


Abstract
DNA topoisomerase II modulates DNA topology by relieving supercoil stress and by unknotting or decatenating entangled DNA. During its reaction cycle, the enzyme creates a transient double-strand break in one DNA segment, the G-DNA. This break serves as a gate through which another DNA segment is transported. Defined topoisomerase II cleavage sites in genomic and plasmid DNA have been previously mapped. To dissect the G-DNA recognition mechanism, we studied the affinity and reactivity of a series of DNA duplexes of varied sequence under conditions that only allow G-DNA to bind. These DNA duplexes could be cleaved to varying extents ranging from undetectable (<0.5%) to 80%. The sequence that defines a cleavage site resides within the central 20 bp of the duplex. The DNA affinity does not correlate with the ability of the enzyme to cleave DNA, suggesting that the binding step does not contribute significantly to the selection mechanism. Kinetic experiments show that the selectivity interactions are formed before rather than subsequent to cleavage. Presumably the binding energy of the cognate interactions is used to promote a conformational change that brings the enzyme into a cleavage competent state. The ability to modulate the extent of DNA cleavage by varying the DNA sequence may be valuable for future structural and mechanistic studies that aim to determine topoisomerase structures with DNA bound in pre- and post-cleavage states and to understand the conformational changes associated with DNA binding and cleavage.
INTRODUCTION
Various topological problems arise for DNA during cellular processes. These problems are rooted in the double helical nature of DNA and its enormous length. The advancing replication and transcription machinery, for example, generates superhelical tension in the adjacent double helical region, and DNA replication and recombination can generate knotted and entangled DNA structures. Topoisomerase II directly modulates the topology of DNA by relieving supercoil stress and by unknotting or decatenating entangled DNA, making it indispensable for metabolic processes involving DNA (1).
DNA topoisomerase II modifies DNA topology by allowing two DNA chains to freely pass through each other by physically breaking one of the chains. Biochemical and structural studies have strongly suggested a plausible model for the overall mechanism of DNA topoisomerase II (Figure 1A) (2,3). During the reaction cycle, the enzyme binds two segments of DNA. It creates a transient double-strand break in one DNA helix, the gated or G-segment, using two active site tyrosines that covalently attach to a staggered pair of 5′-phosphoryl groups (Figure 1B). This break in the G-segment widens and acts as a gate through which the other segment, the transported or T-segment is passed. The DNA transport step is coupled to the hydrolysis of ATP (4,5). The double-strand break is religated and the T-segment exits the enzyme, completing the DNA transport reaction.
Figure 1.

The reaction catalyzed by DNA topoisomerase II. (A) Overview of the reaction cycle. Two ATPs, G- and T-DNA bind (Steps 1 and 2). G-DNA is cleaved and T-DNA is transported through the break in the G-DNA (Step 3). The break in the G-DNA is religated and the T-DNA is released from the enzyme (Step 4). See the Introduction section for details. (B) Schematic of the cleavage complex between enzyme and G-DNA. The active site tyrosines (Tyr782) of the homodimeric enzyme are covalently attached to a staggered pair of 5′-phophoryl groups on the G-DNA. 5′-32P-labeling of the duplex allows quantification of DNA cleavage by denaturing gel electrophoresis (see the Materials and Methods section).
To fulfill its cellular tasks, topoisomerase II must be able to act at numerous locations throughout the genome. An ability to bind and cleave DNA of any sequence would presumably serve the enzyme best for this task. Contrary to this expectation however, the enzyme does not indiscriminately cleave DNA. Defined cleavage sites have been identified in plasmid and genomic DNA. By mapping several hundred of these sites, degenerate consensus sequences of up to 20 bp have been derived for topoisomerases from a number of organisms (6–13). To better characterize the cleavage specificity of topoisomerase II from Drosophila melanogaster, DNA sequences that contain preferred cleavage sites were selected for in an in vitro evolution experiment. The evolved sequence was AT-rich, had alternating purines and pyrimidines and was cleaved 6-fold better than the starting sequences (14). Although it is possible that the observed discrimination is a consequence of the molecular idiosyncrasies of the DNA-binding site, it has been suggested that the DNA specificity of topoisomerases has evolved as a means for the DNA sequence to directly regulate the activity of the enzyme (14–17).
Dissecting the mechanism of cleavage site recognition necessitates that DNA binding to the cleavage sites can be isolated from DNA binding elsewhere on the enzyme. We recently showed that the affinity of the DNA cleavage domains exceeds the affinity of the other DNA-binding site on the enzyme, the T-DNA site (18). Thus, by using DNA duplexes that are not long enough to contact both binding sites simultaneously and by using enzyme in excess of DNA, DNA duplexes can be preferentially bound at the DNA cleavage domains. Moreover, we demonstrated that the presence of T-DNA is not required for the enzyme to cleave and religate DNA. Therefore, these simplified conditions that prevent T-DNA binding can be employed to follow DNA binding and cleavage solely at the G-DNA-binding site (18).
To explore the G-DNA recognition process, we used a series of DNA duplexes of varied sequences, and we determined their affinities and reactivities using thermodynamic and transient kinetic measurements. This approach allowed us to probe the contributions of individual base pairs on the duplex to binding and reactivity, revealed when cognate interactions are formed along the reaction coordinate and exposed a conformational change of the enzyme–DNA complex that brings the enzyme into a cleavage competent state.
MATERIALS AND METHODS
Enzyme expression and purification
Saccharomyces cerevisiae topoisomerase II was expressed and purified as described (18).
DNA duplex substrates
A series of duplexes with palindromic sequences were created based on a strong cleavage site located between 87 and 126 bp in pBR322 (Dup134 bp and Dup234 bp and their derivatives; Table 1A and B) (19). With the intention of creating DNA duplexes that are not cleaved by the enzyme we also designed palindromic duplexes Dup334 bp and Dup434 bp from DNA regions in the human c-myc gene (bp 3201–3217) and pBR322 (bp 21–37) where no cleavage sites were previously detected (13,20). The cleavage specificity of the main cleavage site of the palindromic DNA duplexes was >90%, except for Dup134 bp; G2T, C6T (87%), Dup134 bp; G2T,C6T, C9T (79%) and Dup434 bp; A3C,A5T, G6C (79%; data not shown).
