

Abstract
To create cytotoxic hybrid analogs of somatostatin (SST), octapeptides RC-160 (d-Phe-C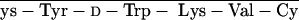 s-Trp-NH2) and RC-121 (d-Phe-C
s-Trp-NH2) and RC-121 (d-Phe-C s-Thr-NH2) were linked to doxorubicin (DOX) or its superactive derivative, 2-pyrrolino-DOX (AN-201). The conjugation was performed by coupling N-9-fluorenylmethoxycarbonyl (N-Fmoc)-DOX-14-O-hemiglutarate or 2-pyrrolino-DOX-14-O-hemiglutarate to the amino terminus of [Lys(Fmoc)5]RC-160 yielding AN-163 and AN-258, respectively, after deprotection. The respective cytotoxic conjugates of RC-121 (AN-162 and AN-238) were prepared similarly. In vitro tests on human cancer cell lines—MKN-45 gastric cancer, MDA-MB-231 breast cancer, PC-3 prostate cancer, and MIA PaCa-2 pancreatic cancer—demonstrated that the antiproliferative activity of the cytotoxic radicals in these conjugates was virtually retained. In H-345 human small cell lung carcinoma cell line, conjugates of RC-121 preserved the cytotoxic activity of their radicals, but the hybrids with RC-160 showed ≈10 times lower activity. The ability of the carriers and the hybrids to inhibit the binding of 125I-labeled RC-160 to receptors for SST on rat pituitary membrane preparation was also determined. The cytotoxic conjugates inhibited 50% of the specific binding of the radioligand in the nanomolar concentration range (IC50 < 80 nM). When SST-like activities of AN-238 and its carrier, RC-121, were compared in the rat pituitary superfusion system, both compounds were found to suppress a stimulated growth hormone release at nanomolar concentrations. Preliminary studies in animal models of breast and prostate cancers showed that AN-238 is less toxic than AN-201 and more potent in inhibiting tumor growth. These highly active cytotoxic analogs of SST have been designed as targeted antitumor agents for the treatment of various cancers expressing receptors for SST octapeptides.
s-Thr-NH2) were linked to doxorubicin (DOX) or its superactive derivative, 2-pyrrolino-DOX (AN-201). The conjugation was performed by coupling N-9-fluorenylmethoxycarbonyl (N-Fmoc)-DOX-14-O-hemiglutarate or 2-pyrrolino-DOX-14-O-hemiglutarate to the amino terminus of [Lys(Fmoc)5]RC-160 yielding AN-163 and AN-258, respectively, after deprotection. The respective cytotoxic conjugates of RC-121 (AN-162 and AN-238) were prepared similarly. In vitro tests on human cancer cell lines—MKN-45 gastric cancer, MDA-MB-231 breast cancer, PC-3 prostate cancer, and MIA PaCa-2 pancreatic cancer—demonstrated that the antiproliferative activity of the cytotoxic radicals in these conjugates was virtually retained. In H-345 human small cell lung carcinoma cell line, conjugates of RC-121 preserved the cytotoxic activity of their radicals, but the hybrids with RC-160 showed ≈10 times lower activity. The ability of the carriers and the hybrids to inhibit the binding of 125I-labeled RC-160 to receptors for SST on rat pituitary membrane preparation was also determined. The cytotoxic conjugates inhibited 50% of the specific binding of the radioligand in the nanomolar concentration range (IC50 < 80 nM). When SST-like activities of AN-238 and its carrier, RC-121, were compared in the rat pituitary superfusion system, both compounds were found to suppress a stimulated growth hormone release at nanomolar concentrations. Preliminary studies in animal models of breast and prostate cancers showed that AN-238 is less toxic than AN-201 and more potent in inhibiting tumor growth. These highly active cytotoxic analogs of SST have been designed as targeted antitumor agents for the treatment of various cancers expressing receptors for SST octapeptides.
Keywords: targeted chemotherapeutic agents, hybrid molecules, receptor binding, antiproliferative activity
Somatostatin (SST) is a hormonal neuropeptide that was isolated from ovine (1) and later from porcine hypothalami (2) and was found to inhibit growth hormone secretion from the pituitary gland (1–3). Many other physiological functions of SST also have been identified including inhibition of release of gastrin, insulin, glucagon, and various intestinal hormones, suppression of gastric and pancreatic exocrine secretion, and modulation of biliary and gastrointestinal motility (1–3). SST also plays a role as a neurotransmitter (1–3). SST was found to exist in two molecular forms, SST-14 and SST-28, that have similar physiological activities (3). In view of the short half-life of SST-14 in circulation and many diverse effects it can exert, it was important to develop more stable and selective synthetic analogs (3–5). A significant milestone in these efforts was the design and synthesis of Sandostatin (octreotide), an octapeptide analog of the native SST (5). This compound was found to be 50 times more potent than SST-14 in suppressing the release of growth hormone (GH) and more selective in its effects (5). Various synthetic octapeptides were subsequently synthesized, such as RC-160 (vapreotide) and RC-121, which were developed in our institute (3, 6). Numerous studies on potential applications in oncology were carried out with some of these compounds including octreotide and RC-160 (3, 7–13). Investigations of the binding of the octapeptides and SST-14 or SST-28 to the receptors on various normal and malignant cells led to the eventual discovery of five SST receptor subtypes (SSTR) and the cloning of their corresponding mRNAs (9).The potential role of these SSTR in cancer therapy has also been the subject of numerous studies (9–21). Although SST-14 or SST-28 show binding to SSTR with similar affinity, the octapeptide analogs display high affinity to SSTR2 and SSTR5, moderate affinity to SSTR3, and have only poor binding to SSTR1 and SSTR4 (9, 14, 18). Based on the findings that various neuroendocrine malignancies and many other solid tumors such as breast, lung, renal, pancreatic, gastric, colon cancers, and brain tumors as well as androgen-independent metastatic adenocarcinoma of the prostate express SSTR2 and/or SSTR5 (3, 9, 13–22), radiolabeled octapeptide analogs were designed for autoradiographic detection of such tumors and their metastases (3, 7, 23, 24). Thus, [111In-diethylenetriaminepentaacetic acid (DTPA)-d-Phe1]octreotide was successfully used for the diagnosis and scintigraphic imaging of various cancers in more than 1,000 patients (25). These results, and our previous promising experience with the antitumor activity of an early cytotoxic SST analog containing methotrexate (7, 26), prompted us to try to develop modern, more active targeted cytotoxic analogs of SST.
