

Abstract
The NF-κB2 gene is recurrently mutated in human lymphoid malignancies. However, a causal relationship between NF-κB2 mutation and lymphomagenesis has not been established. It is also unclear how the mutation may lead to lymphoid malignancies. We report the generation of transgenic mice with targeted expression of p80HT, a lymphoma-associated NF-κB2 mutant, in lymphocytes. The transgenic mice display a marked expansion of peripheral B cell populations and develop predominantly small B cell lymphomas. p80HT expression has no apparent effect on the proliferation of B cells, but renders them specifically resistant to apoptosis induced by cytokine deprivation and mitogenic stimulation. Lymphocytes and lymphoma cells from p80HT mice express high levels of TRAF1, an antiapoptotic protein also implicated in lymphoid malignancies. p80HT binds the TRAF1 promoter in vivo and activates TRAF1 transcription. Moreover, TRAF1 knockdown abrogates the antiapoptotic activity of p80HT and TRAF1 deficiency reestablishes B cell homeostasis in p80HT mice. These findings demonstrate NF-κB2 mutation as an oncogenic event in vivo and suggest a molecular pathway for TRAF1 activation in the pathogenesis of lymphomas.
Introduction
The mammalian NF-κB family consists of 5 structurally related proteins, including RelA, RelB, c-Rel, NF-κB1 (p50 and its precursor p105), and NF-κB2 (p52 and its precursor p100). The active forms of NF-κB are dimers composed of various combinations of the family members, which bind a common DNA sequence motif known as the κB site and regulate the expression of genes crucial to the development and functions of lymphocytes. NF-κB activity is controlled by IκB (inhibitor of κB) proteins and the IκB-like ankyrin-repeat domain in the C-terminal region of NF-κB2 p100. IκB proteins interact with NF-κB dimers composed of NF-κB1 p50 and RelA or c-Rel, and NF-κB2 p100 is primarily associated with RelB. The interactions prevent NF-κB dimers from translocating to the nucleus. On stimulation by certain cytokines, IκBs and the C-terminal region of p100 are phosphorylated by IκB kinase and degraded by the proteasome. The freed p50-RelA/c-Rel or resulting p52-RelB dimers then translocate to the nucleus and transactivate their target genes.1
Constitutive NF-κB activation plays an important role in tumorigenesis by promoting cell proliferation and survival. Several mechanisms have been identified by which activation of NF-κB is uncoupled from its normal modes of regulation in cancer cells. Most of these mechanisms target IκB kinase for activation of NF-κB.2,3 Sustained NF-κB activation can also be caused by genetic alterations that affect the activity and expression of NF-κB proteins.4 The first gene of the family found to be mutated in human lymphoid malignancies is NF-κB2.5 Subsequent studies revealed that chromosomal rearrangements at the NF-κB2 locus occur in a variety of B and T cell lymphoid malignancies.6–9 A cardinal feature of these genetic alterations is the generation of C-terminally truncated NF-κB2 mutants that lack various portions of the ankyrin-repeat domain (Figure 1A) and are constitutive transactivators.10 Some of these mutants have been shown to be capable of transforming immortalized mouse fibroblasts (Balb/3T3),11 indicating their oncogenic potential. However, expression of these mutants had an apparent cytotoxic effect, which may explain their low transformation efficiency in mouse fibroblasts and failure to transform human lymphoblastoid cell lines.11 These findings raise the question of whether NF-κB2 mutation can directly initiate lymphomagenesis.
Figure 1.

Characterization of p80HT transgenic mice. (A) Schematic diagram of NF-κB2 p100, p52, and representative tumor-derived mutants. The arrow indicates the cleavage site in p100 that gives rise to p52. RHD, Rel-homology domain; DD, death domain. (B) Immunoblot analysis of tissue-specific expression of p80HT and p52 using an antibody against the N-terminal region of human NF-κB2. The star indicates a degraded p80HT product. Levels of α-tubulin are shown as loading control. BM, bone marrow; LN, lymph node; Sp, spleen; Th, thymus; H, heart; K, kidney; Li, liver; Lu, lung; St, stomach; T, splenic T cells; B, splenic B cells. (C) Electrophoretic mobility shift assay for κB-binding activity in nuclear extracts of splenic lymphocytes from p80HT transgenic (Tg) and wild-type (WT) mice. Two κB-binding complexes containing either NF-κB2 or NF-κB1 are indicated based on antibody-mediated supershift. The NF-κB2 complex was disrupted by anti-RelA and, to a lesser degree, by anti-c-Rel. Preimmune rabbit IgG was used as control.
In this report, we show that transgenic mice expressing the human lymphoma-associated NF-κB2 mutant p80HT in lymphocytes develop lymphomas, demonstrating directly the tumorigenic capacity of an NF-κB2 mutant in vivo. Furthermore, our study reveals that p80HT promotes lymphomagenesis by inducing TRAF1, which in turn suppresses specific apoptotic responses critical for the maintenance of lymphocyte homeostasis.
Materials and methods
Mice
The human p80HT coding sequence was amplified by polymerase chain reaction (PCR) using a human fetus Marathon-ready cDNA library (Clontech, Mountain View, CA) as the template and specific primers based on the published p80HT sequence (GenBank U09609)8 and cloned into pHSE3′, a vector containing an H-2Kb promoter and an immunoglobulin μ chain enhancer for transgene expression in lymphocytes.12,13 The construct was linearized by PvuI and microinjected into fertilized (C57BL/6J x SJL/J) F2 eggs (University of Michigan Transgenic Animal Model Core). Fourteen transgenic founders were identified by Southern blotting of BamHI digested tail DNA using p80HT cDNA as the probe and by PCR amplification of a 1.3-kb product using the primers 5′-GCGGTCGACATGGAGAGTTGCTACAACCCAG-3′ and 5′-GCGGGATCCTCATCGCTGCAGCATCTCCGGGGC-3′. Two independent lines were established by mating no. 808 and no. 815 male founders to C57BL/6J × SJL/J F1 females. p80HT+/− mice were also mated to TRAF1−/− mice (C57BL/6J)14 to generate wild-type, p80HT+/−, TRAF1−/−, and p80HT+/−/TRAF1−/− mice. All animal studies were preapproved by the Institutional Animal Care and Use Committee of University of Toledo Health Science Campus.
Electrophoretic mobility shift assay
Nuclear extracts were prepared from mouse splenocytes and analyzed for κB-binding activity as described.15 For supershifting, 3 μg of extracts were incubated with 2 μg of either preimmune rabbit IgG or antibodies against NF-κB2 (06-413; Upstate, Temecula, CA), NF-κB1 (06-886; Upstate), RelA (SC-109x; Santa Cruz Biotechnology, Santa Cruz, CA), RelB (SC-226x; Santa Cruz Biotechnology), or c-Rel (SC-71x; Santa Cruz Biotechnology) in binding buffer for 30 minutes at 4°C before addition of the 32P-labeled κB probes: 5′-CAGGGCTGGGGATTCCCCATCTCCACAGTTTCACTTC-3′16; TRAF1-κB1, 5′-CACTGTGGGAATCTCCACAGAG-3′; TRAF1-κB5, 5′-GCAACAAAGGGTAATTCCTGCTCC-3′; or TRAF1-κB5m, 5′-GCAACAAAGCTCGAGTCCTGCTCC-3′ (mutated bases are italicized).
