

Abstract
Biological functions of lysozyme, including its antimicrobial, antitumor, and immune-modulatory activities have been suggested to be largely determined by the lipid binding properties of this protein. To gain further insight into these interactions on a molecular level the association of lysozyme to liposomes composed of either 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine or its mixtures with 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-rac-glycerol, 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-rac-phosphatidylserine, or bovine heart cardiolipin was studied by a combination of fluorescence techniques. The characteristics of the adsorption of lysozyme to lipid bilayers were investigated using fluorescein 5′-isothiocyanate labeled protein, responding to membrane association by a decrease in fluorescence. Upon increasing the content of anionic phospholipids in lipid vesicles, the binding isotherms changed from Langmuir-like to sigmoidal. Using adsorption models based on scaled particle and double-layer theories, this finding was rationalized in terms of self-association of the membrane-bound protein. The extent of quenching of lysozyme tryptophan fluorescence by acrylamide decreased upon membrane binding, revealing a conformational transition for the protein upon its surface association, resulting in a diminished access of the fluorophore to the aqueous phase. Steady-state fluorescence anisotropy of bilayer-incorporated probe 1,6-diphenyl-1,3,5-hexatriene was measured at varying lipid-to-protein molar ratios. Lysozyme was found to increase acyl-chain order for liposomes with the content of acidic phospholipid exceeding 10 mol %. Both electrostatic and hydrophobic protein-lipid interactions can be concluded to modulate the aggregation behavior of lysozyme when bound to lipid bilayers. Modulation of lysozyme aggregation propensity by membrane binding may have important implications for protein fibrillogenesis in vivo. Disruption of membrane integrity by the aggregated protein species is likely to be the mechanism responsible for the cytotoxicity of lysozyme.
INTRODUCTION
The structure and physicochemical properties of lysozyme have been thoroughly characterized and this protein continues to be widely employed as a model in fundamental studies assessing protein adsorption at interfaces (1), lipid-protein interactions (2–6), membrane fusion (7–10), and structural requirements for bactericidal action (11–13), as well as protein folding, aggregation, and amyloid fibrillogenesis (14–16). There are a number of reports on the lipid-binding properties of lysozyme (5–9,17,18). In brief, membrane association of lysozyme is thought to involve both electrostatic and hydrophobic interactions (4,9,17). Lysozyme (pI ∼11.0) bears a net positive charge over a broad pH range and, accordingly, has a high affinity for anionic phospholipids. The primary role of electrostatic interactions in the membrane association of lysozyme has been demonstrated by monitoring changes in the electrophoretic mobility of lipid vesicles after protein adsorption (6,17), examining the effects of pH, ionic strength, and charge state of chemically modified lysozyme on its lipid binding (6,7), and comparing the effects of lysozyme on the aggregation and fusion of negatively charged and neutral liposomes (9). There is also evidence demonstrating the involvement of hydrophobic interaction in the membrane association of lysozyme. Accordingly, fluorescence polarization of fluorescamine-labeled lysozyme showed comparable affinity for neutral and negatively charged vesicles, both at low and high ionic strengths (9). Lysozyme was found also to induce the release of the aqueous contents of uncharged vesicles, presumably resulting from a disruption of the membrane integrity due to the insertion of lysozyme into the lipid bilayer (4,9). Measurements of surface pressure for lipid monolayers revealed a pH and ionic-strength-dependent penetration of lysozyme into the lipid phase (6). Cumulatively, the available data suggest that the membrane binding of lysozyme is a multistep process involving 1), initial protein adsorption to the lipid bilayer surface driven by the formation of ionic and/or hydrogen bond-mediated contacts; 2), conformational alterations of the protein; 3), structural reorganization of the lipid phase; and 4), partial insertion of lysozyme into the membrane acyl-chain region. Although these distinct steps have been identified, the accompanying molecular events have not been characterized in detail.
In addition to its hydrolytic activity, lysozyme displays antimicrobial, antitumor, and immunomodulatory properties, which have been extensively studied. Interestingly, accumulating evidence indicates that not only the catalytic but also the lipid-binding properties of lysozyme may account for its bactericidal action (13). Lysozyme catalyzes hydrolysis of the β-(1,4)-glycosidic linkage between n-acetylglucosamine and muramic acid of the peptidoglycan layer in the bacterial cell wall, thereby promoting cell aggregation and loss of their viability. The antimicrobial activity of lysozyme is directed mainly against Gram-positive bacteria, with much lesser effect on Gram-negative species, featured by the outer membrane preventing protein diffusion to the target site. It was demonstrated that the bactericidal action of lysozyme can be augmented by modifying its cationic charge and hydrophobicity (12,15). More specifically, lysozyme, partially unfolded by heat treatment, proved to exert catalytically independent antimicrobial action against both Gram-negative and Gram-positive bacteria (13). The enhancement of the bactericidal activity of lysozyme against Gram-negative bacteria was observed also after partial reduction of its disulfide bonds (19). These data were interpreted in terms of the altered protein conformation minimizing the energetic cost of diffusion through the outer membrane of Gram-negative bacteria and enhanced insertion into the inner membrane, which is thought to be the target of bacterial cells. A specific helix-loop-helix domain penetrating into lipid bilayers was suggested to be responsible for the bactericidal action of lysozyme (12,13). In view of the above findings, identification of the factors controlling the antimicrobial activity of lysozyme and lysozyme-lipid interactions can be regarded to bear major biological relevance.
Another intriguing property of lysozyme relates to its recently reported propensity for conversion into amyloidlike fibrils in a proper membrane environment (20). Along these lines, protein fibrillization has attracted particular attention because of the critical role of this process in the pathogenesis of a number of diseases involving the deposition of aggregated proteins in various tissues (21). These aggregates are composed of specific filamentous structures, amyloid fibrils, featured by a core cross-β-sheet structure in which polypeptide chains are oriented in such a way that the β-strands run perpendicularly to the long axis of the fibril, while the β-sheets propagate in its direction (22). It has been found that point mutations giving rise to the reduced structural stability and altered folding kinetics of human lysozyme render this protein amenable to amyloid fibrillization in vivo, associated with a familial nonneuropathic systemic amyloidosis, a disease in which aggregated protein tends to form deposits in liver, kidneys, and spleen (23). Lysozyme amyloidogenesis has been extensively studied in vitro, with fibril growth being induced at acidic pH, elevated temperature, or upon addition of organic solvents (16,24,25). In accord with the generally recognized concept, structural transition of lysozyme to partially unfolded, aggregation-prone state has been suggested to be a critical prerequisite for fibrillization (25). Importantly, in vitro protein assembly into protofibrillar and fibrillar structures can be triggered by membrane binding (20,26,27). Despite considerable progress achieved in understanding the mechanisms and pathways of protein fibrillization, the molecular details of membrane-mediated fibrillogenesis remain poorly understood (28). In this regard, studies on lysozyme-liposome model system are likely to provide new insights.
The present study was undertaken to obtain further information on the mechanisms of lysozyme-lipid interactions. In accord with the membrane binding being a multistep process, our goals were several-fold: 1), to characterize the adsorption of lysozyme to neutral and negatively charged model membranes with different surface charge densities and different anionic phospholipid species; 2), to explore the effect of lipids on protein conformation; and 3), to monitor changes in the structural dynamics of the lipid bilayer due to protein adsorption. We utilized fluorescence spectroscopy for lysozyme covalently labeled with fluorescein, quenching of the intrinsic tryptophan fluorescence by acrylamide, and assessment of lipid acyl-chain order by steady-state fluorescence anisotropy of the membrane-incorporated probe diphenylhexatriene (DPH). Neat phosphatidylcholine vesicles or mixtures of this lipid with phosphatidylglycerol, phosphatidylserine, or cardiolipin were used as model membranes.
MATERIALS AND METHODS
Materials
Chicken egg white lysozyme, fluorescein 5′-isothiocyanate (FITC), and HEPES were purchased from Sigma (St. Louis, MO). Dimethyl sulfoxide was of Uvasol grade from Merck (Whitehouse Station, NJ). Bovine heart cardiolipin (CL), 1-palmitoyl -2-oleoyl-sn-glycero-3-phosphocholine, 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-rac-glycerol, and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-rac-phosphatidylserine were from Avanti Polar Lipids (Alabaster, AL). 1,6-Diphenyl-1,3,5-hexatriene (DPH) was from EGA Chemie (Steinheim, Germany). All other chemicals were of analytical grade.
Lysozyme labeling with fluorescein isothiocyanate
Labeling of lysozyme with FITC was performed according to Kok et al. (29). FITC and lysozyme were dissolved simultaneously in 100 mM borate buffer, pH 9.1 to give equimolar concentrations of the protein and label (110 μM). After incubation of the sample for 90 min at 25°C under continuous stirring in the dark, pH was adjusted to 7.4. Subsequently, the solution was dialyzed at 4°C against 20 mM HEPES, 0.1 mM EDTA, pH 7.4. The degree of labeling was estimated using extinction coefficients of 73,000 M−1 cm−1 for FITC at 494 nm and 45,200 M−1 cm−1 at 280 nm. Lysozyme concentration was calculated after subtracting FITC absorbance using extinction coefficient of 37,800 M−1 cm−1 at 280 nm, and revealed a fluorescein-protein molar ratio of 0.9.
