

Abstract
Although the recently identified Pirh2 protein is known as a p53-induced ubiquitin-protein E3 ligase, which negatively regulates p53, the detailed mechanism underlying the regulation of Pirh2 remains largely unknown. Here, we demonstrate that while Pirh2 is mostly detected in the phosphorylated form in normal tissues, it is predominantly present in the unphosphorylated form in majority of tumor cell lines and tissues examined. Phosphorylated Pirh2 is far more unstable than its unphosphorylated form. We further identified that Calmodulin-dependent kinase II (CaMK II) phosphorylates Pirh2 on residues Thr-154 and Ser-155. Phosphorylation of Pirh2 appears to be regulated through cell cycle-dependent mechanism. CaMK II-mediated Pirh2 phosphorylation abrogates its E3 ligase activity toward p53. Together, our data suggest that phosphorylation of Pirh2 may act as a fine-tuning to maintain the balance of p53-Pirh2 autoregulatory feedback loop, which facilitates the tight regulation of p53 stability and tumor suppression.
Keywords: CaMK II, p53, phosphorylation, Pirh2, ubiquitination
Introduction
The tumor suppressor p53 is known to exert its antiproliferative effects through induction of cell cycle arrest and apoptosis (Vogelstein et al, 2000; Vousden, 2000). p53 can mediate apoptosis by transcriptionally activating pro-apoptotic genes like bax, puma, noxa or reversibly, repressing antiapoptotic genes such as bcl-2 and survivin (Hoffman et al, 2002; Nakano and Vousden, 2001; Ryan et al, 2001; Wu et al, 2001). Besides its transcription-dependent functions, p53 can also promote apoptosis through transcription-independent mechanisms (Chipuk et al, 2003, 2004; Mihara et al, 2003). Tight regulation of p53 activity is crucial for maintaining normal cell growth and preventing tumorigenesis and occurs at multiple levels, including protein post-translational modification, stability and subcellular localization (Giaccia and Kastan, 1998). Maintaining stability of p53 is mainly through the ubiquitin–proteasome system. Several E3 ubiquitin ligases for p53 have been identified, including Mdm2, Pirh2 and COP1 (Corcoran et al, 2004; Dornan et al, 2004). Despite the well-established role of Mdm2, recent studies showed that Pirh2 is another critical negative regulator of p53 function. Pirh2 was initially cloned by Beitel et al (2002) as a novel androgen receptor N-terminal-interacting protein (ARNIP), and later, it was identified as an E3 ligase to promote p53 degradation (Leng et al, 2003). Pirh2 is itself a p53-inducible gene product, and this dependence creates an autoregulatory feedback loop in which both the activity of p53 and the expression of Pirh2 are tightly regulated. As a short-lived protein, Pirh2 is a target for RING domain-dependent proteasomal degradation. Binding of Pirh2 to either histone acetylase TIP60 or measles virus phosphoprotein (MV P protein) was reported to efficiently increase its stability by inhibiting polyubiquitination of Pirh2 (Logan et al, 2004; Chen et al, 2005). Collectively, the activities of Pirh2 are controlled at multiple levels, enabling its quick and accurate adjustment in protein level depending on specific needs.
Previous studies suggest that increased Pirh2 expression might affect lung tumorigenesis by reducing p53 activity (Duan et al, 2004), indicating that the disruption of interaction between these two proteins may be a potential target for anticancer strategies. In the case of Mdm2 and p53, post-translational modifications on either protein can profoundly affect their interactions, and subsequently, their functions (Alarcon-Vargas and Ronai, 2002; Meek and Knippschild, 2003; Moll and Petrenko, 2003). Compared with Mdm2, considerably less has been known for the post-translational modifications occurring on Pirh2. Here, we report that Calmodulin-dependent kinase II (CaMK II) is involved in catalyzing phosphorylation of Pirh2 at amino-acid residues Thr154 and Ser155. We also showed that the phosphorylation status of Pirh2 determines its E3 ligase activity toward p53. Majority of Pirh2 is phosphorylated in normal tissues, whereas in tumor tissues examined, Pirh2 is present predominantly in the unphosphorylated form. In addition, we determined that unphosphorylated, but not phosphorylated, Pirh2 was able to associate with p53 and mediate its degradation. Thus, our results define a coordinately controlled network involving CaMK II, Pirh2 and p53. CaMK II-involved phosphorylation of Pirh2 impairs its E3 ligase activity toward p53, leading to increase of p53 stability. This may explain why in most of tumor cells majority of Pirh2 has been found in their unphosphorylated form, resulting in a relatively low level of p53.
Results
Phosphorylation of Pirh2 at Ser/Thr was detected in tumor cell lines
Analysis of endogenous Pirh2 in a panel of tumor cell lines by immunoblotting often reveals two distinct forms of this protein (Figure 1A, top panel). Although the lower band was the dominant form, the ratio of the two forms varies among the examined cell lines; for example, the upper band could hardly be detected in MCF-7 cells (lane 1). To investigate whether the slower migrating isoform represents phosphorylated Pirh2, an immunoprecipitation experiment using phospho-specific antibodies was performed. As shown in Figure 1B, endogenous phosphorylated Pirh2 could be precipitated by anti-phosphoSer/Thr and anti-phosphoThr antibodies, but not by anti-phosphoTyr antibody (P-Tyr-100; Cell Signaling). These data suggest that Pirh2 undergoes phosphorylation at serine and/or threonine.
Figure 1.
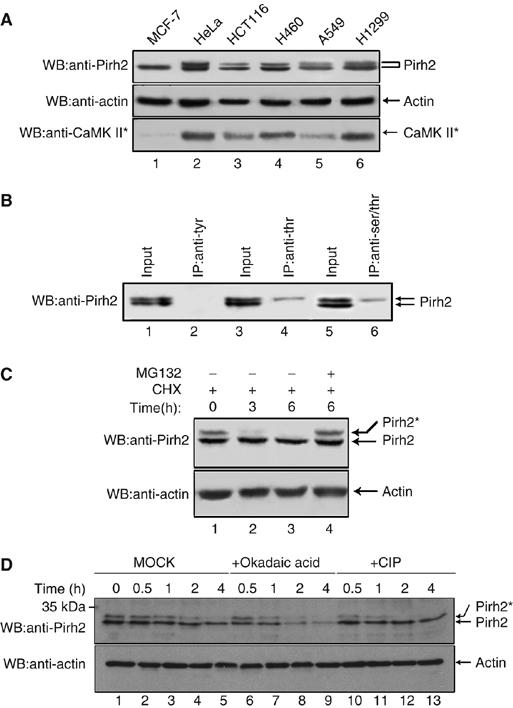
Pirh2 is subjected to phosphorylation in human cancer cells. (A) Western analysis of endogenous Pirh2 expression in the indicated cell lines (top panel). Beta-actin was used as loading control (middle panel). The level of active CaMK II (CaMK II*) was also determined by Western blotting (bottom panel). (B) HeLa cell lysates were immunoprecipitated with anti-phosphoTyrosine antibody (lane 2), anti-phosphoThreonine antibody (lane 4) or anti-phosphoSer/Thr antibody (lane 6), and the resulting immunocomplexes were analyzed by immunoblotting with anti-Pirh2 antibody. (C) A549 cells were treated with CHX (25 μg/ml) in the absence of MG132 (lanes 1–3). Cells were incubated for 0, 3 and 6 h before cell lysates were prepared. A549 cells were treated with CHX for 6 h in the presence of MG132 (20 μM) (lane 4). Levels of phosphorylated Pirh2 were assessed by Western blot analysis using anti-Pirh2 antibody. Beta-actin was used as loading control (bottom panel). (D) A549 cell lysates were either mock treated, or incubated with 10 U CIP (New England Biolabs Inc.), or the protein phosphatases inhibitor Okadaic acid (20 μM), at 37°C for the indicated times, before immunoblot analysis. ‘*' indicates phosphorylation throughout this text, unless notification is given.
Pirh2 has been reported as a short-lived protein (Logan et al, 2004) and is targeted to proteasomal degradation. To examine whether phosphorylation of Pirh2 affects its stability, A549 cells were treated with cycloheximide (CHX) to inhibit de novo protein synthesis. Cells were collected at the indicated times and equal amounts of cell extract were analyzed by Western blotting. As shown in Figure 1C, level of phosphorylated Pirh2 quickly decreased after CHX treatment (lanes 1–3); however, this reduction could be rescued by the proteasome inhibitor MG132 (lane 4). Comparing with phosphorylated Pirh2, levels of unphosphorylated Pirh2 remain more or less unchanged, suggesting that phosphorylated Pirh2 is less stable than its unphosphorylated form.