Table 1.
(A) 34 bp DNA duplexes; (B) 46 bp DNA duplexes and their thermodynamic parameters for binding and cleavage
 |
aBase changes of only the 5′-half of the duplexes are indicated in the names of the duplexes. To keep the DNA duplexes palindromic, corresponding mutations were also introduced in the 3′-half.
bSequences of only one strand of the duplexes are shown from 5′ to 3′. All duplexes are palindromic. The center of the molecule is indicated (dot). The numbering of the bases is relative to the center of the duplex (see ruler) with the base immediately to the left having the position −1 and that to the right the position 1. The enzyme covalently attaches to the −2 position of each strand (arrowhead).
cBest fit parameters from Figure 1 and analogous titrations (see figure legend and Results section for details).
dKd,obs and Kclvg,obs are defined in the Results section. Except where indicated, values calculated from K1/2 and Kclvg,obs (see the Results section). Values for Kd,obs vary <2 two-fold, for Kclvg,obs < 50% in independent experiments (see the Materials and Methods section).
eValue in parenthesis measured in competition experiments (Figure 2).
fN.d.: not determined.
DNA duplexes were assembled from DNA oligonucleotides (IDT, Coralville, IA). DNA oligonucleotides were purified by denaturing polyacrylamide gel electrophoresis (PAGE) and annealed in annealing buffer (10 mM Tris–HCl, pH 8, 50 mM NaCl, 1 mM Na-EDTA) by heating the mixture to 90°C and cooling it to room temperature over 2 h. As necessary, the two strands of the annealed DNA duplexes were 5′-phosphorylated using γ-[32P]ATP (MP Biomedicals, Solon, OH) and T4-polynucleotide kinase (NEB, Ipswich, MA) according to the supplier's instructions. Radiolabeled and unlabeled DNA duplexes were further purified by PAGE on a 15% non-denaturing gel.
DNA cleavage assay
Equilibrium levels of DNA cleavage were measured in the absence of nucleotides and under conditions that prevent T-DNA from binding (18) such that the ATPase and T-DNA transport reactions do not complicate the interpretation of the DNA cleavage data. Enzyme, 5′-32P-labeled DNA and, as indicated, unlabeled competitor DNA were mixed in reaction buffer (50 mM potassium HEPES, pH 7.5, 150 mM potassium acetate, 0.1 mM Na-EDTA, 20% sucrose, 0.25 mg/ml BSA, 0.01% Tween 20, 5 mM 2-mercaptoethanol and 10 mM calcium acetate) and incubated for 10 min before quenching with the same volume of 1 M NaOH. All assays were carried out at 30°C. Control experiments showed that doubling or halving the incubation time gave identical levels of cleavage. The quenched reactions were adjusted to pH 9 with Tris–HCl, pH 7, three volumes of loading buffer (9 M urea, 20% sucrose, 5 mM Na-EDTA) were added and the mixture was heated at 90°C for 1 min. The denaturing conditions ensure that the 32P-labeled, non-covalently bound DNA fragment dissociates from the covalent enzyme–DNA complex (Figure 1B). The cleaved, 32P-labeled DNA strands were then separated from the uncleaved strands by denaturing PAGE as described (18).
The kinetics of DNA religation was measured by first incubating trace amounts of 32P-labeled DNA with the indicated enzyme concentration for 2 min in reaction buffer. Then, 0.2 mg/ml (final concentration) of unlabeled pBR322 plasmid DNA was added, aliquots were removed from the reaction at specified times and quenched as above. Control experiments showed that the observed DNA religation rate constants are not sensitive to increasing the chase DNA concentration up to at least 1 mg/ml. Moreover, the observed rate constants are the same within error if the cleavage complexes are chased by a rapid dilution instead of unlabeled DNA (data not shown). DNA cleavage was analyzed by denaturing PAGE as described above.
Quantitative analysis of DNA cleavage data
The DNA cleavage data were analyzed in terms of a minimal reaction framework describing the DNA cleavage reaction (Scheme 1). According to Scheme 1, the enzyme (E) binds double-stranded DNA (SS, where each ‘S’ refers to one strand in the uncleaved substrate DNA) and undergoes a conformational change (Kiso; see below and our unpublished data) before it cleaves the first and the second DNA strand (Kclvg,1 and Kclvg,2, respectively). The assays herein cannot distinguish all species in Scheme 1, so the available data are not sufficient to define values for all equilibrium constants in Scheme 1. We therefore considered a simplified reaction framework (Scheme 2) that consists of a single binding and a single chemical step with which we can analyze the data, albeit at a lower resolution.
Scheme 1.

E: Enzyme E′: Enzyme in an alternative conformation
Scheme 2.

Reduction of Scheme 1 to Scheme 2 entails grouping together the two non-covalent enzyme-DNA species [(ESS) and (E’SS); Scheme 1] and the two covalent species [(E’SP) and (E’PP)]. We termed the equilibria in Scheme 2 the apparent binding and the apparent cleavage equilibria (Kd,app and Kclvg,app). Kd,app can be interpreted as an approximation of the difference in thermodynamic stability between the most stable non-covalent enzyme–DNA complex in Scheme 1 and the free reactants. Analogously, Kclvg,app approximates the difference in thermodynamic stability between the most stable covalent and the most stable non-covalent complex.