Preparation of lipid vesicles
Large unilamellar vesicles were made by extrusion using 1-palmitoyl -2-oleoyl-sn-glycero-3-phosphocholine (PC) and its mixtures with 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-rac-glycerol (PG), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-rac-phosphatidylserine (PS), or CL, as indicated. A thin lipid film was first formed of the lipid mixtures in chloroform by removing the solvent under a stream of nitrogen. The dry lipid residues were subsequently hydrated with 20 mM HEPES, 0.1 mM EDTA, pH 7.4 at room temperature to yield lipid concentration of 1 mM. Thereafter, the sample was subjected to 15 passes through a 100-nm pore-size polycarbonate filter (Millipore, Bedford, MA), yielding liposomes' desired composition. In this way, 13 types of lipid vesicles containing PC and 0, 5, 10, 20, and 40 mol % PG or PS, and 2.6, 5.3, 11.1, or 25 mol % CL were prepared, with the content of phosphate being identical for all liposome preparations.
Fluorescence measurements
Steady-state fluorescence spectra were recorded with LS-50B spectrofluorometer equipped with a magnetically stirred, thermostated cuvette holder (Perkin-Elmer, Beaconsfield, UK). Fluorescence measurements were performed at 25°C and using 10 mm path-length quartz cuvettes. Emission spectra of fluorescein-labeled lysozyme were recorded in buffer and in the presence of liposomes with excitation wavelength of 470 nm. Excitation and emission band-passes were set at 5 nm.
Fluorescence quenching experiments were carried out with the neutral water soluble quencher, acrylamide. Emission spectra of lysozyme were recorded with excitation at 296 nm, using 5-nm band-passes for both excitation and emission. Small aliquots (10 μl) of acrylamide stock solution (4 M) were added to a stirred and temperature-controlled protein solution or protein-lipid mixtures. Fluorescence intensity measured in the presence of quencher was corrected for reabsorption and inner filter effects using the coefficient
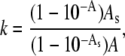 |
(1) |
where A is the protein absorbance in the absence of a quencher, and As is the total absorbance of the sample at excitation or emission wavelengths. The data of the quenching experiments were analyzed according to Stern-Volmer equation (30),
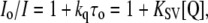 |
(2) |
where Io and I are fluorescence intensities recorded in the absence and presence of a quencher, respectively, kq is the bimolecular quenching rate constant, τo is the fluorophore lifetime in the absence of the quencher, KSV is the Stern-Volmer constant, and [Q] is the quencher concentration.
Steady-state fluorescence anisotropy for DPH was measured using excitation and emission wavelengths of 360 and 450 nm, respectively, with excitation and emission band-passes set at 10 nm. Stock solution of DPH was prepared in tetrahydrofurane. To incorporate the probe into lipid bilayers, liposomal suspensions were incubated with DPH for 60 min at 25°C to achieve a lipid/probe molar ratio 400:1.
THEORETICAL BACKGROUND
Monomodal adsorption model
Lysozyme binding to model membranes has been analyzed in terms of the adsorption models allowing for area exclusion and electrostatic effects. These models are based on Gouy-Chapman double-layer theory and scaled particle theory (SPT), which is currently regarded as providing most adequate description of excluded area interactions between the adsorbing protein molecules. In the simplest case of nonassociating ligand and monomodal adsorption the scaled particle theory isotherm is given by (31)
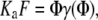 |
(3) |
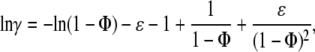 |
(4) |
where Ka is the association constant, F is the concentration of the protein free in solution, γ is the activity coefficient of adsorbed ligand, Φ is the fraction of surface area occupied by adsorbed protein, Φ = nB/La, B is the concentration of bound protein, n is the number of lipid molecules covered by a single protein, La is the concentration of accessible lipids related to total lipid concentration (L) as La = 0.5 L, and ɛ is a shape parameter (for disklike ligand ɛ = 1).
The equilibrium binding constant can be represented as consisting of electrostatic (Kel) and intrinsic or nonelectrostatic (Ko) terms: (Ka = KelKo). Electrostatic component of binding constant, dependent on electrostatic surface potential, environmental conditions (pH, ionic strength), and degree of surface coverage by a protein is given by (32)
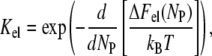 |
(5) |
where T is the temperature, kB is Boltzmann's constant, and ΔFel is the total gain in electrostatic free energy, being a function of the number of adsorbed protein molecules, Np = BNA,
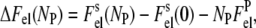 |
(6) |
where 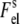 and
and 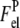 are the electrostatic free energies of a membrane surface and a protein, respectively. The electrostatic free energy of a spherical protein molecule with effective charge +ze and uniform charge distribution can be written as (33)
are the electrostatic free energies of a membrane surface and a protein, respectively. The electrostatic free energy of a spherical protein molecule with effective charge +ze and uniform charge distribution can be written as (33)
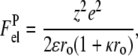 |
(7) |
where ro is the protein radius, and κ is the reciprocal Debye length
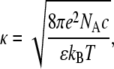 |
(8) |
with e being the elementary charge, NA Avogadro's number, ɛ the dielectric constant, and c the molar concentration of monovalent ions. In terms of the Gouy-Chapman double-layer theory, the electrostatic free energy of a membrane of area Sm = SLLa is given by (34)
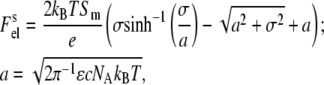 |
(9) |
where SL is the mean area per lipid molecule, SL = (fPCSPC + fASA), fPC, and fA are the mole fractions of PC and acidic phospholipids (PG, PS, or CL), SPC and SA are mean areas per PC and acidic phospholipid headgroups, respectively (taken as 0.65 nm2 for PC, PG, and PS, and 1.2 nm2 for CL (35)), and σ is the surface charge density given by
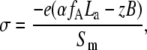 |
(10) |
where α is the degree of anionic phospholipid ionization, which in the case of one-step deprotonation can be written as
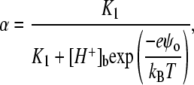 |
(11) |
where K1 is ionization constant, [H+]b is the bulk proton concentration, and ψo is electrostatic surface potential of a membrane related to the surface charge density, defined as
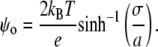 |
(12) |
Numerical solution of the set of Eqs. 3–12 yields theoretical isotherms that can be fitted to experimental binding curves.
Two-state model for the adsorption of a large self-associating ligand
The model is based on the assumption that the adsorbed protein exists in only two states, as a monomer and za-mer. This model can be represented by (31)
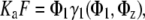 |
(13) |
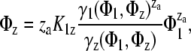 |
(14) |
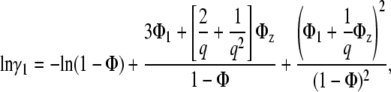 |
(15) |
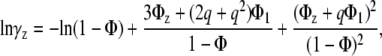 |
(16) |
where Φ1, Φz are fractions of surface area occupied by monomers and za-mers, respectively, Φ = Φ1 + Φz, q = Rz/R1, Rz, and R1 are the radii of the circles representing za-mer and monomer, and K1z is the equilibrium constant for the formation of za-mer. For the case of conserved area upon self-association 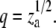
Cluster model
In contrast to the two-state model, the cluster model considers highly heterogeneous populations of aggregated protein (protein clusters of arbitrary size and shape). The activity coefficients of individual adsorbate species can be derived from scaled particle theory of a mixture of hard convex particles in two dimensions (36),
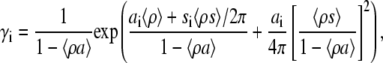 |
(17) |
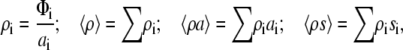 |
(18) |
where ρi, ai, and si are the surface number density, area, and circumference of the footprint of species i, respectively. The equilibrium constants for monomer association with the surface (K1) and formation of species i (Kci) are defined as
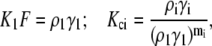 |
(19) |
where mi is the degree of oligomerization of species i. The equilibrium constant Kci is considered as a function of the number of intermolecular contacts in cluster species i (Nci). In particular, when all cluster footprints are represented by circles and mi = i for species i, the empirical relationship holds (36) as
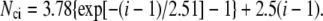 |
(20) |
Multimodal adsorption model
This model allows for the possibility of multiple interconvertible adsorbate conformations or protein association with heterogeneous surface, which contains binding sites differing in their size and free energy of adsorption. The activity coefficient of the protein adsorbed in a particular conformation i or associated with the site of ith type is given by Eq. 17. More specifically, if the membrane in question contains two types of protein binding centers (i.e., there exist two populations of the adsorbed protein differing in free energy of adsorption) and adsorbed protein has a circular footprint, Eqs. 17 and 18 can be rewritten as (37)
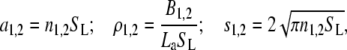 |
(21) |
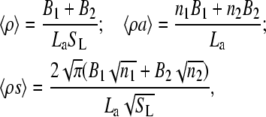 |
(22) |
and adsorption isotherm can be described by the following set of equations:
 |
(23) |
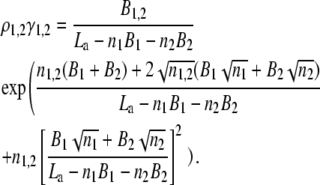 |
(24) |
RESULTS AND DISCUSSION
Binding studies
We first studied the adsorption of lysozyme to liposomes with different compositions and contents of anionic phospholipids. To differentiate between the lipid-bound and free protein in solution, lysozyme was labeled with fluorescein (Fl), an environment-sensitive fluorophore. As illustrated in Fig. 1, the binding of Fl-lysozyme to liposomes was accompanied by a decrease in fluorescein emission. This effect can be explained by changes in the microenvironment on the fluorophore upon its transfer to the interfacial membrane region whose pH is lower than that of the bulk phase due to proton accumulation near negatively charged surfaces (38). Accordingly, fluorescein emission displays a complex pH dependence, reflecting equilibrium between the various ionic forms, of which only monoanion and dianion forms are fluorescent (30). In aqueous solution, the pKa values for stepwise fluorescein deprotonation are known to be ∼2.1, 4.4, and 6.6, with the highest pKa corresponding to the deprotonation of the monoanionic form. The shift of equilibrium between the fluorescent and nonfluorescent fluorescein species after the association of labeled protein with liposomal membranes is likely to be the reason for the observed fluorescence decrease. Notably, the lowest interfacial pH reached in the membrane systems used here can be estimated at ∼5.2.
