


Abstract
This article presents a new method for analyzing microarray time courses by identifying genes that undergo abrupt transitions in expression level, and the time at which the transitions occur. The algorithm matches the sequence of expression levels for each gene against temporal patterns having one or two transitions between two expression levels. The algorithm reports a P-value for the matching pattern of each gene, and a global false discovery rate can also be computed. After matching, genes can be sorted by the direction and time of transitions. Genes can be partitioned into sets based on the direction and time of change for further analysis, such as comparison with Gene Ontology annotations or binding site motifs. The method is evaluated on simulated and actual time-course data. On microarray data for budding yeast, it is shown that the groups of genes that change in similar ways and at similar times have significant and relevant Gene Ontology annotations.
INTRODUCTION
An obvious approach to studying a biological processes, such as the reaction of cells to a stimulus, is to measure the activity of the cell at a sequence of time points. However, when the measurements consist of high-throughput gene expression microarrays, it is not obvious how to extract biologically meaningful results. We describe a new computational method, called StepMiner, the primary goal of which is to assist biologists in understanding the temporal progression of genetic events and biological processes following a stimulus, based on gene expression microarray data.
At the most basic level, StepMiner identifies genes which undergo one or more binary transitions over short time courses. It directly addresses the one of the more basic questions one can ask of time course data: ‘Which genes are up-regulated or down-regulated as a result of the stimulus?’ and ‘When does the gene transition to up- or down-regulated?’
MATERIALS AND METHODS
StepMiner extracts three types of binary temporal patterns. The first type, shown in Figure 1(a and b), describe ‘one-step’ transitions, where the expression level of a gene transitions from a high to a low value or from a low to a high value. The second type, shown in Figure 1(c and d), describes two-step transitions. Genes in this category turn on then back off or vice versa. The third type consists of genes for which the one- or two-step patterns do not fit appreciably better than a constant mean value (the null hypothesis). This can result when the gene expression level is genuinely constant, or when the other patterns fit no better than the constant because the behavior of the gene is complex. The expression levels for up- and down-regulated are chosen that best fit the data.
Figure 1.

Signals of interest. Different types of binary temporal patterns that need to be extracted from the time course microarray data. (a) Gene expressions transition from a low value to a high value. (b) Gene expressions transition from a high value to a low value. (c) Gene expressions transition from low to high and return to the same low value. (d) gene expressions transition from high to low and return to the same high value.
Fitting the patterns of one- and two-step transitions requires an algorithm that evaluates every possible placement of the transitions (or step) between time points, and chooses the one that gives the best fit. This process is called adaptive regression.
Fitting one- or two-step functions
The objective of StepMiner is to find a one- or two-step function that best fits n time points, X1, X2, … Xn. The algorithm evaluates all possible step positions. For each position, it finds the values of constant segments using linear regression. The fitted values 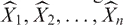 for each step give the square error (SSEstep). The adaptive regression scheme chooses the step positions that minimize the square error. For the two-step curve, the first and third constant segments are assumed to have the same value.
for each step give the square error (SSEstep). The adaptive regression scheme chooses the step positions that minimize the square error. For the two-step curve, the first and third constant segments are assumed to have the same value.
Let be the fitted value from the adaptive regression and X be the mean of the n original time points. The total sum of squares is defined to be
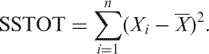 |
The degrees of freedom for SSTOT is n − 1 (the calculation of degrees of freedom appears subsequently). The sum of squares error SSE is defined to be
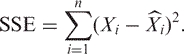 |
Let the degrees of freedom for SSE be n−m. The regression sum of squares SSR is
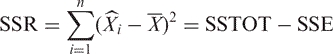 |
Therefore, the degrees of freedom for SSR is (n−1) − (n−m) = m−1. We define the regression mean square MSR as
and the error mean square MSE as
The regression test statistic is
This F-statistic follows an F-distribution with (m−1, n−m) degrees of freedom. Let 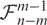 be a random variable that has this distribution. The P-value corresponding to the tail probability of this distribution is computed as
be a random variable that has this distribution. The P-value corresponding to the tail probability of this distribution is computed as
A low P-value represents a good fit of the curve to the data.