The apparent binding and the apparent cleavage equilibria (Kclvg,app and Kd,app, Scheme 2) cannot be precisely determined from the data. This limitation is rooted in the way DNA cleavage is measured. We assay only the total amount of cleaved DNA strands. Single-strand breaks introduced by the enzyme (species E’SP, Scheme 1) produce only half the cleavage signal of double-strand breaks, and a given amount of cleaved product can correspond to different combinations of singly versus doubly cleaved species (E′SP and E′PP). Fortunately, independent experiments using a DNA substrate with the sequence of Dup134 bp; G2T have shown that there is little accumulation of the [E’SP] species [(E′PP)/(E′SP) = 5 and (E′SP)/(ESS) + (E′SS)] = 0.26; Scheme 1; manuscript in preparation). We therefore approximated Kclvg,app and Kd,app in the analysis by neglecting that single-strand breaks result in a lower signal than double- strand breaks. We termed these approximations Kd,obs and Kclvg,obs in the Results section. The deviations of the observed from the apparent equilibria are <2-fold (Supplementary Data), small enough so that conclusions drawn herein are not affected.
Experimental errors were estimated by independent repetitions of experiments with the duplexes Dup134 bp, Dup146 bp, Dup134 bp; G2T, Dup146 bp; G2T, Dup134 bp; G2T, C9T, Dup134 bp; G2T, 11CTAGGAT, Dup234 bp, Dup246 bp. Values obtained for Kd,obs varied <2-fold and for Kclvg,obs < 1.5-fold.
The DNA competition data in Figure 4 below were used to extract the dissociation constants of the competing, unlabeled DNA. To this end, systems of linear equations were constructed based on the equilibria describing Scheme 3. The system of linear equations was solved for the fraction of radio-labeled DNA that is cleaved by the enzyme [(EPP1)/(SS1)tot], dependent on the total enzyme and DNA concentrations and the four equilibrium constants in Scheme 3 in Mathematica 5.0 [Equation (1); Wolfram Research, Champaign, IL].
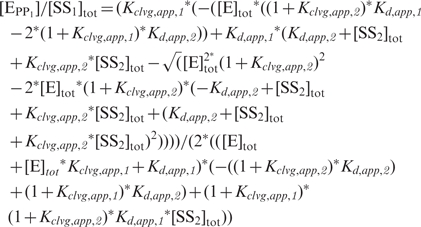 |
1 |
Figure 4.

Dup434 bp, a DNA duplex for which no DNA cleavage was observed, binds to the DNA cleavage domains with a similar affinity as Dup234 bp, a weakly cleavable duplex. Radiolabeled Dup234 bp (0.3 nM) was mixed with enzyme (17 nM) in the presence of varying concentrations of unlabeled competitor DNA (black squares: Dup234 bp; red circles: Dup434 bp). Lines are fits of the data to a simple competition scheme (Scheme 3). Estimates for the DNA duplex affinities determined in this way are summarized in Table 1A.
Scheme 3.

[SS1]tot and [SS2]tot in the equation represent the total concentrations of radio-labeled and unlabeled DNA duplex, and [E]tot the total enzyme concentration. In addition to the total concentrations of DNA and enzyme, three of the four equilibrium constants are known: the cleavage equilibria for both DNA duplexes and the affinity of the radio-labeled DNA were obtained independently by measuring cleavage with varying enzyme concentrations and are tabulated in Table 1A. The remaining unknown variable in Equation (1), the affinity of the competing unlabeled DNA, Kd,app, 2, was obtained through a fit of the competition data to the equation.
RESULTS
The goal of this study was to gain insights into the recognition of cognate DNA by topoisomerase II. By assaying DNA binding and cleavage at equilibrium for a series of DNA duplexes of varying sequences we identified base pair positions along the duplex that are recognized by the enzyme. Equilibrium and transient kinetic measurements were used to distinguish the timing of formation of these cognate interactions with respect to individual steps in the cleavage reaction. In principle, discrimination favoring specific cleavage sequences could arise in the DNA-binding step, in the DNA cleavage step, or after DNA cleavage. These three models are depicted in the panels A, B and C in Scheme 4. For each model, free energy profiles for ‘good’ and ‘poor’ substrates are in black and red, respectively (E: enzyme, SS: DNA, where the two S's designate the two strands in one DNA duplex in their uncleaved substrate form, PP: DNA in which both strands are in the cleaved product form).
Scheme 4.

To facilitate this investigation, our reaction buffer contained Ca2+ in place of the physiological Mg2+. Ca2+ significantly increases the DNA cleavage signal (21), thereby allowing accurate quantification, but does not alter the preferred cleavage sites (13, 14, 20, 22). The DNA duplexes we used had palindromic sequences (Table 1). Because the two DNA strands in a palindrome are identical it is not necessary to distinguish the extent of cleavage of the two strands, greatly simplifying analysis. Most of the palindromes we created were derived from a previously characterized strong DNA cleavage site for topoisomerase II (Dup134 bp and Dup234 bp and their derivatives; Table 1A) (19). In addition, we designed two palindromes from regions in the plasmid pBR322 and the human c-myc gene that were not cleaved according to studies that mapped cleavage sites in long stretches of DNA (Dup334 bp and Dup434 bp, Table 1A; see the Materials and Methods section) (13,20,23). One of these two duplexes, Dup334 bp, unexpectedly contained a very strong DNA cleavage site. Eleven of the 14 duplexes studied were 34-bp long, long enough to cover the 28-bp footprint of topoisomerase II (24,25). To probe for possible length dependences of binding and reactivity, we extended three of these duplexes by 6 bp on both ends, resulting in 46 bp duplexes (Table 1B).
DNA binding and reactivity data were interpreted with the help of a simplified reaction scheme for the DNA cleavage reaction (Scheme 2; see the Materials and Methods section). This scheme consists of three states: the free reactants [enzyme (E) and DNA (SS)], the non-covalent enzyme–DNA complex (ESS) and the cleavage complex (EPP). We termed the equilibria in the Scheme 2 the ‘apparent’ DNA affinity (Kd,app) and the ‘apparent’ cleavage equilibrium (Kclvg,app), because ESS and EPP each consist of subspecies (see the Materials and Methods section). The apparent equilibria cannot be directly determined from the data (see the Materials and Methods section). However, our data allowed us to obtain estimates or ‘observed’ values (Kd,obs and Kclvg,obs) for the apparent equilibria in Scheme 2 (see the Materials and Methods section). These observed values deviate from the apparent equilibria by at most 2-fold (Supplementary Data).
