneath the K* activation threshold, K* levels rapidly fall, causing R* degradation to slow down. At the final stage, K* and R* activation levels slowly decline until reaching steady state. The initial and final phases are dominated by R kinetics, whereas the intermediate stage is dominated by K* deactivation kinetics. At lower doses, R* levels are not sufficient to fully activate K*, and the initial phase is missing. The second effect is a delay in the onset of signaling observed at low stimulus doses because of the time it takes R to reach K's activation threshold. This phenomenon is not exclusive to Model IV A and often occurs when elements with ultrasensitive response curves are involved in signaling.
The addition of intermediate steps in Models IV, A and B, can be accomplished in two ways: 1), extra steps can be placed between the upstream activator and the signaling species K (Fig. 7 A); or 2), extra steps can be placed in the feedback loop (Fig. 7 B). Both architectures add new features to the models and increase the likelihood of generating oscillations. In the first case, the addition of an extra step again introduces a transient memory in the system that delays the downstream effect of the feedback desensitization (or degradation) of R*. As clearly illustrated in Fig. 7 C, this effect allows the system to achieve better sensitivity at low doses. This figure compares the maximum response for Model IV B and the model shown in Fig. 7 A. The figure was produced using similar parameter values for both models with the exception of the feedback strength (k5 in Eq. 21), because the delay produced by the extra step allows a stronger feedback without the loss of the ability to produce a strong response (values for the parameters are listed in Table 1). Note that this increased sensitivity does not require the signal to be amplified by the pathway. The introduction of an intermediate step in the feedback loop (Fig. 7 B) allows a separation of timescales between signal initiation and attenuation. In general, the introduction of intermediate components endows these systems with more flexibility and permits the dynamics of species K to differ from that of R, producing different response profiles with potentially interesting biological implications (see Discussion).
FIGURE 7.

Intermediate pathway components. (A) An intermediate step between stimulus activation of R and the response element K. (B) An intermediate step in the negative feedback loop. (C) Maximum response amplitude for Model IV B (solid curve) and the model shown in panel A (dashed curve).
For both models, recovery is slow—because good adaptation requires slow protein production (Model IV A) or slow recovery from the desensitized state (Model IV B). The recovery time can be improved in Model IV A by a proportional increase in both the production and degradation rates. However, the timescales for these processes are constrained in living cells. Model IV B recovery can be accelerated if the feedback desensitization not only acts on R* but also on the inactive form R. This additional depletion of R permits a faster R− to R turnover, allowing for quicker recovery. When restimulated after the removal of the signal, we observed that significant signaling was still possible even before recovery was complete. This effect is strongly dose-dependent and depends on the amount of activator still available as well as the production and resensitization rates.
Response to time-dependent stimulus levels
To test the ability of each model to respond to a changing environment, we exposed each system to a series of stepped increases in the stimulus level and observed the resulting response. Not surprisingly, each model's behavior depended on both the duration and amplitude of the steps in stimulus level. We observed that when the increases are sufficiently strong and occur on timescales long compared to the adaptation time, most systems produce a train of discrete peaks of varying amplitude (Fig. 8 A). In general, models based on deactivation (I and III) responded to more steps than models based on degradation and desensitization (IV), provided that the maximum stimulation level remained in the region where adaptation is possible. The degradation or desensitization of a pathway component resulting from the initial challenge severely reduced Model IV's ability to respond to subsequent increases in the stimulus level. When the time interval between stimulus changes is short compared to the adaptation timescale, the systems effectively see one challenge and for strong enough stimuli the response is a single, complex-shaped signal (Fig. 8 B), usually of lower amplitude than the response the system generates when exposed to a single dose of maximum strength. Our experiments also showed that the magnitude of the stimulus increase plays a fundamental role. If the dose increase at each step is small, and the interval between steps is long enough to allow for adaptation, then the stimulation level can be increased substantially without a response being produced (Fig. 8 C). These results can be extrapolated to ramped stimuli indicating that feedback-based adapting systems can only generate responses for a limited range of temporal variations in the stimulus strength. This represents a serious limitation for pathways that must respond and adapt to stimulus levels that fluctuate over a wide range of timescales (see Discussion).