Recently, we published the synthesis and in vitro and in vivo evaluation of a series of targeted cytotoxic analogs of luteinizing hormone-releasing hormone (LH-RH) and bombesin antagonists containing doxorubicin (DOX) or its superactive derivative, 2-pyrrolino-DOX (AN-201) (27–32). Here we report the synthesis of cytotoxic SST analogs consisting of cytotoxic radicals DOX and 2-pyrrolino-DOX linked to carriers RC-160 or RC-121 (Table 1). These cytotoxic conjugates were tested in vitro to determine their antiproliferative effects, binding properties, and SST-like activities. Some very promising results of preliminary in vivo oncological tests are also presented.
Table 1.
Structures of cytotoxic SST analogs and carriers and their inhibitory effects on [125I]RC-160 binding to SSTR on rat pituitary membranes
| Structure | IC50,* nM |
|---|---|
| RC-121 | 0.31 |
| AN-162 = DOX-14-O-glt-RC-121 | 2.96 |
| AN-238 = 2-pyrrolino-DOX-14-O-glt-RC-121 | 23.8 |
| RC-160 | 1.74 |
| AN-163 = DOX-14-O-glt-RC-160 | 7.88 |
| AN-258 = 2-pyrrolino-DOX-14-O-glt-RC-160 | 80.1 |
glt, Glutaryl. RC-121 = d-Phe-C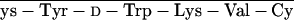 s-Thr-NH2, and RC-160 = d-Phe-C
s-Thr-NH2, and RC-160 = d-Phe-C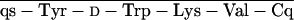 s-Trp-NH2.
s-Trp-NH2.
Binding affinities of the analogs to SSTR on rat pituitary were determined in competitive binding experiments by using [125I] labeled RC-160 as radio ligand (33). The binding affinities were expressed by IC50 values, the concentration of unlabeled analog required to inhibit 50% of the specific binding of the radioligand. Values shown are means of two or three determinations.
MATERIALS AND METHODS
Synthesis.
SST octapeptide carriers RC-160 and RC-121, with N-(9-fluorenylmethoxycarbonyl)-(N-Fmoc) side chain protection, ([Lys(Fmoc)5]RC-160 and [Lys(Fmoc)5]RC-121, respectively), as well as RC-121 were synthesized as described (6, 26). RC-160 was provided by Debiopharm (Lausanne, Switzerland).
Preparation of 2-pyrrolino-DOX-14-O-hemiglutarate (AN-206).
N-Fmoc-DOX-14-O-hemiglutarate (28, 29) (88 mg, 100 μmol) was dissolved in 5.4 ml of N,N-dimethylformamide (DMF), and 0.6 ml of piperidine was added. After 7 min, the reaction was quenched at 4°C with a mixture of 0.6 ml of trifluoroacetic acid (TFA) and 1.5 ml of pyridine in 3 ml of DMF. The solvents were evaporated in vacuo, and the residual oil was purified by HPLC yielding 58 mg (75 μmol) of DOX-14-O-hemiglutarate TFA salt. Thirty-nine milligrams (50 μmol) of this compound was dissolved in 400 μl of DMF, and 130 μl (200 mg, 1 mmol) of 4-iodobutyraldehyde (27) was added, followed by 70 μl (0.4 mmol) of diisopropylethylamine. After 30 min, the DMF solution was added slowly to 10 ml of 70% aqueous acetonitrile (vol/vol) containing 100 μl of TFA. This solution was then diluted with 10 ml of 0.1% aqueous TFA (vol/vol) and applied on semipreparative HPLC. After purification, 31 mg (yield, 75%) of 98% pure AN-206 was obtained.
Preparation of cytotoxic somatostatin analogs containing 2-pyrrolino-DOX or DOX.
