

Abstract
Finding good drug leads de novo from large chemical libraries, real or virtual, is not an easy task. High-throughput screening is often plagued by low hit rates and many leads that are toxic or exhibit poor bioavailability. Exploiting the secondary activity of marketed drugs, on the other hand, may help in generating drug leads that can be optimized for the observed side-effect target, while maintaining acceptable bioavailability and toxicity profiles. Here, we describe an efficient computational methodology to discover leads to a protein target from safe marketed drugs. We applied an in silico “drug repurposing” procedure for identification of nonsteroidal antagonists against the human androgen receptor (AR), using multiple predicted models of an antagonist-bound receptor. The library of marketed oral drugs was then docked into the best-performing models, and the 11 selected compounds with the highest docking score were tested in vitro for AR binding and antagonism of dihydrotestosterone-induced AR transactivation. The phenothiazine derivatives acetophenazine, fluphenazine, and periciazine, used clinically as antipsychotic drugs, were identified as weak AR antagonists. This in vitro biological activity correlated well with endocrine side effects observed in individuals taking these medications. Further computational optimization of phenothiazines, combined with in vitro screening, led to the identification of a nonsteroidal antiandrogen with improved AR antagonism and marked reduction in affinity for dopaminergic and serotonergic receptors that are the primary target of phenothiazine antipsychotics.
Keywords: androgen receptor, drug design, prostate cancer
Current approaches for discovery of novel chemical leads against a molecular target rely heavily on high-throughput screening (HTS) and to a lesser extent on virtual ligand screening (VLS) techniques. HTS has provided rapid lead identification for numerous drug targets (1–8); however, HTS also has major drawbacks, including a significant level of false positives and false negatives and low hit rates for many targets (9). Successful leads from HTS can also suffer from poor bioavailability and unwanted toxicity profiles of compounds. These problems result partially from the nature of the chemical libraries used for HTS. Furthermore, because the pharmacological properties of most compound libraries are largely unknown, there is an additional high risk that optimization of hits identified with HTS will not be sufficient for their evolution into real drugs.
In contrast, retrospective analysis of marketed drugs reveals that their physicochemical and structural properties are clustered around preferred values and scaffolds (10). In addition, some chemical motifs are associated with high biological activity and often confer activity against more than one target/receptor (11–16). These motifs have been referred to as “privileged structures” (11). These observations lead to an assumption that the chemical space of potential drugs is limited. Consequently, currently marketed drugs, which by definition have run the gauntlet of drug optimization, are an attractive collection of compounds for lead identification against new targets.
All drugs have activity that is distinct from that elicited by interaction with their primary (or chosen) target. This activity generally underlies the side-effect profile of the drug. It is this action at secondary or “off” targets that may potentially be harnessed for novel drug development, through optimization of the pharmacological profile of the drug to enhance the secondary activity and effectively eliminate activity at the original target. This procedure has been referred to as drug repurposing. The principal benefit of this approach is inheritance of a chemical scaffold with favorable bioavailability and toxicity profiles. Thus, the repurposed compounds are likely to enter clinical trials more rapidly and at less cost, in contrast to new chemical entities derived “from scratch.” Successful examples of drug repurposing include development of the potassium channel blocker levocromakalim from β-blockers (15) and development of an inhibitor of fibrin transthyretine amyloid formation from the nonsteroidal antiinflammatory drug, flufenamic acid (16).
A key to repurposing drugs is the capacity to identify potential secondary targets of therapeutic value. Recently, we have demonstrated that computational approaches can be used to develop predictive models of antagonist-bound states of nuclear hormone receptors (17). Using the ICM ligand docking and scoring algorithm, these models enabled enrichment of hit lists of sample ligand libraries 33- to 100-fold for 9 of 10 nuclear hormone receptors studied. Furthermore, there was a good correlation between docking scores and relative specificity across nuclear receptors for 78 known nuclear receptor ligands (17). We also identified antagonists to two nuclear receptors, retinoic acid receptor (17, 18) and thyroid hormone receptor (19), by using predicted antagonist-bound models.
The human androgen receptor (AR) is critical for the development and progression of prostate cancer. However, the disease is poorly serviced by the existing clinical repertoire of AR antagonists. Here, we describe and validate a computational method to identify secondary antiandrogen activity of known drugs. By capitalizing on several critical contributions to the understanding of the AR structure and function (20–24), multiple models for antagonist-bound conformations of the AR were developed, and suitable models for ligand screening were selected based on docking scores for known antagonists. These models were subsequently screened against marketed drugs to identify compounds likely to have secondary antiandrogen activity. One of three families identified was confirmed to interact with the AR by in vitro assays and further optimized to improve antiandrogen activity and decrease activity at the primary targets for the lead drug.
Results
Model Optimization and Refinement.
As described in Materials and Methods, two initial models of the antagonist-bound conformation of the AR ligand binding domain (LBD) were developed: one bicalutamide-biased (B-model) and one flutamide-biased (F-model). Structural conformers of these preliminary models, docked to 24 known AR antagonists, were generated and subsequently used to determine their ability to identify AR antagonists [supporting information (SI) Fig. 5]. SI Fig. 6 details the capacity of individual conformers to identify AR antagonist binders and discriminate AR binders from other nuclear hormone receptor ligands. Two structural variants, B5 and F17, demonstrated both a high level of enrichment of AR binders and low cross-reactivity with other nuclear receptor ligands. These were subsequently used for VLS experiments.
Closer examination of variants B5 and F17 revealed a rmsd difference of 0.8 Å within the ligand binding site, resulting primarily from alternative packing of side chains. However, this side-chain rearrangement caused substantial differences in the volume and geometry of the ligand binding pockets, as predicted by the ICM pocket finder algorithm (25). The volume of the B5 model pocket was 689.0 A3 with a surface area of 650.0 A2, whereas the corresponding geometric features of the F17 pocket were 585.0 A3 and 639.0 A2, respectively (SI Fig. 7).
To further validate the selected models, ligand poses, after redocking of each of the 24 known AR antagonists, were manually inspected, with special attention to interactions of the ligands with receptor amino acids previously shown to be important for high-affinity binding by the AR LBD (22, 26–30). In particular, the formation of a hydrogen bond between ligands and R752 is speculated to be essential for high-affinity binding. This residue is highly conserved among nuclear receptors, and recent crystallographic data demonstrated its importance for ligand binding for estrogen and glucocorticoid receptors. Both the B5 and F17 model variants formed this interaction with the majority of the known AR antagonists (SI Fig. 8).
VLS.
The marketed oral drugs database, described in ref. 10, was used for ligand docking experiments. The database was docked into the B5 and F17 structural variants as described in SI Text, and the top binders, with ICM docking scores ranging from −60.0 to −32.0, were examined manually. From the top binders, 11 compounds scoring highest in both structural model variants were selected and purchased for in vitro biological experiments (SI Table 1).
AR Binding and Transactivation.
In AR competitive ligand binding assays, only three of the compounds, fluphenazine (FPZ; KI ≈0.8 μM), acetophenazine (APZ; KI ≈0.8 μM), and periciazine (PCZ; KI ≈3.0 μM), demonstrated competition for [3H]mibolerone binding, each of these belonging to the phenothiazine class of drugs (SI Table 1). These three ligands were subsequently examined for their ability to antagonize dihydrotestosterone (DHT)-stimulated AR transactivation, using transcription of chloramphenicol acetyl transferase (CAT) as the read-out, in CV-1 cells transiently expressing the AR, as described in SI Text. Consistent with the binding data, APZ and PCZ weakly, but significantly, reduced DHT-stimulated CAT activity, while having minimal effect on basal CAT activation (Fig. 1; FPZ trended toward a reduction in CAT activity (Fig. 1A). Because of the low affinity of the drugs for the receptor, no clear dose–response relationship could be established with the concentrations used. Nonetheless, the data suggested that phenothiazines and their derivatives could be considered as a potential scaffold for development of ligands with antiandrogen properties.
Fig. 1.