FIGURE 8.

The response of Model I (feedback deactivation) to stepped increases in the stimulus. (A) When the steps in stimulus level (shaded curve) are sufficiently long and of sufficient amplitude, the model responds with a series of pulses (solid curve) of roughly equal amplitude until adaptation is lost. (B) If the duration of the stimulus steps is short (shaded curve), a complex response is generated (solid curve) whose maximum amplitude is less than the response produced by exposure to constant stimulus at the final concentration level (dashed curve). (C) If the amplitude of the stimulus increases is too small, the system cannot see the stimulus and does not respond (solid curve). For comparison, the response produced by a constant stimulus of at the final concentration level is also shown (dashed curve).
DISCUSSION
Separating timescales
Signaling systems that adapt to sustained stimuli must meet two requirements. The pathway must generate a response of sufficient strength and duration to elicit the correct response, while at the same time returning to basal levels upon continued exposure to the stimulus. We analyzed different strategies of adaptation that revealed several general principles. First, when the signaling molecule is also a direct negative regulator of an upstream pathway component, the timescales for signaling and feedback inhibition are linked limiting the dynamic properties of the pathway. This intrinsic connection is the reason Model II, which relies on feed back deactivation, cannot adapt or signal well. The near irreversible nature of feedback mechanisms based on desensitization and/or degradation (Model IV, A and B) allows strong signaling and good adaptation without requiring a strong sustained negative feedback. The removal of the signaling species from the signaling pool means that a weak feedback is sufficient for adaptation. Even for these models, the addition of a feedback intermediary greatly adds flexibility by allowing different temporal dynamics at different levels in the signaling cascade. This could have interesting implications for the locations of branching points where signaling pathways feed into secondary pathways to elicit a complex cellular response. Different dynamics at different points along the pathway could allow for a variety of responses, depending on where the secondary branches are connected.
Increased sensitivity
It has been recognized that multiple-level signaling cascades can amplify weak signals (26–29), and it has been suggested such cascades provide a mechanism for increasing the rate of signal propagation (26,30). Here we have shown a novel way in which intermediate steps in a signaling cascade can improve sensitivity to low stimulus levels. In this mechanism, a long-lived intermediate step can store information about the activity level of an upstream component after feedback inhibition has terminated it. The result is prolonged and increased activity of any downstream components. An advantage of placing the delay downstream of the feedback target is that this architecture allows a rapid decrease in the activity of promiscuous upstream elements without a rapid attenuation of the response. In contrast, mechanisms that rely on slow intermediary steps in the feedback loop to decouple the signal and feedback timescales (e.g., Model I) must wait until the feedback acts, for the upstream target activation level to subside. This could have adverse effects potentially leading to cross talk, if the upstream component is involved in multiple pathways.
A switch from transient to sustained signaling and dose-duration transduction
It has been suggested that response duration plays a role in developmental decisions. For example, Sabbagh et al. (3) proposed that, in yeast, sustained activation of the MAPK Kss1 leads to invasive growth, whereas transient activation is required for proper mating. We have demonstrated that feedback deactivation provides a mechanism capable of switching between a transient response at low stimulus levels and a sustained response at high levels (Fig. 9 A). The model we presented was based on feedback dephosphorylation. However, the switch behavior is not specific to this mechanism of deactivation and arises in situations when the feedback inhibition dominates at low stimulus doses but saturates before stimulus-dependent activation. A related feature is the ability of feedback deactivation to translate stimulus strength into response duration. This is especially evident in the models that include a multistep feedback loop that allows the buildup of a transient response (e.g., Models I and III A). The ability to convert stimulus strength to response duration is important because it provides an alternative to response amplitude for transmitting information. This has implications for avoiding cross talk in pathways that share common components. That is, in one pathway the signaling elements below the common component might detect amplitude, while in the other pathway, the duration of the response would determine whether the signal is transmitted (Fig. 9 B).
