

Abstract
Rad26, the yeast homologue of human Cockayne syndrome group B protein, and Rpb9, a nonessential subunit of RNA polymerase II, have been shown to mediate two subpathways of transcription coupled DNA repair in yeast. Here we show that Rad26 and Rpb9 mediated repair in the yeast GAL1 gene are differently modulated by different promoter elements. The initiation site and efficiency of Rad26 mediated repair in the transcribed strand are determined by the upstream activating sequence (UAS), but not by the TATA or local sequences. The role of UAS in determining the Rad26 mediated repair is not through loading of RNA polymerase II or the transcriptional regulatory complex SAGA. However, both the UAS and TATA sequences are essential for confining Rad26 mediated repair to the transcribed strand. Mutation of the TATA sequence, which greatly reduces transcription, or deletion of the TATA or mutation of the UAS, which completely abolishes transcription, causes Rad26 mediated repair to occur in both strands. Rpb9 mediated repair only occurs in the transcribed strand, and is efficient only in the presence of both TATA and UAS sequences. Our results suggest that Rad26 mediated repair can be either transcription-coupled, provided that a substantial level of transcription is present, or transcription-independent, if the transcription is too low or absent. In contrast, Rpb9 mediated repair is strictly transcription-coupled, and is efficient only when the transcription level is high.
Nucleotide excision repair (NER) is a conserved DNA repair mechanism that removes a wide range of bulky DNA lesions including ultraviolet (UV) light induced cyclobutane pyrimidine dimers (CPDs) and 6–4 photoproducts [for a recent review, see (1)]. One NER pathway, the so called global genomic repair (GGR), removes lesions throughout the genome including those in the nontranscribed strand (NTS) of an active gene. In mammalian cells, GGR is dependent on Xeroderma pigmentosum complementation group C (2,3) and damage-specific DNA-binding proteins (4). In Saccharomyces cerevisiae, GGR is dependent on Rad7 and Rad16 (5). Another NER pathway, termed transcription-coupled repair (TCR), is responsible for rapid repair in the transcribed strand (TS) of an active gene. An RNA polymerase stalled at a DNA lesion in the TS may serve as the initial signal for TCR (6). The TCR mechanism in Escherichia coli is best understood (7,8). However, in eukaryotes, the detailed biochemical mechanism of TCR has been elusive. In mammalian cells, it has been shown that the Cockayne syndrome group A (CSA) and B (CSB) proteins are required for TCR (9–12). In S. cerevisiae, Rad26, the yeast homologue of mammalian CSB (13), and Rpb9, a nonessential subunit of RNA polymerase II (Pol II) (14,15), have been shown to mediate two subpathways of TCR, respectively.
It seems that the Rad26 and Rpb9 mediated TCR subpathways have different efficiencies in different regions of a gene. Rpb9 mediated TCR operates more effectively in the coding region than in the region upstream of the transcription start site, whereas Rad26 mediated TCR operates equally well in the two regions contributions of Rad26 and Rpb9 to TCR may be different from one gene to another. In the constitutive URA3 gene, TCR seems to be exclusively mediated by Rad26, except for a short region close to the transcription start site (16). In the constitutive gene RPB2, Rad26 is partially required (15,17–19), and both Rad26 and Rpb9 contribute to TCR (15). For the galactose induced GAL1-10 genes, Rad26 is almost dispensable, especially in the coding region, indicating that TCR in these genes is primarily mediated by Rpb9 (14,15).
Both Rad26 and Rpb9 mediated TCR seem to be confined to the TS of the galactose induced GAL1-10 genes, initiating at upstream sites that are ~ 100 nucleotides from the upstream activating sequence (UAS) (14). Interestingly, the initiation sites of the Rad26 and Rpb9 mediated TCRs are not correlated with either the transcription start sites or the key promoter elements, the TATA boxes (14).
At present, it is largely unknown how initiation and efficiency of Rad26 and Rpb9 mediated repairs are regulated in a gene. In this paper, we present evidence that the initiation site and efficiency of Rad26 mediated repair in the TS of the GAL1 gene are determined by the UAS, but not by TATA, local sequences or even active transcription. However, the UAS, TATA, and a substantial level of transcription are essential for confining the Rad26 mediated repair to the TS. In contrast, the Rpb9 mediated repair is always confined to the TS, and is efficient only in the presence of UAS, TATA and a high level of transcription.
EXPERIMENTAL PROCEDURES
Yeast strains and plasmids
The wild-type yeast strain Y452 (MATα, ura3-52, his3–1, leu2-3, leu2-112, cir°) and its isogenic rad16, rad16 rpb9 and rad16 rad26 deletion mutants were created as described previously (15). The rad7, rad7 spt7, rad7 rad26 spt7 deletion mutants are derivatives of wild type strain BJ5465 (MATa ura3-52 trp-1 leu2Δ1 his3Δ200 pep4::HIS3 prb1Δ1.6R can1) (20). Nucleotides (with respect to the starting codon ATG) +214 to +1454, +58 to +2297 and +11 to +4900 were deleted for the RAD7, RAD26 and SPT7 genes, respectively. (14,15). Moreover, in log-phase cells, the relative
PCR primers used for amplifying different GAL1-10 fragments are listed in Table 1. A 2kb normal GAL1-10 fragment, encompassing the UAS and 5′ portions (0.7 kb) of each of the genes, was PCR amplified using primers 1 and 2. Primer pairs 1 and 4, and 2 and 3 were used to amplify two fragments, which were digested with Sty I and ligated to create a GAL1-10 fragment with the GAL1 TATA mutated from ATATAAA (21) to CCATGGA. Primer pairs 1 and 6, and 2 and 5 were used to amplify another two fragments, which were digested with Sph I and ligated to create a GAL1-10 fragment with the UAS mutated (Table 2). Primer 1 was paired with primers 7, 8, 9 and 10 to amplify the GAL1-10 fragments with deletion from the GAL1 gene down to +14, −72, −111 and −185, respectively. All the GAL1-10 fragments were digested with Hind III and inserted in the Hind III site of shuttle vector pRS415 (22) (Fig. 1A). The plasmid constructs were propagated in E. coli and transformed into different yeast strains for transcription and repair analysis.
Table 1.