To further confirm that Pirh2 undergoes phosphorylation modification, which in turn affects its stability, A549 cell lysates were either mock treated, or incubated with calf intestine phosphatase (CIP) or the protein phosphatase inhibitor Okadaic acid for the indicated times. As shown in Figure 1D, the slower migrated band of Pirh2 quickly disappeared upon CIP treatment (lanes 10–13), compared with mock treatment (lanes 2–5), indicating that phosphorylation of Pirh2 did occur. Consistently, incubation with the protein phosphatase inhibitor Okadaic acid which suppresses the dephosphorylation of Pirh2, led to shortened half-life of Pirh2 (lanes 6–9), further supporting the conclusion that phosphorylation of Pirh2 affects its stability.
Pirh2 interacts with Calmodulin in vitro and in vivo
In an effort to identify novel Pirh2-interacting proteins, we utilized a yeast two-hybrid system. Full-length Pirh2 fused in frame with the GAL4-BD proved unsuitable because of self-activation of the bait protein. We therefore used the N-terminal region of Pirh2 (aa 1–186) as bait. About one-third of the positive prey clones (13 out of 42) were identified to encode human Calmodulin (CaM). The interaction between Pirh2 and CaM in mammalian cells was verified by immunoprecipitation experiment. As shown in Figure 2Aa, endogenous Pirh2 was detected in anti-GFP immunoprecipitates of cells expressing GFP-CaM (lane 4, top panel) but not GFP mock (lane 2, top panel). A reciprocal immunoprecipitation/blotting experiment was also performed and this time anti-Pirh2 and irrelevant anti-IgG antibody (negative control) were used. As shown in Figure 2Ab, Pirh2 is able to co-precipitate GFP-CaM (lane 4). These data indisputably demonstrated that Pirh2 interacts with CaM. Their binding sites have been mapped to N-terminus aa 1–94 for Pirh2 and N-terminus aa 1–58 for CaM (Supplementary Figure S1).
Figure 2.
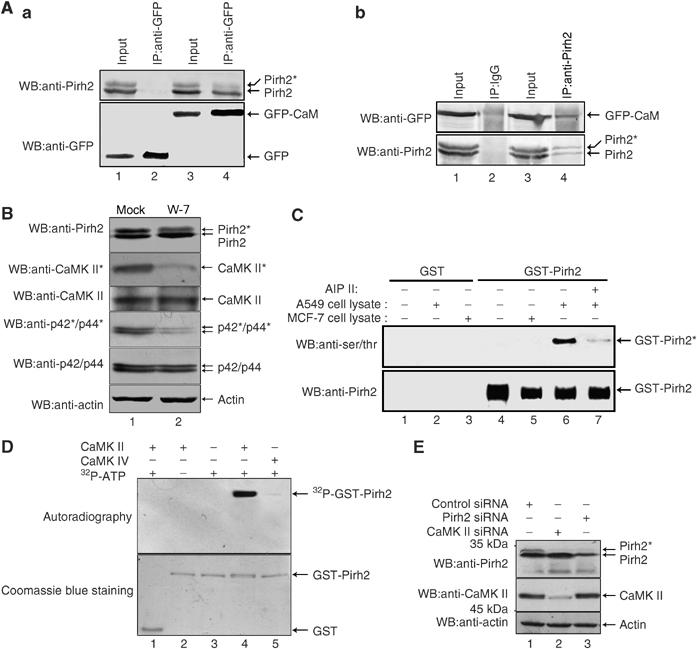
Identification of CaMK II as a kinase for Pirh2. (A) (a) HeLa cells transfected with pEGFP-CaM or the control vector were immunoprecipitated with anti-GFP antibody and the resulting immunocomplexes were analyzed by immunoblotting with both anti-Pirh2 and anti-GFP antibodies. (b) HeLa cells were transfected with pEGFP-CaM. The cell lysates were incubated with either irrelevant anti-rabbit IgG or anti-Pirh2, and the resulting immunoprecipitates were analyzed by Western blotting with anti-GFP. The blots were stripped and reprobed with anti-Pirh2 (bottom panel). (B) A549 cells were treated with either DMSO carrier or W-7 (30 μM) for 6 h, and then harvested and analyzed by immunoblotting using the indicated antibodies. CaMK II* indicates active CaMK II. p44/p42 was used as a known substrate to examine the inhibition of CaMK II with W-7. Beta-actin was used as loading control. (C) Bacterially expressed GST (lanes 1–3) or GST-Pirh2 (lanes 4–7) were bound to glutathione–Sepharose and incubated with cell lysate prepared from A549 or MCF-7 cells supplemented with ATP. The CaMK II-specific inhibitor AIP II was added as indicated (lane 7). Bead-bound proteins were washed and analyzed by Western blotting using anti-phosphoSerine/Threonine, and anti-Pirh2 antibodies. (D) GST alone (lane 1) or GST-Pirh2 (lanes 2–5) was incubated with purified active CaMK II or CaMK IV. At the end of kinase assay, samples were separated on 12% SDS–PAGE gel and analyzed by autoradiography (upper panel). The same gel was stained with Coomassie blue to show equal loading (lower panel). (E) 293T cells were transfected with control siRNA (lane 1), CaMK II-specific siRNA (lane 2) or Pirh2-specific siRNA (lane 3). Whole-cell lysates were analyzed by immunoblotting with anti-Pirh2 and anti-CaMK II antibodies. Beta-actin was used as a loading control.
Pirh2 is a substrate of CaMK II
Treatment of cells with the CaM antagonist W-7 markedly diminished Pirh2 phosphorylation, suggesting the involvement of CaM-dependent kinase activities (Figure 2B, top panel). Since Pirh2 is only sensitive to CaMK II inhibitor AIP II, but not other kinase inhibitors such as the PKC inhibitor Bisindolylmaleimide, the MEK inhibitor U0126, the ERK inhibitor PD 98059 and the PI3K/Akt inhibitor LY 294002 (data not shown), we therefore hypothesize that CaMK II may serve as a potential candidate kinase for Pirh2. To verify this hypothesis, an in vitro kinase assay was performed. A549 and MCF-7 cells, respectively, were pretreated with or without AIP II, a highly specific and cell permeable inhibitor of CaMK II. Equal amounts of each cell lysate were prepared and further incubated with bacterially purified glutathione-S-transferase (GST) or GST-Pirh2. As shown in Figure 2C, incubation with A549 cell lysate leads to phosphorylation of GST-Pirh2 (lane 6), whereas treatment of A549 cells with AIP II abrogates this phosphorylation activity (lane 7). Cell lysate from MCF-7 cells was unable to phosphorylate Pirh2 in vitro (lane 5), since CaMK II activity was found to be extremely low in this cell line (Figure 1A, bottom line). We next performed in vitro kinase assay using purified active CaMK II and CaMK IV (negative control) as enzymes and recombinant Pirh2 purified from bacteria as substrate. CaMK II, but not CaMK IV, was found to efficiently phosphorylate the GST-Pirh2 protein (Figure 2D, lane 4 versus 5). To exclude the possibility that both bands of the Pirh2 doublets shown in immunoblots of Figures 1 and 2 show nonspecific cross-reactivity from the antibody, we use RNA interference to knock down the expression of Pirh2. As shown in Figure 2E top panel, lane 1 versus 3, the intensity of both bands of the doublet was markedly reduced, indicating the doublet was bona fide Pirh2. To further confirm that the Pirh2 upper band results from the phosphorylation of endogenous CaMK II, CaMK II expression was knocked down by the application of small interfering RNA (siRNA) (middle panel, lane 2). Knock down of endogenous CaMK II led to non-appearance of the upper band representing phosphorylated Pirh2 (top panel, lane 2 versus 1). These data strongly suggest that phosphorylation of Pirh2 is specifically mediated by CaMK II.