FIGURE 9.

(A) A transient to sustained switch. An adapting pathway based on saturable negative feedback (e.g., Model I) can produce transient or sustained signals in a dose-dependent fashion. Transient or sustained activation of a kinase (or transcription factor) results in the activation of a different subset of genes, thereby eliciting alternative responses. (B) The ability of this architecture to encode stimulus concentration as signal duration provides a mechanism for preventing cross-talk. The response on the left (RL) is initiated only when MAPK activation is sustained, whereas activation of the response on the right (RR) requires the MAPK activation to transiently exceed a threshold. The upstream adaptive system of the right pathway prevents inappropriate activation of RL by stimulus SR and regulates response LR by transforming the concentration of the stimulus SL into signal duration.
Dose-independent signals
Feedback deactivation has the ability to produce responses in which the response duration depends on the stimulus strength. In contrast, for sufficiently strong stimulus levels mechanisms that rely on desensitization or degradation, the response amplitude and duration become independent of dose. This effect is due to the finite amount and/or dose-independent production rate of the upstream activator that is the target of the feedback. Because of this feature, Models IV, A and B, are well suited for situations in which the stimulus strength is irrelevant for the response and/or an all-or-none response is desired. An interesting observation is that with the appropriate choice of parameters these models can be made into signal repositories with signal potency (area under the peak) regulated by the amount of activator burned (i.e., degraded or desensitized) by the feedback in each event. No signal will result once the pool has been depleted, potentially avoiding multiple reactions to the same event.
Reaction and recovery timescales
Our study suggests that systems that adapt through feedback regulation are inherently slow to recover, often resulting in a refractory period much longer than the adaptation time. The notable exception is Model III B, which relies on an intermediate step delaying the effect of the feedback, to produce a transient response. This model is capable of fast recovery because the slow pathway component returns to its prestimulus level during the adaptation process. However, fast recovery comes at the price of a very limited range of stimulus strengths for which good adaptation is achievable. Obviously, slow recovery times may render these models unsuitable for pathways that must respond to time-dependent stimuli. Interestingly, the responses observed when the models are exposed to stepped increases in the stimulus level demonstrate that, in general, models based on feedback deactivation (Models I–III) do a better job responding to subsequent increases in the stimulus level than models based on degradation or desensitization (Models IV, A and B). However, this effect is strongly dose-dependent, and for a limited range of doses, any of the systems can generate a response to this type of stepped increase in the stimulus level. Interestingly, when the stimulation level increases faster than the system can adapt, the result are often complex responses of lower amplitude than the response corresponding to the same final stimulus level applied all at once, potentially resulting in a suboptimal cellular response. Furthermore, feedback-based adapting systems can produce strong responses only for stimuli that increase fast relative to the adaptation timescale, with slow rising stimuli becoming invisible. Taken together, these observations mean that adapting systems not only must be tailored to elicit a response from their downstream targets, but also to receive particular temporal profiles from upstream activators. This limitation raises the interesting possibility that the redundancy present at the upper levels on some signaling networks (e.g., yeast's osmotic stress response) (31) may have evolved to provide signaling capabilities at multiple timescales and also leads to the intriguing possibility that the parallel pathways found in many signaling systems are designed to deal with different temporal patterns of stimulation.