Selecting the best step function
The P-values for the three different patterns can be computed, using the statistic mentioned in the previous subsection. Let F1 and F2 be the F-statistic described in Fitting One- or Two-step Functions section for the one-step and two-step patterns. The algorithm selects the best step positions adaptively for patterns. Let SSE1 and SSE2 be the sum of squares error for one-step and two-step, and let n−m1 and n−m2 be their corresponding degrees of freedom.
F12 represents the relative goodness of fit of a one-step versus a two-step pattern. This is an F-distribution whose P-value represents the probability of the same result on random data.
StepMiner uses the following algorithm to select the best patterns for each gene:
 |
This algorithm was found in simulation to be superior to the standard forward stepwise and backward stepwise algorithms (see Supplementary Data—S2). It first selects the genes for which a one-step pattern fits well and a two-step pattern does not fit significantly better, based on whether the appropriate P-values fall under the specified threshold. Next, genes are selected from those remaining where two-step patterns fit very well according whether the P-value F2 is under the threshold. Genes that do not fall into any of the above categories are added to the ‘other’ category of genes for which the previous two patterns fit no better than the mean.
Degrees of freedom
The construction of a regression test statistic involves estimating degrees of freedom for the fitted pattern, which adjusts the statistic to eliminate the advantage that a more complex curve has over a simpler curve in fitting a given set of points. The degrees of freedom is estimated using random simulation, since it is non-trivial to derive it analytically in an adaptive framework. Gaussian 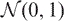 data for 10 000 simulated genes with 15 time points for each gene was generated. The SSR for both the one-step and the two-step pattern was calculated and the tail probabilities (# {SSR > α}/ 10000 for different α) were plotted as shown in Figure 2 χ2-distribution with different degrees of freedom was also plotted in the figure. As can be easily seen from the figures the degrees of freedom for SSR1 and SSR2 can be approximated as 3 and 4. The estimated degrees of freedom is in the range 2–3 for one step and 3–4 for two step. (This is consistent with the results of Owen (1), who estimated that a broken line uses 2–3 degrees of freedom.)
data for 10 000 simulated genes with 15 time points for each gene was generated. The SSR for both the one-step and the two-step pattern was calculated and the tail probabilities (# {SSR > α}/ 10000 for different α) were plotted as shown in Figure 2 χ2-distribution with different degrees of freedom was also plotted in the figure. As can be easily seen from the figures the degrees of freedom for SSR1 and SSR2 can be approximated as 3 and 4. The estimated degrees of freedom is in the range 2–3 for one step and 3–4 for two step. (This is consistent with the results of Owen (1), who estimated that a broken line uses 2–3 degrees of freedom.)
Figure 2.

Estimating degrees of freedom. (Left) Estimating degrees of freedom for the one-step model. (Right) Estimating degrees of freedom for the two-step model.
False discovery rate
A ‘false discovery’ occurs when the algorithm finds a one-step or two-step pattern, but the data contains no steps; a ‘true discovery’ occurs when the algorithm finds a one-step or two-step pattern, when the data contains a one-step or two-step pattern (the algorithm does not have to find the correct number of steps or a step at the correct time to produce a true discovery as defined here). The ‘false discovery rate’ (FDR) in StepMiner is the ratio of false discoveries to true discoveries.
To estimate the FDR, many random permutations of the time points are computed, and StepMiner is run on all of them. The number of significant genes in the original order and the average number of significant genes in the random permutations are computed. The ratio of the average number of significant genes to the original number of significant genes is an estimate of the FDR (2). The FDR can be adjusted by setting the P-value threshold used in the matching algorithm.
RESULTS
Analysis of simulated data
The algorithm was evaluated on simulated time course microarray data with 15 non-uniform time points. Noise-free data was generated for both one-step and two-step categories; Gaussian noise was then added to the original data, and then StepMiner was used to recover the original behavior, with a P-value threshold of 0.05. A total of 4000 genes with 15 time points were artificially created, with 2000 one-step genes and 2000 two-step genes.
Figure 3A describes the proportion of correctly identified gene expression patterns as a function of different step heights, where the position of the steps are fixed at certain time points. All single steps are fixed at the fifth position and all binary two steps are fixed at the fifth and nineth positions. As can be seen in the figure, when the step height is 5σ, StepMiner identifies genes correctly over 90% of the time. As the step height is reduced relative to the noise level, the proportion of correct identifications drops dramatically (as expected). The drop in accuracy is higher for two-step signals because of the greater degrees of freedom for those signals.
Figure 3.