Phosphorylation of Pirh2 is in a cell cycle-dependent manner
CaMK II was reported to play an important role in the signal transduction during cell cycle (Patel et al, 1999). We then inquired whether phosphorylation of Pirh2 would be subjected to cell cycle regulation. A549 cells were arrested at G1/S or G2/M transition of the cell cycle, by treating the cells for 24 h with hydroxyurea (Figure 3Aa, lane 2) or nocodazole (lane 3). FACS analysis confirmed that both treatments resulted in cell cycle arrest. Analysis of the Pirh2 phosphorylation status showed that cells arrested at G1 phase with hydroxyurea predominantly contained unphosphorylated Pirh2 (lane 2). In contrast, in cells arrested at G2/M by nocodazole, concurrently with the maximal activities of CaMK II in G2/M phase, Pirh2 was excessively phosphorylated (lane 3).
Figure 3.
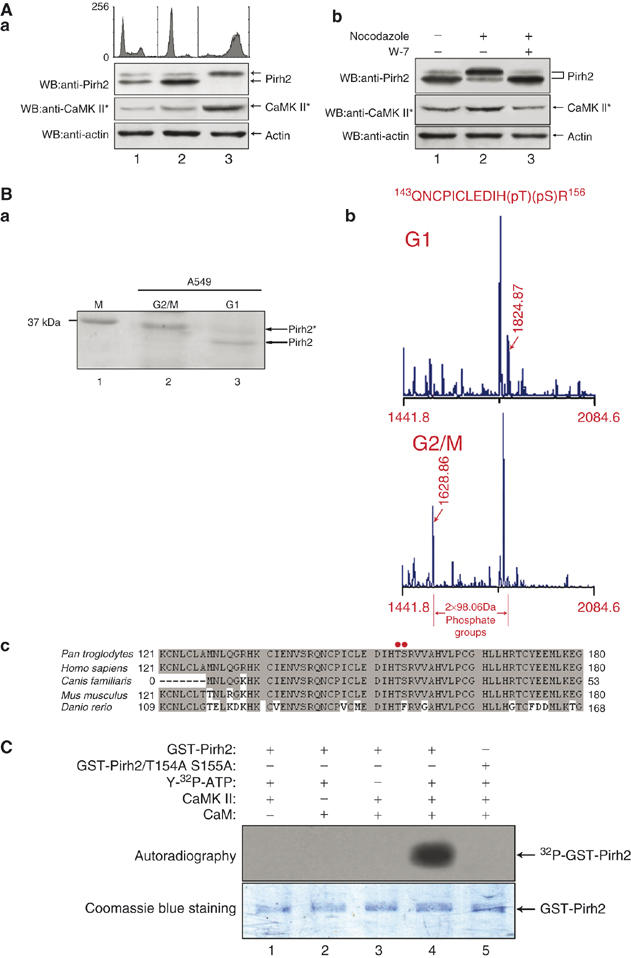
Pirh2 is phosphorylated by CaMK II on Thr154 and Ser155 residues in a cell cycle-dependent manner. (A) (a) A549 cells were left untreated (lane 1) or treated with hydroxyurea (lane 2) or nocodazole (lane 3) individually for 24 h. Cells were harvested and divided into two portions for analysis by flow cytometry and Western blotting, respectively. The cell cycle distributions of the cells were analyzed using a flow cytometer, after staining with propidium iodide (upper panel). The phosphorylation status of Pirh2 was measured by Western blotting. The kinase activity of CaMK II was tested using an anti-active CaMK II antibody. (b) HeLa cells were either untreated (lane 1) or treated with nocodazole, in the absence (lane 2) or presence of W-7 (lane 3), for 24 h. Levels of phosphorylated Pirh2 and active CaMK II were analyzed by Western blotting. (B) (a) A549 cells were either arrested at G2/M phase by nocodazole treatment or arrested at G1 phase by serum deprivation. Cell lysates were prepared for immunoprecipitation with anti-Pirh2 antibody. Proteins were separated by SDS–PAGE and stained with Coomassie blue. Two gel bands representing different forms of Pirh2 were excised, purified and subjected to mass spectrometric analysis. (b) Data from the MALDI-MS analysis. The location of the phosphorylation sites were determined by comparing the two sets of data. Arrows indicate a pair of peaks with a separation of 2 × 98 Da, which represents two phosphate groups. (c) Alignment of amino-acid sequences of Pirh2 among the five species. Gray shading indicates identical residues, and the red dots indicate the conserved phosphorylation sites in Pirh2. (C) Escherichia coli-derived, GST-wt-Pirh2, or GST-Pirh2/T154AS155A fusions were subjected to in vitro phosphorylation reactions with the indicated components of the kinase assay. The samples were separated on 12% SDS–PAGE and analyzed by autoradiography (upper panel). The same gel was stained with Coomassie blue to show equal loading (lower panel).
To confirm the preferentially phosphorylation of Pirh2 at the G2/M transition was due to enhanced CaMK II activity, cells were treated with nocodazole in the presence or absence of the CaM antagonist W-7. As shown in Figure 3Ab, phosphorylation of Pirh2 was much weakly detected in W-7-treated cells (lane 3), compared with non-treated cells (lane 2). Together, these data strongly suggest that the phosphorylation status of Pirh2 displays in a cell cycle-dependent manner and is in accordance with CaMK II activity.
Pirh2 is phosphorylated by CaMK II at T154S155
To identify the potential phosphorylation residues on Pirh2, we employed mass spectrometry analysis. Pirh2 was immunoprecipitated from serum-starved (G1 arrest) and nocodazole-treated (G2/M arrest) A549 cells separately, and subjected to MS/MS peptide sequence analysis (Figure 3Ba). The resulting sequence data revealed one phospho-peptide corresponding to amino-acid residues 143–156, which contains two phosphate groups (Thr154-P and Ser155-P) (Figure 3Bb). Noticeably, these two phosphorylation sites in Pirh2 were highly conserved among mammals (Figure 3Bc), implying phosphorylation on T154 and S155 may be critical in regulating Pirh2 activity.
To determine whether T154S155 is a bona fide target for CaMK II, an in vitro kinase assay was performed. As shown in Figure 3C, CaMK II effectively catalyzed phosphorylation of GST-wt-Pirh2 (lane 4), but not GST-Pirh2/T154AS155A (lane 5), a double mutant in which T154 and S155 were replaced by alanine (A), suggesting CaMK II-mediated phosphorylation occurs exclusively on T154 and S155.
Phosphorylation of Pirh2 at T154S155 by CaMK II enhances its self-ubiquitination
As mentioned above, phosphorylated Pirh2 is less stable. Therefore, it is reasonable to ask: what is the role of the individual sites of phosphorylation of Pirh2 on the stability of itself? The stability of wild-type Pirh2, single mutant Thr154 Ala, Ser155 Ala and double mutant Thr154 Ala/Ser155 Ala were compared and their stabilities, in decreasing order, were as follows: FLAG-Pirh2 T154A S155A>FLAG-Pirh2 T154A, FLAG-Pirh2 S155A>wild-type FLAG-Pirh2 (Figure 4Aa). Wild-type Pirh2 (wt-Pirh2) has a half-life of about 3.5 h (Figure 4Ab), which is consistent with those previously reported (Logan et al, 2004). We further determined that increased stability of Pirh2/T154AS155A resulted from the decrease in polyubiquitination (Figure 4Ba, lane 2 versus lane 3). To demonstrate that phosphorylation of Pirh2 alters its self-ubiquitination more directly, both Pirh2 and Pirh2/T154AS155A were phosphorylated in vitro by CaMK II, or left unphosphorylated, and the resultant products were used in a subsequent in vitro ubiquitination assay. Ubiquitination of Pirh2 appeared to be dependent on phosphorylation at T154S155, as shown in Figure 4Bb, phosphorylated Pirh2 displayed a more significant increase in self-ubiquitination than the unphosphorylated form (lane 2 versus 3), whereas no such effect was observed with the mutant GST-Pirh2/T154AS155A (lane 4 and 5). To further substantiate that CaMK II-mediated phosphorylation of Pirh2 affects its self-ubiquitination, siRNA against CaMK II was employed. As shown in Figure 4Bc, self-ubiquitination of Pirh2 in cells treated with CaMK II-specific siRNA was markedly reduced (top two panels, lane 2 versus 3).
Figure 4.