FIGURE 1.

Fluorescence spectra of fluorescein-labeled lysozyme (Fl-lysozyme) recorded in 20 mM HEPES, pH 7.4 and in the presence of PC/PG (20 mol % PG) liposomes. Protein concentration was 0.4 μM.
This fluorescence response of Fl-lysozyme to membrane binding was exploited to obtain information on the protein adsorption to model membranes. To determine the adsorption isotherms the maximum fluorescence emission intensity (ΔI) was measured as a function of lipid concentration for different types of liposomes (Fig. 2). Several important features of the observed binding curves are evident, such as 1), as the surface charge of liposomal membranes increases, the shape of adsorption isotherm tends to change from Langmuir-like (Fig. 2 A) to sigmoidal (Fig. 2 D); 2), the adsorption of lysozyme to PG and PS containing bilayers is virtually identical; and 3), the differences between the adsorption isotherms obtained for CL and PG or PS containing liposomes become more pronounced with increasing molar percent of anionic phospholipid. In an attempt to interpret these peculiarities of the binding process, we made a semiquantitative analysis of the adsorption isotherms. To this end, the SPT adsorption models developed by Minton (31) and further extended by Heimburg (32) to allow for electrostatic effects employed (i.e., for dependence of association constant on surface coverage). To fit the adsorption models to experimental data it was assumed that the decrease of fluorescein emission observed on titration of the labeled lysozyme by liposomes is related to the concentration of bound protein (Be),
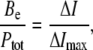 |
(25) |
where Ptot is the total protein concentration and ΔImax is the limit decrease in fluorescence. The fitting procedure involved minimization of the error function
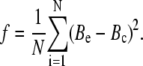 |
(26) |
Here, N is the number of experimental data points and Bc is the concentration of bound protein calculated numerically from the equations underlying the adsorption model employed. Importantly, our goal was not to recover the unique set of model parameters providing the best agreement between the theory and experiment—as, due to strong cross-correlation between the adjustable parameters and limited number of data points, this goal was considered unachievable within the scope of this study. Therefore, our strategy in analyzing the adsorption behavior of lysozyme was to demonstrate the adequacy of a certain adsorption model for a given liposomal system and to gain meaningful (but not unique) estimates of the model parameters.
FIGURE 2.

Decrease of Fl-lysozyme fluorescence measured at 514 nm (ΔI514) as a function of lipid concentration for PC, PC/PG, PC/PS, and PC/CL liposomes. Protein concentration was 0.15 μM. Solid lines represent theoretical curves providing the fit of monomodal adsorption model (A, B) or two-state model of adsorbate self-association (C, D) to the experimental data with the following parameters: PC (n = 16, z = 0.1, Ko = 0.3 μM−1, ΔImax = 17, f = 7.6 × 10−5); 5 mol % PG (n = 16, z = 0.1, Ko = 2.2 μM−1, ΔImax = 45, f = 7.3 × 10−5); 5 mol % PS (n = 16, z = 0.4, Ko = 1.8 μM−1, ΔImax = 24, f = 3.9 × 10−5); 2.6 mol % CL (n = 16, z = 0.4, Ko = 1.7 μM−1, ΔImax = 30, f = 6.9 × 10−6); 10 mol % PG (n = 16, z = 4.2, Ko = 1.9 × 10−8 μM−1, ΔImax = 56, f = 2.5 × 10−5); 10 mol % PS (n = 16, z = 4.2, Ko = 1.1 × 10−7 μM−1, ΔImax = 29, f = 6.9 × 10−5); 5.3 mol % CL (n = 16, z = 4.2, Ko = 2.4 × 10−4 μM−1, ΔImax = 39, f = 5.2 × 10−5); 20 mol % PG (n = 14, Ko = 0.2 μM−1, z = 4.2, za = 4, K1z = 20, ΔImax =118, f = 5.2 × 10−6); 20 mol % PS (n = 14, Ko = 4.0 × 10−3 μM−1, z = 4.2, za = 4, K1z = 20, ΔImax =104, f = 1.9 × 10−6); 11.1 mol % CL (n = 14, Ko = 0.9 μM−1, z = 4.2, za = 4, K1z = 20, ΔImax =152, f = 3.1 × 10−6); 40 mol % PG (n = 8.8, Ko = 4 μM−1, z = 4.2, za = 4, K1z = 0.05, ΔImax =149, f = 3.4 × 10−6); 40 mol % PS (n = 9.7, Ko = 1 μM−1, z = 4.2, za = 4, K1z = 0.04, ΔImax =132, f = 2.8 × 10−6); 25 mol % CL (n = 5.4, Ko = 54 μM−1, z = 4.2, za = 4, K1z = 0.02, ΔImax =209, f = 6.2 × 10−6).
The monomodal adsorption model given by Eqs. 3–12 proved to provide satisfactory fit of the Langmuir-like experimental isotherms obtained at relatively low contents of anionic phospholipid (5 or 10 mol % PG or PS, and 2.6 or 5.3 mol % CL). The data fitting involved optimization of the two parameters, intrinsic association constant (Ko) and effective protein charge (z), with ΔImax being slightly varied in the limits consistent with the saturation level of the binding curves and n being taken as 16 (approximate number of phospholipid headgroups that occupy the cross-sectional area of unperturbed lysozyme molecule). The validity of this choice for the n-value is supported by the finding that lysozyme retains its globular form upon associating with lipids (39).
As illustrated in Fig. 3, the association constants tend to increase with lipid concentration. This tendency reflects the Kel behavior on decreasing surface coverage (Φ = nB/La) coupled with a reduction in the extent of membrane charge neutralization by the adsorbed protein. However, electrostatically controlled increase of association constant proved insufficient to explain the steepening of the adsorption isotherms that occurs upon increasing the mole fractions of the anionic phospholipid to 20 or 40 and 11 or 25 mol % of PG/PS or CL, respectively (Fig. 2, C and D). Neither the monomodal nor multimodal (Eqs. 21–24) adsorption model was adequate in describing the sigmoidal shape of the binding curves (Fig. 4 A). Accordingly, we assumed that the sigmoidal character of the isotherms stems from self-association of the membrane-bound protein. The phenomenon of protein self-association on a surface was treated by the two SPT adsorption models, i.e., a two-state model (Eqs. 13–16) and a cluster model (Eqs. 17–20). Both models were tested for their ability to predict the trend of ΔI(L) plots; however, only the two-state model was capable of reproducing the experimental data. Moreover, as shown in Fig. 4, B and D, sigmoidal dependence on lipid concentration was recovered only for protein monomers, while oligomers exhibited a different behavior. This finding led us to assume that the observed decrease in fluorescein emission arises from the membrane binding of monomeric protein species, with fluorescence of aggregated protein remaining virtually unchanged because of substantially reduced label exposure to the interfacial microenvironment. Notably, simulation results demonstrate that, at fixed protein and lipid concentrations, protein monomers exist in an equilibrium with oligomers. Change in the lipid concentration provokes shift of this equilibrium, so that at a certain surface coverage the amount of protein monomers, forming aggregates, reaches its maximum. The La/B ratios corresponding to maximum degree of the protein oligomerization were found to be ∼24 (40 mol % PG), 33 (40 mol % PS), and 22 (25 mol % CL). Interestingly, these estimates are close to theoretical saturation coverage of the membrane surface with lysozyme, which equals to ∼20 lipid molecules per protein (taking protein cross-section as ∼13.5 nm2 and molecular area per phospholipid headgroup as 0.65 nm2). It is also worthy of mention that a minimum degree of the protein oligomerization required to ensure sigmoidal shape of the binding curves was found to be 4, suggesting that tetramers represent a preferential form of Fl-lysozyme aggregates. These results are of interest in the context of amyloidogenic properties of lysozyme since monomer association into a critical oligomeric nucleus is thought to be a key step in protein fibrillization (40).
FIGURE 3.