Evaluating StepMiner on artificial data. (A) Proportion of correctly classified steps using 15 time points and different step heights with the step position fixed at 5 for one-step. For two-step patterns, the step positions were ‘up’ at 5 and ‘down’ at 9. σ is the SD of the 0-mean additive Gaussian noise. The number of false steps found in a random Gaussian data was 10%. (B) Proportion of correctly classified steps using different step heights with random step position and 15 time points. σ is the SD of the 0-mean additive Gaussian noise added. Ten percent of the steps are false. (C) Sensitivity of StepMiner to the number of time points, using random step positions and step height 5σ. A total of 2000 one-step and 2000 two-step functions were used in the analysis. (D) Sensitivity of StepMiner to the spacing between steps. The first step is fixed at the fourth position and the second step is varied according to the spacing. The height of the step is varied from 1σ to 5σ in a data set of 15 time points.
Figure 3B describes the proportion of correctly identified time courses using the same setting as Figure 3A except that the steps are placed between random time points. As the figure shows, there is a small reduction in the accuracy compared to Figure 3A. The behavior of StepMiner is similar in both Figure 3A and B. Higher confidence matches occur if all constant segments in a curve have several time points. This result shows that most matches where the steps are not at the beginning or end of the time course are reasonably high confidence. Hence, it would be desirable to design experiments so that there are several points before the first interesting transition and after the last interesting transition.
Figure 3C shows the sensitivity of StepMiner to the number of time points and the P-value threshold. As can be seen from the figure, accurate matching of two-step signals requires more time points than matching of one-step signals. The proportion of matches can be increased by increasing the P-value threshold, but only at the cost of an increased FDR (which can be measured and adjusted as described in False Discovery Rate section).
The number of time points between the steps is an important factor in determining the accuracy of extraction. Intuitively, a few consecutive measurements that are higher or lower than average could represent noise instead of a real change in gene expression level.
Figure 3D describes the proportion of two-step genes correctly identified when the number of time points between steps is varied. This figure is based on 2000 genes with 15 time points. The first step is fixed at fourth position and the spacing between steps is varied from 1 to 9. The height of the step is varied from 1σ to 5σ to observe the desired effect. As can be seen from the figure, a spacing of at least three time points is required for over 95% accuracy, when the step height is > 3σ. Also, as the second step approaches the end of the time points, the proportion of correct identifications decreases. The steps are also required to be placed at least three time points from the end points to achieve 95% accuracy.
Analysis of diauxic shift data
It is important to demonstrate the value of the method on real microarray data for at least two reasons: the true signals may not be step functions, and the noise from the actual experiment may not be Gaussian. Hence, StepMiner was applied to a publicly available time course of microarrays monitoring gene expression levels in yeast during the diauxic shift in a glucose-limited culture http://genomics-pubs.princeton.edu/DiauxicRemodeling/data.shtml. In this experiment, the yeast utilizes fermentative metabolism when glucose is abundant. As the glucose is depleted, the metabolism shifts abruptly to oxidative metabolism. RNA samples were collected approximately every 15 min and measured with microarrays.
An analysis of the results was published in 2005 [Brauer et al. (3)]. In that article, the data were analyzed using hierarchical clustering by gene [Gollub et al. (4)]. Of the many clusters generated, the authors picked seven clusters that had fairly high correlations and that, by visual inspection of the dendrogram, appeared to consist of genes with temporal behavior related to the diauxic shift.
In the original article, the sets of genes in the selected clusters were examined using GO-TermFinder (5) to identify GO annotations of genes that are enriched. The article lists several GO annotations that had extremely small P-values according to GO-TermFinder. Many of the annotations are obviously related to diauxic shift. Based on these annotations, three of the clusters of genes appeared to be highly relevant to diauxic shift, three were enriched with annotations of unknown relevance to diauxic shift and one cluster was not significantly enriched with any GO annotations.
For comparison, it is possible to reanalyze the data using gene sets derived from StepMiner. Binary signals were extracted from the diauxic shift data, using a P-value cutoff of 0.05, resulting in an FDR of 15%. Out of a total of 2284 genes in the diauxic shift data, 1088 were matched to single steps, 267 were matched to binary two steps and 929 did not match anything. The fitting step functions are shown for three genes in Figure 4. A heat map of the genes expression profiles appears in Figure 4. In the heat map, the top genes are those that change once, the rising genes first, and falling genes second. Lower, there is a group of genes that go up then down, and last, the genes that go down then up. Each of these groups is sorted by the time of first change. The ordered response of genes to stimuli is immediately evident when so depicted. The heat map also makes apparent two discontinuities, at 8.25 h and 9.25 h. These correspond to observed changes in the growth rate of the yeast around 9 h.