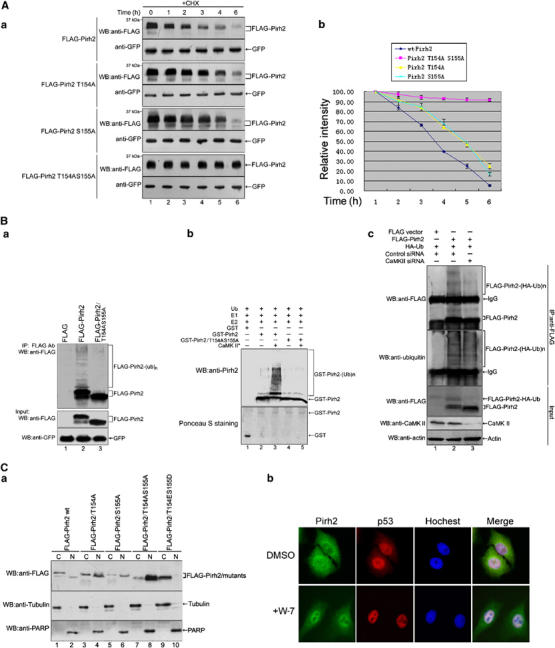
Phosphorylation of Pirh2 at T154S155 affects its stability and subcellular localization. (A) H1299 cells were transiently transfected with the plasmids expressing FLAG-tagged Pirh2 or its mutant Pirh2 proteins, as indicated. The pEGFP-C1 vector was cotransfected as an internal control. Twenty-four hours after transfection, cells were treated with MG132 for 3 h, washed and then cultured in fresh medium with CHX for the indicated periods of time. (a) Protein levels were analyzed by Western blot analysis using anti-FLAG and anti-GFP antibodies. (b) The intensities of bands indicated by Western blot were semi-quantified by densitometry and represented as a line graph. (B) (a) H1299 cells were cotransfected with pCW7-myc-ubiquitin, and either p3XFLAG-wt-Pirh2, p3XFLAG-Pirh2/T154AS155A or p3XFLAG (empty vector), as indicated. The pEGFPC1 vector was also transfected as a transfection efficiency control. Ubiquitinated Pirh2 was detected by Western blotting after purification with anti-FLAG M2-agarose beads (upper panel). The middle and lower panels show equal expression of the transfected proteins. (b) Bacterially expressed GST, GST-wt-Pirh2 and GST-Pirh2/T154AS155A were treated either with (lanes 3 and 5) or without (lanes 1, 2 and 4) CaMK II in vitro, before being subjected to an in vitro ubiquitination assay. The degree of Pirh2 ubiquitination was detected by immunoblotting using anti-Pirh2 antibody (upper panel). Equal amount of added GST-Pirh2 and GST-Pirh2/T154AS155A was detected by Ponceau S staining (lower panel). (c) H1299 cells transfected with control siRNA (lanes 1 and 2) or CaMK II siRNA (lane 3) for 30 h were cotransfected with the plasmids, as indicated. Twelve hours after transfection, cells were treated with MG132 for 4 h and cell lysates were immunoprecipitated with anti-FLAG antibody. High-molecular-weight ubiquitinated Pirh2 was detected with anti-FLAG antibody (upper panel) and anti-ubiquitin antibody (second panel from the top). A total of 10% of total cell lysate (input) was analyzed to examine the protein levels of FLAG-tagged Pirh2 and endogenous CaMK II in each transfection. (C) (a) H1299 cells were transfected with the plasmids expressing FLAG-tagged Pirh2 or its mutant Pirh2 proteins as indicated. Twenty-four hours after transfection, cells were harvested, and cytoplasmic (C) and nuclear (N) fractions were prepared and analyzed by immunoblotting using indicated antibodies. Tubulin (a cytoplasmic-specific protein) and nuclear protein PARP were examined as cell fraction markers as well as loading controls. (b) A549 cells were treated with DMSO or W-7 (30 μM) for 12 h. Intracellular localizations of endogenous Pirh2 (green fluorescence) and p53 (red) were analyzed by indirect immunofluorescence with anti-Pirh2 and anti-p53 antibodies, respectively. Nuclei were visualized by staining with Hoechst 33342 (blue).
Phosphorylation of Pirh2 regulates its subcelluar localization
Next, we sought to investigate whether the phosphorylation of Pirh2 affects its subcelluar localization. Levels and phosphorylation status of wt-Pirh2, Pirh2/T154A, Pirh2/S154A, Pirh2/T154AS155A and a phospho-mimic mutant Pirh2/T154ES155D from cytosolic and nuclear fractions were compared. As shown in Figure 4Ca, blocking Pirh2 phosphorylation at Thr154 and Ser155 singly or in combination increased the nuclear/cytosolic Pirh2 ratio (lane 2 versus 1; 4 versus 3; 6 versus 5 and 8 versus 7); conversely, Pirh2/T154ES155D was found to be present predominantly in the cytoplasmic fraction (lane 9 versus 10), suggesting phosphorylated Pirh2 prefer to remain outside the nucleus. Consistently, inhibition of endogenous Pirh2 phosphorylation by treatment with the CaM antagonist W-7 resulted in significant accumulation of Pirh2 in the nucleus where it colocalized with p53 (Figure 4Cb).
The Pirh2–p53 interaction is affected by Pirh2 phosphorylation
The above results strongly indicate that the T154 and S155 phosphorylation is crucial for stability and localization of Pirh2, and moreover, Pirh2 has been reported to be an E3 ligase towards p53. We therefore examined whether phosphorylation of Pirh2 interferes with its association with p53. Co-immunoprecipitation experiments showed that the association between p53 and Pirh2/T154ES155D was much reduced (Figure 5Aa, lane 6 versus 8). Consistently, nullifying the phosphorylation of Pirh2 on both T154 and S155 increases the association between p53 and Pirh2/T154AS155A (lane 7 versus 8). Similar results were obtained by in vitro GST pull-down assay using purified proteins (Figure 5Ab, lane 3 or 4 versus 2).
Figure 5.
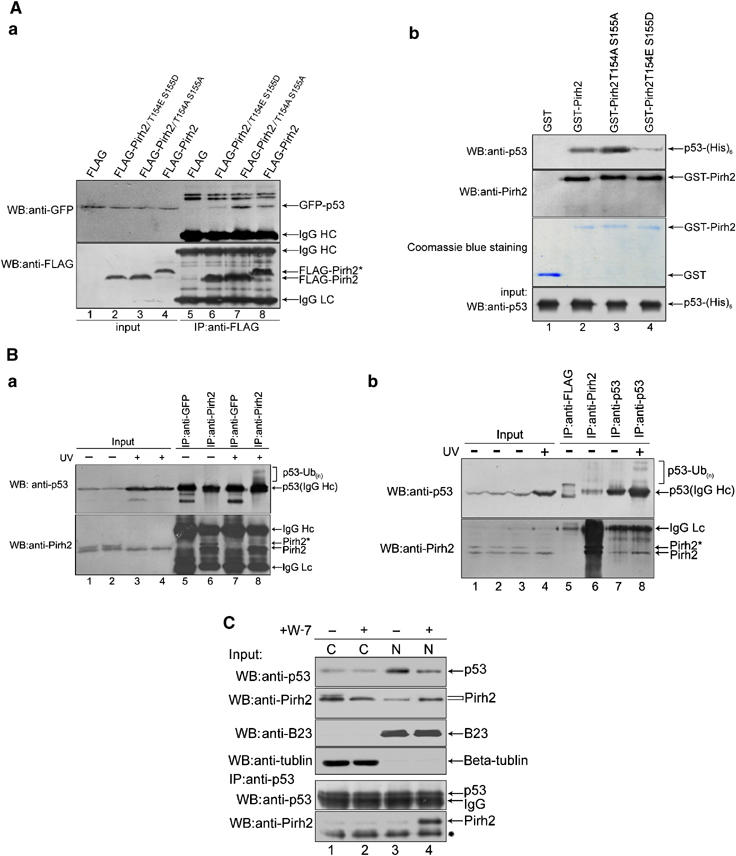
Phosphorylation of Pirh2 interferes with its association with p53. (A) (a) H1299 cells expressing GFP-p53 were transfected with plasmids p3XFLAG, p3XFLAG-wt-Pirh2, p3XFLAG-Pirh2/T154AS155A or p3XFLAG-Pirh2/T154ES155D, as indicated. Equal amounts of cell extract were immunoprecipitated with anti-FLAG antibody. The amounts of co-precipitated p53 and Pirh2 were evaluated by immunoblotting with anti-GFP and anti-FLAG antibody, respectively (lanes 5–8). Input was also analyzed to ensure equal expression of p53 and Pirh2 in each sample (lanes 1–4). (b) GST-Pirh2, GST-Pirh2/T154AS155A and GST-Pirh2/T154ES155D fusions were incubated separately with His-tagged p53 protein. The amount of p53 that physically interacted with wild-type or mutant Pirh2 was evaluated by immunoblotting of the glutathione–agarose complexes, using anti-p53 antibody (top panel). GST-Pirh2 wild-type or mutant fusion proteins were visualized by immunoblotting with anti-Pirh2 antibody (second panel from the top) or Coomassie blue staining (third panel from the top). GST alone was used as a negative control (lane 1). Equal amounts of p53 used for the GST pull-down assays were confirmed by immunoblotting (bottom panel). (B) (a) A549 cells were either left untreated (lanes 1 and 2) or exposed to UV irradiation (2 J/m2, lanes 3 and 4) and cell lysates were immunoprecipitated with anti-Pirh2 antibody (rabbit polyclonal antibody, lanes 6 and 8). The anti-GFP antibody (rabbit polyclonal antibody, lanes 5 and 7) was used as a negative anti-IgG control. p53-Ub(n) indicates ubiquitinated p53. (b) A549 cells were either left untreated (lanes 1–3) or exposed to UV irradiation (2 J/m2, lane 4), and cell lysates were immunoprecipitated with anti-p53 antibody (mouse monoclonal antibody, lanes 7 and 8) or anti-Pirh2 antibody (lane 6). The anti-FLAG antibody (mouse monoclonal, lane 5) was used as a negative anti-IgG control. (C) Cytoplasmic (C) and nuclear (N) extracts were prepared from A549 cells treated with DMSO and W-7 (30 μM), separately for 12 h. The precipitated p53 derived from each fraction was adjusted to equal amounts (second panel from bottom) before immunoblotting with antibodies against Pirh2 and p53, individually. B23 and beta-tubulin served as markers for the purity of the nuclear and cytoplasmic fractions. ‘*' in the bottom panel denotes an unspecific band.