Association constants recovered from the fitting of monomodal adsorption model (A) or two-state model of adsorbate self-association (B) to the experimental binding curves presented in Fig. 2, B and D.
FIGURE 4.

Fitting of different adsorption models to the experimental isotherms obtained for PC/CL (25 mol % CL) liposomes (A): dash line, monomodal model (n = 12, z = 4.2, Ko = 3.3 μM−1, and f = 3.6 × 10−5); dot line, multimodal model (n1 = 14, n2 = 8, z = 4.2, Ko1 = 800 μM−1, Ko2 = 84, and f = 4.7 × 10−5); dash-dot line, cluster model (n = 10, K1 = 20 μM−1, and f = 1.2 × 10−4); solid line, two-state model of adsorbate self-association (n = 5.4, Ko = 54 μM−1, z = 4.2, za = 4, K1z = 0.02, and f = 6.2 × 10−6); and ΔImax = 209 in all cases. (B–D) Plots illustrating the shift of equilibrium between monomeric and oligomeric protein species with lipid concentration.
Fluorescence quenching studies
As the next step of the study, it was of importance to define changes in lysozyme structure, related to the augmented protein aggregation upon membrane binding. This issue was approached by examining the quenching of tryptophan fluorescence by acrylamide. Lysozyme contains six tryptophan residues, of which Trp62, Trp63, Trp108, and Trp123 are exposed to solvent, while Trp28 and Trp111 are buried into the hydrophobic interior. It has been shown that Trp62 and Trp108 account for 80% of lysozyme fluorescence (41), with their emission maxima being observed at 352 and 342 nm, respectively (42). The remaining fluorescence is due to Trp28 and Trp111 whose emission maximum is ∼323 nm (43). Superposition of spectral contributions from several fluorophores differing in their local environment results in a relatively broad fluorescence spectrum for lysozyme. Acrylamide quenching was found to be accompanied by a blue shift of emission maximum, indicating that the individual Trp residues are differently accessible to the quencher. Accordingly, Stern-Volmer plots show dependence on emission wavelength (Fig. 5). In this case, rigorous analysis of quenching data must involve estimation of the set of multiple parameters including not only Stern-Volmer constants, but also fractional contributions by the different fluorophores to the measured overall fluorescence intensity (30). For multi-tryptophan proteins such as lysozyme, unambiguous determination of these parameters for either free protein in solution or the membrane-associated protein would be ambiguous without selective substitution of Trp residues with nonfluorescent residues. In this study, we did not pursue rigorous quantitative interpretation of the quenching data, and confined ourselves to establishing the main tendencies in the changes of fluorescence quenching efficiency upon membrane binding of lysozyme. For this purpose, the results of quenching experiments were treated in terms of the simplest model (Eq. 2), which yields wavelength-dependent Stern-Volmer constants averaged over all Trp residues contributing to lysozyme fluorescence.
FIGURE 5.

Typical plots for the quenching of lysozyme fluorescence by acrylamide. Lipid concentration was 50 μM, protein concentration 0.4 μM.
As seen in Table 1, lysozyme association with lipid vesicles was followed by a decrease in Stern-Volmer constants for acrylamide quenching. This effect was found to become more pronounced with increasing surface charge of the liposomal membranes. Since acrylamide is a water-soluble collisional quencher that is reported not to penetrate into the hydrophobic core of the bilayer (30), the observed changes in KSV could be interpreted in terms of reduced accessibility of tryptophan to solvent. However, while examining fluorescence quenching of membrane-bound protein one should also bear in mind that bimolecular quenching constant depends not only on the fluorophore microenvironment but also on the rates of translational and rotational diffusion of the protein molecule (44,45). In other words, immobilization of soluble protein on the surface of lipid vesicle may lead to KSV decrease unrelated to any changes in the protein conformation. Using the approach of Shoup et al. (44) described in more detail in the Appendix, we approximated the magnitude of diffusion-controlled effects in the systems under study. It appears that the slower translational and rotational diffusion of the protein associated with liposomes could account for KSV decrease by a factor of up to 1.3. The KSV decrement observed experimentally at anionic phospholipid contents >10 mol % (Table 1) appeared to exceed the above estimate, suggesting the reduction of Trp exposure to a solvent for the membrane-bound lysozyme. The finding that the magnitude of changes in KSV is virtually independent of wavelength allowed us to attribute all the observed effects to a modified local environment of the two dominant long-wavelength fluorophores, Trp62 and Trp108. The reduced solvent accessibility of these fluorophores could result from the transfer of tryptophan into a membrane environment with a lower polarity. However, this contradicts our observation that lysozyme emission maximum does not exhibit a blue shift upon membrane binding (data not shown). Alternatively, the decreased quenching efficiency of Trp62 and Trp108 could be explained by these residues residing in a region with intermolecular contacts between lysozyme molecules aggregating in a membrane environment. This view is corroborated by the early calorimetric, circular dichroism, and 13C-NMR data indicating that Trp62 and other residues located in the active site are involved in lysozyme self-association (46,47).
TABLE 1.
Stern-Volmer constants for the quenching of lysozyme fluorescence by acrylamide (KSV, M−1)
| Emission wavelength, nm
|
||||||
|---|---|---|---|---|---|---|
| System | 320 | 330 | 340 | 350 | 360 | 370 |
| Buffer | 4.8 ± 0.1 | 5.5 ± 0.11 | 6.8 ± 0.07 | 7.2 ± 0.06 | 7.6 ± 0.06 | 7.8 ± 0.05 |
| PC | 2.3 ± 0.02 | 2.7 ± 0.16 | 3.4 ± 0.02 | 3.6 ± 0.02 | 4.1 ± 0.03 | 4.0 ± 0.03 |
| PC/PG, 5 mol % | 3.9 ± 0.11 | 4.5 ± 0.09 | 5.5 ± 0.08 | 5.8 ± 0.08 | 6.0 ± 0.09 | 6.0 ± 0.08 |
| PC/PG, 10 mol % | 3.7 ± 0.08 | 4.2 ± 0.08 | 5.2 ± 0.06 | 5.3 ± 0.05 | 5.5 ± 0.06 | 5.7 ± 0.07 |
| PC/PG, 20 mol % | 2.5 ± 0.02 | 2.7 ± 0.02 | 3.3 ± 0.03 | 3.6 ± 0.04 | 3.9 ± 0.03 | 4.2 ± 0.05 |
| PC/PG, 40 mol % | 1.9 ± 0.14 | 2.1 ± 0.02 | 2.5 ± 0.02 | 2.7 ± 0.02 | 2.8 ± 0.02 | 3.0 ± 0.01 |
| PC/PS, 5 mol % | 3.6 ± 0.08 | 4.2 ± 0.08 | 5.0 ± 0.06 | 5.4 ± 0.06 | 5.6 ± 0.07 | 5.6 ± 0.08 |
| PC/PS, 10 mol % | 3.4 ± 0.06 | 4.0 ± 0.07 | 4.9 ± 0.04 | 5.1 ± 0.05 | 5.3 ± 0.04 | 5.5 ± 0.05 |
| PC/PS, 20 mol % | 2.6 ± 0.03 | 2.9 ± 0.02 | 3.4 ± 0.02 | 3.7 ± 0.02 | 3.9 ± 0.03 | 4.1 ± 0.02 |
| PC/PS, 40 mol % | 1.9 ± 0.01 | 2.2 ± 0.01 | 2.6 ± 0.02 | 2.8 ± 0.02 | 3.0 ± 0.01 | 3.1 ± 0.01 |
| PC/CL, 2.6 mol % | 3.9 ± 0.09 | 4.6 ± 0.09 | 5.6 ± 0.08 | 5.8 ± 0.07 | 6.0 ± 0.08 | 6.3 ± 0.08 |
| PC/CL, 5.3 mol % | 2.9 ± 0.04 | 3.5 ± 0.04 | 4.2 ± 0.03 | 4.5 ± 0.03 | 4.6 ± 0.03 | 4.7 ± 0.04 |
| PC/CL, 11 mol % | 2.0 ± 0.01 | 2.3 ± 0.01 | 2.7 ± 0.01 | 2.9 ± 0.02 | 3.0 ± 0.03 | 3.0 ± 0.02 |
| PC/CL, 25 mol % | 1.7 ± 0.01 | 1.9 ± 0.01 | 2.3 ± 0.01 | 2.3 ± 0.02 | 2.5 ± 0.01 | 2.5 ± 0.02 |
Enhanced aggregation of water-soluble proteins is generally ascribed to structural transformation of polypeptide chain into a partially unfolded conformation (47). Accumulating evidence substantiates the idea that the lipid bilayer can lower the activation energy barrier for protein unfolding (49,50). Nevertheless, to the best of our knowledge, definitive evidence for the unfolding of lysozyme upon membrane association is still lacking. Likewise, the fluorescence quenching experiments described here do not reveal transition of lysozyme into a partially unfolded state upon membrane binding. Yet, one should bear in mind that detection of increase in solvent accessibility of the buried tryptophans, which is expected to take place in the course of protein unfolding, may be hampered by relatively small contributions to overall lysozyme fluorescence by Trp28 and Trp111, occluded from solvent in the unperturbed protein state. Notably, lysozyme is a very stable protein whose secondary and tertiary structures have been reported not to undergo significant changes in acidic solution down to pH 0.6 (51). Therefore, if lysozyme adopts a more loose conformation in the membrane-associated state, this could not be exclusively attributed to the reduced interfacial pH, because, as indicated above, the lowest pH experienced by the protein bound to the model membranes used here is ∼5.2 (at 40 mol % PG). It seems probable that other factors such as formation of ionic contacts with anionic phospholipids and maximizing the interaction between hydrophobic amino acid residues and lipid acyl chains play essential roles in determining the structural features of bound protein. Clearly, the changes in lysozyme conformation and aggregation state on membrane binding must be coupled with structural reorganization of the lipid bilayer.