Figure 4.

Application of StepMiner to real microarray time course data. Comparison of StepMiner to hierarchical clustering for the analysis of diauxic shift time course microarray data on glucose-limited budding yeast. The expression level of each gene in StepMiner is centered around the midpoint of the step to display the transitions clearly. Fitted steps for three example genes are shown on the right.
The genes are then automatically collected into five generic gene sets: ‘up’, ‘down’, ‘up then down’, ‘down then up’. The generic gene sets are further divided into specific gene sets based on the position and the direction of the transition. This process resulted in 80 different generic and specialized gene sets, which were analyzed using GO-TermFinder with a P-value cutoff of 0.001. A table of the 120 low P-value GO annotations, in ascending order, is included in the Supplementary Data S3. Many of the GO annotations are directly related to metabolism.
The GO annotations and FDR-corrected P-values for the clusters reported in Brauer et al. were recomputed with the latest yeast gene annotations from the Gene Ontology Consortium website (6). To compare with the results of Brauer et al., Table 1 shows the GO annotations from that article that had low P-values, and shows the corresponding P-values from the StepMiner groups. The annotations that had the lowest P-values in Brauer et al. had even lower P-values in the StepMiner groups. Further, the GO annotations are obtained fully automatically using StepMiner — it is not necessary to select interesting clusters manually. In most cases, the P-values in the reanalysis are lower than Brauer et al.'s, which suggests that grouping by time-of-change is at least as effective as hierarchical clustering at identifying relevant genes.
Table 1.
GO annotations of different groups and P-values according to GO-TermFinder perl module
| GO Annotations | Group | P-value | P-value1 |
|---|---|---|---|
| Protein biosynthesis | Down-9.25 | 3.4E–51 | 9.7E–33 |
| Ribosome biogenesis and assembly | Down | 1.2E–39 | 1.4E–33 |
| Generation H of M precursor metabolites and energy | Up | 7.4E–24 | 6.1E–14 |
| Oxidative phosphorylation | Up | 4.9E−14 | 6E−08 |
| Amino acid and derivative metabolism | Up-Down | 1.7E−11 | 6.2E−25 |
| Amine biosynthesis | U-D-9 | 1.7E−12 | 1.1E−24 |
| Hexose catabolism | U-8.25-D | 0.00046 | 0.044 |
| Monosaccharide catabolism | U-8.25-D | 0.0012 | 0.091 |
| Siderophore transport | – | – | 0.013 |
| Intracellular transport | – | – | 1.6E−08 |
| Secretory pathways | – | – | 1.5E−06 |
‘P-value1’ is the P-value using the list of genes from the clusters reported by Brauer et al. ‘Down-9.25’ uses all the genes that turn off significantly at 9.25h time step. ‘Up’ uses all the genes that turn on at some time step. ‘U-D-9’ uses the list of genes that turn on at some point before 9 h but turn off at 9 h. ‘U-8.25-D’ genes turn on at 8.25 h and turn off later.
Four GO annotations had significant results in Brauer et al.'s analysis, but not in the StepMiner analysis: ‘siderophore transport’, ‘intracellular transport’, and ‘secretory pathways.’ Interestingly, these GO annotations were associated with clusters that, in the words of Brauer et al. were ‘less interpretable in terms of diauxic shift’.
DISCUSSION
Comparing StepMiner to other tools
Even though many tools are available for analyzing microarray time course microarray data, StepMiner is the only one that directly identifies the time and direction of step-wise temporal transitions in a statistically rigorous manner. While other tools may be more suitable for their intended purpose, they do not identify expression-level transitions as conveniently as StepMiner.
Other tools developed for the analysis of time course microarray data can be classified broadly as being either clustering or model based. In time course studies, clustering-based techniques partition genes into sets based on their proximity according to some measure of distance between gene expression profiles (7–15). Some of these methods take into account the temporal ordering of measurements, but most do not. A user may be able to select clusters of genes that appear to be up- or down-regulated at a particular time, but doing so is a hit-or-miss process that requires additional effort and is likely to yield uncertain results. Unlike StepMiner, these methods do not directly identify the time and direction of step-wise changes in the gene expression temporal profile.