To determine endogenously whether the binding of Pirh2 to p53 is regulated by phosphorylation of Pirh2, immunoprecipitation experiments in the presence or absence of UV irradiation were performed. Although the phosphorylation of Pirh2 was reduced upon UV irradiation (Figure 5Ba, lane 4 versus 2), the level of p53 in the anti-Pirh2 immunoprecipitates was nonetheless increased (Figure 5Ba, lane 8 versus 6), indicating that p53 interacts with unphosphorylated but not phosphorylated Pirh2. Interestingly, after UV irradiation, increased ubiquitination of p53 in co-precipitated Pirh2 was observed (lane 8 versus 6), suggesting that p53 may be ubiquitinated by unphosphorylated Pirh2 during exposure to UV radiation. Similarly, results from a reciprocal immunoprecipitation revealed that p53 associates with unphosphorylated Pirh2, but not with its phosphorylated form (Figure 5Bb, bottom panel, lanes 7, 8 versus 6). It is worthwhile to emphasize that the diminishment of phosphorylation of Pirh2 from UV irradiation is likely caused by UV-induced G1 arrest.
As mentioned above, unphosphorylated Pirh2 was mainly localized in the nucleus (Figure 4Ca and b); we therefore asked whether unphosphorylated Pirh2 interacts with p53 in the nucleus. As shown in Figure 5C, much greater binding of Pirh2 to p53 was observed in the nuclear fraction of W-7-treated cells (bottom panel, lane 4). It is interesting to note that the level of p53 was decreased in the nuclear fraction after W-7 treatment (upper panel, lane 4 versus 3). One reasonable explanation is that W-7 treatment resulted in Pirh2 accumulation in the nucleus; thereby the level of p53 was decrease through Pirh2-mediated degradation.
Phosphorylation of Pirh2 impairs its E3 ligase activity toward p53
Since a physical interaction between Pirh2 and its target p53 is essential for proteasome-mediated protein degradation, we therefore tested whether phosphorylation of Pirh2 reduces polyubiquitination of p53. First, an in vitro ubiquitination assay was performed. As shown in Figure 6A, addition of active CaMK II notably reduced the ubiquitination of p53 by wt-Pirh2, but not by its mutant counterpart T154AS155A (lanes 4, 5 versus 2, 3). To further confirm that Pirh2-mediated ubiquitination of p53 was inhibited by CaMK II-mediated phosphorylation of Pirh2, endogenous CaMK II was knocked down by RNA interference. As shown in Figure 6B, polyubiquitinated p53 was much increased in cells treated with siRNA against CaMK II (top two panels, lane 3 versus 2). Levels of immunoprecipitated p53 from three transfections were comparable (upper panel). Moderate ubiquitination of p53 in the absence of FLAG-Pirh2 was also detected, which could be resulted from endogenous yet-unknown E3 ligases activity.
Figure 6.
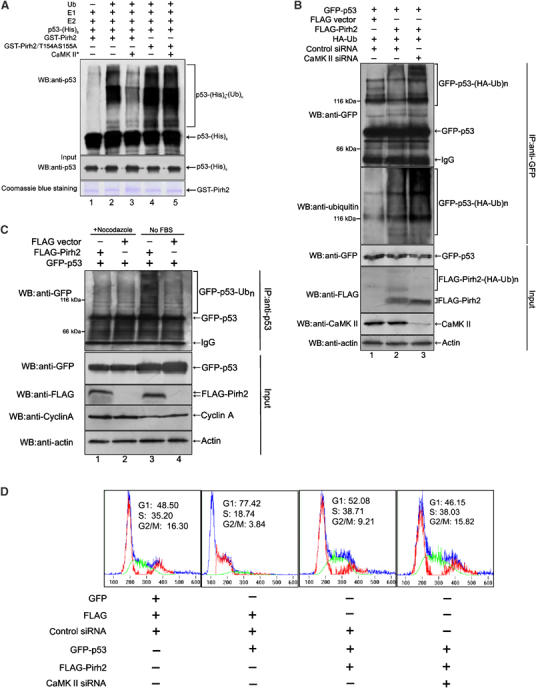
Effect of phosphorylation on Pirh2 E3 ligase activity toward p53. (A) GST-tagged wt-Pirh2 and mutant Pirh2/T154AS155A were first subjected to in vitro kinase assay in the presence or absence of active CaMK II. The resultant products were then evaluated for their E3 activities toward His-tagged p53, via in vitro ubiquitination assay. Ubiquitinated p53 were analyzed by immunoblotting using anti-p53 antibody (upper panel). Equal amount of purified His-p53 (middle panel), or GST-wt-Pirh2 and Pirh2/T154AS155A (bottom panel), used in each reaction, was confirmed by Western blotting and Coomassie blue staining, respectively. (B) H1299 cells treated with control siRNA (lanes 1 and 2) or CaMK II siRNA (lane 3) for 30 h were further transfected with the indicated plasmids. Twelve hours after transfection, cells were treated with MG132 for 4 h and ubiquitinated p53 was detected by immunoprecipitation with anti-GFP antibody under denaturing conditions and immunoblotting with anti-GFP antibody (upper panel) and anti-Ubiquitin antibody (second panel from the top), respectively. GFP-p53-(HA-Ub)n indicates polyubiquitinated GFP-p53. A total of 10% of total cell lysate (input) was analyzed by immunoblotting, to examine the indicated protein levels in each sample. (C) H1299 cells were cotransfected with pEGFP-p53, and either p3XFLAG-Pirh2 or the empty vector. Cells were treated with nocodazole (lanes 1 and 2) or cultured in medium deprived of serum (no FBS) (lanes 3 and 4). Cell lysates prepared from differently treated cells were subjected to immunoprecipitation with anti-p53 antibody. Ubiquitinated p53 was detected by immunoblotting with anti-GFP antibody (top panel). A total of 10% of total cell lysate (input) was analyzed for successful expression of transfected plasmids, by using the indicated antibodies. Upregulation of cyclin A was detected as a marker of G2/M phase arrest caused by nocodazole treatment. (D) H1299 cells treated with control siRNA or CaMK II-specific siRNA were transfected with plasmids in various combinations, as indicated. DNA content and cell cycle analysis (1 × 104 cells) were measured by FACS after PI staining, and the percentage of cells in each phase of cell cycle is labeled in the figure.