Immunoblotting
Cells were directly suspended in sodium dodecyl sulfate sample buffer, and 50 μg of proteins were separated on 10% or 12% sodium dodecyl sulfate–polyacrylamide gels, transferred to nitrocellulose membranes, probed with antibodies, and visualized by electrochemiluminescence. The antibodies (all from Santa Cruz Biotechnology unless indicated) were used: mouse anti-NF-κB2, 1:500 (05-361; Upstate); rabbit anti-Bcl-2, 1:100 (ΔC21); rabbit anti-Bcl-XL, 1:200 (S-18); rabbit anti-cIAP2, 1:200 (H-85); rabbit anti-XIAP, 1:500 (no. 2042; Cell Signaling, Danvers, MA); rabbit anti-TRAF1, 1:200 (H-132); rabbit anti-TRAF2, 1:200 (H-249); and mouse anti-α-tubulin, 1:2000 (B-5-1-2; Sigma). Horseradish peroxidase–conjugated antimouse and antirabbit were used as secondary antibodies (ICN, Solon, OH).
Flow cytometry
Single-cell suspensions were prepared from lymphoid organs of 6- to 8-week-old mice according to standard procedures. Red blood cells were lysed in ACK buffer (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA, pH 7.3), and dead cells were removed by passing through Lympholyte-M (Cedarlane, Burlington, ON). Lymphocytes were then stained with fluorescein isothiocyanate-conjugated rat antimouse B220 (RA3-6B2) and CD4 (GK1.5); allophycocyanin-conjugated hamster antimouse CD3e (145-2C11); R-phycoerythrin-conjugated rat antimouse CD8a (53-6.7) and IgM (R6-60.2) (all from BD Pharmingen, San Jose, CA) and analyzed by flow cytometry (Epics Elite instrument; Beckman-Coulter, Fullerton, CA).
Histopathology and immunohistochemistry
Tumors and tissue samples were fixed in 10% neutral-buffered formalin, embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin. For immunohistochemistry, the sections were deparaffinized, rehydrated, and boiled in 10 mM citrate buffer (pH 6.0) or 1 mM EDTA (pH 8.0) for 10 minutes for retrieval of B220 or CD3 antigen. After quenching endogenous peroxidase activity with H2O2 and blocking with normal serum, the sections were incubated for 60 minutes with rat anti-B220 (RA3-6B2, 5 μg/mL; BD Pharmingen), rat anti-CD3 (CD3-12, 10 μg/mL; Serotec, Raleigh, NC), or an isotype control antibody (10 μg/mL; BD Pharmingen). After washing, a biotinylated rabbit antirat secondary antibody (Vector Laboratories, Burlingame, CA) was applied for 30 minutes. The sections were then incubated for 30 minutes with ABC reagent (Vector Laboratories), and the immunostaining was visualized with 3,3′-diaminobenzidine (Sigma, St. Louis, MO). The sections were counterstained with hematoxylin. An Olympus BHSZ (Tokyo, Japan) microscope with 40×/0.95 objective was used; mounting medium, Eukitt, Calibrated Instruments, Hawthorne, NJ; H&E staining solution, Richard-Allan Scientific, Kalamazoo, MI; Evolution MP Color Camera, Media Cybernetics, Bethesda, MD; and Qcapture Pro S.01 image acquisition software, Media Cybernetics.
Southern blot analysis of antigen-receptor gene rearrangements
Genomic DNA was prepared from tails and tumor samples, and 10 μg of DNA was digested with EcoRI, resolved by 0.8% agarose gel electrophoresis, transferred to nylon membranes, and hybridized with a JH4 probe to detect IgH-μ gene rearrangements17 or with a Cβ1 probe to detect TCRβ gene rearrangements.18
In vitro lymphocyte proliferation and survival assays
For cell cycle analysis, splenic B cells were purified from 6- to 8-week-old mice using mouse B immunocolumns (Cedarlane), cultured in Dulbecco Modified Eagle Medium supplemented with 10% fetal bovine serum, 250 μM L-asparagine and 50 μM 2-mercaptoethanol, and stimulated with 20 μg/mL of F(ab′)2 goat antimouse IgM (Jackson ImmunoResearch, West Grave, PA) or 20 μg/mL of lipopolysaccharide (LPS; Sigma). After 48 hours, cells were harvested for cell cycle analysis on an Epics Elite flow cytometer (Beckman-Coulter). The data were analyzed with MultiCycle AV (Phoenix Flow Systems, San Diego, CA). For 3H-thymidine incorporation assays, purified splenic B cells (105/well, 96-well plate) were stimulated with LPS for 48 hours and then pulsed for 12 hours with 3H-thymidine (1 μCi/well). Incorporation of 3H-thymidine was measured using a scintillation counter. For survival assays, purified splenic B cells and B lymphoma cells were either untreated or treated with recombinant mouse tumor necrosis factor-α (10 ng/mL; Calbiochem, San Diego, CA), Fas ligand (100 ng/mL; PeproTech, Rocky Hill, NJ), LPS (20 μg/mL), or doxorubicin (0.5 μg/mL; Ben Venue Laboratories, Bedford, OH) and viable cells were determined daily by trypan blue exclusion assays.
Ribonuclease protection assay
Total RNA was extracted from cells using an RNeasy kit (Qiagen, Valencia, CA). The human apoptosis template set hAPO-5 (BD Pharmingen) was used for in vitro generation of RNA probes labeled with α-32P-UTP. The labeled RNA probes were hybridized to the isolated RNA (10 μg) and treated with ribonuclease. The protected probes were resolved on a 5% urea–polyacrylamide gel. The dried gel was then exposed to a PhosphorImager screen (Molecular Dynamics, Sunnyvale, CA).
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation assays were performed as described19 using either preimmune rabbit serum or antiserum against human NF-κB2 (06-413; Upstate). The human TRAF1 promoter (GenBank Y10284) region −704 to −495 was amplified by PCR using the primers 5′-GAGTCTGTGGTGTAGGCCTTGG-3′ and 5′-GCTTTTGCTCTGCTCTGTTTGG-3′. For control, the human telomerase reverse transcriptase promoter region −235 to + 3 was amplified by PCR using the primers 5′-CGGGCTCCCAGTGGATTC-3′ and 5′-TGCCTGAAACTCGCGCCG-3′. The PCR products were resolved on a 2% agarose gel and stained with ethidium bromide.
Site-directed mutagenesis, transient transfection, and luciferase assay
Mutations of the TRAF1 promoter κB1 and κB5 sites were performed as described.20 HT1080 cells in 6-well plates were transfected with 0.6 μg of the TRAF1 luciferase reporter construct, 0.3 μg of pSV-β-galactosidase plasmid, and 0.3 μg of pcDNA3, pcDNA3-p80HT, or pcDNA3-p52 using a LipofectAmine Plus kit (Invitrogen, Carlsbad, CA). Cells were lysed 40 hours after transfection, and luciferase and β-galactosidase activities were assayed using a luciferase and β-galactosidase assay kit (Promega, Madison, WI). Luciferase activity was normalized to β-galactosidase activity to account for differences in the transfection efficiency.