Many tools are based on matching models of gene behavior to time-course data. For example, the models could be piecewise linear models(16), rising/falling (17), transition intervals (18) or hidden Markov models (HMMs) (19,20), differential equations(21), Bayesian models (22), or Boolean models (23).
StepMiner is also a model-based method, but the one- and two-step patterns are different from the models of other methods. The transition interval method from Hottes et al. (18) is perhaps the most similar, but their models have a transition interval segment between constant-level segments. The transition interval in their model is defined as the change from 25 to 75% of the maximum. The Boolean model proposed by Shmulevich et al. (23) binarizes genes without considering the time component. These methods do not provide P-values, FDR or other statistically justifiable measures of confidence.
Other methods for analyzing time courses are not easily categorized, including identification of differentially expressed genes (24–28) and alignment of time series (29,30). It is unclear how these methods could be used to identify the direction and times of expression level transitions.
For a more concrete view of the differences among tools, StepMiner and four other widely used publicly available programs were run on the same publicly available microarray time course, tracing the response of fibroblasts to the addition of serum (31,32). The time course consists of 13 arrays, taken at the time 0, 1, 2, 3, 4, 6, 8, 10, 12, 16, 20, 24 and 36 h. The data for all of the 5,289 genes with no missing time points were used. The time course was analyzed using hierarchical clustering (8), SAM (2), EDGE (25), STEM (12) and StepMiner. There is a more detailed discussion, with examples, in the Supplementary Data S1, including figures showing the results of each program on the above mentioned data set.
A side-by-side comparison of these algorithms does not necessarily show one to be superior, since the algorithms were developed for different purposes, but it does clarify the differences between them. For example, it is tempting to try to use SAM to find transition points in genes by looking for significant differences in average expression before and after a specified time point. However, many of the genes selected by this method do not, in fact, have a transition at the specified time point.
Hierarchical clustering sometimes finds clusters of genes that seem to transition at the same time point. However, using hierarchical clustering to find transitions involves subjective and time-consuming manual search through the clusters, and the selected clusters only imperfectly capture the genes with transitions at a particular time. EDGE retrieves the list of differentially expressed genes over the time course, which answers a question that is different from finding the seems to be totally unrelated to finding direction and times of transitions. STEM provides model profiles and their significance; but the profiles generally look nothing like step functions, and are not helpful for locating transitions.
Strengths and limitations of StepMiner
StepMiner is an appropriate tool for users who are interested in binary models of gene expression time courses. Although a binary model abstracts away from many complexities of gene expression, it has several advantages: it is easy to understand; it has few parameters; and, in many cases, the details of the behavior between transitions may not be as biologically interesting as the transition. Moreover, StepMiner is very fast. It can process 15 microarrays of 40 000 genes each in < 15 s. (The optional FDR calculation in StepMiner for this microarray data using 100 permutations takes ∼ 12 min.)
Even when the gene expression level over time is only approximately binary, we find that the results produced by StepMiner are sensible. For example, consider the measurements for the genes in Figure 4. In each case, the behavior of the gene may be complex or noisy, but StepMiner reports reasonable (and objective) results about when each gene becomes up-regulated.
The P-value for an individual gene captures the degree to which the binary model fits the temporal variation in gene expression. Large variations in the supposedly down-regulated and up-regulated intervals will lead to worse P-values than approximately constant behavior. Signals that transition between two levels, but transition slowly, will have worse P-values than signals that transition rapidly. For a slowly transitioning signal, the best placement of the transition is not obvious; StepMiner will tend to put it in the middle of the transition. In the extreme case of purely linear behavior, StepMiner will place a transition in the middle—but the P-value will be poor and the gene is likely to end up in the ‘other’ category depending on the user-specified P-value cutoff.
The current version of StepMiner is most appropriate for experiments that measure the transcriptional response to a stimulus, and for time courses with 10 – 30 measurements (however, a time course of five time points with three replicated arrays at each time point gives the confidence of 15 measurements).
There are two ways that a low P-value match can occur: (1) there could be several consecutive points that are consistently low or high, or (2) there could be one or two measurements that deviate greatly from the others. In practice, a low P-value from multiple points is more trustworthy than a low P-value from large differences, because a single deviant measurement could be an outlier resulting from non-Gaussian measurement error.