The DNA-binding step does not contribute significantly to the selection of the cognate DNA
We first compared the binding affinity of DNA duplexes that are good cleavage substrates (so-called ‘cognate’ duplexes) and DNA duplexes that are poorly or imperceptibly cleaved. For the subset of duplexes that are cleaved by the enzyme, Kd,obs and Kclvg,obs can be determined by monitoring the extent of cleavage at equilibrium in titrations with constant, subsaturating concentrations of DNA and varying concentrations of enzyme (Figure 2). These curves yield two constants, the concentration of enzyme that promotes half maximal DNA cleavage (K1/2) and the fraction of DNA cleavage with saturating concentrations of enzyme (‘maxcleavage’). From this fraction, it is possible to calculate Kclvg,obs [Equation (2)].
| 2 |
With K1/2 and Kclvg,obs, Kd,obs is obtainable using Equation (3), which is derived from Scheme 2.
| 3 |
Values for Kd,obs and Kclvg,obs for individual duplexes determined in this way are summarized in Table 1. For Dup134 bp and its 34-bp long derivatives the maximal extent of DNA cleavage varies between 10 and 76% (Figure 2 and Table 1A). However, the DNA-binding affinity is largely unaffected within the series of duplexes (average affinity 48 ± 17 nM; Table 1A; Figure 3). Moreover, 34-bp duplexes of unrelated DNA sequences (Dup234 bp, Dup334 bp and Dup434 bp; A3C,A5T, G6C) bind DNA with affinities that are within a factor of two of the average affinity (23–100 nM; Table 1A; Figure 3). We conclude from these results that topoisomerase II does not discriminate between DNA sequences based on DNA affinity.
Figure 2.

Affinity and cleavage equilibrium determination for DNA duplexes. (A) Subsaturating concentrations of DNA (Dup134 bp; G2T; <0.5 nM) and varying enzyme concentrations were mixed and equilibrated before the reaction was quenched. Cleaved DNA was separated from uncleaved DNA by denaturing gel electrophoresis. Every second lane was loaded on the gel after a time delay to increase the space between the bands for optimal quantification. DNA cleavage data for the DNA palindromes Dup134 bp and Dup134 bp; G2T,C6T, C9T are shown in Figure S1. (B) DNA cleavage from (A) was quantified (blue squares). In addition, results for the palindromes Dup134 bp (red triangles), Dup134 bp; G2T, C9T (green circles), Dup134 bp; G2T, C6T (black diamonds) and Dup134 bp; G2T, C6T, C9T (magenta triangles) are shown. The data were fit to the equation f = [E] * ‘maxcleavage’/(K1/2 + [E]) with f being the observed fraction of cleaved duplex at equilibrium, ‘maxcleavage’ the maximal fraction of cleaved DNA with saturating enzyme concentrations, and K1/2 the concentration of enzyme necessary to achieve half-maximal DNA cleavage. The fit parameters are summarized in Table 1A, along with other results.
Figure 3.

DNA duplex affinity (Kd,obs) does not correlate with the ability of the enzyme to cleave the duplex (Kclvg,obs). The observed affinity and cleavage equilibria of 34-bp duplexes were obtained from Table 1A. The arrow indicates a data point, for which only an upper limit was established. A model in which the fold increase in the observed affinity correlates with the fold increase in the observed cleavage equilibrium fits the data poorly (dashed line; slope of negative one in the double logarithmic plot). The data are best represented by a model in which there is at most weak correlation between the DNA affinity and its propensity to being cleaved (solid line; slope of 0.11 in the double logarithmic plot).
We identified one DNA duplex (Dup434 bp) for which we could not detect cleavage, even with enzyme concentrations up to 900 nM (Table 1A and data not shown). The model proposed above predicts that this duplex binds with affinities indistinguishable from the cleavable duplexes. But because the duplex was not cleaved, we could not use the DNA cleavage assay to determine the affinity of this duplex. Instead, we turned to a competition assay (Scheme 3). In this assay, we incubated subsaturating concentrations of enzyme, trace amounts of a weakly cleavable, radiolabeled duplex (Dup234 bp) and varying concentrations of unlabeled competitor DNA. After equilibration, we determined the fraction of cleaved radiolabeled duplex (Figure 4). As unlabeled competitor DNA, we used either the duplex for which no cleavage was detected (Dup434 bp) or, as a control, a weakly cleavable duplex (Dup234 bp). The two unlabeled DNA duplexes gave similar competition with the radiolabeled duplex. Estimates for the affinities were obtained through fits of the data to a simple competition model, with values of 23 and 25 nM for the two duplexes (Figure 4; Dup234 bp and Dup434 bp, Table 1A). The dissociation constant measured for Dup234 bp by the competition assay was within error of the value obtained from titrating DNA with enzyme (23 and 36 nM, Table 1A).
We conclude from the above data that topoisomerase II does not distinguish cognate from non-cognate DNA during the DNA-binding step (Scheme 4, Model A). Consequently, steps after DNA binding must be used to discriminate cleavage from non-cleavage sites in DNA (Model B or C). Before we distinguish between those models, we examine the length dependence of binding and reactivity.
Additional interactions involved in binding G-DNA
To probe the length dependence of DNA binding, we measured the DNA affinities of three DNA duplexes that were 46 instead of 34-bp long (Table 1B). The 46-bp duplexes bound on average 7.8 ± 1.5-fold more tightly than their shorter relatives (Table 1). The increase of the affinity upon increasing the DNA length beyond 34 bp could indicate that the enzyme can bind in multiple registers when longer DNA is used or that 34-bp duplexes are not sufficiently long to realize all potential binding interactions with the enzyme (26). Most simply, DNA cleavage at the central cleavage site within the duplexes would be expected to decrease if the first model were to hold. Such a decrease is not observed (Table 1). The data therefore support models in which 34-bp duplexes do not provide all of the determinants for binding. Crystal structures of fragments of DNA gyrase and yeast DNA topoisomerase II, however, reveal a DNA-binding domain that is not wide enough to contact >34 bp (27–29). It is possible that there is an additional DNA-binding domain that is not present in the crystal structures. One candidate is the C-terminal domain (CTD), as CTDs of prokaryotic and eukaryotic type II topoisomerases have been suggested to be involved in DNA binding (30–36). Alternatively, the additional flanking sequences could have an indirect effect, rigidifying DNA within the binding site or providing an electrostatic attraction.