PCR primers used to create plasmids bearing different GAL1-10 fragments
| Primer | Sequence (5′ → 3′)a | Location |
|---|---|---|
| 1 | GATGTAAAGCTTCTCGCGCCTAC | GAL10, +700 |
| 2 | AAACAAGCTTAGCCTGATCCATACCGC | GAL1, +700 |
| 3 | ACAGATCCATGGATGCAAAAACTGCATAACCACTTT | GAL1, TATA |
| 4 | TTGCATCCATGGATCTGTTAATAGATCAAAAATCATCGC | GAL1, TATA |
| 5 | CTCCTGCATGCGTCCTCGTCTTCACCGGTCGCGTTCCTGAAACGCAGATGTGCCTTGCGCCGCACTGCT | UAS |
| 6 | AGGACGCATGCAGGAGAGTCTTCCATTGGAGGGCTGTCACCCACTTGGCGGCTTCTAATC | UAS |
| 7 | TTAAAGCTTTTTTGTTGATACTTTTATTACATTTG | GAL1, +14 |
| 8 | TATGAAGCTTTTGCATTTATATATCTGTTAATAGAT | GAL1, −72 |
| 9 | ATCAAAGCTTCGCTGATTAATTACCCCA | GAL1, −111 |
| 10 | TAATAAGCTTCGTTCATTTGAAGGTTTGTGG | GAL1, −185 |
Underlined are native or introduce restrict sites (Hind III for primers 1, 2, 7 – 10, Sph I for primers 5 and 6, and Sty I for primers 3 and 4); letters in bold are introduced mutations.
Table 2.
Mutations in the UAS of plasmid borne GAL1 genea
| Gal4 sites | Normal sequence | Mutated sequence |
|---|---|---|
| I | CGGATTAGAAGCCGCCG | CGGATTAGAAGCCGCCA |
| II | CGGGTGACAGCCCTCCG | TGGGTGACAGCCCTCCA |
| III | AGGAAGACTCTCCTCCG | TGGAAGACTCTCCTGCA |
| IV | CGCGCCGCACTGCTCCG | TGCGCCGCACTGCTCCG |
Gal4 cognitive triplet nucleotide sequences are underlined; nucleotides mutated are shown in bold.
Fig. 1. Transcription in plasmid-born GAL1 gene.
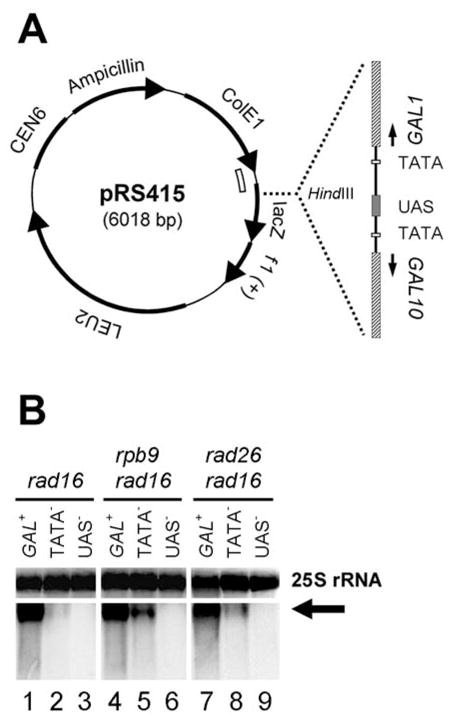
A. Schematic showing the structure of the plasmid constructs bearing the 2 kb GAL1-10 fragments encompassing the shared UAS and ~ 700 bp of each of the genes. The open arch inside of the pRS415 circle indicates the sequence used for generating Northern blot probes. B. Northern blot showing transcription in the constructs with normal GAL1 (GAL+), mutated GAL1 TATA (TATA−) and mutated UAS (UAS−). The arrow indicates transcripts from the constructs. 25S rRNA serves as internal loading control.
UV irradiation, repair incubation and DNA isolation
Yeast cells were grown at 28°C in minimal medium containing 2% galactose to late log phase (OD600 ≈ 1.0), harvested and irradiated with 50 J/m2 of 254 nm UV light. The cells were incubated in the same medium at 28°C for various times in the dark. Total DNA was isolated from the cells following repair incubation, using a glass beads method (15).
Mapping of NER
DNA fragments of interest were strand specifically end labeled with [α-32P]dATP using the procedure described previously (23,24). Briefly, ~ 1 μg of total DNA was digested with restriction endonuclease(s) to release the plasmid-borne GAL1 fragments of interest and incised at CPD sites with an excess amount of purified T4 endonuclease V (Epicentre). To specifically label the plasmid-borne GAL1 fragments, the incised total DNA was mixed with a DNA ladder (e.g., 2 Log DNA Ladder from New England Biolabs), resolved on a native agarose gel. The band corresponding to the sizes of the plasmid-borne GAL1 fragments were purified from the gel using the DNA ladder as carrier. Excess copies of biotinylated oligonucleotides, which are complementary to the 3′ ends of the fragments to be labeled, were mixed with the sample. The mixture was heated to 95°C for 5 min to denature the DNA and then cooled to an annealing temperature. The annealed fragments were attached to streptavidin magnetic beads (Invitrogen) and the other fragments were removed by washing the beads at the annealing temperature. The attached fragments were labeled with [α-32P]dATP (Perkin Elmer), resolved on sequencing gels. The dried gels were exposed to a Phosphorimager screen (GE Healthcare or Bio-Rad).
Northern blot analysis
Cells were cultured to late log phase under the same conditions as those used for NER analysis. Total RNA was isolated using a hot acidic phenol method, as described (25). The RNA was fractionated by electrophoresis on formaldehyde-agarose gels (26), followed by transfer to Hybond N+ membranes (GE Healthcare). For, analysis of transcription driven by the plasmid-borne GAL1 promoter with different mutations or deletions, the pRS415 vector fragment (Fig. 1A, indicated by the open arch inside of the vector circle) was inserted into the multiple cloning site of plasmid pGEM-3Z (Promega). Radioactive RNA probes were made by in vitro transcription of the pRS415 fragment using T7 or SP6 RNA polymerases (26). Membranes were first hybridized with the respective probes. After exposure to Phosphorimager screens, the probes were stripped off by boiling the membranes in 1% SDS. The 25S rRNA on the stripped membranes was re-hybridized with probes generated from a 2.9 kb 25S DNA and exposed to Phosphorimager screens. The signal intensities of the 25S rRNA were served as loading controls.