Fluorescence anisotropy measurements
In the last part of this study, we focused on lysozyme-induced modification of the bilayer physical state. To this end, steady-state fluorescence anisotropy of the bilayer-incorporated probe DPH, a parameter reflecting mainly the molecular order of lipid acyl chains, was measured for the indicated lipid-protein assemblies. Lysozyme association with a PC bilayer or model membranes containing 5 or 10 mol % of PG or PS was not accompanied by noticeable changes in DPH anisotropy, with this parameter taking values ∼0.1. Similarly, anisotropy was unaltered upon the binding of lysozyme to liposomes containing 2.6 or 5.2 mol % CL. However, for membranes with higher contents of anionic phospholipid lowering the lipid/protein molar ratio resulted in a marked increase of DPH anisotropy (Fig. 6).
FIGURE 6.

Changes in DPH fluorescence anisotropy upon lysozyme binding to the model membranes. Lipid concentration was 24 μM, probe concentration, 0.06 μM.
DPH fluorescence anisotropy reflects the lipid acyl-chain order or, more correctly, the bilayer free volume characterizing the difference between effective and van der Waals volumes of lipid molecules (52,53). Packing constraints and thermal motion cause trans-gauche isomerization of acyl chains, thereby producing dynamic, short-lived defects (kinks) in the hydrocarbon core. A local increment in free volume is generated in a bilayer due to lateral displacement of hydrocarbon chain on the kink formation. Our previous study employing pyrene excimer formation provided data in favor of reduction of the bilayer free volume upon lysozyme binding to PC and PC/CL model membranes (54). The results presented here are consistent with the above findings because lysozyme-induced increase of DPH anisotropy can be interpreted as reflecting restriction of the probe rotational mobility due to decreased rate of trans-gauche isomerization of hydrocarbon chains. This ordering effect is most likely to result from partial insertion of lysozyme into the nonpolar membrane core, giving rise to modification of the profile of acyl-chain order around the rough protein surface.
Although the binding data presented here are indicative of hydrophobic interactions of lysozyme with uncharged (PC) membranes, DPH anisotropy proved insensitive to protein-induced perturbation of PC bilayer. In keeping with this observation are the results of Arcuri et al. (10) on the association of lysozyme with PC/PE/Chol bilayers. Using the above values of binding parameters we approximated lysozyme concentrations yielding similar L/B ratios for uncharged and charged (fA = 0.4 or 0.25 (CL)) liposomes. It appeared that at L/B ratio ∼100, corresponding to saturating conditions for lysozyme adsorption onto PC membranes, DPH anisotropy in charged bilayers increased by 10–20%, with no statistically significant anisotropy changes being observed for PC bilayers. These findings suggest the extent and character of bilayer modification by lysozyme to depend on relative contributions of electrostatic and hydrophobic binding components. Protein location in a lipid bilayer is dictated by minimizing the interfacial free energies of interactions between polar and nonpolar portions of the protein and lipids that undergo interdependent conformational alterations. Evidently, orientation of lysozyme with respect to lipid-water interface, the depth of bilayer penetration, and the structure of the membrane-anchoring domain may differ for uncharged and charged membranes. Furthermore, acidic phospholipids may represent a key determinant for the protein adsorption behavior. Our data indicate that lysozyme binding to PG-, PS-, and CL-containing model membranes is accompanied by similar changes of all the examined parameters. However, at molar fractions of acidic phospholipid exceeding 10 mol %, the magnitude of the observed changes was markedly greater for PC/CL bilayers. This finding cannot be interpreted as arising from enhanced electrostatic binding because, as follows from our estimates, surface potential of PC/CL membranes is slightly lower than that of PC/PG (PS) membranes (Table 2). CL, a structurally unique phospholipid composed of two phosphate groups and four acyl chains, is known as having two widely separated pK values, one at 2.8 and a second between 7.5 and 9.5 (55). The peculiar ionization properties of CL are attributed to intramolecular hydrogen bonding between the free hydroxyl of the central glycerol and protonated phosphate. For this reason, at neutral pH there exist two populations of CL molecules representing both deprotonated and partially protonated species. Another distinctive feature of CL is associated with its ability to form nonbilayer structures, inverted micelles, and hexagonal HII phase. Such a rearrangement of lipid molecules has been observed upon cyt c association with PC/CL membranes (57). Once the nonbilayer structures are formed, the charged protein molecules may become trapped into the interior of aqueous cylinders, spanning the lipid bilayer. In principle, one cannot rule out this possibility for lysozyme, which does resemble cyt c in its size and charge. However, this assumption is difficult to reconcile with the propensity of lysozyme for self-association in a membrane environment. In addition, formation of nonbilayer structures in PC/CL bilayers might be expected to manifest in peculiar behavior of the parameters under study. Since this is not the case, we are prone to hypothesize that CL-induced structural alterations of lysozyme are different from those caused by PG or PS. Likewise, the extent of the protein bilayer penetration and concomitant perturbation of acyl-chain packing seem to be more pronounced in CL-containing membranes. Comparison of the L/P ratios at which lysozyme oligomerization is assumed to occur (5–50) with those coupled with ordering of hydrocarbon chains (40–300) suggests that protein insertion into membrane interior precedes its aggregation.
TABLE 2.
Electrostatic surface potential of the model membranes under study
|
ψ0, mV
|
|||
|---|---|---|---|
| Proportion of PG, PS, (CL) mol % | PC/PG | PC/PS | PC/CL |
| 5 (2.6) | −35 | −34 | −34 |
| 10 (5.3) | −60 | −56 | −55 |
| 20 (11) | −92 | −80 | −79 |
| 40 (25) | −126 | −101 | −105 |
Concluding remarks
Overall, the above findings strongly suggest that the observed enhanced tendency of lysozyme to aggregate in membranes with increasing membrane surface charge cannot be explained exclusively by electrostatically driven protein accumulation at the lipid-water interface. Modification of the protein self-associating properties may reflect the ability of acidic lipids to alter the conformational state of lysozyme, its interfacial orientation, and the extent of bilayer penetration. Interestingly, recent studies revealed a relationship between the protein aggregation propensity and its structural instability arising from the underwrapping of backbone amide-carbonyl H-bonds (27). A structural motif such as partial exposure of H-bonds to water was supposed to be an important determinant of protein reactivity. It was demonstrated that a lower extent of H-bond protection from water correlates with higher amyloidogenic propensity and higher affinity of a series of soluble proteins for PC bilayers. Along this line, energetically favorable screening of underwrapped H-bonds must be regarded as an additional factor that may account for the observed differences in lysozyme aggregation behavior in neutral and negatively charged membranes. More specifically, if one assumes that the orientation and location of lysozyme in a PC bilayer is governed by minimizing the H-bond exposure to solvent, some reduction of the protein reactivity, and, as a consequence, decrease of the aggregation propensity might be anticipated. In the meantime, the orientation of lysozyme relative to the negatively charge bilayer is likely to be different, being dictated to a large extent by electrostatic factors. In this case, the number of the exposed H-bonds is expected to be higher, thereby enhancing this protein's tendency to aggregate. All this notwithstanding, one should bear in mind that aggregation behavior of a membrane-bound protein reflects a complex interplay between a variety of factors, differing in their nature and relative significance.
Notably, theoretical analyses of the energetics of lipid-lipid interactions compared to lipid-helix and helix-helix interactions showed that even a small increase in hydrocarbon chain order could favor oligomerization of transmembrane α-helices (58). Hydrophobic mismatch is assumed to promote lipid-mediated protein-protein attraction (59). Attractive interactions between interfacially adsorbed (partially inserted) α-helical amphipathic peptides were theoretically predicted on the basis of mean-field chain packing theory (60). It cannot be excluded that similar driving forces play a role in the observed aggregation of membrane-bound lysozyme. Other membrane-related determinants of lysozyme aggregation behavior include conformational changes of the protein, increase of its local concentration at lipid-water interface, specific orientation of aggregating species, neutralization of the protein surface charges by anionic lipid headgroups, and particular arrangement of the inserted and solvent exposed segments of the protein molecule. Of interest in this regard is the viewpoint that helix-loop-helix domain (87–114 residues of hen egg white lysozyme) located at the upper lip of the active site cleft may serve as a membrane anchor (13). The hydrophobic portion of this domain (residues 87–95) is likely to insert into a nonpolar bilayer region, with terminal basic residues (Arg112, Arg114) being in contact with anionic headgroups of phospholipids, and tryptophans (Trp108, Trp111) residing at lipid-water interface (Fig. 7). In such an orientation, the portion of lysozyme molecule reported to possess high aggregation propensity (residues 49–101 (16)) appears to embrace both solvent-exposed and membrane-embedded protein segments. Thus, in keeping with the above considerations, it seems probable that membrane-mediated self-association of lysozyme proceeds via formation of intermolecular contacts between protein species partially inserted into the lipid bilayer.