Signatures of adaptation
Our results allow us to list the distinctive features of each model that can be used to distinguish feedback mechanisms of adaptation. The most obvious distinguishing characteristic of feedback deactivation is the loss of adaptation at high stimulus levels. This loss of adaptation is generally preceded by an increase in response duration. In contrast, adaptation and response duration in mechanisms that rely on degradation or desensitization tend to become dose-independent at moderate to high stimulus levels. In these systems, response duration at low doses frequently decreases with increasing stimulus concentration maintaining (or even improving) the adaptation level. The recovery time can also help identify feedback mechanisms. As discussed above, most mechanisms are slow to recover with the notable exception of Model III B. Unlike other features that are sensitive to the choice of parameter values (such as amplitude or response duration for a given dose) the properties described above are robust features of the different models and exist for a wide range of parameter values. Alternatively, these properties can be used as predictions to support candidate mechanisms suspected to be responsible for adaptation in actual signaling systems. In most real scenarios, adaptation is not based solely on a single mechanism, but rather relies on multiple feedback and feed-forward mechanisms. Nevertheless, when combined with experimental analysis the results presented here provide powerful clues for determining the biochemical mechanisms that underlie adaptation in signal transduction pathways.
TABLE 1.
Model parameter values
| Model I | Model II | ||
| k1 | 1 | k1 | 2 × 10−1 |
| k1m | 5 × 10−3 | k1m | 1 |
| v2 | 3 × 10−4 | v2 | 1 × 10−6 |
| k2m | 1 × 10−2 | k2m | 5 × 10−1 |
| k3 | 2.5 × 10−1 | k3 | 1.5 × 10−2 |
| k3m | 2 × 10−3 | K3m | 1 × 10−2 |
| k4 | 1 × 10−3 | v4 | 6 × 10−4 |
| k4m | 5 × 10−1 | k4m | 1 × 10−2 |
| v5 | 1 × 10−5 | k5 | 3.2 × 10−2 |
| k5m | 1 | k5m | 1 × 10−3 |
| Model III A | Model III B | ||
| k1 | 1 × 10−1 | k1 | 5 × 10−1 |
| k1m | 1.5 × 10−1 | k1m | 1.5 |
| v2 | 3 × 10−3 | v2 | 1 × 10−6 |
| k2m | 1 | k2m | 8 × 10−2 |
| k3 | 4.8 × 10−4 | k3 | 1.5 × 10−1 |
| k3m | 1 | k3m | 6 × 10−1 |
| v4 | 5 × 10−6 | v4 | 3 × 10−3 |
| k4m | 1 | k4m | 6 × 10−1 |
| k5 | 3 × 10−2 | k5 | 8 × 10−2 |
| k5m | 2 × 10−3 | k5m | 3 × 10−3 |
| k6 | 10 | k6 | 1 × 10−2 |
| k6m | 0.1 | k6m | 1 |
| v7 | 1 | v7 | 5 × 10−3 |
| k7m | 1 × 10−1 | k7m | 1 |
| Model IV A | Model IV B | ||
| k1 | 5 × 10−2 | k1 | 6 × 10−1 |
| k1m | 1 | k1m | 5 × 10−1 |
| v2 | 1 × 10−3 | v2 | 2 × 10−2 |
| k2m | 1 × 10−2 | k2m | 1 × 10−1 |
| k3 | 7.5 × 10−1 | k3 | 1 × 10−2 |
| k3m | 5 × 10−2 | k3m | 5 × 10−2 |
| v4 | 1.9 × 10−1 | v4 | 4 × 10−3 |
| k4m | 5 × 10−3 | k4m | 1 × 10−1 |
| k0 | 7.5 × 10−5 | k5 | 4 × 10−3 |

|
7.5 × 10−5 | k5m | 1 |
|
9.6 × 10−3 | k6 | 2 × 10−5 |
| Model Fig. 7 A | |||
| k1 | 6 × 10−1 | k5 | 8 × 10−3 |
| k1m | 5 × 10−1 | k5m | 1 |
| v2 | 2 × 10−2 | k6 | 2 × 10−5 |
| k2m | 1 × 10−1 | k8 | 2.5 × 10−3 |
| k3 | 1 × 10−2 | k8m | 1 × 10−2 |
| k3m | 5 × 10−2 | k9 | 5 × 10−5 |
| v4 | 4 × 10−3 | k9m | 1 × 10−2 |
| k4m | 1 × 10−1 |
APPENDIX A: MODEL EQUATIONS
This Appendix lists the equations for each model described in the text.