[Lys(Fmoc)5]RC-121 (13.8 mg, 10 μmol) and AN-206 (8.3 mg, 10 μmol) plus 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (Calbiochem-Novabiochem, La Jolla, CA) (4 mg, 10.5 μmol) were dissolved in 300 μl of DMF, and diisopropylethylamine (30 μl, 172 μmol) was added. After 15 min, the coupling is completed and 700 μl of DMF was added, followed by 100 μl of piperidine. Seven minutes later, the reaction was quenched at 4°C with a mixture of 100 μl of TFA and 300 μl of pyridine in 300 μl of DMF. This solution was added slowly to 6 ml of 70% aqueous acetonitrile containing 0.1% TFA (vol/vol) and further diluted with 10 ml of 0.1% TFA in water and applied on semipreparative HPLC. After purification, 6 mg of 97% pure AN-238 was obtained (overall yield, 30%). AN-258 was prepared in the same way by using [Lys(Fmoc)5]RC-160. Analogs AN-162 and AN-163, containing DOX, were synthesized in a similar fashion by using N-Fmoc-DOX-14-O-hemiglutarate as described (28).
Analytical HPLC.
A Beckman analytical HPLC system equipped with model 168 diode array detector and System Gold chromatography software (Beckman) was employed to monitor the chemical reactions and check the purity. The column used was a Phenomenex (Torrance, CA) “Jupiter” C18 (250 × 4.6 mm; pore size, 300 Å; particle size, 5 μm).The solvent system consisted of two components: 0.1% TFA in water and 0.1% TFA in 70% aqueous acetonitrile.
Purification.
The final purification of all the peptide conjugates was carried out on a Beckman model 342 semipreparative HPLC system by using Phenomenex “Jupiter” (250 × 10 mm; pore size, 300 Å; particle size, 15 μm) column. The solvent system was the same as described above.
Analysis.
Electrospray mass spectrometer Finnigan-MAT (San Jose, CA) TSQ 7000 was used for the structural identification of the peptide conjugates.
Cytotoxicity Assay.
Several cancer cell lines, which grow attached to tissue culture plates, were used for the determination of the antiproliferative activity of the cytotoxic SST conjugates. The MDA-MB-231 human breast cancer, PC-3 human prostate cancer, and MIA PaCa-2 human pancreatic cancer cell lines were obtained from the American Type Culture Collection. The MKN-45 human gastric cancer cell line was a gift from J. Primrose (University of Leeds, Leeds, U.K.). For the evaluation of the cytotoxic activity of the analogs in these cell lines, a colorimetric cytotoxicity assay in microtitration plates was used, based on quantification of biomass by staining cells with crystal violet (34). The activity of the analogs on H-345 human small cell lung carcinoma (SCLC), obtained from the American Type Culture Collection, which grows in a suspension, was determined by a cytotoxicity assay based on tetrazolium dye [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazoloum bromide (MTT)] (35). MKN-45 cells were incubated in RPMI 1640 medium containing 10% fetal bovine serum. MDA-MB-231 cells were grown in Dulbecco’s modified essential medium containing 10% newborn calf serum. PC-3 cells were incubated in RPMI 1640 medium: F-12 (1:1) medium containing 1 mM pyruvate, 1 μM FeSO4, and 0.5% BSA. MIA PaCa-2 cells were cultured in Dulbecco’s modified essential medium containing 10% fetal bovine serum. H-345 cells were incubated in RPMI 1640 medium containing 5% fetal bovine serum.
Receptor Binding.
Binding affinities of the analogs to SSTR on rat pituitary membrane preparations were determined in competitive binding experiments by using 125I-labeled RC-160 as radio ligand (33). When AN-238 and AN-258 were tested in comparison with their respective carriers, a binding buffer (25 mM Tris, pH 7.4) containing 5% mannitol (wt/vol) was used to improve the solubility.
GH-Release Inhibiting Activities.
GH-release inhibiting activities of cytotoxic SST analog AN-238 and carrier RC-121 were tested in vitro in a dispersed rat pituitary superfusion system. The method was described in detail elsewhere (36). Briefly, anterior pituitaries of young male Sprague–Dawley rats were digested with collagenase, and the mechanically dispersed cell clusters were sedimented together with Sephadex G-10 into superfusion columns. The columns then were perfused with tissue culture medium (medium 199) at a rate of 0.33 ml/min. After an overnight recovery period, the cells were exposed to human GH-releasing hormone (hGH-RH)(1–29)NH2, a specific GH secretagogue, and adenylate-cyclase activator forskolin, a nonspecific stimulator of GH, in the presence or absence of an SST analog. The concentration of GH in 1 ml (3 min) fractions of the effluent medium was determined by RIA. The RIA results were processed with the aid of a special computer program to calculate the “net integral” values of the responses that were used for the determination of the inhibitory effects of the analogs (36).
In Vivo Experiments.
Preliminary in vivo experiments on MDA-MB-231 estrogen-independent human mammary carcinoma in nude mice, MXT estrogen-independent mouse mammary carcinoma in BDF mice, and Dunning AT-1 androgen-independent rat prostate cancers in Copenhagen rats were carried out as described (31, 37), the analogs being injected i.v.
RESULTS
Design and Synthesis.
To create targeted cytotoxic analogs of SST containing DOX, N-Fmoc-DOX-14-O-hemiglutarate was activated by 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate and coupled covalently to the amino terminals of [Lys(Fmoc)5]RC-160 and [Lys(Fmoc)5]RC-121. The reaction was carried out in DMF; after 15 min, cleavage of the Fmoc protecting groups was effected by 5% piperidine in DMF (vol/vol) to yield hybrid analogs with DOX, AN-163, and AN-162, respectively.