Chromatin immunoprecipitation (ChIP) assay
ChIP was carried out as described previously with minor modifications (27,28). Cells were grown in minimal medium containing 2% galactose to late log phase (A600 ≈ 1.0). Chromatin crosslinking was achieved by addition of 1.1 ml of 36.5% formaldehyde to 40 ml of culture (final concentration 1%). After 10 minutes of incubation, the action of formaldehyde was quenched with glycine (125 mM final concentration). Cells were resuspended in buffer A (50 mM HEPES pH 7.5, 140 mM NaCl, 1% Triton X-100, 0.1% sodium deoxycholate, 1mM EDTA, and phosphatase and protease inhibitors) and lysed by vortexing with glass beads. The cell lysates were sonicated to shear the DNA to an average size of 500 bp and clarified by centrifugation at 16,000 × g for 10 minutes at 4°C. The supernatant was collected and subjected to repeated centrifugation until no visible pellet can be seen at the bottom of tube. An aliquot from each of the clarified lystes was saved as input control. The rest of the clarified lysates were incubated with or without affinity-purified anti-Rpb1 antibody 8WG16 (Neoclone) at 4°C overnight. Protein A-coated agarose beads (Sigma) were added to the samples and the incubation was continued for 3 hr at 4°C. The beads were washed twice with buffer A, twice with buffer B (50 mM HEPES pH 7.5, 500 mM NaCl, 1% Triton X-100, 0.1% sodium deoxycholate, 1mM EDTA, and phosphatase and protease inhibitors), twice with buffer C (10 mM Tris-Cl pH 8.0, 250 mM LiCl, 0.5% NP-40, 0.5% sodium deoxycholate, 1mM EDTA, and phosphatase and protease inhibitors) and twice with TE (10 mM Tris-Cl, 1mM EDTA, pH 8.0). Bead-bound chromatin fragments were eluted by incubating the beads in buffer D (50 mM Tris-Cl pH 8.0, 1% SDS and 10 mM EDTA) at 65 °C for 30 minutes. The elution was repeated one more time and the eluates were pooled. Crosslinks were reversed by incubating the eluates at 65°C overnight. The inputs and the eluates were treated with proteinase K and extracted once with phenol, once with phenol/chloroform/isoamylalcohol (25/24/1) and once with chloroform/isoamylalcohol (24/1). The DNA in the eluates was precipitated with ethanol.
The input and immunoprecipitated DNA were amplified by PCR. The pRS415 sequence of 210 bp located immediately downstream of the GAL1 sequence in the different plasmid constructs (Fig. 1A, indicated by the open arch inside of the plasmid circle) was amplified using primers 5′-GGGGGATCCACTAGTTCTAGAGC-3′ and 5′-TGAGGTACCTCACTCATTAGGCACCCC-3′. To provide an internal control of Pol II loading onto a gene, a 109 bp fragment in the coding region of the ACT1 gene was amplified using primers, 5′-CTGAGGTTGCTGCTTTGGTTATTG-3′ and 5′-CTTGGTCTACCGACGATAGATGGG-3′. The PCR products were resolved on an agarose gel and stained with ethidium bromide. The band intensities on the gel were quantified using AlphaEase 4.0 software (Innotech). The signal intensities of the amplified pRS415 sequence of 210 bp were normalized to those of the ACT1 sequence.
RESULTS
Modulation of Rad26 and Rpb9 mediated repair in the TS of the GAL1 gene by TATA and UAS elements
The GAL1 gene is essential for yeast cells to grow in galactose media, because it encodes a galactokinase which phosphorylates galactose to galactose-1-phosphate in the first step of galactose catabolism (29,30). To examine the modulation of Rad26 and Rpb9 mediated repair by different gene elements, we created a number of plasmids bearing the GAL1 gene with different mutations or deletions. A 2 kb GAL1-10 region encompassing the shared UAS and ~700 bp coding region of each of the genes (Fig. 1A) was amplified by PCR. The GAL1 TATA mutation was achieved by changing the original sequence ATATAAA (21) to CCATGGA. The UAS mutation was achieved by changing the cognitive triplet sequences of CGG, AGG or CGC located at both ends of the four palindromic binding sites of the transcriptional activator Gal4 (Table 2) (31). The GAL1-10 fragments with normal, mutated GAL1 TATA or UAS sequences were inserted in the Hind III site of plasmid pRS415 (22) (Fig. 1A). The plasmid constructs were transformed into GAL+ rad16 cells, to eliminate GGR and specifically analyze Rad26 and Rpb9 mediated repairs in the plasmid-borne GAL1 gene.
Total RNA was isolated from the cells cultured in the galactose medium to induce the GAL1 gene (29,30). Transcription driven by the plasmid-borne GAL1 promoter was analyzed by Northern blots. To distinguish transcription on the plasmid constructs from that on the genomic GAL1 gene, the pRS415 sequence located immediately downstream of the GAL1 sequence (Fig. 1A, indicated by the open arch inside of the plasmid circle) was used as a Northern blot probe. A transcript of ~ 3 kb can be seen from the normal GAL1 construct (Fig. 1B, arrow; lanes 1, 4 and 7). The transcription level in the mutated GAL1 TATA construct is much lower than that in the normal GAL1 construct (Fig. 1B. compare lanes 1 and 2, 4 and 5, and 7 and 8). Quantification of the data (not shown) suggests that mutation of the GAL1 TATA reduced the transcription level 50 – 100 fold. Essentially no transcription can be detected in the UAS mutated construct (Fig. 1B, lanes 3, 6 and 9). These results indicate that the GAL1 TATA is required for high levels of transcription, while the UAS is indispensable for transcription.
Total DNA was isolated from the transformed cells following UV irradiation and repair incubation in the galactose medium. To specifically analyze Rad26 and Rpb9 mediated repairs in the plasmid-borne GAL1 fragments, the 2 kb plasmid-borne GAL1-10 fragments (Fig. 1A) were gel purified following digestion of the total DNA with Hind III and incised at CPDs with excess amount of T4 endonuclease V (32). The gel purified fragments were further restricted to release the GAL1 fragment of interest and strand specifically end labeled with [α-32P]dATP (23,24). The labeled fragments were resolved on a DNA sequencing gel and exposed to a Phosphorimager screen.
In rad16, rad16 rpb9, rad16 rad26 and rad16 rad26 rpb9 cells, the repair patterns in the plasmid-borne normal GAL1 gene (Fig. 2A – D) were very similar to those in the genomic GAL1 gene (14,15). These results indicate that the transcription levels and repair patterns are similar in the plasmid-borne and genomic GAL1 genes.
Fig. 2. Rad26 and Rpb9 mediated repairs in the TS of plasmid-borne GAL1 gene.