Very short time courses are problematic, because reliable low P-value matches are unlikely to occur. There is simply too little evidence to support the matching of steps, even when steps exist. On the other hand, very long time courses are problematic because the data may actually have more than two steps, and neither the one-step nor two-step patterns will match well. There is currently an upper limit of two steps in StepMiner because the running time of adaptive regression algorithm increases exponentially with the number of steps.
The StepMiner algorithm can deal gracefully with missing measurements, which are common in microarray data. Omission of one or two measurements for a gene simply degrades the confidence in the results for that gene. However, in practice, it is probably better to fill in missing data points using one of a variety of existing imputation algorithms for microarrays (33).
Optimizing time course experiments for StepMiner
Simulations suggest several guidelines for experimental design that can lead to more meaningful results with StepMiner. There should be enough time points, spaced closely enough, so that there will be multiple points during the constant segments of the step patterns. In particular, there should be several time points before a transition that is expected—otherwise, there will be little evidence to distinguish the first responses to a stimulus from noise.
Replicated measurements at the same time point should not be averaged. Instead, they should be handled using the same matching algorithm as sequential measurements, except that the algorithm should not try to put a step between simultaneous measurements. With this processing, they can directly improve the P-values of extracted signals.
If the only concern is getting the most accurate results from a given number of microarrays, it is better to take more frequent measurements than to follow the common practice of repeating several microarrays at the same time, if the results are to be analyzed with StepMiner. For example, given 10 h time course, it is better to use 30 arrays by using one every 20 min than to use three arrays simultaneously every hour. Since StepMiner tries inserting steps between every pair of transitions, the time resolution of the results nearly triples, at the cost of a small loss of accuracy in recognizing the correct kind of step.
This conclusion is supported by simulation results shown in Table 2. Each of the four different step types was simulated, with time of each step ts from a uniform distribution over the entire interval. As discussed above, the measurements at each time point were taken, and Gaussian noise was added so that the step height is 5σ. When a step is found between time points ti and ti+1, the time of the step is estimated to be (ti + ti+1)/2. The ‘time error’ of the step is |ts − (ti + ti+1)/2|. The number of correctly classified steps is shown.
Table 2.
Identification of steps and average deviation from the true step positions by StepMiner with replication versus the addition of more time points
| Type | True | Missed | False | Average |
|---|---|---|---|---|
| Step | Step | Step | Deviation(Min) | |
| Addition | 99% | 1% | 8% | 11 |
| Replication | 100% | 0% | 8% | 34 |
The Addition method uses 30 different time points. The Replication method uses 10 time points with three replicates. The analysis was performed on artificial data for 1000 genes with 500 single steps (Step height = 5σ) placed uniformly randomly across the time-course of 10 h. ‘True Step’ is the number of correctly identified steps. ‘Missed Step’ is number of steps missed. ‘False Step’ is the number of steps detected in random data.
Combining StepMiner with other tools
Once StepMiner is run on a given data set, the genes that are identified as undergoing binary transitions can easily be partitioned into sets based on the number, direction, and timing of transitions. Using other tools, these sets can be merged at the user's discretion (e.g., the set of one-step genes that rise at time 3 could be merged with the two-step genes that rise at time 3).
The sets can be placed in a specific order for visualization in a heat map using a tool such as TreeView (34). First, genes are categorized by the direction of change and number of steps into five generic gene sets: ‘up’, ‘down’, ‘up then down’, and ‘down then up’ and ‘other’. The one-step sets are further subdivided into more specific sets by time of change, and the two-step categories were divide by time of the first change, and, secondarily, by the time of the second change.
The resulting gene sets also facilitate analysis by other tools that can compare different kinds of gene sets for unexpectedly large overlaps. Many programs perform this kind of analysis (5,35–38).
The basic gene sets found by StepMiner can be combined into larger sets of genes with common characteristics. For example, a user might be interested in the set of all genes that contain a step up during a range of time points, regardless of how many steps there are.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
The authors thank Mathew J. Brauer and Howard Chang for providing the necessary data for the analysis and Prof. Trevor Hastie for his useful advice on the StepMiner algorithm. This work, including Open Access publication charges for this article, was supported by the National Institute of Health as part of the Integrative Cancer Biology Program, under grant number NIH 5U56CA112973-02. The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Conflict of interest statement. None declared.