For the synthesis of analogs with 2-pyrrolino-DOX, 2-pyrrolino-DOX-14-O-hemiglutarate (AN-206) was first prepared from DOX-14-O-hemiglutarate by a reaction with an excess of 4-iodobutyraldehyde (27–29). Coupling AN-206 to the amino terminals of [Lys(Fmoc)5]RC-160 and [Lys(Fmoc)5]RC-121 was followed by deprotection to yield cytotoxic conjugates AN-258 and AN-238, respectively. The compounds were purified by HPLC, and the structures were confirmed by mass spectrometry.
Cytotoxicity.
Antiproliferative activities of the cytotoxic hybrid molecules and their corresponding cytotoxic radicals were evaluated in MKN-45 human gastric cancer, MDA-MB-231 human breast cancer, PC-3 human prostate cancer, MIA PaCa-2 human pancreatic cancer, and H-345 human SCLC cell lines in vitro. Drug concentrations that inhibited cell growth by 50% (IC50 values) are shown in Table 2. The results indicate that the cytotoxic activity of the antineoplastic radicals was virtually preserved in the conjugates. Thus, analogs AN-162 and AN-163 with DOX showed IC50 values in the 10−7 M concentration range, whereas analogs containing AN-201 had IC50 values in the 10−10 M concentration range. Interestingly, in H-345 human SCLC cell line, analogs AN-163 and AN-258 with carrier RC-160 showed ≈10 times lower antiproliferative activity than their respective cytotoxic radicals.
Table 2.
Inhibition of growth of MKN-45 human gastric cancer, MDA-MB-231 human breast cancer, PC-3 human prostate cancer, MIA PaCa-2 human pancreatic cancer, and H-345 human SCLC cell lines by DOX, 2-pyrrolino-DOX (AN-201), and their corresponding cytotoxic SST analogs
| Compound | IC50,* 10−10 M
|
||||
|---|---|---|---|---|---|
| MKN-45 at 100 hr | MDA-MB-231 at 120 hr | PC-3 at 120 hr | MIA PaCa-2 at 94 hr | H-345 at 96 hr | |
| DOX | 1,900 | 1,200 | 920 | 440 | 35,000 |
| Analogs with DOX | |||||
| AN-162† | 2,300 | 1,300 | 1,800 | 560 | 35,000 |
| AN-163‡ | 1,900 | 1,600 | 2,100 | 700 | 650,000 |
| AN-201 | 1.8 | 1.9 | 5 | 3.5 | 24 |
| Analogs with AN-201 | |||||
| AN-238† | 3.6 | 3.2 | 5 | 3.3 | 40 |
| AN-258‡ | 4.2 | 3.5 | 22 | 5.7 | 166 |
Cell growth inhibition data, determined at three different concentrations, were used to calculate the drug concentration that inhibited cell growth by 50% as compared to untreated control cultures. All data was derived from an average of three determinations each in eight replicates. The different cell lines were grown as described. The carrier peptides had no effect on cell proliferation in these assays at 10−6 M and lower concentrations.
Analogs with carrier RC-121.
Analogs with carrier RC-160.
Receptor Binding Affinities.
The carrier peptides and their cytotoxic analogs containing DOX or 2-pyrrolino-DOX were tested for their ability to inhibit the binding of [125I]RC-160 to SSTR on rat pituitary membrane homogenates. As shown in Table 1, cytotoxic SST conjugates AN-162 and AN-163 containing DOX displayed ≈10 times lower binding affinities than their respective carriers (IC50 < 10 nM). Analogs with cytotoxic radical 2-pyrrolino-DOX also showed lower binding affinity than their parent peptides, displaying IC50 values of 23.8 nM in the case of AN-238 and 80.1 nM for AN-258.
GH-Release Inhibiting Activities.
GH-release inhibiting activities of analog AN-238 (Fig. 1), which was selected for further evaluation, and its carrier peptide RC-121 were tested in vitro by using a dispersed rat pituitary superfusion system (36). Administration of hGH-RH(1–29)NH2 or forskolin for various durations (3- to 30-min exposures) in this dynamic bioassay system greatly increased GH secretion, producing sharp peaks of GH release (Table 3, control values). Simultaneous administration of AN-238 with either of these secretagogues significantly reduced or completely blocked the GH response (Table 3). SST analog RC-121 also blocked GH release, induced by hGH-RH(1–29)NH2 or forskolin. Results of these experiments demonstrate that cytotoxic SST analog AN-238 fully preserves the GH-release inhibitory potency of RC-121.
Figure 1.

Molecular structure of cytotoxic SST analog AN-238. 2-Pyrrolino-DOX-14-O-hemiglutarate is linked to the amino terminus of RC-121.
Table 3.
Effects of cytotoxic SST hybrid AN-238 and its carrier analog RC-121 on hGH-RH(1-29)NH2 or forskolin-induced GH-release in dispersed rat pituitary superfusion system in vitro
| Compound | Exposure time, min | GH response, ng
|
Inhibition (%) | GH response, ng
|
Inhibition (%) | ||
|---|---|---|---|---|---|---|---|
| Control | RC-121 | Control | AN-238 | ||||
| 1 nM GH-RH + 1 nM analog | 3 | 560 | 22 | 96 | 493 | 80 | 84 |
| 1 nM GH-RH + 1 nM analog | 30 | 2,598 | 0 | 100 | 1,939 | 101 | 95 |
| 0.2 nM GH-RH + 0.1 nM analog | 6 | 260 | 0 | 100 | 214 | 23 | 91 |
| 0.2 nM GH-RH + 0.5 nM analog | 6 | 277 | 0 | 100 | 215 | 18 | 92 |
| 4 μM forskolin + 0.5 nM analog | 6 | 760 | 90 | 88 | 760 | 230 | 70 |
| 10 μM forskolin + 2 nM analog | 6 | 696 | 294 | 58 | 554 | 258 | 54 |
Cells were exposed to the secretagogues alone or in combination with the SST analogs for different duration and at various concentrations. The GH-release inhibitory activities of the analogs are expressed as relative GH-releasing potencies (ng, “net integral” values) of the secretagogues administered with or without the analogs.