It is interesting to know whether the cell cycle-regulated phosphorylation of Pirh2 (Figure 3) correlates with its E3 ligase activity toward p53. To address this question, H1299 cells cotransfected with plasmids pEGFPC1-p53 and p3xFLAG-Pirh2 were either treated with nocodazole (to induce G2/M arrest) or cultured in serum-free medium (to cause G1 arrest). As shown in Figure 6C, comparable ubiquitinations of p53 were detected in cells both with and without Pirh2 overexpression during G2/M phase (lane 1 versus 2). However, expression of Pirh2 led to an increased ubiquitination of p53 in G1 phase (lane 3 versus 4), suggesting that Pirh2-mediated p53 degradation may occur in G1, other than G2 phase.
Since p53 is a crucial regulator of cell cycle progression, we next examined whether the inhibition of endogenous CaMK II affects p53-induced G1 arrest. As shown in Figure 6D, expression of p53 in H1299 cells (p53−/−) blocked the cell cycle progression and arrested cells in G1 phase (left-second panel). Coexpression of Pirh2 and p53 partly led to an alleviation of p53-induced G1 arrest (right-second panel). Moreover, when siRNA against CaMK II was additionally introduced into cells coexpressing Pirh2 and p53, p53-mediated G1 arrest was significantly derepressed (right panel). Together, these data indicated that the inhibition of endogenous CaMK II affects p53 activity involving the regulation of p53-mediated checkpoint control.
Next, we investigated the effect of phosphorylation of Pirh2 on the p53-dependent transactivation. As shown in Supplementary Figure S2, in either PG-13-luciferase or p21-luciferase reporter assay, Pirh2/T154AS155A inhibits the transcriptional activity of p53 more efficiently than wt-Pirh2 (right and left panels, column 3 versus 2), conversely, phospho-mimic mutant Pirh2/T154ES155D leads to less inhibition than in the wt-Pirh2 (right and left panels, column 4 versus 2), indicating that Pirh2 represses p53-dependent transactivation and this repression is regulated by phosphorylation of Pirh2.
Phosphorylation of Pirh2 at T154S155 inhibits tumorigenicity in vivo
Western blot analysis of Pirh2 in several types of human carcinomas and non-cancerous surrounding tissues showed that unphosphorylated form of Pirh2 was predominant in the cancerous tissues, whereas phosphorylated Pirh2 was present predominantly in all four non-cancerous surrounding tissues examined (Figure 7A, upper panel). Phosphorylation/unphosphorylation status of Pirh2 is consistent with CaMK II kinase activity in these tissues (bottom panel).
Figure 7.
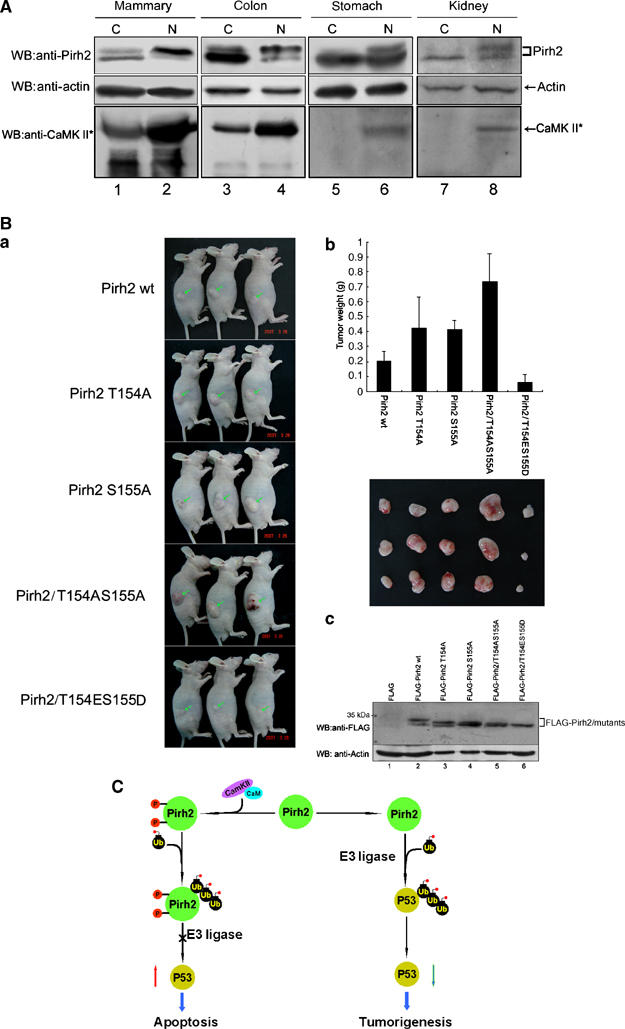
Phosphorylation of Pirh2 at Thr154Ser155 impairs tumorigenicity in vivo. (A) Protein extracts were prepared from four indicated human cancer tissues (C) and the non-cancerous surrounding tissues (N). Total cellular proteins were separated by SDS–PAGE and immunoblotted with anti-Pirh2 and anti-CaMK II (active) antibodies. ‘*' denotes active CaMK II. Beta-actin was used as a loading control. (B) NIH3T3 cells stably expressing wt-Pirh2 or various Pirh2 mutant proteins, as indicated, were injected subcutaneously into athymic nude mice. (a) Representative pictures of tumor bearing nude mice. Photographs were taken 5 weeks after inoculation. Inoculated tumors are pointed with arrows. (b) Histograms show data points representing average tumor weights in each group. The data were represented by the mean values. The volume of tumors excised from the mice was compared among five groups (n=3). (c) Tumor samples recovered 5 weeks after inoculation were analyzed by Western blotting to determine the expression of Pirh2 and various Pirh2 mutant proteins. Actin was examined as a loading control. (C) A diagram illustrating the potential role of CaMK II-dependent phosphorylation of Pirh2 in modulating its E3 ligase activity toward p53.
To investigate the impact of Pirh2 phosphorylation on tumorigenesis in vivo, NIH3T3 cells stably expressing wt-Pirh2 or mutant Pirh2 were injected into nude mice. As shown in Figure 7Ba and b, expression of Pirh2/T154AS155A in NIH3T3 cells developed rapidly growing tumors; whereas cells expressing phospho-mimic mutant Pirh2/T154DS155E only formed relatively small tumors. All tumors examined in this study expressed similar level of the transfected proteins, as indicated by Western blot analysis (Figure 7Bc). These results strongly suggest that Pirh2 could promote tumorigenesis, and this ability is compromised by the phosphorylation modification on Thr154 and Ser155.
Discussion
The data presented here demonstrate a link between CaMK II-dependent phosphorylation of Pirh2 and the regulation of p53 activity, and suggest that the phosphorylation status of Pirh2 may be used as a molecular indicator of tumorigenesis.
Mass spectrometry analysis of Pirh2 revealed that it is a phosphoprotein. Using a yeast two-hybrid system, we have identified CaM as a novel Pirh2 binding partner. CaM is a ubiquitous intracellular second messenger, known to activate calcium/CaM-dependent protein kinases. Our experiments showed that Pirh2 is a direct substrate of CaMK II, a serine/threonine kinase that has been shown to be involved in a number of cellular activities (Yang et al, 2003; Yadav et al, 2004; Nutt et al, 2005). Consistently, the level of phosphorylated Pirh2 detected in different cell lines and tissues matches remarkably well with the CaMK II activity, further indicating CaMK II regulates Pirh2. These findings, together with the previous report that CaMK II was downregulated in human tumor cells (Tombes et al, 1999), might explain why in most malignant tissues examined, majority of Pirh2 is in its less phosphorylated form.
We further identified two functionally relevant CaMK II phosphorylation sites Thr154 and Ser155 located in the RING domain of Pirh2. It is noteworthy that these sites are well conserved among mammals. Phosphorylation of these sites leads to accelerated degradation of Pirh2 through yet-unknown mechanisms. One possible explanation is that phosphorylation at these sites may cause conformational change that makes Pirh2 more susceptible to degradation. Inhibition of Pirh2 phosphorylation alters its localization from cytosol to nucleus, where majority of p53 is located. This coincidence raises the possibility that unphosphorylated Pirh2 may be associated with p53 in the nucleus, and provides new insight into the mechanism of how Pirh2-mediated ubiquitination of p53 is regulated spatially and temporally in response to cellular signals such as UV irradiation and serum starvation.