Mapping cognate enzyme/DNA interactions
The DNA sequence in one family of DNA duplexes, Dup134 bp and its eight derivatives, was systematically varied to probe the effects of base-pair mutations on DNA cleavage. The extent of DNA cleavage within this series varies between 10 and 80% (Figure 2 and Table 1). The effects of varying the DNA sequence on Kclvg,obs allowed us to infer contributions of different regions of the duplex in the recognition process.
Varying the duplex length from 34 and 46 bp has only a small effect on the observed cleavage equilibrium (on average 1.2 ± 0.1-fold corresponding to 0.11 ± 0.06 kcal/mol; Table 1), suggesting that the enzyme does not make specific contacts to the flanking 6 bp in the three 46 bp duplexes. It cannot be ruled out, however, that the enzyme could form specific interactions to nucleotides within this region if different flanking sequences were used; it is also possible that cognate interactions could form with regions outside the central 46 bp. Varying the outer 7 bp on both sides of the 34 bp duplexes also has only small effects on the observed DNA cleavage equilibrium (<1.3-fold, corresponding to <0.06 kcal/mol per side of the duplex; compare Dup134 bp; G2T with Dup134 bp; G2T, 11CTAGGAT and Dup134 bp; G2T, 11GATTTCA; Table 1A and Figure 5). Thus, the results suggest that this region of the duplex is not specifically recognized in the cleavage process. In contrast, changing single base pairs within the central 20 bp of the duplexes affected the cleavage equilibrium by up to 0.6 kcal/mol (Table 1A and Figure 5), suggesting that the enzyme forms cognate interactions with these base pairs.
Figure 5.

Deleterious effect of base-pair changes along the DNA duplex on the cleavage equilibrium Kclvg,obs. Numbering of base pairs as in Table 1. Fold differences in Kclvg,obs (Table 1) upon changing base pairs in Dup134 bp were calculated from which the energy of this perturbation was calculated (see the Materials and Methods section). Positions along the duplex that were not mutated remain blank in the graph. The dashed line circle designates the center of the palindrome. Each base-pair change in one half side was also introduced in the other half side of the duplex. In the graph, we assume that the observed energetic effects of the mutations in the half sides are additive. The outer 7 nt of the duplex were changed en bloc (compare Dup134 bp; G2T with Dup134 bp; G2T, 11CTAGGAT and Dup134 bp; G2T, 11GATTTCA; Table 1). Substitution of this region had a <30% effect on Kclvg,obs (Table 1). We assume in the graph that this modest effect did not arise through compensatory large positive and large negative effects of changing multiple base pairs at a time. The arrowhead marks the base to which the active site tyrosine (Y782) covalently attaches.
To gain further insights into the importance of individual base pairs for the recognition process we attempted to transform the non-cleavable DNA duplex Dup434 bp into a cleavable one. To this end, we compared the sequences of the Dup434 bp with the sequences of the cleavable substrates. Base pairs at three positions (3, 5 and 6) within one half-site of the central 20 bp of the non-cleavable duplex differed from the corresponding base pairs in the cleavable duplexes (Table 1A). The enzyme gained the ability to cleave this duplex upon mutation of these 3 bp in both half-sites (Table 1A, compare Kclvg,obs of Dup434 bp with that of Dup434 bp; A3C,A5T, G6C). Per half-site, these 3 bp collectively contribute >1 kcal/mol during the recognition process. We have not deconvoluted the relative contributions of these 3 bp in cleavage site selection.
Topoisomerase forms specific interactions to cognate DNA before DNA cleavage
Above, we identified interactions between the DNA and the enzyme that contribute to recognition of cognate DNA. The observation that DNA cleavage, but not binding, is affected by DNA sequence suggests that the contacts important for cleavage site selection are formed only after association of enzyme and DNA. Here we ask whether the contacts are formed before the cleavage step—to promote it or after the DNA cleavage step—to stabilize the cleaved complex (Scheme 4, Models B and C). These models can be distinguished by measuring the religation rate constant for good and poor DNA cleavage substrates. If formation of these contacts prior to cleavage is responsible for the different extents of cleavage (Model B) then the religation rate would be unaffected by the DNA sequence. If, on the other hand, the contacts are formed only after cleavage and ‘pull’ the equilibrium toward cleaved products (Model C) then the religation rate would be slower with cognate DNA sequences. As DNA religation, not DNA dissociation, is rate limiting for the overall religation reaction (manuscript in preparation), DNA religation can be directly probed by pulse chase experiments in which the enzyme is first allowed to cleave a radio-labeled DNA duplex before the mixture is chased with excess unlabeled DNA to allow irreversible religation.
We measured DNA religation for the duplex Dup134 bp; G2T and a variant with a mutation at the positions −6 and 6 (Dup134 bp; G2T, C6T). This mutation reduces the cleavage equilibrium by 8-fold (Table 1A). As a control, we monitored DNA religation of a third duplex in which the bases at the positions −9 and 9 are altered (Dup134 bp; G2T, C9T). The C9T mutation has only a small (1.5-fold) effect on DNA cleavage (Table 1A Figure 5). The DNA religation time courses are shown in Figure 6. The three DNA duplexes are religated with indistinguishable rate constants. These results provide strong evidence that the enzyme has interactions with the regions around the base pairs at position −6 and 6 in the transition state for DNA cleavage (Scheme 4, Model B) and not just after DNA cleavage (Model C). Additional kinetic data show that the transition state for overall DNA cleavage is associated with cleavage of the first DNA strand and not the second (manuscript in preparation). The cognate contacts are therefore formed prior to cleavage of the first strand.