Preliminary in Vivo Oncological Results.
Because of its high in vitro activity, AN-238 was selected for in vivo studies. In preliminary tests with MXT mouse mammary carcinoma in female BDF mice, AN-238 was found to inhibit tumor growth, in a dose-dependent fashion, in the dose range from 150 to 300 nmol/kg, the latter being the maximum tolerated dose (MTD) in this experimental model. Cytotoxic radical AN-201 was effective at its MTD (250 nmol/kg) (31) but toxic at 300 nmol/kg. AN-238 also significantly reduced tumor volume in nude mice bearing MDA-MB-231 estrogen-independent human breast cancers after 2 weeks of treatment at a dose of 250 nmol/kg (MTD), whereas AN-201 at the same dose did not inhibit tumor growth. Determination of the receptor status for SSTR on these tumors is in progress. In preliminary experiments in Dunning AT-1 androgen-independent rat prostate cancers, previously found to be positive for SSTR (37), AN-238 showed strong tumor growth inhibition, which was dose-dependent in the range from 150 to 300 nmol/kg. No toxic side effects were observed with AN-238 at a dose of 300 nmol/kg, whereas cytotoxic radical AN-201 at a dose of 115 nmol/kg did not inhibit tumor growth and led to the death of one of six animals due to toxicity. Detailed and complete studies in these models will be the subject of subsequent publications.
DISCUSSION
SST is a peptide hormone with a wide spectrum of activities that are in nearly all the cases inhibitory (3, 8). Extensive investigations on the receptors and the antiproliferative effects of SST octapeptide analogs revealed that ≈30% of human tumors express subtypes of SSTR2 or SSTR5. These findings, and the successful localization of various human tumors by scintigraphy with radiolabeled SST octapeptides (23–25), strongly suggest that SST analogs could be used as carriers for targeted chemotherapy for those tumors that express SSTR2 or SSTR5. Our previous promising results on the antitumor effects of an early cytotoxic SST conjugate, comprising carrier RC-121 and methotrexate, also support this view (26). Recently, we developed cytotoxic conjugates of LH-RH and bombesin antagonists containing DOX, the most widely used anti-cancer antibiotic, and 2-pyrrolino-DOX (AN-201), a derivative that is 500–1,000 times more potent than DOX in vitro. Cytotoxic LH-RH analogs AN-152 and AN-207 containing DOX and 2-pyrrolino-DOX were shown to inhibit the growth of LH-RH receptor positive tumors more effectively than their respective cytotoxic radicals (28–32). These cytotoxic hybrids were also found to be much less toxic than the radicals alone. These very promising results encouraged us to create targeted cytotoxic hybrids of SST octapeptides containing DOX or AN-201. SST octapeptide analogs, RC-160 and RC-121, that were developed at our institute, were selected as carriers because these analogs have high binding affinities to SSTR on various tumors (13). Thus, N-Fmoc-DOX-14-O-hemiglutarate was coupled to the amino terminals of [Lys(Fmoc)5]RC-160 or [Lys(Fmoc)5]RC-121 and deprotected, to result in hybrid analogs AN-163 and AN-162, respectively, containing DOX. Similarly, the conjugation of 2-pyrrolino-DOX-14-O-hemiglutarate to the amino terminals of [Lys(Fmoc)5]RC-160 or [Lys(Fmoc)5]RC-121, followed by deprotection, yielded cytotoxic analogs AN-258 and AN-238, respectively, with AN-201. In earlier efforts to synthesize cytotoxic analogs of LH-RH and bombesin antagonists, containing daunosamine-modified DOX derivative AN-201, an excess amount of 4-iodobutyraldehyde was reacted with peptide conjugates containing DOX. The reagent was found to react specifically with the amino alcohol function of the daunosamine moiety in DOX, without affecting the amino acid residues, including Arg, Ser, Trp, and His, of the peptides. However, our attempts to use this reaction for the conversion of AN-162 and AN-163 to AN-238 and AN-258, respectively, in an acceptable yield have failed. Alkylation of the sulfur atoms and/or the unprotected amino group of the Lys-5 side chain of the carriers may be responsible for the formation of several byproducts, making the reaction unsuitable for the preparation of larger quantities of these analogs. To overcome this problem, 2-pyrrolino-DOX-14-O-hemiglutarate (AN-206) was prepared, in good yield, by reacting 4-iodobutyraldehyde with DOX-14-O-hemiglutarate. Because AN-206 can withstand the conditions required for the removal of the N-Fmoc protecting group, it was coupled to the amino terminals of [Lys(Fmoc)5]RC-160 or [Lys(Fmoc)5]RC-121. A moderate 30% overall yield was achieved by this method for the coupling, deprotection, and purification steps.