It is interesting to note that Pirh2 is predominantly phosphorylated in G2/M phase of cell cycle, and during that time, the activity of CaMK II is also peaked, suggesting the phosphorylation of Pirh2 by CaMK II occurs mainly in G2/M phase. CaMK II has been observed in the mitotic apparatus of dividing cells and was suggested as a regulator of the G2/M transition (Ohta et al, 1990; Matsumoto and Maller, 2002). Whether the phosphorylation of Pirh2 actively participates in the cell cycle regulation at G2/M phase, or is just a consequence of cell cycle arrest, is currently being under investigation. Importantly, expression of Pirh2 led to an increase in ubiquitination of p53 in G1 phase, but not G2/M phase (Figure 6C), suggesting that Pirh2-mediated p53 degradation preferentially occurs during G1 phase.
Previous studies established an autoregulatory feedback loop between p53 and Pirh2 (Leng et al, 2003). We have demonstrated that the phosphorylation status of Pirh2 affects its E3 ligase activity toward p53. Phosphorylated Pirh2 fails to bind p53, and as a result, Pirh2 lost its ability to ubiquitinate p53. Hence, data presented in this study further support the following scenario: Pirh2 is under dual regulation by p53 and CaMK II. On one hand, Pirh2 is transcriptionally upregulated by p53, and on the other hand, it can be post-translationally modified by CaMK II via phosphorylation. Once Pirh2 is phosphorylated, Pirh2 becomes less stable and the association between Pirh2 and p53 is weakened. As a result, Pirh2 no longer functions as an E3 ligase toward p53, thereby enhancing the stability of p53. Reversibly, if Pirh2 remains less phosphorylated, as shown in the cancerous tissues, more p53 will be degraded and cells could become more susceptible to tumor formation. We further utilized mouse models to examine the ability of wt-Pirh2 or mutant Pirh2 to promote tumorigenicity in vivo. As expected, Pirh2/T154AS155A double mutant induced tumors with large volume, whereas phosphor-mimic mutant Pirh2/T154ES155D could only form tumors of small size, demonstrating that lack of Pirh2 phosphorylation could be one of the causes of tumorigenesis. Thus CaMK II-mediated phosphorylation acts as a fine-tuning in adjusting the level of Pirh2, which in turn, modulates polyubiquitination and degradation of p53 (Figure 7C).
Overall, these observations added knowledge to a delicate and tightly coordinated mechanism, which regulates the Pirh2–p53 interplay. Studies on regulation of Pirh2 may help identify valuable therapeutic targets for maximizing the p53 response to stress and sensitizing cancer cells to chemotherapeutic drugs.
Materials and methods
Cell culture and transfection
Cell lines HeLa, H1299, A549 and MCF-7 were cultured in DMEM containing 10% FBS at 37°C under an atmosphere of 5% CO2 in air. Transfection of cells with various mammalian expression constructs by Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) was according to the methods provided by manufacturer's specification.
Reagents and antibodies
The following antibodies were used in this study: rabbit polyclonal Pirh2 antibody (Bethyl, Montgomery, TX), anti-active CaMK II pAb (Cell Signaling, Danvers, MA), anti-phosphoSerine and anti-phosphoThreonine antibodies (Qiagen, Valencia, CA), anti-phosphoThreonine and anti-phosphoTyrosine antibodies (Cell Signaling, Danvers, MA), mAb-p53 (Ab-6, Oncogene Science, Manhasset, NY), mAb-FLAG M2 (Sigma-Aldrich, St Louis, MO, USA), mAb-GFP (Clontech), mAb-beta-actin (Abcam, Cambridge, MA), mAb-B23 (Sigma-Aldrich) antibodies. MG132 was purchased from Calbiochem (La Jolla, CA), and protease-inhibitor cocktail was from Roche (Mannhein, Germany). Staurosporine, CHX and nocodazole were purchased from Sigma-Aldrich. Pharmacological inhibitors W-7, Autocamtide-2-related inhibitory peptide II (Ant-AIP II), LY 294002 and Bisindolylmaleimide (GF109203X) were purchased from Calbiochem. The MEK inhibitor U0126 and the MAPK/ERK inhibitor PD 98059 were purchased from Promega (Promega, Madison, WI).
In vitro kinase assay
Glutathione beads (Amersham Biosciences) were used to purify the GST fusions of Pirh2 and its mutants. Purified fusion proteins (1–2 μg) were incubated with activated CaMK II (New England Biolabs) in 1 × CaMK II reaction buffer supplemented with 200 μM ATP, in a total volume of 30 μl, for 90 min at 30°C. After the end of the reaction, bead-bound GST fusions of Pirh2 and its mutants were washed, and then either used for subsequent in vitro ubiquitination assay or boiled with 2 × SDS loading buffer. For detection of phosphorylated proteins by autoradiography, the reaction mixture was supplemented with 200 μM ATP and [γ-32P]-ATP, to a final concentration of 500 μCi/μmol. To test the inhibition of Pirh2 phosphorylation by CaMK II inhibitors, cells were treated with or without AIP II (5 μg/ml), overnight, before being harvested. Cell lysates were purified by centrifugation, before incubation with purified GST-Pirh2 in 1 × reaction buffer and the kinase reactions were initiated by addition of 100 μM ATP and 10 mM MgCl2.
In vivo ubiquitination assay
H1299 cells were transfected with expression plasmids encoding p53, Pirh2 and ubiquitin in various combinations, as indicated. Sixteen hours after transfection, MG132 was added for a further 6 h. Cells were pretreated with W-7, wherever required, before transfection. In vivo ubiquitination assay was performed according to the procedure described elsewhere (Zhang et al, 2004).
In vitro ubiquitination assay
The bead-bound GST alone, GST-wt-Pirh2, or GST-Pirh2/T154AS155A were phosphorylated by CaMK II, or mock treated, and the resultant products were then washed and incubated for 1 h at 37°C with rabbit E1 (3 pmol), UbcH5b (16 pmol) and human recombinant Ubiquitin (3 nmol), in ubiquitination buffer (50 mM Tris–HCl, pH 7.5, 2 mM NaF, 0.5 mM DTT, 20 μM ZnCl2) containing 1 × energy regeneration solution (ERS). Ubiquitin, E1, UbcH5b and ERS were purchased from Boston Biochem. For p53 ubiquitination assays, GST-Pirh2 or GST-Pirh2/T154AS155A were subjected to in vitro kinase assay, before incubation with purified His-p53 for at least 2 h at 4°C to form GST-Pirh2/p53 complexes. These bead-bound complexes were subsequently incubated with the indicated ubiquitination components at 30°C for 90 min. The reactions were terminated by adding equal amount of 2 × SDS loading buffer, boiled and resolved on 8% SDS–PAGE gel.
Cell cycle analysis
Cells were harvested, washed and fixed with 70% (v/v) ethanol at 4°C, overnight. After fixation, cells were centrifuged for 5 min at 1200 r.p.m., washed twice with PBS and incubated in PBS containing 40 μg/ml propidium iodide (Sigma), 200 μg/ml RNase A (Sigma) for 30 min at room temperature. Cell cycle distribution was examined by flow cytometry using a FACScan flow cytometer (Becton Dickinson, Oxford, UK).
Mapping phosphorylation sites in Pirh2 by mass spectrometry
A549 cells were either serum-starved or treated with nocodazole, to induce G1 or G2/M arrest, respectively. Pirh2 was immunoprecipitated individually from two differently treated cells, and after SDS/PAGE and Coomassie staining, two different forms of Pirh2 were excised and digested with trypsin. The phosphorylated residues and the amino-acid sequence of the phosphopeptide of interest were further determined by MALDI-TOF mass spectrometric analysis.