REFERENCES
- 1. The serum time course data can be directly downloaded from SMD at http://genome-www5.stanford.edu/cgibin/publication/viewPublication.pl?pub_no=293.
- 2.Aach J, Church GM. Aligning gene expression time series with time warping algorithms. Bioinformatics. 2001;17:495–508. doi: 10.1093/bioinformatics/17.6.495. [DOI] [PubMed] [Google Scholar]
- 3.Amato R, Ciaramella A, Deniskina N, Del Mondo C, di Bernardo D, Donalek C, Longo G, Mangano G, Miele G, et al. A multi-step approach to time series analysis and gene expression clustering. Bioinformatics. 2006;22:589–596. doi: 10.1093/bioinformatics/btk026. [DOI] [PubMed] [Google Scholar]
- 4.Antoniotti M, Ramakrishnan N, Kumar D, Spivak M, Mishra B. Remembrance of experiments past: analyzing time course datasets to discover complex temporal invariants. NYU-CS-TR858. 2005 [Google Scholar]
- 5.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, et al. Gene ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.BarJoseph Z. Analyzing time series gene expression data. Bioinformatics. 2004;20:2493–2503. doi: 10.1093/bioinformatics/bth283. [DOI] [PubMed] [Google Scholar]
- 7.BarJoseph Z, Gerber G, Simon I, Gifford DK, Jaakkola TS. Comparing the continuous representation of time-series expression profiles to identify differentially expressed genes. PNAS. 2003;100:10146–10151. doi: 10.1073/pnas.1732547100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.BarJoseph Z, Gerber GK, Gifford DK, Jaakkola TS, Simon I. Continuous representations of time-series gene expression data. J. Comput. Biol. 2003;10:341–356. doi: 10.1089/10665270360688057. [DOI] [PubMed] [Google Scholar]
- 9.Brauer MJ, Saldanha AJ, Dolinski K, Botstein D. Homeostatic adjustment and metabolic remodeling in glucose-limited yeast cultures. Mol. Biol. Cell. 2005;16:2503–2517. doi: 10.1091/mbc.E04-11-0968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang HY, Sneddon JB, Alizadeh AA, Sood R, West RB, Montgomery K, Chi JT, van de Rijn M, et al. Gene expression signature of fibroblast serum response predicts human cancer progression: Similarities between tumors and wounds. PLoS Biology. 2004;2:E7. doi: 10.1371/journal.pbio.0020007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Costa IG, Schonhuth A, Schliep A. The Graphical Query Language: a tool for analysis of gene expression time-courses. Bioinformatics. 2005;21:2544–2545. doi: 10.1093/bioinformatics/bti311. [DOI] [PubMed] [Google Scholar]
- 12.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. PNAS. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ernst J, Nau GJ, Bar-Joseph Z. Clustering short time series gene expression data. Bioinformatics. 2005;21(1):i159–i168. doi: 10.1093/bioinformatics/bti1022. [DOI] [PubMed] [Google Scholar]
- 14.Filkov V, Skiena S, Zhi J. Analysis techniques for microarray time-series data. J. Comput. Biol. 2002;9:317–330. doi: 10.1089/10665270252935485. [DOI] [PubMed] [Google Scholar]
- 15.Gollub J, Ball CA, Binkley G, Demeter J, Finkelstein DB, Hebert JM, Hernandez-Boussard T, Jin H, Kaloper M, et al. The Stanford Microarray Database: data access and quality assessment tools. Nucleic Acids Res. 2003;31:94–96. doi: 10.1093/nar/gkg078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grant GR, Liu J, Stoeckert CJ., Jr A practical false discovery rate approach to identifying patterns of differential expression in microarray data. Bioinformatics. 2005;21:2684–2690. doi: 10.1093/bioinformatics/bti407. [DOI] [PubMed] [Google Scholar]
- 17.Hottes AK, Shapiro L, McAdams HH. Dnaa coordinates replication initiation and cell cycle transcription in caulobacter crescentus. Mol. Microbiol. 2005;58:1340–1353. doi: 10.1111/j.1365-2958.2005.04912.x. [DOI] [PubMed] [Google Scholar]
- 18.Lee HK, Braynen W, Keshav K, Pavlidis P. Erminej: tool for functional analysis of gene expression data sets. BMC Bioinformatics. 2005;6:269. doi: 10.1186/1471-2105-6-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leng X, Müller HG. Time ordering of gene co-expression. Biostatistics. 2006;7:569–84. doi: 10.1093/biostatistics/kxj026. [DOI] [PubMed] [Google Scholar]