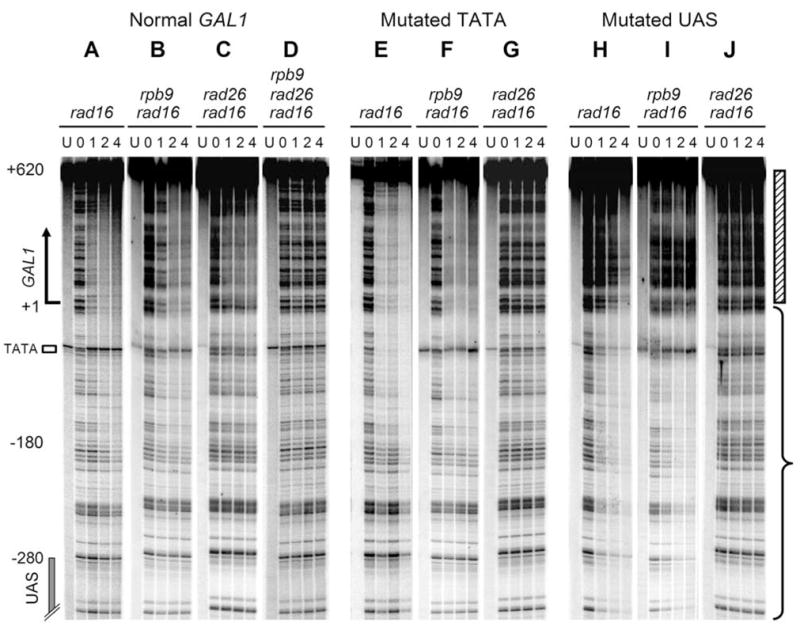
Lanes U represent unirradiated samples. Lanes 0, 1, 2 and 4 indicate different times (hour) of repair incubation following UV irradiation. Hatched bar and brace on the right of the gels mark the transcribed and upstream regions, respectively.
In rad16 cells, where both the Rad26 and Rpb9 mediated repairs are active, and in rad16 rpb9 cells, where only the Rad26 mediated repair is active (14,15), mutation of the GAL1 TATA resulted in no significant repair deficiency in the TS of the GAL1 gene (Fig. 2, compare panels A and E, and B and F). Furthermore, the mutation did not change the initiation site of repair, which is ~ 180 nucleotides upstream of the transcription start site and ~ 100 nucleotides from the UAS (Fig. 2A, B, E and F). In contrast, in rad16 rad26 cells, where only the Rpb9 mediated repair is active (14,15), mutation of the GAL1 TATA essentially abolished repair in the TS of the GAL1 gene (Fig. 2, compare panels C and G). These results indicate that the initiation site and efficiency of Rad26 mediated repair in the TS of the GAL1 gene are largely independent of the TATA element and transcription level. In contrast, the Rpb9 mediated repair is dependent on the gene element and a high level of transcription.
Repair was observed in the UAS-mutated GAL1 gene in rad16 and rad16 rpb9 cells, although it was much less efficient in the transcribed region (Fig. 2H and I, marked by hatched bar on the right of the gels) of the gene. Furthermore, the UAS mutation abolished an apparent initiation site for this repair, as repair seemed to occur throughout the GAL1-10 region analyzed (Fig. 2H and I, region marked with brace; data not shown). However, the UAS mutation essentially eliminated repair in rad16 rad26 cells (Fig. 2J). These results suggest that the UAS and transcription may not be absolutely required for Rad26 mediated repair, but may be required for Rpb9 mediated repair. Furthermore, the UAS may determine the initiation site of the Rad26 mediated repair in the TS of the GAL1 gene.
Modulation of Rad26 and Rpb9 mediated repair in the NTS of the GAL1 gene by TATA and UAS elements
In all the rad16 cells examined, no repair can be seen in the NTS of the normal GAL1 construct (Fig. 3A, B and C). However, mutation of either TATA or UAS resulted in repair in the NTS of the GAL1 gene in rad16 and rad16 rpb9 cells (Fig. 3, compare panels A, D and G, and B, E and H), but not in rad16 rad26 cells (Fig. 3, compare panels C, F and I). These results suggest that the TATA and UAS elements and a substantial level of transcription are essential for preventing the Rad26 mediated repair from occurring in the NTS of the GAL1 gene. However, the Rpb9 mediated repair does not operate in the NTS of the gene in all the conditions.
Fig. 3. Rad26 and Rpb9 mediated repairs in the NTS of plasmid-borne GAL1 gene.
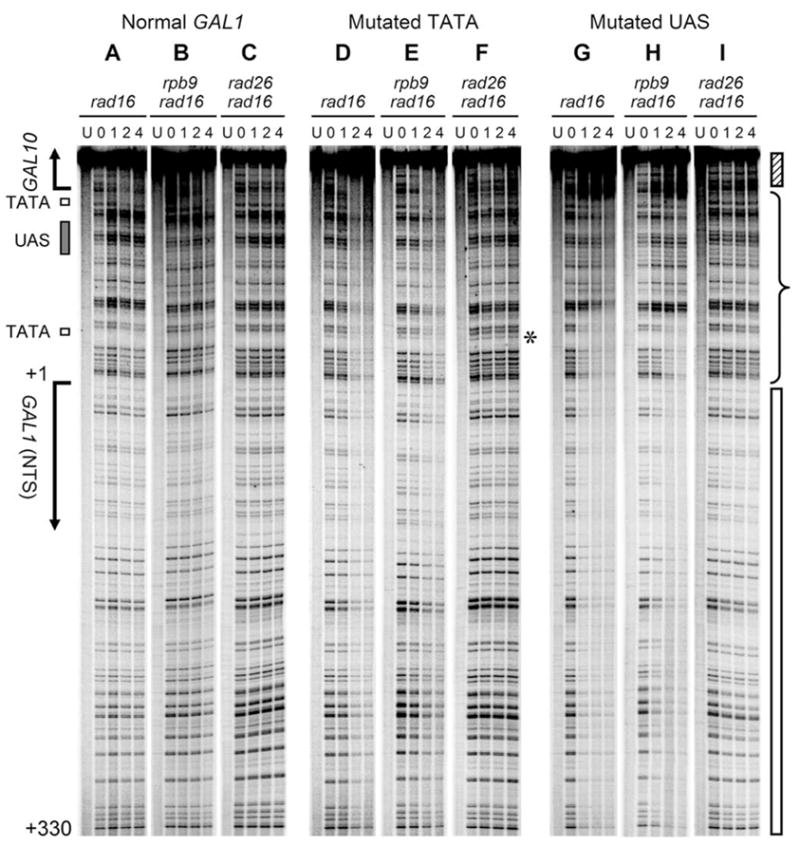
Lanes U represent unirradiated samples. Lanes 0, 1, 2 and 4 indicate different times (hour) of repair incubation following UV irradiation. The asterisk on the right of panel F indicates a CPD site (CC) present only in the mutated (CCATGGA) (panels D, E and F) but not in the normal (ATATAAA) GAL1 TATA. Hatched and open bars on the right of the gels mark the transcribed regions of the GAL10 (TS) and GAL1 (NTS), respectively. Brace indicates the upstream region.