Retroviral infection and anti-IgM-induced apoptosis of WEHI-231 cells
Murine WEHI-231 B lymphoma cells (ATCC CRL-1702) were cultured in high glucose-Dulbecco Modified Eagle Medium supplemented with 10% fetal bovine serum and 50 μM 2-mercaptoethanol. The retroviral constructs MSCV-IRES-GFP, MSCV-p80HT-IRES-GFP, and MSCV-TRAF1-IRES-GFP were used in overexpression studies. For down-regulation of TRAF1, synthesized 60-bp oligonucleotides encoding mouse TRAF1 siRNA (5′-CAGCTTTCTACACTGCCAA-3′ and 5′-GCTACTTGACCAGAACAAC-3′, positions 859 to 877 bp and 1022 to 1040 bp relative to the start codon, respectively; GenBank NM_009421) were cloned into pSuper-retro/puro for producing retroviruses as described.21 Retroviral infection of WEHI-231 cells was performed according to the “2× spin infection” protocol.22 One day after infection, cells were cultured in the presence of puromycin (1 μg/mL) for 3 days, and drug-resistant cells were pooled. For B cell antigen receptor ligation-induced apoptosis, WEHI-231 cells were treated with F(ab′)2 goat antimouse IgM (1 or 10 μg/mL) for 48 hours and analyzed for apoptosis by annexin-V staining and trypan blue exclusion assay.
Results
p80HT transgenic mice display high levels of constitutive NF-κB2 activity in lymphocytes
To determine whether NF-κB2 mutation has a causal role in lymphomagenesis, we generated transgenic mice with targeted expression in lymphocytes of p80HT, an NF-κB2 mutant originally identified in the human cutaneous T-lymphoma cell line HUT78.8,9 Among the 131 founder mice, 14 (7 males and 7 females) were found to carry various copy numbers of the p80HT transgene (Figure S1, available on the Blood website; see the Supplemental Materials link at the top of the online article). Two transgenic founder mice and a wild-type littermate were killed for immunoblot analysis of tissue-specific expression of p80HT (Figure 1B). High-level expression of p80HT and p52 (probably as a result of p80HT processing) was observed in lymphoid organs of transgenic mice. We also confirmed p80HT expression in purified splenic B and T cells as well as in bone marrow cells.
We next performed electrophoretic mobility shift assay using nuclear extracts from unstimulated p80HT and wild-type splenocytes, which revealed 2 distinct κB-binding complexes (Figure 1C). Both extracts contained similar amounts of the faster migrating complex that could be supershifted by an antibody against NF-κB1 (Figure 1C; NF-κB1). By contrast, p80HT extracts contained a significantly higher amount of the slower migrating complex compared with wild-type extracts, and the complex could be supershifted by an antibody against NF-κB2 (Figure 1C; NF-κB2), demonstrating that p80HT expression resulted in high levels of constitutive NF-κB2 activity in lymphocytes. The NF-κB2 complex apparently contained RelA and c-Rel, because preincubation of the extracts with an antibody against either RelA or c-Rel significantly inhibited its formation (Figure 1C).
p80HT transgenic mice develop lymphomas with multiorgan metastases
The remaining 12 transgenic founders were monitored for tumor development (Table S1). Half of them died between 41 and 89 weeks, whereas only one of the 6 wild-type littermates died at the age of 87 weeks. Histopathologic examination revealed that all of the 6 dead p80HT founders had developed lymphomas with extensive metastases in the liver, lungs, and/or kidneys. The deceased wild-type mouse had localized lymphoma. The rest of the p80HT founders (n = 6) and their wild-type littermates (n = 5) were killed at 96 weeks of age, and histopathologic examination showed the development of disseminated lymphomas in 5 p80HT mice and of localized lymphoma in one wild-type mouse. The significantly higher tumor incidence in p80HT founders provides direct evidence that p80HT has an oncogenic activity in vivo. The observation that 11 of the 12 independent p80HT founders developed lymphomas also rules out the possibility that the tumorigenesis might result from insertional effects of the transgene.
To confirm the tumorigenic activity of p80HT in a large-scale study and also to determine the effect of its expression levels on tumor incidence, we monitored the F2 offspring of 2 independent p80HT lines for lymphoma development. Mice of the 808 line expressed higher levels of p80HT in lymphocytes than those of the 815 line (Figure 2A). Correlating with the p80HT expression levels, the 808 line had a significantly higher mortality rate compared with the 815 line (79% versus 24%; Figure 2B). All of the wild-type littermates were alive and apparently healthy during the same period. Autopsy examinations revealed that 75% of the dead p80HT mice had lymphadenopathy and splenomegaly (Figure 2C,D).
Figure 2.

p80HT transgenic mice develop lymphomas. (A) Immunoblot analysis of p80HT (and p52) expression in splenic B cells (815B and 808B) and T cells (815T and 808T) from the 808 and 815 lines of transgenic mice. Levels of α-tubulin are shown as loading control. (B) Survival curve of p80HT mice and their wild-type littermates. Numbers of mice for each group are indicated. (C) Autopsy examination of a dead p80HT mouse showing markedly enlarged lymph nodes (arrows). (D) Representative examples of lymph nodes and spleens from a dead p80HT mouse and an age-matched wild-type littermate.
All of the dead p80HT mice had developed lymphomas with metastases in other organs characterized by complete effacement of normal organ architecture by massive infiltration of small- to medium-sized lymphocytes (Figure 3A). Among the 22 dead mice of the 808 line, only 2 showed apparent bone marrow involvement, indicated by extensive infiltration of small lymphocytes (data not shown). Immunohistochemical staining of lung metastasis sections revealed that the malignant lymphocytes expressed either B220 (a B cell marker) or CD3 (a T cell marker) (Figure 3B), indicating that p80HT mice developed either B or T cell lymphomas. Of the 10 samples examined, 8 stained strongly for B220 and 2 for CD3. Flow cytometry analysis showed that B220 + lymphoma cells also expressed surface IgM (data not shown). Thus, the majority of p80HT mice developed lymphomas with histologic and immunophenotypic features of small B cell lymphoma based on the Bethesda proposals for classification of murine lymphoid malignancies.23
Figure 3.

p80HT transgenic mice develop disseminated B or T cell lymphomas. (A) Histopathologic analysis of lymphomas in p80HT mice by hematoxylin and eosin staining. The normal architecture shown in the wild-type organs are completely effaced by extensive lymphocyte infiltrations in the organs from a dead p80HT mouse. (B) Immunohistochemical examination of p80HT mouse lung sections with malignant lymphocyte infiltration. The sections stained strongly either for B220 (a B cell marker) or CD3 (a T cell marker), indicative of B or T cell lymphomas. Scale bars in panels A and B, 50 μm. (C) Southern blot analysis of IgH (top) and TCR (bottom) gene rearrangements in representative lymphoma samples. EcoRI-digested DNA was hybridized with an IgH-μ JH4 probe or with a TCR Cβ1 probe. Tail DNA from a p80HT mouse was used as control for the germ line (GL) IgH and TCR loci. Arrowheads indicate rearrangements at either the IgH-μ or the TCR-β locus. Size markers in kilobases are shown to the right.