Figure 6.

The DNA duplexes Dup134 bp; G2T (red circles), Dup134 bp; G2T, C6T (black triangles) and Dup134 bp; G2T, C9T (blue squares) are religated with similar rate constants. DNA religation was measured in a pulse chase experiment. First, the enzyme was incubated with radiolabeled DNA duplex to allow DNA cleavage to take place. Then, the reaction mixture was chased with excess, unlabeled DNA, and the disappearance of cleaved DNA was followed over time. The data are fit to a single exponential expression (lines). Best fit values: kobs = 0.037 ± 0.003 s−1 (red), kobs = 0.042 ± 0.011 s−1 (black), kobs = 0.038 ± 0.004 s−1 (blue).
Although in principle the cognate interactions could arise concomitant with formation of the transition state for cleavage, the direct coupling of a physical process of formation of binding interactions and the bond vibrations that lead to cleavage is a physically unreasonable interpretation. This scenario would require the coupling of physical events that typically take place on disparate time scales and an enormously complex (and thus highly improbable) reaction coordinate (37,38). Thus, we conclude that interactions involved in recognizing cognate DNA sequences occur subsequent to binding but prior to cleavage of the first DNA strand.
DISCUSSION
More than a century ago, Fisher (39) proposed that biological recognition is analogous to the fit between a lock and key. Since then, this proposal has been refined to include dynamic motions that the enzyme and substrate undergo upon binding (38, 40–42) and it has been recognized that binding energy can be used to promote these conformational changes (43). Such recognition processes become still more complex for enzymes that couple different processes, such as the coupling of the conformational changes for DNA strand passage and the hydrolysis of two ATPs by type II topoisomerases.
We have investigated the binding process between topoisomerase II and its DNA cleavage substrate, the G-DNA. We used a series of DNA duplexes of related and unrelated sequences and determined their affinity and reactivity with thermodynamic and transient kinetic measurements. Our results indicate that the recognition process occurs in at least two phases. The affinities for duplexes are similar and do not correlate with the ability of the enzyme to cleave them. Thus, specific interactions between the enzyme and the elements on the DNA that define the cleavage site are not yet formed in this phase. The similar DNA affinities of the tested DNA duplexes are consistent with a model in which DNA poses an isoenergetic surface for the enzyme. Such an isoenergetic surface may facilitate sliding of the enzyme along the DNA (44), a property that has been suggested for other DNA-binding enzymes to accelerate formation of the cognate enzyme–DNA complex (45) and may increase the processivity of topoisomerase II (46).
The second phase of the recognition process takes place after DNA binding. During this phase, cognate interactions between the enzyme and the DNA cleavage site are formed. The contacts provide a specificity of >600-fold, corresponding to >3.8 kcal/mol of recognition energy (Table 1A; compare Kclvg,obs of Dup134 bp with that of Dup434 bp). Changes in the DNA sequence outside the central 20 bp did not significantly affect the enzyme's ability to cleave the bound duplex. We therefore suggest that the sequence information defining a cleavage site of yeast topoisomerase II is contained within the central 20 bp. Such a length for the recognition site is consistent with earlier studies that derived consensus sequences of up to 20 bp (6–12). In the light of the modest energetic contributions of individual base pairs within the cleavage site for the selection process, the long recognition sequence and the limited number of known cleavage sites, it is not surprising that the predictive value of existing consensus sequences has been limited (14).
Using the strongest cognate contact that we have identified (Figure 5, positions −6 and 6) we probed the timing of cognate contact formation relative to DNA cleavage and showed that the cognate interaction is formed prior to DNA cleavage (Figure 6). This interaction therefore does not provide specificity by merely stabilizing the cleaved state. Presumably the cognate interaction is formed upon a transient, energetically uphill conformational rearrangement of the enzyme–DNA complex that takes place before DNA cleavage. A hypothetical free energy profile for a model that is consistent with our data is shown in Scheme 5 (coloring as in Scheme 4). To account for the similar DNA binding of DNA duplexes that vary in their capacity to being cleaved, the isomerization step must be thermodynamically unfavorable. The binding energy of the cognate contact is used to promote this conformational change, stabilizing E’SS relative to ESS. In Scheme 5, the chemical step, not the binding or isomerization step, limits the rate of DNA cleavage as suggested from additional kinetic data (manuscript in preparation).
Scheme 5.

A thermodynamically unfavorable isomerization step of the enzyme–DNA complex that occurs prior to DNA cleavage was independently identified by a kinetic dissection of the DNA cleavage reaction, and the results suggested that the closure of the ATPase domains is part of this conformational change (manuscript in preparation). We therefore speculate that the conformational step that examines the identity of DNA is also induced by closure of the ATPase domains. The ability to adjust the extent of DNA cleavage by varying the DNA sequence may be a valuable tool for future structural studies aimed at distinguishing different complexes and conformational states that are involved in the complex and fascinating reaction cycle of DNA topoisomerase II.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We would like to thank Janet Lindsley (University of Utah) for providing expression plasmids and for initial advice on this project, and we thank members of the Herschlag lab for critically reading the manuscript. This work was supported by a grant from the National Institutes of Health to D.H. (GM64798), and F.M.-P. was supported in part by a fellowship from the Boehringer Ingelheim Fonds. Funding to pay the Open Access publication charges for this article was provided by an NIH grant (GM64798) to D.H.
Conflict of interest statement. None declared.