Antitumor effects of SST octapeptide analogs have been shown previously in various experimental tumor models, including breast, prostate, gastric, pancreatic, and lung cancers (3, 9, 26, 37, 38). Although the demonstration of binding of SST octapeptide analogs to receptors on various cell lines and tumors is still subject to controversy, more and more evidence suggests the involvement of SSTR2 and SSTR5 in the antitumor activity of these peptides (3, 9, 11–21). Various human cancer cell lines, that are regularly used in our institute for tumor growth studies in animal models in vivo, and known to express mRNAs for SSTR2 and/or SSTR5, were selected for the determination of the antiproliferative activities of the cytotoxic SST hybrids in vitro. Such cells, including MKN-45 human gastric cancer (9), MDA-MB-231 human breast cancer (39), PC-3 human prostate cancer (38), MIA PaCa-2 human pancreatic cancer (9), and H-345 human SCLC cells (33), have the potential to express SSTR. Our results on the antiproliferative effects of the cytotoxic SST conjugates indicate that the activity of the cytotoxic radicals is essentially preserved in the hybrid analogs. However, AN-163 and AN-258, containing RC-160 as a carrier, showed ≈10 times weaker effect than their corresponding cytotoxic radicals on H-345 SCLC cell lines. Because analogs AN-162 and AN-238 with carrier RC-121 showed activities similar to their respective cytotoxic radicals, a lower binding affinity of AN-163 and AN-258 to the SSTR on this cell line could possibly account for this finding.
When we evaluated the binding affinities of the carrier peptides and their cytotoxic analogs containing DOX or 2-pyrrolino-DOX to rat pituitary membrane homogenates, all analogs were found to inhibit the binding of [125I]RC-160 in the nanomolar concentration range (IC50 < 80 nM), demonstrating high specific affinity to these receptors. However, RC-160 and its cytotoxic conjugates showed somewhat lower binding affinities than their respective counterparts with RC-121.
AN-238 (Fig. 1) was further evaluated in vitro for its SST-like activity. SST and its octapeptide analogs are known to inhibit the stimulated GH release from pituitary cells, induced by GH-RH (36) or adenylate-cyclase activator, forskolin (40) through receptor-mediated action. Thus, the GH-release inhibitory activities of AN-238 and carrier RC-121 were compared in superfused rat pituitary cell system. Our results demonstrate that cytotoxic conjugate AN-238 has an inhibitory effect on the stimulated GH secretion similar to that of RC-121 (Table 3). These results also indicate that conjugation of cytotoxic radical AN-201 with the carrier RC-121 does not alter significantly the binding affinity of RC-121 to SSTR on living cells, as those in a superfusion system. Twenty-four hours after a long-term exposure (30 min) of the pituitary cells to the cytotoxic conjugate in vitro, no permanent damage of the pituitary somatotrope function was observed (data not shown). This finding is in agreement with our previous findings that the pituitary gonadotrope cells fully recover from the effects of long-term exposure to cytotoxic LH-RH conjugate (AN-207) containing 2-pyrrolino-DOX (41).
It is very encouraging that in preliminary in vivo studies, using i.v. administration, cytotoxic SST analog AN-238 inhibited the growth of MXT estrogen-independent mouse mammary cancers, MDA-MB-231 estrogen-independent human mammary cancers in nude mice, and Dunning AT-1 androgen-independent rat prostate cancers. In all these tumor models, cytotoxic SST analog AN-238 was found to be much more effective and less toxic than AN-201, the cytotoxic radical it contains. The difference between the tolerance of AN-201 and the conjugate AN-238 by mice and rats is conspicuous. It is well known that mice tolerate treatment with cytotoxic agents better than rats. Accordingly, cytotoxic agent AN-201 was found to be more toxic in rats than in mice. Surprisingly, cytotoxic conjugate AN-238 was better tolerated by rats than by mice. Because AN-201 is linked covalently to its carrier by an ester bond, deconjugation of the radical by nonspecific esterase enzyme activity can occur. This enzyme activity may be different in the two species and could account for the different toxicity of AN-201 and AN-238 in mice and rats. In fact, we investigated the deconjugation of DOX from one of our cytotoxic LH-RH conjugates (AN-152), which contains a similar ester bond, in vitro by incubating the analog for different time periods in sera from mice and rats at 37°C (32). The approximate half-life of this analog in mouse serum was found to be 10 min, whereas 60–70% of the conjugate was still intact in rat sera after 1 hr, as assessed by HPLC.
We previously have synthesized cytotoxic LH-RH analogs that were intended for the treatment of tumors that express receptors for LH-RH, including prostatic, mammary, ovarian, and endometrial cancers. The cytotoxic analogs of SST reported herein were designed to be targeted to pancreatic, gastric, colorectal, renal, and lung cancers, both SCLC and non-SCLC, and brain tumors. In addition, our work shows that cytotoxic SST analogs could be also targeted to breast and prostatic cancers.
In conclusion, our results strongly indicate that cytotoxic SST analogs could be used as targeted antitumor agents for the treatment of cancers that possess receptors for SSTR2 and/or SSTR5. Indications for such therapy could be assessed by Octreoscan scintigraphy, which is presently used for tumor localization and as a predictive tool in determining whether patients should receive SST octapeptide hormone therapy.