FIGURE 7.

Schematic representation for possible disposition of lysozyme relative to a lipid-water interface.
Within the context of this reasoning, it is important to analyze to what extent the membrane binding and self-associating propensities of lysozyme may be influenced by its labeling with FITC, employed in the present study to monitor lysozyme-lipid interactions. The covalent attachment of fluorescein moiety of FITC to a protein occurs in the amino groups of lysine residues. Lysozyme contains six lysines, Lys1, Lys13, Lys33, Lys96, Lys97, and Lys116. In principle, each of these residues represents a potential site for covalent labeling with FITC. However, lysozyme lysine residues have been reported to differ in their chemical properties (61). Mass-spectrometric peptide-mapping analyses showed that the relative reactivities of these lysines directly correlate with their surface accessibilities (determined from x-ray structural data for a 1.4 Å van der Waals sphere). Accordingly, the highest chemical reactivity was found for Lys97 and Lys33 whose surface accessibilities (47% and 40%, respectively) are twice as large as those for the other lysine residues (∼20%). These findings allow us to assume that at the labeling ratio of 0.9, characteristic of the Fl-lysozyme preparation used in our experiments, the fluorescein moiety is attached primarily to either Lys97 or Lys33. If lysozyme is anchored to membrane via helix-loop-helix domain (Fig. 7), both of these residues would reside in the interface favoring the formation of ionic contacts with phospholipid headgroups. In this case, lysozyme labeling with FITC might be expected to lower the affinity of this protein for negatively charged lipid bilayers. Indeed, it appeared that unlabeled lysozyme can displace Fl-lysozyme from the membrane binding sites, as judged by the observed increase of FITC fluorescence (Fig. 8). Importantly, one of the most reactive lysine residues, Lys97, is located in the protein active site—which has been suggested to participate in the head-to-tail lysozyme self-association implicating the formation of asymmetric intermolecular contacts (62,63). Fluorescent labeling may therefore be anticipated to modify the aggregation properties of lysozyme. For instance, if the fluorescein moiety covalently attached to Lys97 forms ionic contacts with the neighboring Lys96, charge neutralization would result in the enhancement of the protein self-associating propensity. However, because of the low labeling ratio (one dye molecule per protein), the principal features of lysozyme adsorption behavior (especially when relating to the effect of surface charge on the protein oligomerization state) seem to be similar for the native and labeled proteins. All these issues, together with the above assumption about the reduced label exposure to interfacial microenvironment in oligomeric protein species, give ample grounds for believing that, in the Fl-lysozyme preparation employed in our experiments, fluorescein is attached to Lys97, located in the active site, and exhibited highest surface accessibility and chemical reactivity.
FIGURE 8.

Competitive binding of Fl-lysozyme and unlabeled lysozyme to lipid vesicles. Fractional displacement of Fl-lysozyme was calculated as (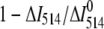 ),
), 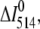 and ΔI514 are the changes of Fl-lysozyme fluorescence intensity in the absence and presence of unlabeled protein, respectively. Lipid concentration was 45 μM.
and ΔI514 are the changes of Fl-lysozyme fluorescence intensity in the absence and presence of unlabeled protein, respectively. Lipid concentration was 45 μM.
It seems also of significance to emphasize that the above adsorption models describe the equilibria between free protein in solution and the interfacially adsorbed protein species, ignoring the possibility of intercalation of lysozyme into the lipid phase. Evidently, in this case the concentration of lipids involved in protein binding (La) cannot be simply taken as 0.5 L, because of the disruption of membrane integrity by the inserted protein species. Direct evidence for the ability of lysozyme to destabilize membrane structure by producing local fluctuations in lipid packing comes from the leakage experiments demonstrating that lysozyme can induce extensive release of aqueous liposomal contents in the absence of vesicle aggregation or fusion (6,9,10). The SPT-based model describing the competition between the processes of surface adsorption and bilayer insertion has been recently proposed by Zuckermann and Heimburg (64). This model allows for factors such as 1), reduction of the membrane charge density resulting from the increase of the total interfacial area upon protein insertion; and 2), the edge tension, which can oppose or favor membrane penetration of the protein, depending on the bilayer elastic properties and spatial configuration of the membrane-buried protein domain. Correct application of this model would require global analysis of multiple data sets obtained with several experimental variables (for instance, at varying ionic strength, protein, and lipid concentrations). For the lysozyme-lipid system, this is problematic because of the rather narrow ranges of the protein and lipid concentrations at which the measured quantity is not distorted by the contributions from vesicle aggregation and fusion. In the present study we restricted ourselves to the quantitative interpretation of the limited amount of binding data in terms of the simplified SPT models. Our goal was to demonstrate that sigmoidal shape of the adsorption isotherms obtained under conditions of decreasing surface coverage can be explained only when assuming protein oligomerization. No other processes such as coverage-dependent variations in surface charge density or a shift of equilibrium between different protein species could account for the observed adsorption behavior of lysozyme. The experiments aimed at gaining extended data sets appropriate for more adequate analysis of the surface adsorption and membrane insertion processes are currently planned in our laboratory.
To summarize, the present study provides further insight into several important aspects of lysozyme-membrane interactions. By monitoring fluorescence changes of fluorescein-labeled lysozyme, we explored lysozyme adsorption to model membranes composed of PC in mixtures with PG, PS, or CL, with varying content of these anionic phospholipids. The shape of adsorption isotherms was found to change from Langmuir-like to sigmoidal upon increasing the surface charge of the lipid bilayers. Analysis of the binding data in terms of electrostatically controlled adsorption of a large self-associating ligand allowed us to interpret this behavior as arising from aggregation of the adsorbed protein. Fluorescence quenching studies revealed that the accessibility of dominating fluorophores Trp62 and Trp108 to a solvent decreases upon membrane binding of lysozyme. These tryptophan residues were assumed to be involved in the formation of intermolecular contacts between the aggregating protein species. Measurements of steady-state fluorescence anisotropy of bilayer-incorporated probe DPH provided evidence for lysozyme-induced increase of acyl-chain order at molar fractions of acidic phospholipid exceeding 10 mol %. This effect was attributed to the insertion of lysozyme into the nonpolar membrane core. Based on the dependencies of the examined parameters on lipid/protein molar ratio we hypothesized that bilayer penetration of lysozyme occurs before its self-association. Taken together, the results obtained emphasize the importance of both electrostatic and hydrophobic protein-lipid interactions in determining the aggregation properties of membrane-bound lysozyme. In this context, it is tempting to assume that lysozyme oligomerization in membrane environment represents an essential prerequisite for its assembly into amyloidlike structures described recently by Zhao et al. (20). Furthermore, bactericidal action of lysozyme may be related to enhanced membrane permeabilization ability of the aggregated protein species (20,65).
Acknowledgments
The authors thank Dr. Juha-Matti Alakoskela for helpful discussions, and Kristiina Söderholm, Kaija Tiilikka, and Kaija Niva for excellent technical assistance.
G.G. gratefully acknowledges a visiting scientist award by the Sigrid Juselius Foundation. H.B.B.G. is supported by the Finnish Academy and Sigrid Juselius Foundation.
APPENDIX
Protein binding to lipid vesicles is followed by the reduction of bimolecular rate constant for collisional quenching due to orientational constraints and slower translational and rotational diffusion of immobilized fluorophore (45). This may lead to KSV decrease independent of modification of the protein structure or geometrical masking factors. Possible contribution of this phenomenon to KSV changes can be approximated by calculating the ratio of the kq values expected for free Trp residues in solution (kMQ) and in a lipid-bound (kVQ) protein. According to the equations by Shoup et al. (44) for the rate of reaction between asymmetric molecules, kMQ can be evaluated from
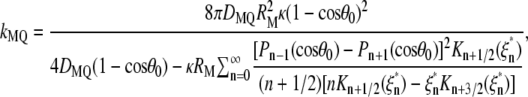 |
(A1) |
where 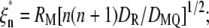 DMQ is a sum of translational diffusion coefficients of the quencher (DQ) and the protein (DM), which for a spherical protein of radius RM is given by the Stokes-Einstein expression DM = kT/6πηRM (cm2/s); T is the temperature; k is the Boltzmann's constant; η is the solvent viscosity; DR is rotational diffusion coefficient;
DMQ is a sum of translational diffusion coefficients of the quencher (DQ) and the protein (DM), which for a spherical protein of radius RM is given by the Stokes-Einstein expression DM = kT/6πηRM (cm2/s); T is the temperature; k is the Boltzmann's constant; η is the solvent viscosity; DR is rotational diffusion coefficient; 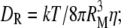 κ is a parameter related to the probability of reaction upon encounter, which approaches a value of infinity for a probability of 1; K is a modified spherical Bessel function of the third kind; and Pn(cosθ0) is the nth order Legendre polynomial. The reactive area is determined by the cone angle 2θ0 related to the fraction of the fluorophore's area exposed on the protein surface (fMQ) and fluorophore radius (RF),
κ is a parameter related to the probability of reaction upon encounter, which approaches a value of infinity for a probability of 1; K is a modified spherical Bessel function of the third kind; and Pn(cosθ0) is the nth order Legendre polynomial. The reactive area is determined by the cone angle 2θ0 related to the fraction of the fluorophore's area exposed on the protein surface (fMQ) and fluorophore radius (RF), 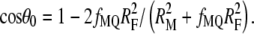 Parameter fMQ can vary from 0 (for fluorophore inaccessible to a quencher) to 1 (for completely exposed fluorophore).