REFERENCES
- 1.Wang JC. DNA topoisomerases. Annu. Rev. Biochem. 1996;65:635–692. doi: 10.1146/annurev.bi.65.070196.003223. [DOI] [PubMed] [Google Scholar]
- 2.Corbett KD, Berger JM. Structure, molecular mechanisms, and evolutionary relationships in DNA topoisomerases. Annu. Rev. Biophys. Biomol. Struct. 2004;33:95–118. doi: 10.1146/annurev.biophys.33.110502.140357. [DOI] [PubMed] [Google Scholar]
- 3.Schoeffler AJ, Berger JM. Recent advances in understanding structure-function relationships in the type II topoisomerase mechanism. Biochem. Soc. Trans. 2005;33:1465–1470. doi: 10.1042/BST0331465. [DOI] [PubMed] [Google Scholar]
- 4.Lindsley JE, Wang JC. On the coupling between ATP usage and DNA transport by yeast DNA topoisomerase II. J. Biol. Chem. 1993;268:8096–8104. [PubMed] [Google Scholar]
- 5.Baird CL, Harkins TT, Morris SK, Lindsley JE. Topoisomerase II drives DNA transport by hydrolyzing one ATP. Proc. Natl Acad. Sci. USA. 1999;96:13685–13690. doi: 10.1073/pnas.96.24.13685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sander M, Hsieh TS. Drosophila topoisomerase II double-strand DNA cleavage: analysis of DNA sequence homology at the cleavage site. Nucleic Acids Res. 1985;13:1057–1072. doi: 10.1093/nar/13.4.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spitzner JR, Muller MT. A consensus sequence for cleavage by vertebrate DNA topoisomerase II. Nucleic Acids Res. 1988;16:5533–5556. doi: 10.1093/nar/16.12.5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spitzner JR, Chung IK, Muller MT. Eukaryotic topoisomerase II preferentially cleaves alternating purine-pyrimidine repeats. Nucleic Acids Res. 1990;18:1–11. doi: 10.1093/nar/18.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cornarotti M, Tinelli S, Willmore E, Zunino F, Fisher LM, Austin CA, Capranico G. Drug sensitivity and sequence specificity of human recombinant DNA topoisomerases IIalpha (p170) and IIbeta (p180) Mol. Pharmacol. 1996;50:1463–1471. [PubMed] [Google Scholar]
- 10.Pommier Y, Capranico G, Orr A, Kohn KW. Local base sequence preferences for DNA cleavage by mammalian topoisomerase II in the presence of amsacrine or teniposide. Nucleic Acids Res. 1991;19:5973–5980. doi: 10.1093/nar/19.21.5973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Capranico G, Kohn KW, Pommier Y. Local sequence requirements for DNA cleavage by mammalian topoisomerase II in the presence of doxorubicin. Nucleic Acids Res. 1990;18:6611–6619. doi: 10.1093/nar/18.22.6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marsh KL, Willmore E, Tinelli S, Cornarotti M, Meczes EL, Capranico G, Fisher LM, Austin CA. Amsacrine-promoted DNA cleavage site determinants for the two human DNA topoisomerase II isoforms alpha and beta. Biochem. Pharmacol. 1996;52:1675–1685. doi: 10.1016/s0006-2952(96)00516-3. [DOI] [PubMed] [Google Scholar]
- 13.Strumberg D, Nitiss JL, Rose A, Nicklaus MC, Pommier Y. Mutation of a conserved serine residue in a quinolone-resistant type II topoisomerase alters the enzyme-DNA and drug interactions. J. Biol. Chem. 1999;274:7292–7301. doi: 10.1074/jbc.274.11.7292. [DOI] [PubMed] [Google Scholar]
- 14.Burden DA, Osheroff N. In vitro evolution of preferred topoisomerase II DNA cleavage sites. J. Biol. Chem. 1999;274:5227–5235. doi: 10.1074/jbc.274.8.5227. [DOI] [PubMed] [Google Scholar]
- 15.Razin SV, Vassetzky YS, Hancock R. Nuclear matrix attachment regions and topoisomerase II binding and reaction sites in the vicinity of a chicken DNA replication origin. Biochem. Biophys. Res. Commun. 1991;177:265–270. doi: 10.1016/0006-291x(91)91977-k. [DOI] [PubMed] [Google Scholar]
- 16.Kas E, Laemmli UK. In vivo topoisomerase II cleavage of the Drosophila histone and satellite III repeats: DNA sequence and structural characteristics. EMBO J. 1992;11:705–716. doi: 10.1002/j.1460-2075.1992.tb05103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miassod R, Razin SV, Hancock R. Distribution of topoisomerase II-mediated cleavage sites and relation to structural and functional landmarks in 830 kb of Drosophila DNA. Nucleic Acids Res. 1997;25:2041–2046. doi: 10.1093/nar/25.11.2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mueller-Planitz F, Herschlag D. Interdomain communication in DNA topoisomerase II: DNA binding and enzyme activation. J. Biol. Chem. 2006;281:23395–23404. doi: 10.1074/jbc.M604119200. [DOI] [PubMed] [Google Scholar]
- 19.Corbett AH, Zechiedrich EL, Osheroff N. A role for the passage helix in the DNA cleavage reaction of eukaryotic topoisomerase II. A two-site model for enzyme-mediated DNA cleavage. J. Biol. Chem. 1992;267:683–686. [PubMed] [Google Scholar]
- 20.Strumberg D, Nitiss JL, Dong J, Kohn KW, Pommier Y. Molecular analysis of yeast and human type II topoisomerases. Enzyme-DNA and drug interactions. J. Biol. Chem. 1999;274:28246–28255. doi: 10.1074/jbc.274.40.28246. [DOI] [PubMed] [Google Scholar]
- 21.Osheroff N, Zechiedrich EL. Calcium-promoted DNA cleavage by eukaryotic topoisomerase II: trapping the covalent enzyme-DNA complex in an active form. Biochemistry. 1987;26:4303–4309. doi: 10.1021/bi00388a018. [DOI] [PubMed] [Google Scholar]
- 22.Lee MP, Sander M, Hsieh TS. Single strand DNA cleavage reaction of duplex DNA by Drosophila topoisomerase II. J. Biol. Chem. 1989;264:13510–13518. [PubMed] [Google Scholar]