Acknowledgments
We thank Prof. J. Engel, Dr. M. Bernd, and Dr. E. Busker (Degussa AG and Asta Medica AG, Frankfurt am Main, Germany) for mass spectra analyses and for their help in preparation of this manuscript. We also thank Katalin Halmos, Elena Glotser, Ágnes Koppán, and Dr. Ilona Hoffmann for their excellent technical assistance. Some work described in this paper was supported by the Medical Research Service of the Veterans Affairs and CaP CURE.
ABBREVIATIONS
- DOX
doxorubicin
- GH
growth hormone
- hGH-RH
human GH-releasing hormone
- LH-RH
luteinizing hormone-releasing hormone
- SCLC
small cell lung carcinoma
- SST
somatostatin
- SSTR
somatostatin receptor subtypes
- DMF
N,N-dimethylformamide
- TFA
trifluoroacetic acid
References
- 1.Brazeau P, Vale W, Burgus R, Ling N, Butcher M, Rivier J, Guillemin R. Science. 1973;179:77–79. doi: 10.1126/science.179.4068.77. [DOI] [PubMed] [Google Scholar]
- 2.Schally A V, Coy D H, Meyers C A. Annu Rev Biochem. 1978;47:89–128. doi: 10.1146/annurev.bi.47.070178.000513. [DOI] [PubMed] [Google Scholar]
- 3.Pollak M N, Schally A V. Proc Soc Exp Biol Med. 1998;217:143–152. doi: 10.3181/00379727-217-44216. [DOI] [PubMed] [Google Scholar]
- 4.Veber D F, Freidlinger R M, Perlow D S, Paleveda W J, Jr, Holly F W, Strachan R G, Nutt R F, Arison B H, Homnick C, Randall W C, Glitzer M S, Saperstein R, Hirschmann R. Nature (London) 1981;292:55–58. doi: 10.1038/292055a0. [DOI] [PubMed] [Google Scholar]
- 5.Bauer W, Briner U, Doepfner W, Haller R, Huguenin R, Petcher T J, Pless J. Life Sci. 1982;31:1133–1140. doi: 10.1016/0024-3205(82)90087-x. [DOI] [PubMed] [Google Scholar]
- 6.Cai R-Z, Szoke B, Lu R, Fu D, Redding T W, Schally A V. Proc Natl Acad Sci USA. 1986;83:1896–1900. doi: 10.1073/pnas.83.6.1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schally A V, Comaru-Schally A-M. In: Cancer Medicine. 4th Ed. Holland J F, Frei E I, Bast R C J, Kufe D E, Morton D L, Weichselbaum R R, editors. Baltimore: Williams & Wilkins; 1997. pp. pp.1067–1085. [Google Scholar]
- 8.Schally A V. Cancer Res. 1988;48:6877–6885. [Google Scholar]
- 9.Weckbecker G, Raulf F, Stolz B, Bruns C. Pharmacol Ther. 1993;60:245–264. doi: 10.1016/0163-7258(93)90009-3. [DOI] [PubMed] [Google Scholar]
- 10.Lamberts S W, van der Lely A, Deherder W W, Hofland L J. N Engl J Med. 1996;334:246–254. doi: 10.1056/NEJM199601253340408. [DOI] [PubMed] [Google Scholar]
- 11.Reubi J-C, Laissue J A. Trends Pharmacol Sci. 1995;16:110–115. doi: 10.1016/s0165-6147(00)88992-0. [DOI] [PubMed] [Google Scholar]
- 12.Cordelier P, Estève J-P, Bousquet C, Delesque N, O’Carroll A-M, Schally A V, Vaysse N, Susini C, Buscail L. Proc Natl Acad Sci USA. 1997;94:9343–9348. doi: 10.1073/pnas.94.17.9343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Srkalovic G, Cai R-Z, Schally A V. J Clin Endocrinol Metab. 1990;70:661–669. doi: 10.1210/jcem-70-3-661. [DOI] [PubMed] [Google Scholar]
- 14.Buscail L, Estève J-P, Saint-Laurent N, Bertrand V, Reisine T, O’Carroll A-M, Bell G I, Schally A V, Vaysse N, Susini C. Proc Natl Acad Sci USA. 1995;92:1580–1584. doi: 10.1073/pnas.92.5.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reubi J-C, Waser B, Schaer J C, Markwalder R. J Clin Endocrinol Metab. 1995;80:2806–2814. doi: 10.1210/jcem.80.9.7673428. [DOI] [PubMed] [Google Scholar]
- 16.Reubi, J.-C., Schaer, J. C., Laissue, J. & Waser, B. (1996) Metabolism 45, Suppl. 1, 39–41. [DOI] [PubMed]
- 17.Buscail L, Delesque N, Estève J-P, Saint-Laurent N, Prats H, Clerc P, Robberecht P, Bell G I, Liebow C, Schally A V, Vaysse N, Susini C. Proc Natl Acad Sci USA. 1994;91:2315–2319. doi: 10.1073/pnas.91.6.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Patel Y C. J Endocrinol Invest. 1997;20:348–367. doi: 10.1007/BF03350317. [DOI] [PubMed] [Google Scholar]
- 19.Patel Y C, Greenwood M T, Panetta R, Demchyshyn L, Niznik H, Srikant C B. Life Sci. 1995;57:1249–1265. doi: 10.1016/0024-3205(95)02082-t. [DOI] [PubMed] [Google Scholar]