Model I
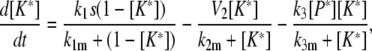 |
(2) |
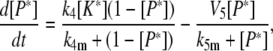 |
(3) |
 |
(4) |
Model II
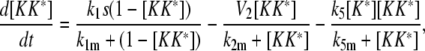 |
(5) |
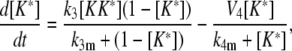 |
(6) |
 |
(7) |
Model III A
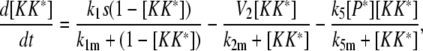 |
(8) |
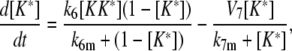 |
(9) |
 |
(10) |
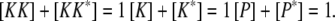 |
(11) |
Model III B
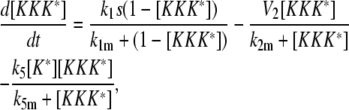 |
(12) |
 |
(13) |
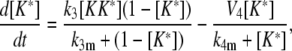 |
(14) |
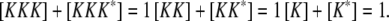 |
(15) |
Model IV A
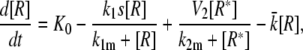 |
(16) |
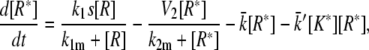 |
(17) |
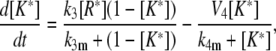 |
(18) |
 |
(19) |
Model IV B
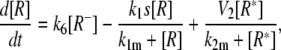 |
(20) |
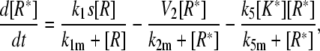 |
(21) |
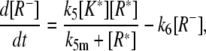 |
(22) |
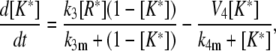 |
(23) |
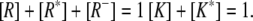 |
(24) |
Model Fig. 7 A
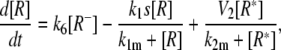 |
(25) |
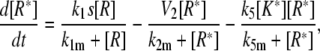 |
(26) |
 |
(27) |
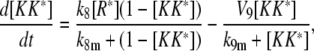 |
(28) |
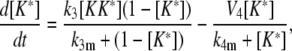 |
(29) |
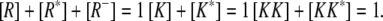 |
(30) |
APPENDIX B: ANALYSIS OF MODEL I
Adaptation in Model I can be further clarified by studying the individual dose response curves for [K*] and [P*]. The dose response curve for [P*] as a function of active [K*] is depicted in Fig. 10. The curve for [K*] as a function of stimulation level s is depicted in Fig. 11 for the cases when [P*] is absent and only basal regulation is acting (shaded line) and when [P*] reaches its maximum theoretical value (solid line). Note that in Fig. 10, the P* dose response, is the [P*] nullcline of Fig. 2 B with the axes rotated. This is not surprising given that P is activated by K and does not directly depend on the stimulus level. This explains why the [P*] nullcline cannot be perfectly horizontal, because this would require an infinite slope. On the other hand, the [K*] dose response curves make it evident how this mechanism produces adaptation: In the basal state, the activation threshold for K lays at a low dose level. Upon stimulation, K is modified to its active form K*, which causes the production of P*. This in turn shifts the K* activation threshold to higher stimulus levels. Eventually, enough P* is produced and the threshold moves above the current the stimulus level, turning off the signal. The feedback loop reaches steady state when the amount of P* is enough to keep the threshold slightly above the applied dose. This analysis can be extended to the other models even for higher dimensional systems, where it can be used to estimate maximum peak amplitudes and dose dependency of the signals.
FIGURE 10.

The [P*] vs. [K*] response curve for Model I.
FIGURE 11.