The role of local DNA sequences in Rad26 and Rpb9 mediated repair
The studies described above suggest that the initiation of Rad26 mediated repair may be determined by the UAS, but is independent of the TATA element. Furthermore, the efficiency of the Rad26 mediated repair does not seem to be significantly affected by active transcription. However, a low level of transcription still occurred in the GAL1 TATA mutated construct (Fig. 1B, lanes 2, 5 and 8). It is possible that the mutated TATA or a nearby local sequence may be able to partially compensate for the TATA element, resulting in a low level of transcription and the initiation of Rad26 mediated repair. To examine this possibility, a series of GAL1-10 fragments, encompassing ~ 700 bp of the GAL10 coding sequence, the UAS and different lengths of the GAL1 sequence (Fig. 4A) were amplified by PCR. These PCR fragments were inserted in the Hind III site of plasmid pRS415 (Fig. 1A). Plasmid constructs bearing the different GAL1-10 fragments were transformed into yeast cells. Total RNA was isolated from the transformed cells cultured in the galactose medium. Transcription driven by the GAL1 gene promoter borne on the plasmid constructs was analyzed by Northern blots, using the vector sequence located immediately downstream of the GAL1 sequence (Fig. 1A, indicated by the open arch inside of the plasmid circle) as a probe. Strong transcription can be seen when the GAL1 gene was deleted down to +14 (relative to the transcription start site) (GAL1Δ +14) (Fig. 4B, lanes 4, 8 and 12). A further deletion, which left the TATA element intact but had the GAL1 transcription start site removed (i.e. deletion down to – 72, GAL1Δ-72), also showed strong transcription (Fig. 4B, lanes 3, 7 and 11). However, deletion down to −111 or −185, each of which removed the TATA element, caused undetectable transcription from the sequence (Fig. 4B, lanes 1, 2, 5, 6, 9 and 10).
Fig. 4. Transcription driven by the promoter of plasmid-born GAL1 gene with different deletions.
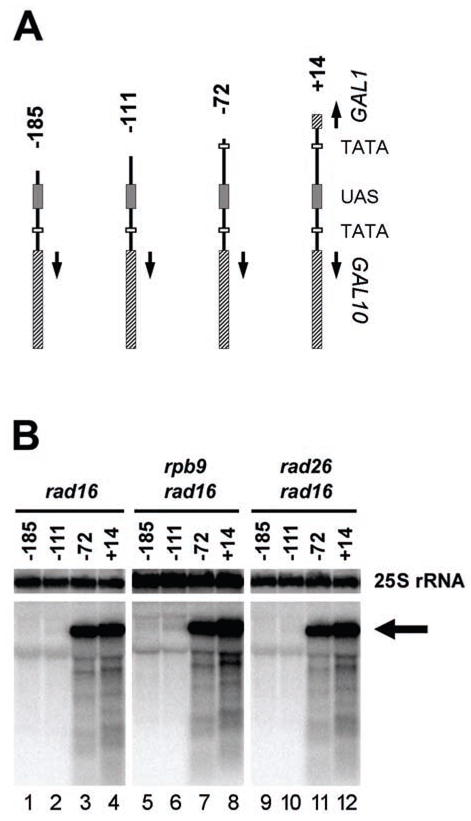
A, structures of the GAL1-10 genes with different deletions from the GAL1 side. These GAL1-10 fragments were inserted into the Hind III site of vector pRS415 (see Fig. 1A) to create different plasmid constructs. B, Northern blot showing transcription driven by the GAL1 promoter. The arrow indicates transcripts from the constructs. The Northern blot probe was made from the pRS415 vector sequence that is immediately downstream of the inserted GAL1-10 fragment (see Fig. 1A, marked by the open arch). 25S rRNA serves as internal loading control.
Total DNA was isolated from the transformed cells following UV irradiation and repair incubation. The plasmid fragments encompassing the UAS, part of the GAL1 sequence and a 740 bp pRS415 sequence immediately downstream of the GAL1 sequence (Figs. 1A and 4A) were released by restriction, and incised at the CPDs with excess amount of T4 endonuclease V. The plasmid fragments were purified from an agarose gel and strand specifically end labeled with [α-32P]dATP.
In rad16 cells, where both Rad26 and Rpb9 mediated repairs are operative, and in rad16 rpb9 cells, where only Rad26 mediated repair is active, efficient repair can be seen in the TS of all the plasmid constructs, even in those without the TATA and detectable transcription (Fig. 5A, B, D, E, G, H, J and K). In all the constructs, repair initiates ~ 100 nucleotides from the UAS (Fig. 5, marked by open arrows). Interestingly, the approximate initiation sites of the repair can be located in the GAL1 sequence (Fig. 5, panels D, E, G, H, J and K; marked by solid lines), or the pRS415 sequences (Fig. 5, panels A and B; marked by the dotted line). These results indicate that the initiation site and efficiency of Rad26 mediated repair in the TS of the GAL1 gene are indeed not significantly affected by the TATA sequence, or even actual transcription. Moreover, the Rad26 mediated repair event may not be significantly influenced by other local sequences (e.g., transcription start site, sequence where Rad26 mediated repair initiates and the sequence adjacent to the TATA box).
Fig. 5. Rad26 and Rpb9 mediated repairs in the TS of plasmid constructs with different deletions from the GAL1 gene.

Lanes U represent unirradiated samples. Lanes 0, 1, 2 and 4 indicate different times (hour) of repair incubation. Solid and dotted lines indicate GAL1 and vector pRS415 sequences on the constructs, respectively (see Figs. 1A and 4A). Open arrows depict the approximate initiation sites of repair. Solid bent arrow on the right of panel L indicates the GAL1 transcription start site borne on construct GAL1Δ+14. Shaded bent arrow on the right of panel I marks the transcription start site driven by the GAL1 promoter borne on construct GAL1Δ-72.
In rad16 rad26 cells, where only the Rpb9 mediated repair is active, efficient repair can be seen in the transcribed regions of the constructs that contain the TATA element [Fig. 5I and L, see the regions above the shaded (panel I) and solid (panel L) arrows]. In contrast, little repair can be seen in the same sequences borne on the constructs that do not have the TATA element (Fig. 5C and F). These results further suggest that the Rpb9 mediated repair is largely dependent on the TATA and active transcription.