Immunoprecipitation, Western blotting and immunofluorescence microscopy
Immunoprecipitation and Western blot analysis were performed as described elsewhere (Barila et al, 2003). For analysis of Pirh2 phosphorylation status in human tissues, samples were obtained from patients who underwent surgical resection. The purity of cancer tissues and the surrounding normal tissues has been evaluated histologically. The protocol of the study was approved by local ethics committees. Tissue samples were rapidly frozen in liquid nitrogen and homogenized on ice in lysis buffer. Immunofluorescence was performed according to procedures described by Zhang and Xiong (1999). Images were obtained using an Olympus IX71 inverted microscope coupled to an Olympus DP70 high-resolution color camera.
siRNA-mediated gene knockdown
siRNAs against human Pirh2 and CaMK II were purchased from Santa Cruz Biotechnology. Cells were transfected with 200 nM of the siRNA oligos by using Oligofectamine (Invitrogen), according to the protocols provided by Santa Cruz Biotechnology.
Tumorigenicity assays
NIH3T3 cells (107) stably expressing wt-Pirh2 or various mutant Pirh2 were injected subcutaneously into 4–5 week old, male athymic nude mice (Shanghai SLAC Laboratory Animal Co. Ltd.). After 5 weeks, tumors were excised, weighed, and proteins extracted from these tumors were used for Western blot analysis. A total of 15 athymic nude mice were used and all animal experiments were performed strictly in accordance with local Animal Care and Use Committee.
Supplementary Material
Supplementary Figures
Supplementary Information
Acknowledgments
We are grateful to Dr Lenore K Beitel (Lady Davis Institute for Medical Research and McGill University) for kindly providing the plasmid pcDNA3-hARNIP; Dr Ron Kopito (Stanford University) for plasmid pCW7-myc-ubiquitin and Dr Donna D Zhang (University of Arizona) for plasmid pMT123-HA-ubiquitin. We also thank Dr Zheng Sun for helpful advice on the ubiquitination assay. This research was supported by grants from the National Natural Science Foundation of China (30530200 and 30121001), grants from the Ministry of Science and Technology of China (2002CB713702, 2006AA02Z101 and 2006CB910300), a grant from Chinese Academy of Sciences (KSCX1-YW-R-57)' to Mian Wu, and a grant from University of Science and Technology of China (KD2005034) to Shanshan Duan.
References
- Alarcon-Vargas D, Ronai Z (2002) p53–Mdm2—the affair that never ends. Carcinogenesis 23: 541–547 [DOI] [PubMed] [Google Scholar]
- Barila D, Rufini A, Condo I, Ventura N, Dorey K, Superti-Furga G, Testi R (2003) Caspase-dependent cleavage of c-Abl contributes to apoptosis. Mol Cell Biol 23: 2790–2799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beitel LK, Elhaji YA, Lumbroso R, Wing SS, Panet-Raymond V, Gottlieb B, Pinsky L, Trifiro MA (2002) Cloning and characterization of an androgen receptor N-terminal-interacting protein with ubiquitin-protein ligase activity. J Mol Endocrinol 29: 41–60 [DOI] [PubMed] [Google Scholar]
- Chen M, Cortay JC, Logan IR, Sapountzi V, Robson CN, Gerlier D (2005) Inhibition of ubiquitination and stabilization of human ubiquitin E3 ligase PIRH2 by measles virus phosphoprotein. J Virol 79: 11824–11836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR (2004) Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303: 1010–1014 [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Maurer U, Green DR, Schuler M (2003) Pharmacologic activation of p53 elicits Bax-dependent apoptosis in the absence of transcription. Cancer Cell 4: 371–381 [DOI] [PubMed] [Google Scholar]
- Corcoran CA, Huang Y, Sheikh MS (2004) The p53 paddy wagon: COP1, Pirh2 and MDM2 are found resisting apoptosis and growth arrest. Cancer Biol Ther 3: 721–725 [DOI] [PubMed] [Google Scholar]
- Dornan D, Wertz I, Shimizu H, Arnott D, Frantz GD, Dowd P, O'Rourke K, Koeppen H, Dixit VM (2004) The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature 429: 86–92 [DOI] [PubMed] [Google Scholar]
- Duan W, Gao L, Druhan LJ, Zhu WG, Morrison C, Otterson GA, Villalona-Calero MA (2004) Expression of Pirh2, a newly identified ubiquitin protein ligase, in lung cancer. J Natl Cancer Inst 96: 1718–1721 [DOI] [PubMed] [Google Scholar]
- Giaccia AJ, Kastan MB (1998) The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev 12: 2973–2983 [DOI] [PubMed] [Google Scholar]
- Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M (2002) Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J Biol Chem 277: 3247–3257 [DOI] [PubMed] [Google Scholar]
- Leng RP, Lin Y, Ma W, Wu H, Lemmers B, Chung S, Parant JM, Lozano G, Hakem R, Benchimol S (2003) Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell 112: 779–791 [DOI] [PubMed] [Google Scholar]
- Logan IR, Sapountzi V, Gaughan L, Neal DE, Robson CN (2004) Control of human PIRH2 protein stability: involvement of TIP60 and the proteosome. J Biol Chem 279: 11696–11704 [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Maller JL (2002) Calcium, calmodulin, and CaMKII requirement for initiation of centrosome duplication in Xenopus egg extracts. Science 295: 499–502 [DOI] [PubMed] [Google Scholar]
- Meek DW, Knippschild U (2003) Posttranslational modification of MDM2. Mol Cancer Res 1: 1017–1026 [PubMed] [Google Scholar]
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM (2003) p53 has a direct apoptogenic role at the mitochondria. Mol Cell 11: 577–590 [DOI] [PubMed] [Google Scholar]
- Moll UM, Petrenko O (2003) The MDM2–p53 interaction. Mol Cancer Res 1: 1001–1008 [PubMed] [Google Scholar]
- Nakano K, Vousden KH (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 7: 683–694 [DOI] [PubMed] [Google Scholar]
- Nutt LK, Margolis SS, Jensen M, Herman CE, Dunphy WG, Rathmell JC, Kornbluth S (2005) Metabolic regulation of oocyte cell death through the CaMKII-mediated phosphorylation of caspase-2. Cell 123: 89–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta Y, Ohba T, Miyamoto E (1990) Ca2+/calmodulin-dependent protein kinase II: localization in the interphase nucleus and the mitotic apparatus of mammalian cells. Proc Natl Acad Sci USA 87: 5341–5345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel R, Holt M, Philipova R, Moss S, Schulman H, Hidaka H, Whitaker M (1999) Calcium/calmodulin-dependent phosphorylation and activation of human Cdc25-C at the G2/M phase transition in HeLa cells. J Biol Chem 274: 7958–7968 [DOI] [PubMed] [Google Scholar]
- Ryan KM, Phillips AC, Vousden KH (2001) Regulation and function of the p53 tumor suppressor protein. Curr Opin Cell Biol 13: 332–337 [DOI] [PubMed] [Google Scholar]
- Tombes RM, Mikkelsen RB, Jarvis WD, Grant S (1999) Downregulation of delta CaM kinase II in human tumor cells. Biochim Biophys Acta 1452: 1–11 [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ (2000) Surfing the p53 network. Nature 408: 307–310 [DOI] [PubMed] [Google Scholar]
- Vousden KH (2000) p53: death star. Cell 103: 691–694 [DOI] [PubMed] [Google Scholar]
- Wu Y, Mehew JW, Heckman CA, Arcinas M, Boxer LM (2001) Negative regulation of bcl-2 expression by p53 in hematopoietic cells. Oncogene 20: 240–251 [DOI] [PubMed] [Google Scholar]
- Yadav M, Roach SK, Schorey JS (2004) Increased mitogen-activated protein kinase activity and TNF-a Production associated with Mycobacterium smegmatis but not Mycobacterium avium-infected macrophages requires prolonged stimulation of the calmodulin/calmodulin kinase and cyclic AMP/protein kinase A pathways. J Immunol 172: 5588–5597 [DOI] [PubMed] [Google Scholar]
- Yang BF, Xiao C, Roa WH, Krammer PH, Hao C (2003) Calcium/calmodulin-dependent protein kinase II regulation of c-FLIP expression and phosphorylation in modulation of Fas-mediated signaling in malignant glioma cells. J Biol Chem 278: 7043–7050 [DOI] [PubMed] [Google Scholar]
- Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M (2004) Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol 24: 10941–10953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xiong Y (1999) Mutations in human ARF exon 2 disrupt its nucleolar localization and impair its ability to block nuclear export of MDM2 and p53. Mol Cell 3: 579–591 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures
Supplementary Information