The [K*] response curve as a function of stimulus strength (s) for Model I in the absence of P* (shaded curve), and maximum P* (solid curve). Intermediate P* levels will shift the shaded curve to the right and turn off the signal.
Editor: Herbert Levine.
References
- 1.Kholodenko, B. N. 2006. Cell-signaling dynamics in time and space. Nat. Rev. Mol. Cell Biol. 7:165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marshall, C. J. 1995. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 80:179–185. [DOI] [PubMed] [Google Scholar]
- 3.Sabbagh, W., Jr., L. J. Flatauer, A. J. Bardwell, and L. Bardwell. 2001. Specificity of MAP kinase signaling in yeast differentiation involves transient versus sustained MAPK activation. Mol. Cell. 8:683–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wormald, S., J. G. Zhang, D. L. Krebs, L. A. Mielke, J. Silver, W. S. Alexander, T. P. Speed, N. A. Nicola, and D. J. Hilton. 2006. The comparative roles of suppressor of cytokine signaling-1 and -3 in the inhibition and desensitization of cytokine signaling. J. Biol. Chem. 281:11135–11143. [DOI] [PubMed] [Google Scholar]
- 5.Hao, N., M. Behar, S. C. Parnell, M. P. Torres, C. H. Borchers, T. C. Elston, and H. G. Dohlman. 2007. A systems-biology analysis of feedback inhibition in the Sho1 osmotic-stress-response pathway. Curr. Biol. 17:659–667. [DOI] [PubMed] [Google Scholar]
- 6.Yi, T. M., Y. Huang, M. I. Simon, and J. Doyle. 2000. Robust perfect adaptation in bacterial chemotaxis through integral feedback control. Proc. Natl. Acad. Sci. USA. 97:4649–4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alon, U., M. G. Surette, N. Barkai, and S. Leibler. 1999. Robustness in bacterial chemotaxis. Nature. 397:168–171. [DOI] [PubMed] [Google Scholar]
- 8.Barkai, N., and S. Leibler. 1997. Robustness in simple biochemical networks. Nature. 387:913–917. [DOI] [PubMed] [Google Scholar]
- 9.Alon, U. 2007. An Introduction to Systems Biology: Design Principles of Biological Circuits. Chapman & Hall/CRC, Boca Raton, FL.
- 10.Mangan, S., S. Itzkovitz, A. Zaslaver, and U. Alon. 2006. The incoherent feed-forward loop accelerates the response-time of the GAL system of Escherichia coli. J. Mol. Biol. 356:1073–1081. [DOI] [PubMed] [Google Scholar]
- 11.Tyson, J. J., K. C. Chen, and B. Novak. 2003. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr. Opin. Cell Biol. 15:221–231. [DOI] [PubMed] [Google Scholar]
- 12.Sasagawa, S., Y. Ozaki, K. Fujita, and S. Kuroda. 2005. Prediction and validation of the distinct dynamics of transient and sustained ERK activation. Nat. Cell Biol. 7:365–373. [DOI] [PubMed] [Google Scholar]
- 13.Errede, B., and Q. Y. Ge. 1996. Feedback regulation of map kinase signal pathways. Philos. Trans. R. Soc. Lond. B Biol. Sci. 351:143–148 (discussion 148–149). [DOI] [PubMed] [Google Scholar]
- 14.Wurgler-Murphy, S. M., T. Maeda, E. A. Witten, and H. Saito. 1997. Regulation of the Saccharomyces cerevisiae HOG1 mitogen-activated protein kinase by the PTP2 and PTP3 protein tyrosine phosphatases. Mol. Cell. Biol. 17:1289–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saito, H., and K. Tatebayashi. 2004. Regulation of the osmoregulatory HOG MAPK cascade in yeast. J. Biochem. (Tokyo). 136:267–272. [DOI] [PubMed] [Google Scholar]