UAS does not load Pol II onto the GAL1 promoter in the absence of TATA
The experiments described above suggest that the initiation and efficiency of Rad26 mediated repair in the GAL1 TS are determined by the UAS, but are largely independent of either TATA or actual transcription. One explanation is that the UAS may be able to load Pol II in the absence of TATA, and the loading of Pol II may be sufficient to initiate Rad26 mediated repair, regardless of actual transcription. To test this idea, we examined loading of Pol II onto the GAL1 promoter in the plasmid constructs bearing different GAL1-10 fragments (Fig. 4A). Yeast cells transformed with the different plasmid constructs and the empty pRS415 vector (as control) were cultured in galactose medium to late log phase, and ChIP analysis was performed using a monoclonal antibody to Rpb1, the largest subunit of Pol II (33). A 210 bp pRS415 sequence immediately downstream of the GAL1 sequence (Fig. 1A, marked with the open arch inside of the plasmid circle) was amplified by PCR. As shown in Fig. 6, the 210 bp pRS415 sequence in the GAL1Δ+14 was immunoprecipitated by the anti-Rpb1 antibody. In contrast, the levels of the pRS415 sequence pulled down by the antibody are similar between the GAL1Δ-185 construct and the empty pRS415 vector (Fig. 6). These results indicate that the UAS element may not be able to recruit Pol II to the GAL1 promoter in the absence of TATA. Furthermore, the modulation of the Rad26 mediated repair by the UAS must be by a mechanism that is independent of Pol II.
Fig. 6. ChIP assay showing loading of Pol II onto a plasmid sequence by a GAL1 promoter.
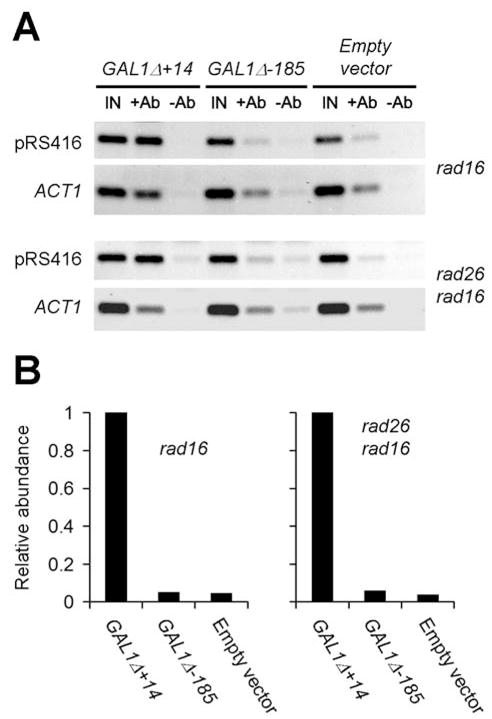
The structures of the empty pRS415 vector and the plasmid constructs GAL1Δ+14 and GAL1-185 are shown in Figs. 1A and 4A, respectively. The pRS415 sequence amplified by PCR was a 210 bp fragment located immediately downstream of the GAL1 sequence (Fig. 1A, marked with the open arch inside of the plasmid circle), using template DNA isolated from ChIP input (IN), immunoprecipitated with anti-Rpb1 (+Ab), or mock immunoprecipitated (−Ab) samples. The 109 bp ACT1 sequence was amplified under the same conditions as the 210 bp pRS415 sequence, to offer an internal control. Plots shown in panel B are relative amounts of the 210 bp pRS415 sequence immunoprecipitated with anti-Rpb1 antibody. The loading was normalized according to the signal intensities of the internal control ACT1 fragment.
Deletion of SPT7 dramatically compromises Rpb9-, but not Rad26-mediated repair
SAGA, a transcriptional regulatory complex, plays an important role in transcription of the GAL1 gene (34,35). At the GAL1 promoter, SAGA is first recruited by the Gal4 activator to the UAS. The UAS-bound SAGA then facilitates the binding of TATA binding protein to the core promoter, thereby stimulating transcription (34,35). It has been proposed that the histone acetyltransferase activity of Gcn5 in the SAGA complex, remodels local chromatin structure and subsequently facilitates binding of the TATA binding protein (36). Spt7 is one of the SAGA subunits that are essential for the recruitment of SAGA to the GAL1 promoter (37).
It is possible that the effect of the UAS on the Rad26 and Rpb9 mediated repair is through the recruitment of the SAGA complex to the GAL1 promoter. To test this hypothesis, we analyzed NER in the genomic GAL1 gene in cells with the SPT7 gene deleted. In spt7 cells, efficient repair occurred in the whole GAL1 fragment, including the upstream regions where TCR did not operate (Fig. 7A). The initiation site and efficiency of repair in rad7 spt7 cells (Fig. 7B) were very similar to those in rad16 (Fig. 2A) or rad7 (not shown) cells, where GGR are inactive. Similar to rad16 rad26 cells (Fig. 2C), rad7 rad26 cells showed very efficient repair in the GAL1 gene (not shown). However, deletion of SPT7 essentially abolished repair in rad7 rad26 cells (Fig. 7C). These results indicate that the recruitment of SAGA complex to the GAL1 promoter plays little role in the Rad26 mediated repair. In contrast, this recruitment is critical for Rpb9 mediated repair.
Fig. 7. Repair in the TS of the genomic GAL1 gene in spt7 cells.
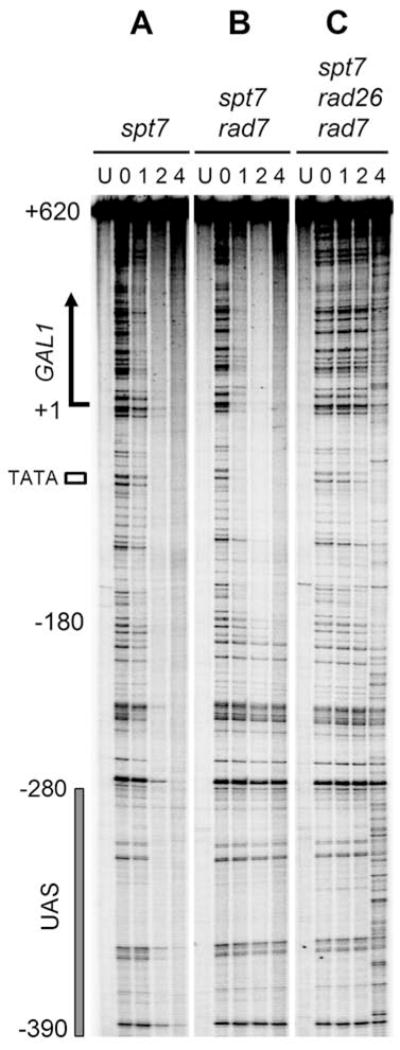
Lanes U represent unirradiated samples. Lanes 0, 1, 2 and 4 indicate different times (hour) of repair incubation.