- 20.Bruns C, Weckbecker G, Raulf F, Kaupmann K, Schoeffter P, Hoyer D, Lubbert H. Ann NY Acad Sci. 1994;733:138–146. doi: 10.1111/j.1749-6632.1994.tb17263.x. [DOI] [PubMed] [Google Scholar]
- 21.Reisine T, Bell G I. Endocr Rev. 1995;16:427–442. doi: 10.1210/edrv-16-4-427. [DOI] [PubMed] [Google Scholar]
- 22.Nilsson S, Reubi J C, Kalkner K-M, Laissue J A, Horisberger U, Olerud C, Westlin J-E. Cancer Res (Suppl) 1995;55:5805s–5810s. [PubMed] [Google Scholar]
- 23.Breeman W A P, Hofland L J, van der Plujim M, van Koetsveld P M, de Jong M, Steyono-Han B, Bakker W H, Kwekkeboom D J, Visser T J, Lamberts S W J, Krenning E P. Eur J Nucl Med. 1994;21:328–335. [PubMed] [Google Scholar]
- 24.Bakker W H, Krenning E P, Reubi J-C, Breeman W A P, Seytono-Han B, de Jong M, Kooij P P M, Bruns C, van Hagen P M, Marbach P, Visser T J, Pless J, Lamberts S W J. Life Sci. 1991;49:1593–1601. doi: 10.1016/0024-3205(91)90053-e. [DOI] [PubMed] [Google Scholar]
- 25.Krenning E P, Kwekkeboom D J, Bakker W H, Breeman W A P, Kooij P P M, Oei H Y, van Hagen M, Postema P T E, de Jong M, Reubi J-C, Visser T J, Reijis A E M, Hofland L J, Koper J W, Lamberts S W J. Eur J Nucl Med. 1993;20:716–731. doi: 10.1007/BF00181765. [DOI] [PubMed] [Google Scholar]
- 26.Radulovic S, Nagy A, Szoke B, Schally A V. Cancer Lett. 1992;62:263–271. doi: 10.1016/0304-3835(92)90105-5. [DOI] [PubMed] [Google Scholar]
- 27.Nagy A, Armatis P, Schally A V. Proc Natl Acad Sci USA. 1996;93:2464–2469. doi: 10.1073/pnas.93.6.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagy A, Schally A V, Armatis P, Szepesházi K, Halmos G, Kovács M, Zarándi M, Groot K, Miyazaki M, Jungwirth A, Horváth J. Proc Natl Acad Sci USA. 1996;93:7269–7273. doi: 10.1073/pnas.93.14.7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagy A, Armatis P, Cai R-Z, Szepesházi K, Halmos G, Schally A V. Proc Natl Acad Sci USA. 1997;94:652–656. doi: 10.1073/pnas.94.2.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jungwirth A, Schally A V, Nagy A, Pinski J, Groot K, Galvan G, Szepesházi K, Halmos G. Int J Oncol. 1997;10:877–884. doi: 10.3892/ijo.10.5.877. [DOI] [PubMed] [Google Scholar]
- 31.Szepesházi K, Schally A V, Nagy A, Halmos G, Groot K. Anti-Cancer Drugs. 1997;8:974–987. doi: 10.1097/00001813-199711000-00009. [DOI] [PubMed] [Google Scholar]
- 32.Miyazaki M, Nagy A, Schally A V, Lamharzi N, Halmos G, Szepesházi K, Groot K, Armatis P. J Natl Cancer Inst. 1997;89:1803–1809. doi: 10.1093/jnci/89.23.1803. [DOI] [PubMed] [Google Scholar]
- 33.O’Byrne K J, Halmos G, Pinski J, Groot K, Szepesházi K, Schally A V, Carney D N. Eur J Cancer. 1994;30A:1682–1687. doi: 10.1016/0959-8049(94)00351-5. [DOI] [PubMed] [Google Scholar]
- 34.Reile H, Birnböck H, Bernhardt G, Spruss T, Schönenberger H. Anal Biochem. 1990;187:262–267. doi: 10.1016/0003-2697(90)90454-h. [DOI] [PubMed] [Google Scholar]
- 35.Plumb J A, Milroy R, Kaye S B. Cancer Res. 1989;49:4435–4440. [PubMed] [Google Scholar]
- 36.Csernus V, Schally A V. In: Neuroendocrine Research Methods. Greenstein B, editor. London: Harwood; 1991. pp. 66–102. [Google Scholar]
- 37.Pinski J, Reile H, Halmos G, Groot K, Schally A V. Cancer Res. 1994;54:169–174. [PubMed] [Google Scholar]
- 38.Pinski J, Schally A V, Halmos G, Szepesházi K. Int J Cancer. 1993;55:963–967. doi: 10.1002/ijc.2910550615. [DOI] [PubMed] [Google Scholar]
- 39.Eden P A, Taylor J E. Life Sci. 1993;53:85–90. doi: 10.1016/0024-3205(93)90614-9. [DOI] [PubMed] [Google Scholar]
- 40.Evans W S, Brannagan T H, Limber E R, Cronin M J, Rogol A D, Hewlett E L, Thorner M O. Am J Physiol. 1985;249:E392–E397. doi: 10.1152/ajpendo.1985.249.4.E392. [DOI] [PubMed] [Google Scholar]
- 41.Kovács M, Schally A V, Nagy A, Koppán M, Groot K. Proc Natl Acad Sci USA. 1997;94:1420–1425. doi: 10.1073/pnas.94.4.1420. [DOI] [PMC free article] [PubMed] [Google Scholar]