We next conducted Southern blot analysis of antigen-receptor gene rearrangements to determine the clonality of lymphomas from p80HT mice. Rearrangements at the IgH μ locus were detected in 7 of the 8 lymphoma samples examined with one of them (no. 471) also showing rearrangements at the T cell receptor β gene locus (Figure 3C), suggesting that the tumor is of mixed lineages. The remaining tumor (no. 458) showed rearrangements only at the T cell receptor β gene locus (Figure 3C). These results demonstrate that lymphomas in p80HT mice resulted from clonal expansion of malignant B or T cells.
Perturbation of B cell homeostasis in p80HT transgenic mice
To gain insights into the mechanism whereby p80HT induces lymphomagenesis, we examined lymphocyte populations in 6- to 8-week-old mice of the 808 line and their wild-type littermates. Flow cytometry analysis revealed no significant difference in the numbers of total thymocytes and splenic T cells between the groups (Figure 4A). In addition, p80HT mice showed normal ratios of the major subsets of thymocytes (CD4−8−, CD4 +8 +, CD4 +8−, and CD4−8 +) and of splenic T cells (CD4 + and CD8 +) (data not shown). However, p80HT mice displayed a marked increase (87%) in the number of total splenic B cells compared with wild-type littermates (Figure 4A), demonstrating that p80HT expression promoted expansion of the B cell population, a finding consistent with the observation that most of the lymphomas in p80HT mice were of B cell origin.
Figure 4.

B lymphocytes and lymphoma cells from p80HT transgenic mice are resistant to certain apoptotic stimuli. (A) The numbers of total lymphocytes in the indicated lymphoid organs. Lymphocytes were stained with fluorescence-conjugated antibodies against B220, Thy-1.2, CD4, and CD8 and analyzed by flow cytometry. (B) Cell cycle analysis of splenic B cells that were either untreated or treated for 48 hours with lipopolysaccharide (20 μg/mL). Percentages of cells in each phase of the cell cycle are shown. (C-E) In vitro survival and apoptosis assays of splenic B cells and B lymphoma cells. Cells were either untreated (D) or treated with 0.5 μg/mL of doxorubicin (C) or with 20 μg/mL of lipopolysaccharide (E). Viability was determined by trypan blue dye exclusion assay. Data in panels A-E represent means (± standard deviation) of cells from 5 mice of each genotype or from 5 B lymphomas samples. **Two-tailed Student t test, P < .01.
B cells of p80HT mice show normal proliferative responses but are resistant to certain apoptotic stimuli
The accumulation of splenic B cells in p80HT mice could result from excess production of B cells in the bone marrow, increased proliferation or survival of peripheral B cells, or a combination of these factors. Flow cytometry analysis revealed no abnormality in the numbers (data not shown) and ratios of bone marrow B cell subsets in p80HT mice (Supplementary Figure 2). In addition, colony-forming unit assays showed no difference in the numbers of bone marrow pre-B cells between p80HT and wild-type mice (data not shown). Thus, p80HT expression has no significant effect on B cell development in the bone marrow.
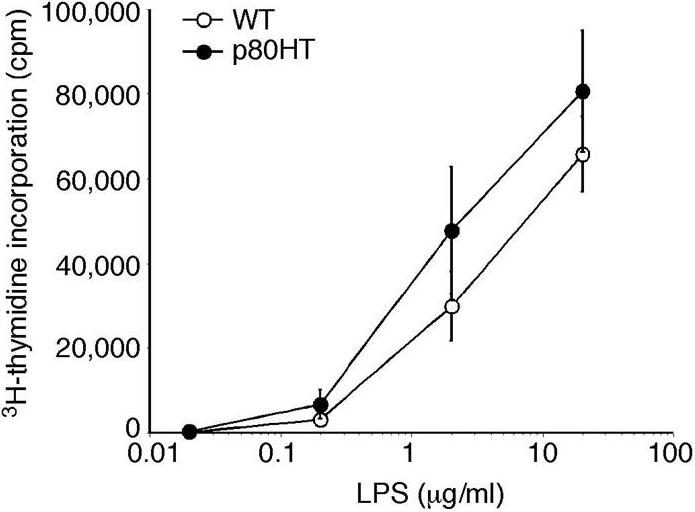
We next examined the growth properties of splenic B cells from 6-week-old p80HT mice. Freshly isolated B cells showed no significant proliferation as determined by cell cycle analysis (Figure 4B). To assess whether p80HT expression enhances B cell proliferative responses to mitogens, purified B cells were treated for 2 days with either LPS or the anti-μ chain antibody F(ab′)2, which induces ligation of the surface IgM. Flow cytometry analysis revealed that the percentages of cells in the S and G2/M phases were similar between p80HT and wild-type B cells (Figure 4B for LPS studies, data not shown for anti-IgM studies). We also performed 3H-thymidine incorporation assays and observed no significant difference in the levels of 3H-thymidine incorporation between LPS-stimulated p80HT and wild-type B cells (Figure S3). Thus, p80HT expression did not cause B cells to grow autonomously or enhance their proliferative response to mitogenic stimuli.
Apoptosis also plays a critical role in maintaining lymphocyte homeostasis.24 We examined the survival of splenic B cells from p80HT and wild-type mice under a variety of conditions. Both wild-type and p80HT B cells were highly resistant to Fas ligand and tumor necrosis factor-α (data not shown). They were also equally sensitive to the DNA damage drug doxorubicin (Figure 4C). However, p80HT B cells showed a markedly enhanced survival in the absence of cytokines (Figure 4D, no treatment). During the course of investigation, we noted that in response to LPS, splenic B cells generally proliferated for 3 days and then underwent extensive apoptosis. Although p80HT B cells displayed normal proliferative response to LPS (Figure 4B), they were highly resistant to activation-induced apoptosis (Figure 4E). Thus, p80HT specifically promotes survival of B cells in the absence of growth cytokines or after mitogenic activation, which likely contributes to the B cell expansion in p80HT mice.
To further assess the role of apoptosis suppression in lymphoma development in p80HT mice, we examined apoptotic responses of B lymphoma cells in comparison with their pretumor counterparts. In contrast to p80HT pretumor B cells, the lymphoma cells were highly resistant to apoptosis induced by doxorubicin (Figure 4C). They also survived better in the absence of cytokines (Figure 4D). These results suggest that secondary genetic or epigenetic alterations may have taken place in p80HT B cells during the tumorigenic process.
Up-regulation of TRAF1 by p80HT
Several antiapoptotic genes have been identified as transcriptional targets of NF-κB, including Bcl-XL, cIAP2, TRAF1, TRAF2, and XIAP.25–28 Because p80HT is a κB-site transcription activator,10 it may up-regulate the same group of genes. Immunoblot analysis revealed no difference in the levels of Bcl-XL, Bcl-2, cIAP2, and XIAP between wild-type and p80HT B cells as well as p80HT lymphoma cells. However, p80HT pretumor B cells and B lymphoma cells showed a significant increase in the levels of TRAF1 and, to a lesser extent, TRAF2 compared with wild-type B cells (Figure 5A). Up-regulation of TRAF1 was also observed in p80HT splenic T cells and T lymphoma cells (Figure 5B). We further examined TRAF1 levels in human fibrosarcoma HT1080 cells overexpressing p80HT and T lymphoma HUT78 cells carrying a mutated NF-κB2 allele encoding p80HT.8,9 The control HT1080/GFP and T leukemia Jurkat cells expressed similar levels of XIAP as HT1080/p80HT and HUT78 cells. However, only HT1080/p80HT and HUT78 cells expressed significant levels of TRAF1 (Figure 5C). These results indicate that up-regulation of TRAF1 by p80HT is not a cell type-specific phenomenon or an artifact of overexpression.