DISCUSSION
We showed that in the TS of the GAL1 gene, the initiation site and efficiency of Rad26 mediated repair are dependent on the UAS, but largely independent of the TATA, other local sequences or even actual transcription. Furthermore, if the transcription level is too low (e.g., if the TATA is mutated) or absent (e.g, if the TATA is deleted or the UAS is mutated), Rad26 mediated repair occurs not only in the TS, but also in the NTS. These results are surprising in view of the widely accepted notion that Rad26 is a “transcription-repair coupling factor”.
The mechanism by which the UAS determines the initiation site of Rad26 mediated repair in the TS of the GAL1 gene remains to be determined. Pol II may not play a role in the initiation, as our ChIP analysis suggests that the UAS is unable to recruit Pol II in the absence of TATA (Fig. 6). Also, SAGA facilitated chromatin remodeling or transcription in the GAL1 gene seems to be dispensable for initiation of the repair, as deletion of SPT7 did not change the initiation (Fig. 7). One possible scenario is that Rad26 is directly or indirectly recruited to the initiation site of the repair, by, for example, interacting with a transcription mediator. Alternatively, Rad26 mediated repair may be intrinsically operative in nontranscribed regions (nontranscribed genes or inter-gene regions). The UAS may simply set up a boundary at about −180 nt in the GAL1 gene. The Rad26 mediated repair may be inhibited beyond the boundary.
A question remains as to how Rad26 mediates repair in both strands of the GAL1 gene when the TATA or UAS is mutated or deleted. It seems that the Rad26 mediated repair will occur in both strands of a gene whenever transcription is very low or absent. The GAL1-10 genes are highly induced by galactose, but completely repressed in glucose media (29,30). We found that efficient repair, which is dependent on Rad26 but not on Rpb9 or Rad16, also occurs in both strands of the glucose repressed GAL1-10 genes in rad16 cells (not shown). Rad26 (38) and its human homologue CSB (39) are DNA-dependent ATPase of the SWI2/SNF2 family proteins, which are implicated in chromatin remodeling during transcription (40). In vitro studies have shown that CSB is able to remodel chromatin structure at the expense of ATP hydrolysis (41). It is possible that Rad26 mediates repair in both strand of a DNA sequence that is not transcribed or very scarcely transcribed, by increasing repair protein access to sites of DNA damage in chromatin. Alternatively, a very low level of non-productive form of Pol II may be gratuitously associated with and “patrols” on both strands of a DNA sequence that is not actively transcribed (e.g., in search for transcription start sites). Rad26 may mediate repair by facilitating the “patrol” process of the non-productive form of Pol II along the DNA.
It has been shown that, in two mutant Chinese hamster ovary cell lines in which the entire APRT promoter region has been deleted, NER is still efficient in both strands of the promoterless APRT gene, even though neither strand appears to be transcribed (42). Rodent cells are profoundly deficient in GGR of CPDs, typically showing efficient TCR in the TS of actively transcribed genes, but little repair in the NTS, or in nontranscribed regions of the genome (4,43,44). Therefore, a transcription-independent non-GGR mechanism may also exist in mammalian cells. It would be interesting to test if the efficient NER in the promoterless APRT gene is dependent on CSB, the human homologue of yeast Rad26.
In contrast to Rad26, Rpb9 seems to mediate a repair mechanism that is strictly coupled to transcription, which is conceivable in view of the fact that Rpb9 itself is a subunit of Pol II. The Rpb9 mediated repair seems to be effective only when a high level of transcription is present, as mutation of the TATA sequence, which dramatically reduces transcription, or deletion of the TATA sequence or mutation of UAS, which eliminated transcription, essentially abolishes the Rpb9 mediated repair. We also found that the Rpb9 mediated repair does not operate in the GAL1-10 genes in glucose cultured GAL4+ cells or in galactose cultured gal4 cells (not shown).
Following analysis of NER in different genes (15,16,18,19), it became obvious that the relative contributions of Rad26 and Rpb9 to TCR are different from gene to gene. This difference could simply be due to different levels of transcription in different genes. For highly expressed genes, such as the galactose induced GAL1-10 genes, the Rpb9 mediated TCR is very effective, making Rad26 less important for TCR (14,15). For slowly transcribed genes, such as the constitutive RPB2 (15,18,19) or URA3 (16) genes, the Rpb9 mediated TCR is less effective or essentially absent and Rad26 contributes much more to the overall TCR.
The abbreviations used are
- CPD
cyclobutane pyrimidine dimmer
- CSA
Cockayne syndrome group A
- CSB
Cockayne syndrome group B
- GGR
global genomic repair
- NER
nucleotide excision repair
- NTS
nontranscribed strand
- PCR
polymerase chain reaction
- TCR
transcription coupled repair
- TS
transcribed strand
- UAS
upstream activating sequence
- UV
ultraviolet
Footnotes
We thank Dr. Steven Barker for helping with ChIP assays, Drs. Feng Gong and John Wyrick for critical comments about the manuscript. This study was made possible by NIH grants ES012718 (to SL) and ES04106 (to MJS) from the National Institute of Environmental Health Sciences (NIEHS).