- 16.Dougherty, M. K., J. Muller, D. A. Ritt, M. Zhou, X. Z. Zhou, T. D. Copeland, T. P. Conrads, T. D. Veenstra, K. P. Lu, and D. K. Morrison. 2005. Regulation of Raf-1 by direct feedback phosphorylation. Mol. Cell. 17:215–224. [DOI] [PubMed] [Google Scholar]
- 17.Kang, T. H., and K. T. Kim. 2006. Negative regulation of ERK activity by VRK3-mediated activation of VHR phosphatase. Nat. Cell Biol. 8:863–869. [DOI] [PubMed] [Google Scholar]
- 18.Shimada, Y., M. P. Gulli, and M. Peter. 2000. Nuclear sequestration of the exchange factor Cdc24 by Far1 regulates cell polarity during yeast mating. Nat. Cell Biol. 2:117–124. [DOI] [PubMed] [Google Scholar]
- 19.Xu, X., M. Meier-Schellersheim, X. Jiao, L. E. Nelson, and T. Jin. 2005. Quantitative imaging of single live cells reveals spatiotemporal dynamics of multistep signaling events of chemoattractant gradient sensing in Dictyostelium. Mol. Biol. Cell. 16:676–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Charest, P. G., and R. A. Firtel. 2006. Feedback signaling controls leading-edge formation during chemotaxis. Curr. Opin. Genet. Dev. 16:339–347. [DOI] [PubMed] [Google Scholar]
- 21.Esch, R. K., and B. Errede. 2002. Pheromone induction promotes Ste11 degradation through a MAPK feedback and ubiquitin-dependent mechanism. Proc. Natl. Acad. Sci. USA. 99:9160–9165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reference deleted in proof.
- 23.Goldbeter, A., and D. E. Koshland, Jr. 1981. An amplified sensitivity arising from covalent modification in biological systems. Proc. Natl. Acad. Sci. USA. 78:6840–6844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kholodenko, B. N. 2000. Negative feedback and ultrasensitivity can bring about oscillations in the mitogen-activated protein kinase cascades. Eur. J. Biochem. 267:1583–1588. [DOI] [PubMed] [Google Scholar]
- 25.Wang, X., N. Hao, H. G. Dohlman, and T. C. Elston. 2006. Bistability, stochasticity, and oscillations in the mitogen-activated protein kinase cascade. Biophys. J. 90:1961–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heinrich, R., B. G. Neel, and T. A. Rapoport. 2002. Mathematical models of protein kinase signal transduction. Mol. Cell. 9:957–970. [DOI] [PubMed] [Google Scholar]
- 27.Legewie, S., N. Bluthgen, and H. Herzel. 2005. Quantitative analysis of ultrasensitive responses. FEBS J. 272:4071–4079. [DOI] [PubMed] [Google Scholar]
- 28.Hornberg, J. J., F. J. Bruggeman, B. Binder, C. R. Geest, A. J. de Vaate, J. Lankelma, R. Heinrich, and H. V. Westerhoff. 2005. Principles behind the multifarious control of signal transduction. ERK phosphorylation and kinase/phosphatase control. FEBS J. 272:244–258. [DOI] [PubMed] [Google Scholar]
- 29.Asthagiri, A. R., and D. A. Lauffenburger. 2001. A computational study of feedback effects on signal dynamics in a mitogen-activated protein kinase (MAPK) pathway model. Biotechnol. Prog. 17:227–239. [DOI] [PubMed] [Google Scholar]
- 30.Rosenfeld, N., M. B. Elowitz, and U. Alon. 2002. Negative autoregulation speeds the response times of transcription networks. J. Mol. Biol. 323:785–793. [DOI] [PubMed] [Google Scholar]
- 31.O'Rourke, S. M., I. Herskowitz, and E. K. O'Shea. 2002. Yeast go the whole HOG for the hyperosmotic response. Trends Genet. 18:405–412. [DOI] [PubMed] [Google Scholar]