Figure 5.

p80HT up-regulates TRAF1 expression. (A) Immunoblot analysis of the expression of indicated antiapoptotic genes in splenic B cells from wild-type (WT) and p80HT mice and in representative B lymphoma samples. Levels of α-tubulin are shown as loading control. (B) Immunoblot analysis of TRAF1 expression in splenic T cells from wild-type (WT) and p80HT mice and in one T lymphoma sample. Levels of α-tubulin are shown as loading control. (C) Immunoblot analysis of TRAF1 and XIAP expression in the human fibrosarcoma HT1080 cells infected with GFP- or p80HT-expressing retroviruses, the T-cell leukemia cell line Jurkat, and the T-cell lymphoma cell line HUT78 that harbors a mutated NF-κB2 allele encoding p80HT. Levels of α-tubulin are shown as loading control. (D) Ribonuclease protection assay for mRNA levels of the indicated antiapoptotic genes in the same human cell lines. The mRNA levels for the housekeeping genes L32 and GAPDH are shown as loading control.
We next performed ribonuclease protection assay for TRAF1 mRNA levels in these human cell lines (Figure 5D). TRAF1 mRNA was essentially undetectable in the control HT1080/GFP and Jurkat cells but was highly induced in HT1080/p80HT and HUT78 cells, suggesting that TRAF1 up-regulation by p80HT occurs at the transcription level. p80HT also induced TRPM2 in HT1080/p80HT cells and cIAP2 and TRAF2 in HUT78 cells.
TRAF1 is a direct transcriptional target of p80HT
Given that TRAF1 up-regulation occurs at the transcription level and that p80HT is a transcription activator,10 we investigated the possibility of TRAF1 as a direct target of p80HT. Chromatin immunoprecipitation was performed to determine whether p80HT binds the TRAF1 promoter in vivo. Crosslinked genomic DNA from the human cell lines was precipitated with antiserum against NF-κB2 and analyzed by PCR using TRAF1 promoter-specific primers. A DNA fragment corresponding to the TRAF1 promoter region from −704 to −495 was detected only in samples from p80HT-expressing cell lines (Figure 6A). As control, we also performed PCR amplification of the anti-NF-κB2 precipitated genomic DNA using primers specific for the human telomerase reverse transcriptase promoter, and no DNA fragment was generated. Together, these results demonstrate that p80HT specifically associates with the TRAF1 promoter in vivo.
Figure 6.

The TRAF1 gene is a direct transcriptional target of p80HT. (A) Chromatin immunoprecipitation assay for binding of p80HT to the human TRAF1 promoter in vivo. Chromatin isolated from the indicated human cell lines was immunoprecipitated either with normal rabbit serum (IgG) or with anti-NF-κB2 serum and then amplified with human TRAF1 or telomerase reverse transcriptase promoter-specific primers. Input control indicates amplified total DNA. (B) Schematic representation of the human TRAF1 promoter structure with sequences of the κB1 and κB5 sites. (C) Electrophoretic mobility shift assay for TRAF1 κB5-binding activity in nuclear extracts of splenic lymphocytes from p80HT and wild-type (WT) mice. Two κB-binding complexes containing either NF-κB2 or NF-κB1 are indicated based on antibody-mediated supershift. The NF-κB2 complex was supershifted by anti-c-Rel or partially disrupted by anti-RelA. Preimmune rabbit IgG was used as control. (D) Reporter assays for luciferase expression under the control of wild-type, κB1-, or κB5-mutant TRAF1 promoter in HT1080 cells. Luciferase values were normalized to β-galactosidase activity to account for differences in the transfection efficiency. Bars represent means (± standard deviation) from 3 independent assays.
We next performed electrophoretic mobility shift assay to determine the specific binding site(s) for p80HT. Previous sequence examination revealed 5 potential κB-binding sites in the TRAF1 promoter region from −1404 to + 1 and further functional analysis demonstrated a major role for the κB1 and κB5 sites in mediating tumor necrosis factor induction of TRAF1 by NF-κB20 (Figure 6B). Nuclear extracts from p80HT splenocytes did not bind the κB1 probe (data not shown) or the mutated κB5 probe, but formed 2 complexes with the wild-type κB5 probe, which could be competed off with 100-fold excess unlabeled wild-type κB5 probe (Figure 6C). The slower migrating complex could be supershifted by an antibody against NF-κB2 or c-Rel or disrupted by an antibody to RelA (Figure 6C), suggesting that the complex contained p80HT (and/or p52), c-Rel, and RelA. These findings indicate that p80HT specifically interacts with the κB5 site.
To assess the functional relevance of the p80HT-κB5 interaction, we performed luciferase assays using reporter constructs in which luciferase expression is under the control of wild-type, κB1- or κB5-mutant TRAF1 promoter spanning from −1404 to + 1.20 HT1080 cells cotransfected with pcDNA3-p80HT and the wild-type TRAF1 promoter reporter construct (TRAF1pWT) showed a 3.5-fold increase in luciferase activity relative to the pcDNA3 vector control (Figure 6D). Mutation of the κB1 site had no significant effect on the ability of p80HT to activate luciferase expression (Figure 6D; TRAF1pκB1m). In contrast, mutation of the κB5 site resulted in an approximately 2-fold reduction in the luciferase activity (Figure 6D; TRAF1pκB5m), suggesting a critical role for the κB5 site in up-regulation of TRAF1 by p80HT. Together, the results of chromatin immunoprecipitation, electrophoretic mobility shift assay, and luciferase reporter assays demonstrate that TRAF1 is a direct transcriptional target of p80HT. Interestingly, overexpression of p52 actually inhibited the TRAF1 promoter-directed luciferase expression (Figure 6D; p52 + TRAF1pWT), indicating that p80HT is functionally distinct from p52, as reported previously.9,10,29
TRAF1 is essential for the antiapoptotic activity of p80HT
We next examined whether TRAF1 is required for the antiapoptotic activity of p80HT in WEHI-231 B lymphoma cells, a commonly used system for activation-induced apoptosis.30,31 We generated 2 TRAF1 siRNA-expressing retroviral constructs that target different regions of the mouse TRAF1-coding sequence, and retroviruses produced from either construct were effective in down-regulating TRAF1 in WEHI-231 cells (Figure 7A, lane 6 for one construct). No such effect was observed in WEHI-231 cells expressing GFP siRNA (Figure 7A, lane 5), demonstrating the specificity of the TRAF1 siRNA constructs. We also generated WEHI-231 cell lines overexpressing either p80HT or TRAF1 (Figure 7A, lanes 1-4). As expected, p80HT overexpression up-regulated TRAF1 in WEHI-231 cells (Figure 7A, lane 2), and p80HT or TRAF1 overexpression significantly protected the cells from apoptosis induced by a crosslinking anti-IgM antibody (Figure 7B). Importantly, TRAF1 knockdown completely abrogated the antiapoptotic activity of p80HT (Figure 7B).
Figure 7.