Parameter fMQ can vary from 0 (for fluorophore inaccessible to a quencher) to 1 (for completely exposed fluorophore).
To estimate kVQ, Eq. A1 was used in the form valid for the limiting case where the vesicle radius (RV) is much greater than the quencher radius (RQ),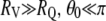 (43):
(43):
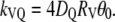 |
(A2) |
Taking RQ = 0.2 nm for acrylamide, RF = 0.2 nm for tryptophan, RM = 1.8 nm, RV = 50 nm, DQ = 1 × 10−5 cm2/s, and fMQ = 1 we found maximum possible value of kMQ/kVQ to be ∼1.3. Thus, Stern-Volmer constant could reduce by a factor of 1.3 at most only because of the slower translational and rotational diffusion of membrane-bound lysozyme.
Editor: Antoinette Killian.
References
- 1.Wertz, C. F., and M. Santore. 2002. Adsorption and reorientation kinetics of lysozyme on hydrophobic surfaces. Langmuir. 18:1190–1199. [Google Scholar]
- 2.Gulik-Krzywicki, T., E. Shechter, V. Luzzati, and M. Faure. 1969. Interactions of proteins and lipids: structure and polymorphism of protein-lipid-water phases. Nature. 223:1116–1120. [DOI] [PubMed] [Google Scholar]
- 3.Sessa, G., and G. Weissmann. 1970. Incorporation of lysozyme into liposomes. A model for structure-linked latency. J. Biol. Chem. 245:3295–3301. [PubMed] [Google Scholar]
- 4.Kimelberg, H. K. 1976. Protein-liposome interactions and their relevance to the structure and function of cell membranes. Mol. Cell. Biochem. 10:171–190. [DOI] [PubMed] [Google Scholar]
- 5.Zschornig, O., G. Paasche, C. Thieme, N. Korb, A. Fahrwald, and K. Arnold. 2000. Association of lysozyme with phospholipid vesicles is accompanied by membrane surface dehydration. Gen. Physiol. Biophys. 19:85–101. [PubMed] [Google Scholar]
- 6.Zschornig, O., G. Paasche, C. Thieme, N. Korb, and K. Arnold. 2005. Modulation of lysozyme charge influences interaction with phospholipid vesicles. Colloids Surf. B Biointerfaces. 42:69–78. [DOI] [PubMed] [Google Scholar]
- 7.Posse, E., A. Vinals, B. de Arcuri, R. Farias, and R. D. Morero. 1990. Lysozyme induced fusion of negatively charged phospholipid vesicles. Biochim. Biophys. Acta. 104:390–394. [DOI] [PubMed] [Google Scholar]
- 8.Arnold, K., D. Hoekstra, and S. Ohki. 1992. Association of lysozyme to phospholipid surfaces and vesicle fusion. Biochim. Biophys. Acta. 1124:88–94. [DOI] [PubMed] [Google Scholar]
- 9.Posse, E., B. F. De Arcuri, and R. D. Morero. 1994. Lysozyme interactions with phospholipid vesicles: relationships with fusion and release of aqueous content. Biochim. Biophys. Acta. 1193:101–106. [DOI] [PubMed] [Google Scholar]
- 10.de Arcuri, B. F., G. F. Vechetti, R. N. Chehin, F. M. Goni, and R. D. Morero. 1999. Protein-induced fusion of phospholipid vesicles of heterogeneous sizes. Biochem. Biophys. Res. Commun. 262:586–590. [DOI] [PubMed] [Google Scholar]
- 11.Ibrahim, H. R., M. Yamada, K. Matsushita, K. Kobayashi, and A. Kato. 1994. Enhanced bactericidal action of lysozyme to Escherichia coli by inserting a hydrophobic pentapeptide into its C terminus. J. Biol. Chem. 269:5053–5063. [PubMed] [Google Scholar]
- 12.Pellegrini, A., U. Thomas, N. Bramaz, S. Klauser, P. Hunziker, and R. von Fellenberg. 1997. Identification and isolation of a bactericidal domain in chicken egg white lysozyme. J. Appl. Microbiol. 82:372–378. [DOI] [PubMed] [Google Scholar]
- 13.Ibrahim, H. R., U. Thomas, and A. Pellegrini. 2001. A helix-loop-helix peptide at the upper lip of the active site cleft of lysozyme confers potent antimicrobial activity with membrane permeabilization action. J. Biol. Chem. 276:43767–43774. [DOI] [PubMed] [Google Scholar]
- 14.Adams, S., A. M. Higgins, and R. A. L. Jones. 2002. Surface-mediated folding and misfolding of proteins at lipid/water interfaces. Langmuir. 18:4854–4861. [Google Scholar]
- 15.Sato, T., K. W. Mattison, P. L. Dubin, M. Kamachi, and Y. Morishima. 1998. Effect of protein aggregation on the binding of lysozyme to pyrene-labeled polyanions. Langmuir. 14:5430–5437. [Google Scholar]
- 16.Frare, E., P. Polverino de Laureto, J. Zurdo, C. Dobson, and A. Fontana. 2004. A highly amyloidogenic region of hen lysozyme. J. Mol. Biol. 340:1153–1165. [DOI] [PubMed] [Google Scholar]
- 17.Bergers, J. J., M. H. Vingerhoeds, L. van Bloois, J. N. Herron, L. H. Jassen, M. J. Fisher, and D. Crommelin. 1993. The role of protein charge in protein-lipid interactions. pH-dependent changes of the electrophoretic mobility of liposomes through adsorption of water-soluble, globular proteins. Biochemistry. 32:4641–4649. [DOI] [PubMed] [Google Scholar]
- 18.Tsunoda, T., T. Imura, M. Kadota, T. Yamazaki, H. Yamauchi, K. Ok Kwon, S. Yokoyama, H. Sakai, and M. Abe. 2001. Effects of lysozyme and bovine serum albumin on membrane characteristics of dipalmitoylphosphatidylglycerol liposomes. Colloids Surf. B Biointerfaces. 20:155–163. [DOI] [PubMed] [Google Scholar]
- 19.Touch, V., S. Hayakawa, and K. Saito. 2004. Relationships between conformational changes and antimicrobial activity of lysozyme upon reduction of its disulfide bonds. Food Chem. 84:421–428. [Google Scholar]
- 20.Zhao, H., E. K. J. Tuominen, and P. K. J. Kinnunen. 2004. Formation of amyloid fibers triggered by phosphatidylserine-containing membranes. Biochemistry. 43:10302–10307. [DOI] [PubMed] [Google Scholar]
- 21.Stefani, M. 2004. Protein misfolding and aggregation: new examples in medicine and biology of the dark side of the protein world. Biochim. Biophys. Acta. 1739:5–25. [DOI] [PubMed] [Google Scholar]
- 22.Serpell, L. C. 2000. Alzheimer's amyloid fibrils: structure and assembly. Biochim. Biophys. Acta. 1502:16–30. [DOI] [PubMed] [Google Scholar]
- 23.Pepys, M. B., P. N. Hawkins, D. R. Booth, D. M. Vigushin, G. A. Tennent, A. K. Souter, N. Totty, O. Nguyen, C. C. F. Blake, C. J. Terry, T. G. Feest, A. M. Zalin, and J. J. Hsuan. 1993. Human lysozyme gene mutations cause hereditary systemic amyloidosis. Nature. 362:553–557. [DOI] [PubMed] [Google Scholar]
- 24.Cao, A., D. Hu, and L. Lai. 2004. Formation of amyloid fibrils from fully reduced hen egg white lysozyme. Protein Science. 13:319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dumoulin, M., D. Canet, A. M. Last, E. Pardon, D. B. Archer, S. Muyldermans, L. Wyns, A. Matagne, C. V. Robinson, C. Redfield, and C. M. Dobson. 2005. Reduced global cooperativity is a common feature underlying the amyloidogenicity of pathogenic lysozyme mutations. J. Mol. Biol. 346:773–788. [DOI] [PubMed] [Google Scholar]
- 26.Bokvist, M., F. Lindstrom, A. Watts, and G. Grobner. 2004. Two types of Alzheimer's β-amyloid (1–40) peptide membrane interactions: aggregation preventing transmembrane anchoring versus accelerated surface fibril formation. J. Mol. Biol. 335:1039–1049. [DOI] [PubMed] [Google Scholar]
- 27.Fernandez, A., and R. S. Berry. 2003. Proteins with H-bond packing defects are highly interactive with lipid bilayers: implications for amyloidogenesis. Proc. Natl. Acad. Sci. USA. 100:2391–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gorbenko, G. P., and P. K. J. Kinnunen. 2006. The role of lipid-protein interactions in amyloid-type protein fibril formation. Chem. Phys. Lipids. 141:72–82. [DOI] [PubMed] [Google Scholar]
- 29.Kok, R. J., M. Haas, F. Moolenar, D. de Zeeuw, and D. K. F. Meijer. 1998. Drug delivery to the kidneys and the bladder with the low molecular weight protein lysozyme. Ren. Fail. 20:211–217. [DOI] [PubMed] [Google Scholar]
- 30.Lakowicz, J. R. 1999. Principles of Fluorescent Spectroscopy. Plenum Press, New York.