- 20.Luan Y, Li H. Clustering of time-course gene expression data using a mixed-effects model with B-splines. Bioinformatics. 2003;19:474–482. doi: 10.1093/bioinformatics/btg014. [DOI] [PubMed] [Google Scholar]
- 21.Ma P, Castillo-Davis CI, Zhong W, Liu JS. A data-driven clustering method for time course gene expression data. Nucleic Acids Res. 2006;34:1261–1269. doi: 10.1093/nar/gkl013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moller CS, Klawonn F, Cho KH, Yin H, Wolkenhauer O. Clustering of unevenly sampled gene expression time-series data. Fuzzy Sets and Systems. 2004:49–66. [Google Scholar]
- 23.Owen A. Discussion: Multivariate adaptive regression splines. Ann. Stat. 1991;19:102–112. [Google Scholar]
- 24.Park T, Yi SG, Lee S, Lee SY, Yoo DH, Ahn J-I, Lee Y-S. Statistical tests for identifying differentially expressed genes in time-course microarray experiments. Bioinformatics. 2003;19:694–703. doi: 10.1093/bioinformatics/btg068. [DOI] [PubMed] [Google Scholar]
- 25.Ramoni MF, Sebastiani P, Kohane IS. From the cover: cluster analysis of gene expression dynamics. PNAS. 2002;99:9121–9126. doi: 10.1073/pnas.132656399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saldanha AJ. Java Treeview – extensible visualization of microarray data. Bioinformatics. 2004;20:3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- 27.Sasik R, Iranfar N, Hwa T, Loomis WF. Extracting transcriptional events from temporal gene expression patterns during Dictyostelium development. Bioinformatics. 2002;18:61–66. doi: 10.1093/bioinformatics/18.1.61. [DOI] [PubMed] [Google Scholar]
- 28.Schliep A, Schonhuth A, Steinhoff C. Using hidden Markov models to analyze gene expression time course data. Bioinformatics. 2003;19(1):i255–i263. doi: 10.1093/bioinformatics/btg1036. [DOI] [PubMed] [Google Scholar]
- 29.Segal E, Shapira M, Regev A, Pe'er D, Botstein D, Koller D, Friedman N. Module networks: identifying regulatorymodules and their condition-specific regulators from gene expression data. Nat. Genet. 2003;34:166–176. doi: 10.1038/ng1165. [DOI] [PubMed] [Google Scholar]
- 30.Sherlock G. GO-TermFinder. The Comprehensive Perl Archive Network. (0.7ed) 2003 [Google Scholar]
- 31.Shmulevich I, Zhang W. Binary analysis and optimization-based normalization of gene expression data. 2002;18:555–565. doi: 10.1093/bioinformatics/18.4.555. [DOI] [PubMed] [Google Scholar]
- 32.Storey JD, Tibshirani R. Statistical significance for genomewide studies. PNAS. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Storey JD, Xiao W, Leek JT, Tompkins RG, Davis RW. Significance analysis of time course microarray experiments. PNAS. 2005;102:12837–12842. doi: 10.1073/pnas.0504609102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, et al. From the cover: gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. PNAS. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tai YC, Speed TP. Techreports 667. Berkeley: University of California; 2004. A multivariate empirical bayes statistic for replicated microarray time course data. [Google Scholar]
- 36.Troyanskaya O, Cantor M, Sherlock G, Brown P, Hastie T, Tibshirani R, Botstein D, Altman RB. Missing value estimation methods for DNA microarrays. Bioinformatics. 2001;17:520–525. doi: 10.1093/bioinformatics/17.6.520. [DOI] [PubMed] [Google Scholar]
- 37.Willbrand K, Radvanyi F, Nadal JP, Thiery J-P, Fink TMA. Identifying genes from up-down properties of microarray expression series. Bioinformatics. 2005;21:3859–3864. doi: 10.1093/bioinformatics/bti549. [DOI] [PubMed] [Google Scholar]
- 38.Zhang B, Kirov S, Snoddy J. Webgestalt: an integrated system for exploring gene sets in various biological contexts. NAR. 2005;33:W741–W748. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.