TRAF1 is essential for the antiapoptotic activity of p80HT. (A) Immunoblot analysis of p80HT, p52, and TRAF1 expression in WEHI-231 B lymphoma cells infected with retroviruses expressing GFP, p80HT, TRAF1, GFP siRNA (GFPsi), or TRAF1 siRNA (TRAF1si). Levels of α-tubulin are shown as loading control. (B) Anti-IgM-induced apoptosis in WEHI-231 cells overexpressing GFP, p80HT, TRAF1, or p80HT and TRAF1 siRNA (p80HT-TRAF1si), as analyzed by annexin-V staining and trypan blue dye exclusion assay. (C) In vitro survival assays of splenic B cells from the mice of indicated genotypes. Cells were either untreated or treated with 20 μg/mL of lipopolysaccharide. Viability was determined by trypan blue dye exclusion assay. (D) The numbers of total splenic B cells from the mice of indicated genotypes. Splenic lymphocytes were stained with fluorescein-conjugated anti-B220 and analyzed by flow cytometry. Data in panels B-D represent means (± standard deviation) from at least 3 independent experiments or from 3 to 5 mice of each genotype. Statistical analysis was performed using the 2-tailed Student t test; *P < .05; **P < .01.
We further assessed whether p80HT can promote the survival of primary TRAF1-deficient B cells. We crossed p80HT transgenic mice with TRAF1−/− mice to generate wild-type, TRAF1−/−, p80HT+/−/TRAF1+/+, and p80HT+/−/TRAF1−/− mice. Splenic B cells were isolated from 6- to 8-week-old mice and cultured in the absence of cytokines with or without LPS. Compared with p80HT+/− B cells, the survival of p80HT+/−TRAF1−/− B cells in the absence of cytokines was significantly reduced (Figure 7C; no treatment). Moreover, TRAF1 deficiency abrogated the ability of p80HT to protect B cells from LPS-induced apoptosis (Figure 7C; LPS). Together, these studies demonstrated that TRAF1 is a critical mediator of the antiapoptotic activity of p80HT.
TRAF1 deficiency reestablishes B cell homeostasis in p80HT mice
We reasoned that if p80HT induction of TRAF1 underlies the B cell expansion in p80HT mice, then TRAF1 deficiency should restore the B cell population to its normal level in these mice. To test the hypothesis, we quantified splenic B cells in 6- to 8-week-old wild-type, TRAF1−/−, p80HT+/−, and p80HT+/−/TRAF1−/− mice by flow cytometry (Figure 7D). TRAF1 deficiency had no significant effect on the number of splenic B cells in agreement with the previous report.14 Also as expected, p80HT mice had a significant increase in the number of splenic B cells relative to their wild-type littermates. In support of our hypothesis, the number of splenic B cells in p80HT+/−/TRAF1−/− mice was very similar to that in the wild-type mice, indicating that the loss of TRAF1 reestablished B cell homeostasis in p80HT mice. So far, 3 of the 12 p80HT + /−/TRAF1+/+ mice have died from lymphoma at the ages of 39, 45, and 51 weeks, whereas all of the 12 p80HT+/−/TRAF1−/− littermates remain alive and healthy. Thus, p80HT promoted B cell expansion and lymphoma development primarily through a TRAF1-dependent antiapoptotic pathway.
Discussion
The present study provides the first demonstration of a causal role for NF-κB2 mutation in lymphomagenesis. Transgenic mice expressing the tumor-derived NF-κB2 mutant p80HT in lymphocytes showed marked expansion of the peripheral B cell population and developed predominantly small B cell lymphomas with multiorgan metastases. p80HT expression had no significant effect on the proliferation of lymphocytes but protected them from apoptosis induced by cytokine deprivation or after mitogenic stimulation, suggesting that the antiapoptotic activity of p80HT is critical for its oncogenic function. Moreover, we identified TRAF1 as a key target of p80HT in the tumorigenic process. p80HT directly up-regulates TRAF1 through the κB5 site in the promoter. Importantly, TRAF1 down-regulation or deficiency largely abrogated the antiapoptotic activity of p80HT and reestablished B cell homeostasis in p80HT mice. It remains possible, however, that other p80HT target genes such as TRAF2 might also contribute to p80HT-induced lymphomagenesis.
The lymphoma development in p80HT mice was characterized by a prolonged latent period. In addition, B lymphoma cells were significantly more resistant to apoptosis induced by doxorubicin and cytokine deprivation compared with pretumor B cells. These observations suggest that additional genetic or epigenetic alterations may have taken place during the tumorigenic process, which may be required for malignant transformation of p80HT lymphocytes and their clonal expansion.
Given that the transgenic mice expressed p80HT in both T and B cells, it is somewhat surprising that they developed predominantly B cell lymphomas. Although the underlying mechanism remains to be defined, we speculate that it may reflect a major role of the NF-κB2 signaling pathway in maintaining the peripheral B cell population. NF-κB2−/− mice presented a marked decrease in the number of splenic B cells.32,33 In addition, B cell-activating factor, a cytokine required for the development and survival of peripheral B cells,34 activated NF-κB2 by inducing p100 processing to p52.35,36 In this regard, it is notable that p80HT mice expressed high levels of p52 in lymphocytes, raising the possibility that the sustained activation of p52 may underlie the B cell expansion and lymphomagenesis in p80HT mice. However, accumulated evidence suggests that it may not be the case. It has been shown previously that p80HT has not only acquired a transactivation activity distinct from p52, but also lost the transcriptional repressor function of p52.9,10,29 Similarly, we found that, in striking contrast to p80HT, p52 overexpression actually inhibited the TRAF1 promoter-directed luciferase expression. Interestingly, the NF-κB2-TRAF1κB5 complex in p80HT nuclear extracts contained RelA and c-Rel, but little RelB, a common partner of p52.37,38 This preferential dimerization with RelA and c-Rel might contribute to the distinct transcriptional activity of p80HT. Clearly, further studies, including investigation using p52 transgenic mice and p80HT-specific antibody, are needed for a molecular understanding of p80HT-induced lymphomagenesis.
TRAF1 is a member of the TRAF family of adapter proteins that regulate diverse cellular processes, including apoptosis.39,40 Several recent studies implicate TRAF1 in the pathogenesis of lymphoid malignancies. TRAF1 is overexpressed in a variety of lymphoma and leukemia cell lines and specimens.41–46 In addition, association with TRAF1 and TRAF2 is critical for the Epstein-Barr virus latent membrane protein 1 to transform primary B cells.47 Furthermore, transgenic mice expressing a TRAF2 mutant lacking the N-terminal RING and zinc finger domains (TRAF2DN), which structurally mimics TRAF1, have polyclonal expansion of B cells,48 and TRAF2DN cooperates with Bcl-2 to induce small B lymphoma in mice.49 These are the phenotypes shared by p80HT mice, suggesting a common pathway or mechanism underlying the lymphoma development in these mice. Together, these findings suggest that TRAF1 is a crucial mediator of diverse oncogenic signals in the development of lymphoid malignancies.
The p80HT transgenic mice are the first animal model for human lymphoma carrying the NF-κB2 mutation. These mice should be useful for the identification of genes that cooperate with NF-κB2 mutation in the pathogenesis of lymphoid malignancies and for the development and testing of therapeutic and prevention drugs that specifically interfere with the pathogenic process.