- 23.Strumberg D, Nitiss JL, Dong J, Walker J, Nicklaus MC, Kohn KW, Heddle JG, Maxwell A, Seeber S, et al. Importance of the fourth alpha-helix within the CAP homology domain of type II topoisomerase for DNA cleavage site recognition and quinolone action. Antimicrob. Agents Chemother. 2002;46:2735–2746. doi: 10.1128/AAC.46.9.2735-2746.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomsen B, Bendixen C, Lund K, Andersen AH, Sorensen BS, Westergaard O. Characterization of the interaction between topoisomerase II and DNA by transcriptional footprinting. J. Mol. Biol. 1990;215:237–244. doi: 10.1016/S0022-2836(05)80342-0. [DOI] [PubMed] [Google Scholar]
- 25.Lee MP, Sander M, Hsieh T. Nuclease protection by Drosophila DNA topoisomerase II. Enzyme/DNA contacts at the strong topoisomerase II cleavage sites. J. Biol. Chem. 1989;264:21779–21787. [PubMed] [Google Scholar]
- 26.Jen-Jacobson L. Protein-DNA recognition complexes: conservation of structure and binding energy in the transition state. Biopolymers. 1997;44:153–180. doi: 10.1002/(SICI)1097-0282(1997)44:2<153::AID-BIP4>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 27.Morais Cabral JH, Jackson AP, Smith CV, Shikotra N, Maxwell A, Liddington RC. Crystal structure of the breakage-reunion domain of DNA gyrase. Nature. 1997;388:903–906. doi: 10.1038/42294. [DOI] [PubMed] [Google Scholar]
- 28.Berger JM, Gamblin SJ, Harrison SC, Wang JC. Structure and mechanism of DNA topoisomerase II. Nature. 1996;379:225–232. doi: 10.1038/379225a0. [DOI] [PubMed] [Google Scholar]
- 29.Fass D, Bogden CE, Berger JM. Quaternary changes in topoisomerase II may direct orthogonal movement of two DNA strands. Nat. Struct. Biol. 1999;6:322–326. doi: 10.1038/7556. [DOI] [PubMed] [Google Scholar]
- 30.Hsieh TJ, Farh L, Huang WM, Chan NL. Structure of the topoisomerase IV C-terminal domain: A broken beta-propeller implies a role as geometry facilitator in catalysis. J. Biol. Chem. 2004;279:55587–55593. doi: 10.1074/jbc.M408934200. [DOI] [PubMed] [Google Scholar]
- 31.Corbett KD, Shultzaberger RK, Berger JM. The C-terminal domain of DNA gyrase A adopts a DNA-bending beta-pinwheel fold. Proc. Natl Acad. Sci. USA. 2004;101:7293–7298. doi: 10.1073/pnas.0401595101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reece RJ, Maxwell A. Tryptic fragments of the Escherichia coli DNA gyrase A protein. J. Biol. Chem. 1989;264:19648–19653. [PubMed] [Google Scholar]
- 33.Reece RJ, Maxwell A. Probing the limits of the DNA breakage-reunion domain of the Escherichia coli DNA gyrase A protein. J. Biol. Chem. 1991;266:3540–3546. [PubMed] [Google Scholar]
- 34.Liu LF, Wang JC. Micrococcus luteus DNA gyrase: active components and a model for its supercoiling of DNA. Proc. Natl Acad. Sci. USA. 1978;75:2098–2102. doi: 10.1073/pnas.75.5.2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu LF, Wang JC. DNA-DNA gyrase complex: the wrapping of the DNA duplex outside the enzyme. Cell. 1978;15:979–984. doi: 10.1016/0092-8674(78)90281-7. [DOI] [PubMed] [Google Scholar]
- 36.McClendon AK, Dickey JS, Osheroff N. Ability of viral topoisomerase II to discern the handedness of supercoiled DNA: bimodal recognition of DNA geometry by type II enzymes. Biochemistry. 2006;45:11674–11680. doi: 10.1021/bi0520838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hammes GG. Multiple conformational changes in enzyme catalysis. Biochemistry. 2002;41:8221–8228. doi: 10.1021/bi0260839. [DOI] [PubMed] [Google Scholar]
- 38.Cannon WR, Singleton SF, Benkovic SJ. A perspective on biological catalysis. Nat. Struct. Biol. 1996;3:821–833. doi: 10.1038/nsb1096-821. [DOI] [PubMed] [Google Scholar]
- 39.Fisher E. Einfluss der Configuration auf die Wirkung der Enzyme. Ber. Dt. Chem. Ges. 1894;27:2985–2993. [Google Scholar]
- 40.Koshland DE. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc. Natl Acad. Sci. USA. 1958;44:98–104. doi: 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wolfenden R. Enzyme catalysis: conflicting requirements of substrate access and transition state affinity. Mol. Cell. Biochem. 1974;3:207–211. doi: 10.1007/BF01686645. [DOI] [PubMed] [Google Scholar]
- 42.Hammes-Schiffer S, Benkovic SJ. Relating protein motion to catalysis. Annu. Rev. Biochem. 2006;75:519–541. doi: 10.1146/annurev.biochem.75.103004.142800. [DOI] [PubMed] [Google Scholar]
- 43.Jencks WP. Utilization of binding energy and coupling rules for active transport and other coupled vectorial processes. Methods Enzymol. 1989;171:145–164. doi: 10.1016/s0076-6879(89)71010-7. [DOI] [PubMed] [Google Scholar]
- 44.Osheroff N. Eukaryotic topoisomerase II. Characterization of enzyme turnover. J. Biol. Chem. 1986;261:9944–9950. [PubMed] [Google Scholar]
- 45.Winter RB, Berg OG, von Hippel PH. Diffusion-driven mechanisms of protein translocation on nucleic acids. 3. The Escherichia coli lac repressor—operator interaction: kinetic measurements and conclusions. Biochemistry. 1981;20:6961–6977. doi: 10.1021/bi00527a030. [DOI] [PubMed] [Google Scholar]
- 46.Strick TR, Croquette V, Bensimon D. Single-molecule analysis of DNA uncoiling by a type II topoisomerase. Nature. 2000;404:901–904. doi: 10.1038/35009144. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.