References
- 1.Reardon JT, Sancar A. Prog Nucleic Acid Res Mol Biol. 2005;79:183–235. doi: 10.1016/S0079-6603(04)79004-2. [DOI] [PubMed] [Google Scholar]
- 2.Venema J, van Hoffen A, Karcagi V, Natarajan AT, van Zeeland AA, Mullenders LH. Mol Cell Biol. 1991;11(8):4128–4134. doi: 10.1128/mcb.11.8.4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Venema J, van Hoffen A, Natarajan AT, van Zeeland AA, Mullenders LH. Nucleic Acids Res. 1990;18(3):443–448. doi: 10.1093/nar/18.3.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang JY, Hwang BJ, Ford JM, Hanawalt PC, Chu G. Mol Cell. 2000;5(4):737–744. doi: 10.1016/s1097-2765(00)80252-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verhage R, Zeeman AM, de Groot N, Gleig F, Bang DD, van de Putte P, Brouwer J. Mol Cell Biol. 1994;14(9):6135–6142. doi: 10.1128/mcb.14.9.6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. 2. ASM Press; Washington D.C: 2006. [Google Scholar]
- 7.Park JS, Marr MT, Roberts JW. Cell. 2002;109(6):757–767. doi: 10.1016/s0092-8674(02)00769-9. [DOI] [PubMed] [Google Scholar]
- 8.Selby CP, Sancar A. Science. 1993;260(5104):53–58. doi: 10.1126/science.8465200. [DOI] [PubMed] [Google Scholar]
- 9.Lommel L, Hanawalt PC. Mutat Res. 1991;255(2):183–191. doi: 10.1016/0921-8777(91)90052-q. [DOI] [PubMed] [Google Scholar]
- 10.Troelstra C, van Gool A, de Wit J, Vermeulen W, Bootsma D, Hoeijmakers JH. Cell. 1992;71(6):939–953. doi: 10.1016/0092-8674(92)90390-x. [DOI] [PubMed] [Google Scholar]
- 11.van Hoffen A, Natarajan AT, Mayne LV, van Zeeland AA, Mullenders LH, Venema J. Nucleic Acids Res. 1993;21(25):5890–5895. doi: 10.1093/nar/21.25.5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Venema J, Mullenders LH, Natarajan AT, van Zeeland AA, Mayne LV. Proc Natl Acad Sci U S A. 1990;87(12):4707–4711. doi: 10.1073/pnas.87.12.4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Gool AJ, Verhage R, Swagemakers SM, van de Putte P, Brouwer J, Troelstra C, Bootsma D, Hoeijmakers JH. EMBO J. 1994;13(22):5361–5369. doi: 10.1002/j.1460-2075.1994.tb06871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li S, Smerdon MJ. J Biol Chem. 2004;279(14):14418–14426. doi: 10.1074/jbc.M312004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li S, Smerdon MJ. EMBO J. 2002;21(21):5921–5929. doi: 10.1093/emboj/cdf589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tijsterman M, Verhage RA, van de Putte P, Tasseron-de Jong JG, Brouwer J. Proc Natl Acad Sci U S A. 1997;94(15):8027–8032. doi: 10.1073/pnas.94.15.8027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhatia PK, Verhage RA, Brouwer J, Friedberg EC. J Bacteriol. 1996;178(20):5977–5988. doi: 10.1128/jb.178.20.5977-5988.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gregory SM, Sweder KS. Nucleic Acids Res. 2001;29(14):3080–3086. doi: 10.1093/nar/29.14.3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verhage RA, van Gool AJ, de Groot N, Hoeijmakers JH, van de Putte P, Brouwer J. Mol Cell Biol. 1996;16(2):496–502. doi: 10.1128/mcb.16.2.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones EW. Methods Enzymol. 1991;194:428–453. doi: 10.1016/0076-6879(91)94034-a. [DOI] [PubMed] [Google Scholar]
- 21.Yocum RR, Hanley S, West R, Jr, Ptashne M. Mol Cell Biol. 1984;4(10):1985–1998. doi: 10.1128/mcb.4.10.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sikorski RS, Hieter P. Genetics. 1989;122(1):19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li S, Waters R. Carcinogenesis. 1996;17(8):1549–1552. doi: 10.1093/carcin/17.8.1549. [DOI] [PubMed] [Google Scholar]
- 24.Li S, Waters R, Smerdon MJ. Methods. 2000;22(2):170–179. doi: 10.1006/meth.2000.1058. [DOI] [PubMed] [Google Scholar]
- 25.Collart MA, Oliviero S. Preparation of yeast RNA. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors. Current Protocols in Molecular Biology. John Wiley & Sons, Inc; New York: 2004. [Google Scholar]
- 26.Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2001. [Google Scholar]
- 27.Strahl-Bolsinger S, Hecht A, Luo K, Grunstein M. Genes Dev. 1997;11(1):83–93. doi: 10.1101/gad.11.1.83. [DOI] [PubMed] [Google Scholar]
- 28.Sugawara N, Wang X, Haber JE. Mol Cell. 2003;12(1):209–219. doi: 10.1016/s1097-2765(03)00269-7. [DOI] [PubMed] [Google Scholar]
- 29.Bash R, Lohr D. Prog Nucleic Acid Res Mol Biol. 2001;65:197–259. doi: 10.1016/s0079-6603(00)65006-7. [DOI] [PubMed] [Google Scholar]
- 30.Lohr D, Venkov P, Zlatanova J. FASEB J. 1995;9(9):777–787. doi: 10.1096/fasebj.9.9.7601342. [DOI] [PubMed] [Google Scholar]
- 31.Giniger E, Varnum SM, Ptashne M. Cell. 1985;40(4):767–774. doi: 10.1016/0092-8674(85)90336-8. [DOI] [PubMed] [Google Scholar]
- 32.Lloyd RS. Mutat Res. 2005;577(1–2):77–91. doi: 10.1016/j.mrfmmm.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 33.Thompson NE, Steinberg TH, Aronson DB, Burgess RR. J Biol Chem. 1989;264(19):11511–11520. [PubMed] [Google Scholar]
- 34.Bhaumik SR, Green MR. Genes Dev. 2001;15(15):1935–1945. doi: 10.1101/gad.911401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larschan E, Winston F. Genes Dev. 2001;15(15):1946–1956. doi: 10.1101/gad.911501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sterner DE, Grant PA, Roberts SM, Duggan LJ, Belotserkovskaya R, Pacella LA, Winston F, Workman JL, Berger SL. Mol Cell Biol. 1999;19(1):86–98. doi: 10.1128/mcb.19.1.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhaumik SR, Green MR. Mol Cell Biol. 2002;22(21):7365–7371. doi: 10.1128/MCB.22.21.7365-7371.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guzder SN, Habraken Y, Sung P, Prakash L, Prakash S. J Biol Chem. 1996;271(31):18314–18317. doi: 10.1074/jbc.271.31.18314. [DOI] [PubMed] [Google Scholar]
- 39.Selby CP, Sancar A. Proc Natl Acad Sci U S A. 1997;94(21):11205–11209. doi: 10.1073/pnas.94.21.11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eisen JA, Sweder KS, Hanawalt PC. Nucleic Acids Res. 1995;23(14):2715–2723. doi: 10.1093/nar/23.14.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Citterio E, Van Den Boom V, Schnitzler G, Kanaar R, Bonte E, Kingston RE, Hoeijmakers JH, Vermeulen W. Mol Cell Biol. 2000;20(20):7643–7653. doi: 10.1128/mcb.20.20.7643-7653.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zheng Y, Pao A, Adair GM, Tang M. J Biol Chem. 2001;276(20):16786–16796. doi: 10.1074/jbc.M010973200. [DOI] [PubMed] [Google Scholar]
- 43.Hwang BJ, Toering S, Francke U, Chu G. Mol Cell Biol. 1998;18(7):4391–4399. doi: 10.1128/mcb.18.7.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mellon I, Spivak G, Hanawalt PC. Cell. 1987;51(2):241–249. doi: 10.1016/0092-8674(87)90151-6. [DOI] [PubMed] [Google Scholar]