Supplementary Material
Acknowledgments
We thank Arnold Rabson for p80HT cDNA, Alan Harris for the IgH μ-chain JH4 probe, Mark Davis for the TCR Cβ1 probe, and Harald Wajant and Frank Henkler for the TRAF1 promoter-luciferase construct. We also thank Yongqing Wang for initial work on chromatin immunoprecipitation and RNase protection assays, Tom Sawyer and Karen Domenico for flow cytometry, and Douglas Pittman for advice on animal studies.
This work was supported by grants from the American Cancer Society (RSG-03-173-01-CCG) and the National Cancer Institute (R01 CA106550) to H.-F.D.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: B.Z. and H.-F.D. designed research and analyzed data; B.Z., Z.W., and T.L. performed research; E.N.T. contributed TRAF1−/− mice; and H.-F.D. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Han-Fei Ding, Department of Biochemistry and Cancer Biology, University of Toledo Health Science Campus, 3035 Arlington Avenue, Toledo, OH 43614; e-mail: han-fei.ding@utoledo.edu.
References
- 1.Gilmore TD. Introduction to NF-κB: players, pathways, perspectives. Oncogene. 2006;25:6680–6684. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 2.Basseres DS, Baldwin AS. Nuclear factor-κB and inhibitor of κB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25:6817–6830. doi: 10.1038/sj.onc.1209942. [DOI] [PubMed] [Google Scholar]
- 3.Karin M, Greten FR. NF-κB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 4.Courtois G, Gilmore TD. Mutations in the NF-κB signaling pathway: implications for human disease. Oncogene. 2006;25:6831–6843. doi: 10.1038/sj.onc.1209939. [DOI] [PubMed] [Google Scholar]
- 5.Neri A, Chang CC, Lombardi L, et al. B cell lymphoma-associated chromosomal translocation involves candidate oncogene lyt-10, homologous to NF-κB p50. Cell. 1991;67:1075–1087. doi: 10.1016/0092-8674(91)90285-7. [DOI] [PubMed] [Google Scholar]
- 6.Fracchiolla NS, Lombardi L, Salina M, et al. Structural alterations of the NF-κB transcription factor lyt-10 in lymphoid malignancies. Oncogene. 1993;8:2839–2845. [PubMed] [Google Scholar]
- 7.Migliazza A, Lombardi L, Rocchi M, et al. Heterogeneous chromosomal aberrations generate 3′ truncations of the NFKB2/lyt-10 gene in lymphoid malignancies. Blood. 1994;84:3850–3860. [PubMed] [Google Scholar]
- 8.Thakur S, Lin HC, Tseng WT, et al. Rearrangement and altered expression of the NFKB-2 gene in human cutaneous T-lymphoma cells. Oncogene. 1994;9:2335–2344. [PubMed] [Google Scholar]
- 9.Zhang J, Chang CC, Lombardi L, Dalla-Favera R. Rearranged NFKB2 gene in the HUT78 T-lymphoma cell line codes for a constitutively nuclear factor lacking transcriptional repressor functions. Oncogene. 1994;9:1931–1937. [PubMed] [Google Scholar]
- 10.Chang CC, Zhang J, Lombardi L, Neri A, Dalla-Favera R. Rearranged NFKB-2 genes in lymphoid neoplasms code for constitutively active nuclear transactivators. Mol Cell Biol. 1995;15:5180–5187. doi: 10.1128/mcb.15.9.5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ciana P, Neri A, Cappellini C, et al. Constitutive expression of lymphoma-associated NFKB-2/Lyt-10 proteins is tumorigenic in murine fibroblasts. Oncogene. 1997;14:1805–1810. doi: 10.1038/sj.onc.1201015. [DOI] [PubMed] [Google Scholar]
- 12.Pircher H, Mak TW, Lang R, et al. T cell tolerance to Mlsa encoded antigens in T cell receptor V beta 8.1 chain transgenic mice. EMBO J. 1989;8:719–727. doi: 10.1002/j.1460-2075.1989.tb03431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y, Schlossman SF, Edwards RA, Ou C-N, Gu J, Wu MX. Impaired apoptosis, extended duration of immune responses, and a lupus-like autoimmune disease in IEX-1-transgenic mice. Proc Natl Acad Sci U S A. 2002;99:878–883. doi: 10.1073/pnas.022326699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsitsikov EN, Laouini D, Dunn IF, et al. TRAF1 is a negative regulator of TNF signaling: enhanced TNF signaling in TRAF1-deficient mice. Immunity. 2001;15:647–657. doi: 10.1016/s1074-7613(01)00207-2. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, Cui H, Schroering A, et al. NF-κB2 p100 is a pro-apoptotic protein with anti-oncogenic function. Nat Cell Biol. 2002;4:888–893. doi: 10.1038/ncb872. [DOI] [PubMed] [Google Scholar]
- 16.Finco TS, Beg AA, Baldwin AS., Jr Inducible phosphorylation of IκB alpha is not sufficient for its dissociation from NF-κB and is inhibited by protease inhibitors. Proc Natl Acad Sci U S A. 1994;91:11884–11888. doi: 10.1073/pnas.91.25.11884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adams JM, Harris AW, Pinkert CA, et al. The c-myc oncogene driven by immunoglobulin enhancers induced lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- 18.Hedrick SM, Cohen DI, Nielsen EA, Davis MM. Isolation of cDNA clones encoding T cell-specific membrane-associated proteins. Nature. 1984;308:149–153. doi: 10.1038/308149a0. [DOI] [PubMed] [Google Scholar]
- 19.Hao H, Qi H, Ratnam M. Modulation of the folate receptor type beta gene by coordinate actions of retinoic acid receptors at activator Sp1/ets and repressor AP-1 sites. Blood. 2003;101:4551–4560. doi: 10.1182/blood-2002-10-3174. [DOI] [PubMed] [Google Scholar]
- 20.Schwenzer R, Siemienski K, Liptay S, et al. The human tumor necrosis factor (TNF) receptor-associated factor 1 gene (TRAF1) is up-regulated by cytokines of the TNF ligand family and modulates TNF-induced activation of NF-κB and c-Jun N-terminal kinase. J Biol Chem. 1999;274:19368–19374. doi: 10.1074/jbc.274.27.19368. [DOI] [PubMed] [Google Scholar]
- 21.Cui H, Schroering A, Ding HF. p53 mediates DNA damaging drug-induced apoptosis through a caspase-9-dependent pathway in SH-SY5Y neuroblastoma cells. Mol Cancer Ther. 2002;1:679–686. [PubMed] [Google Scholar]
- 22.Krebs DL, Yang Y, Dang M, Haussmann J, Gold MR. Rapid and efficient retrovirus-mediated gene transfer into B cell lines. Methods Cell Sci. 1999;21:57–68. doi: 10.1023/a:1009843325770. [DOI] [PubMed] [Google Scholar]
- 23.Morse HC, III, Anver MR, Fredrickson TN, et al. Bethesda proposals for classification of lymphoid neoplasms in mice. Blood. 2002;100:246–258. doi: 10.1182/blood.v100.1.246. [DOI] [PubMed] [Google Scholar]