- 31.Chatelier, R., and A. P. Minton. 1996. Adsorption of globular proteins on locally planar surfaces: models for the effect of excluded surface area and aggregation of adsorbed protein on adsorption equilibria. Biophys. J. 71:2367–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heimburg, T., and D. Marsh. 1995. Protein surface-distribution and protein-protein interactions in the binding of peripheral proteins to charged lipid membranes. Biophys. J. 68:536–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanford, C. 1955. The electrostatic free energy of globular protein ions in aqueous solution. J. Phys. Chem. 59:788–793. [Google Scholar]
- 34.Jahnig, F. 1976. Electrostatic free energy and shift of the phase transition for charged lipid membranes. Biophys. Chem. 4:309–318. [DOI] [PubMed] [Google Scholar]
- 35.Ivkov, V. G., and G. N. Berestovsky. 1981. Dynamic Structure of Lipid Bilayer. Nauka, Moscow.
- 36.Minton, A. 2000. Effects of excluded surface area and adsorbate clustering on surface adsorption of proteins. I. Equilibrium models. Biophys. Chem. 86:239–247. [DOI] [PubMed] [Google Scholar]
- 37.Minton, A. P. 1999. Adsorption of globular proteins on locally planar surfaces. II. Models for the effect of multiple adsorbate conformations on adsorption equilibria and kinetics. Biophys. J. 76:176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gorbenko, G. P., J. G. Molotkovsky, and P. K. J. Kinnunen. 2006. Cytochrome c interaction with cardiolipin/phosphatidylcholine model membranes: effect of cardiolipin protonation. Biophys. J. 90:4093–4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sankaram, M., and D. Marsh. 1993. Protein-lipid interactions with peripheral membrane proteins. In Protein-Lipid Interactions. Elsevier, Dordrecht, The Netherlands.
- 40.Lomakin, A., D. S. Chung, G. B. Benedek, D. A. Kirschner, and D. B. Teplow. 1996. On the nucleation and growth of amyloid beta-protein fibrils: detection of nuclei and quantitation of rate constants. Proc. Natl. Acad. Sci. USA. 93:1125–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Imoto, T., L. S. Forster, J. A. Rupley, and F. Tanaka. 1971. Fluorescence of lysozyme: emission from tryptophan residues 62 and 108 and energy migration. Proc. Natl. Acad. Sci. USA. 69:1151–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nishimoto, E., S. Yamashita, A. G. Szabo, and T. Imoto. 1998. Internal motion of lysozyme studied by time-resolved fluorescence depolarization of tryptophan residues. Biochemistry. 37:5599–5607. [DOI] [PubMed] [Google Scholar]
- 43.Li, S. J., A. Nakagawa, and T. Tsukihara. 2004. Ni2+ binds to active site of hen egg-white lysozyme and quenches fluorescence of Trp62 and Trp108. Biochem. Biophys. Res. Commun. 324:529–533. [DOI] [PubMed] [Google Scholar]
- 44.Shoup, D., G. Lipari, and A. Szabo. 1981. Diffusion-controlled bimolecular reaction rates. The effect of rotational diffusion and orientation constraints. Biophys. J. 36:697–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson, D. A., and J. Yguerabide. 1985. Solute accessibility to n′-fluorescein isothiocyanate-lysine-23 cobra α-toxin bound to the acetylcholine receptor. A consideration of the effect of rotational diffusion and orientation constraints on fluorescence quenching. Biophys. J. 48:949–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Banerjee, S. K., A. Pogolotti, and J. A. Rupley. 1975. Self-association of lysozyme. Thermochemical measurements: effect of chemical modification of Trp62, Trp108 and Glu35. J. Biol. Chem. 250:8260–8266. [PubMed] [Google Scholar]
- 47.Norton, R. S., and A. Allerhand. 1977. Participation of tryptophan 62 in the self-association of hen egg white lysozyme. Application of natural abundance carbon 13 nuclear magnetic resonance spectroscopy. J. Biol. Chem. 252:1795–1798. [PubMed] [Google Scholar]
- 48.Uversky, V. N., and A. L. Fink. 2004. Conformational constraints for amyloid fibrillation: the importance of being unfolded. Biochim. Biophys. Acta. 1698:131–153. [DOI] [PubMed] [Google Scholar]
- 49.Pinheiro, T. J. T., G. A. Elove, A. Watts, and H. Roder. 1997. Structural and kinetic description of cytochrome c unfolding induced by the interaction with lipid vesicles. Biochemistry. 36:13122–13132. [DOI] [PubMed] [Google Scholar]
- 50.Shin, I., D. Kreimer, I. Silman, and L. Weiner. 1997. Membrane-promoted unfolding of acetylcholinesterase: a possible mechanism for insertion into the lipid bilayer. Proc. Natl. Acad. Sci. USA. 94:2848–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Polverino de Laureto, P., E. Frare, R. Gottardo, H. Van Dael, and A. Fontana. 2002. Partly folded states of members of the lysozyme/lactalbumin superfamily: a comparative study by circular dichroism spectroscopy and limited proteolysis. Protein Sci. 11:2932–2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hildebrand, K., and C. Nicolau. 1979. Nanosecond fluorescence anisotropy decays of 1,6-diphenyl-1,3,5-hexatriene in membranes. Biochim. Biophys. Acta. 553:365–377. [DOI] [PubMed] [Google Scholar]
- 53.Kinnunen, P. J. K. 1991. On the principles of functional ordering in biological membranes. Chem. Phys. Lipids. 57:375–399. [DOI] [PubMed] [Google Scholar]
- 54.Ioffe, V. M., and G. P. Gorbenko. 2005. Lysozyme effect on structural state of model membranes as revealed by pyrene excimerization studies. Biophys. Chem. 114:199–204. [DOI] [PubMed] [Google Scholar]
- 55.Kates, M., J. Syz, D. Gosser, and T. H. Haines. 1993. pH-dissociation characteristics of cardiolipin and its 2′-deoxy analogue. Lipids. 28:877–882. [DOI] [PubMed] [Google Scholar]
- 56.Tocanne, J., and J. Teissie. 1990. Ionization of phospholipids and phospholipid-supported interfacial lateral diffusion of protons in membrane model systems. Biochim. Biophys. Acta. 1031:111–142. [DOI] [PubMed] [Google Scholar]
- 57.de Kruijff, B., P. R. Cullis, A. J. Verkleij, M. J. Hope, C. J. A. van Echteld, T. F. Taraschi, P. van Hoogevest, J. A. Killian, A. Rietveld, and A. T. M. van der Steen. 1985. Modulation of lipid polymorphism by lipid-protein interactions. In Progress in Protein-Lipid Interactions. A. Watts and J. J. H. H. M. de Pont, editors. Elsevier Science Publishers, Dordrecht, The Netherlands.
- 58.Lee, A. G. 2004. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta. 1666:62–87. [DOI] [PubMed] [Google Scholar]
- 59.Gil, T., J. H. Ipsen, O. G. Mouritsen, M. C. Sabra, M. M. Sperotto, and M. J. Zuckermann. 1998. Theoretical analysis of protein organization in lipid membranes. Biochim. Biophys. Acta. 1376:245–266. [DOI] [PubMed] [Google Scholar]
- 60.Zemel, A., A. Ben-Shaul, and S. May. 2004. Membrane perturbation induced by interfacially absorbed peptides. Biophys. J. 86:3607–3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Suckau, D., M. Mak, and M. Przybylski. 1992. Protein surface topology-probing by selective chemical modification and mass spectrometric peptide mapping. Proc. Natl. Acad. Sci. USA. 89:5630–5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sophianopoulos, A. J. 1969. Association sites of lysozyme in solution. I. The active site. J. Biol. Chem. 244:3188–3193. [PubMed] [Google Scholar]
- 63.Holloday, L. A., and A. J. Sophianopoulos. 1972. Association sites of lysozyme in solution. II. Concentration-dependence of the near-ultraviolet circular dichroism. J. Biol. Chem. 247:1976–1979. [PubMed] [Google Scholar]
- 64.Zuckermann, M. J., and T. Heimburg. 2001. Insertion and pore formation driven by adsorption of proteins onto lipid bilayer membrane-water interfaces. Biophys. J. 81:2458–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhao, H., R. Sood, A. Jutila, S. Bose, G. Fimland, J. Nissen-Meyer, and P. K. J. Kinnunen. 2006. Interaction of the antimicrobial peptide pheromone plantaricin A with model membranes: implications for a novel mechanism of action. Biochim. Biophys. Acta. 1758:1461–1474. [DOI] [PubMed] [Google Scholar]