- 24.Rathmell JC, Thompson CB. Pathways of apoptosis in lymphocyte development, homeostasis, and disease. Cell. 2002;109(suppl):S97–S107. doi: 10.1016/s0092-8674(02)00704-3. [DOI] [PubMed] [Google Scholar]
- 25.Chu Z-L, McKinsey TA, Liu L, Gentry JJ, Malim MH, Ballard DW. Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis c-IAP2 is under NF-kappaB control. Proc Natl Acad Sci U S A. 1997;94:10057–10062. doi: 10.1073/pnas.94.19.10057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang C-Y, Mayo MV, Korneluk RG, Goeddel DV, Baldwin AS. NF-κB antiapoptosis: induction of TRAF1 and TRAF2 and cIAP1 and cIAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 27.Tang G, Minemoto Y, Dibling B, et al. Inhibition of JNK activation through NF-κB target genes. Nature. 2001;414:313–317. doi: 10.1038/35104568. [DOI] [PubMed] [Google Scholar]
- 28.Chen C, Edelstein LC, Gelinas C. The Rel/NF-kB family directly activates expression of the apoptosis inhibitor Bcl-XL. Mol Cell Biol. 2000;20:2687–2695. doi: 10.1128/mcb.20.8.2687-2695.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Epinat JC, Kazandjian D, Harkness DD, et al. Mutant envelope residues confer a transactivation function onto N-terminal sequences of the v-Rel oncoprotein. Oncogene. 2000;19:599–607. doi: 10.1038/sj.onc.1203376. [DOI] [PubMed] [Google Scholar]
- 30.Benhamou LE, Cazenave PA, Sarthou P. Anti-immunoglobulins induce death by apoptosis in WEHI-231 B lymphoma cells. Eur J Immunol. 1990;20:1405–1407. doi: 10.1002/eji.1830200630. [DOI] [PubMed] [Google Scholar]
- 31.Hasbold J, Klaus GG. Anti-immunoglobulin antibodies induce apoptosis in immature B cell lymphomas. Eur J Immunol. 1990;20:1685–1690. doi: 10.1002/eji.1830200810. [DOI] [PubMed] [Google Scholar]
- 32.Caamano JH, Rizzo CA, Durham SK, et al. Nuclear factor (NF)-κB2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. J Exp Med. 1998;187:185–196. doi: 10.1084/jem.187.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Franzoso G, Carlson L, Poljak L, et al. Mice deficient in nuclear factor (NF)-κB/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. J Exp Med. 1998;187:147–159. doi: 10.1084/jem.187.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schiemann B, Gommerman JL, Vora K, et al. An essential role for BAFF in the normal development of B cells through a BCMA-independent pathway. Science. 2001;293:2111–2114. doi: 10.1126/science.1061964. [DOI] [PubMed] [Google Scholar]
- 35.Claudio E, Brown K, Park S, Wang H, Siebenlist U. BAFF-induced NEMO-independent processing of NF-kB2 in maturing B cells. Nat Immunol. 2002;3:958–965. doi: 10.1038/ni842. [DOI] [PubMed] [Google Scholar]
- 36.Kayagaki N, Yan M, Seshasayee D, et al. BAFF/BLyS receptor 3 binds the B cell survival factor BAFF ligand through a discrete surface loop and promotes processing of NF-kB2. Immunity. 2002;17:515–524. doi: 10.1016/s1074-7613(02)00425-9. [DOI] [PubMed] [Google Scholar]
- 37.Solan NJ, Miyoshi H, Carmona EM, Bren GD, Paya CV. RelB cellular regulation and transcriptional activity are regulated by p100. J Biol Chem. 2002;277:1405–1418. doi: 10.1074/jbc.M109619200. [DOI] [PubMed] [Google Scholar]
- 38.Coope HJ, Atkinson PG, Huhse B, et al. CD40 regulates the processing of NF-κB2 p100 to p52. EMBO J. 2002;21:5375–5385. doi: 10.1093/emboj/cdf542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arch RH, Gedrich RW, Thompson CB. Tumor necrosis factor receptor-associated factors (TRAFs)—a family of adapter proteins that regulates life and death. Genes Dev. 1998;12:2821–2830. doi: 10.1101/gad.12.18.2821. [DOI] [PubMed] [Google Scholar]
- 40.Bradley JR, Pober JS. Tumor necrosis factor receptor-associated factors (TRAFs). Oncogene. 2001;20:6482–6491. doi: 10.1038/sj.onc.1204788. [DOI] [PubMed] [Google Scholar]
- 41.Durkop H, Foss HD, Demel G, Klotzbach H, Hahn C, Stein H. Tumor necrosis factor receptor-associated factor 1 is overexpressed in Reed-Sternberg cells of Hodgkin's disease and Epstein-Barr virus-transformed lymphoid cells. Blood. 1999;93:617–623. [PubMed] [Google Scholar]
- 42.Izban KF, Ergin M, Martinez RL, Alkan S. Expression of the tumor necrosis factor receptor-associated factors (TRAFs) 1 and 2 is a characteristic feature of Hodgkin and Reed-Sternberg cells. Mod Pathol. 2000;13:1324–1331. doi: 10.1038/modpathol.3880243. [DOI] [PubMed] [Google Scholar]
- 43.Zapata JM, Krajewska M, Krajewski S, et al. TNFR-associated factor family protein expression in normal tissues and lymphoid malignancies. J Immunol. 2000;165:5084–5096. doi: 10.4049/jimmunol.165.9.5084. [DOI] [PubMed] [Google Scholar]
- 44.Murray PG, Flavell JR, Baumforth KR, et al. Expression of the tumor necrosis factor receptor-associated factors 1 and 2 in Hodgkin's disease. J Pathol. 2001;194:158–164. doi: 10.1002/path.873. [DOI] [PubMed] [Google Scholar]
- 45.Munzert G, Kirchner D, Stobbe H, et al. Tumor necrosis factor receptor-associated factor 1 gene overexpression in B-cell chronic lymphocytic leukemia: analysis of NF-kappa B/Rel-regulated inhibitors of apoptosis. Blood. 2002;100:3749–3756. doi: 10.1182/blood.V100.10.3749. [DOI] [PubMed] [Google Scholar]
- 46.Savage KJ, Monti S, Kutok JL, et al. The molecular signature of mediastinal large B-cell lymphoma differs from that of other diffuse large B-cell lymphomas and shares features with classical Hodgkin lymphoma. Blood. 2003;102:3871–3879. doi: 10.1182/blood-2003-06-1841. [DOI] [PubMed] [Google Scholar]
- 47.Cahir McFarland ED, Izumi KM, Mosialos G. Epstein-Barr virus transformation: involvement of latent membrane protein 1-mediated activation of NF-kappaB. Oncogene. 1999;18:6959–6964. doi: 10.1038/sj.onc.1203217. [DOI] [PubMed] [Google Scholar]
- 48.Lee SY, Reichlin A, Santana A, Sokol KA, Nussenzweig MC, Choi Y. TRAF2 is essential for JNK but not NF-κB activation and regulates lymphocyte proliferation and survival. Immunity. 1997;7:703–713. doi: 10.1016/s1074-7613(00)80390-8. [DOI] [PubMed] [Google Scholar]
- 49.Zapata JM, Krajewska M, Morse HC, III, Choi Y, Reed JC. TNF receptor-associated factor (TRAF) domain and Bcl-2 cooperate to induce small B cell lymphoma/chronic lymphocytic leukemia in transgenic mice. Proc Natl Acad Sci U S A. 2004;101:16600–16605. doi: 10.1073/pnas.0407541101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}