Regulation of AR transactivation by selected marketed drugs, FPZ and APZ (A) and PCZ (B). The CAT reporter gene assays were conducted as described in Materials and Methods. (−), untreated cells, negative control; (+), 1 nM DHT, positive control; filled bars, 1 nM DHT was added together with ligand at the indicated concentration; empty bars, ligand alone was added at the indicated concentration; dashed bars, raw β-gal activity data. The data are mean ± SEM of three independent experiments. CAT data are normalized against β-gal activity. a.u., arbitrary units. ∗, P < 0.05.
To test this assumption, a phenothiazine core substructure search was performed in available chemical databases (Chembridge, Chemdiv, and others), and a set of 10 phenothiazine derivatives was purchased and screened for their antiandrogen activity in vitro using the CAT reporter assay at a constant ligand concentration of 500 nM (SI Table 2). The D4 ligand was found to be the most potent antiandrogen molecule, inhibiting almost 50% of the DHT-stimulated CAT reporter gene transactivation (SI Table 2). Further evaluation of this compound over a range of concentrations demonstrated a dose-dependent inhibition of DHT-stimulated AR activity (Fig. 2A), with a pIC50 value of 6.60 ± 0.25 (n = 3; IC50 = 0.25 μM), consistent with its binding affinity for the AR (KI ≈ 1.0 μM). Intriguingly, the D4 ligand had greater efficacy than the prototypic AR antagonist, Casodex (bicalutamide), in inhibiting DHT-stimulated CAT activity (D4 Emax, 76 ± 7% inhibition, n = 3; Casodex Emax, 44 ± 6% inhibition, n = 3), albeit that Casodex was more potent. The D4 ligand had minimal effect on β-gal activity (Fig. 2C) and did not exhibit non-AR-related cytotoxicity in PC3 prostate cancer cells at concentrations up to 10 μM (data not shown).
Fig. 2.

Antiandrogen activity of the D4 phenothiazine derivative. The CAT reporter gene assays were conducted as described in Materials and Methods. (A) Inhibition of DHT (1 nM)-stimulated CAT activity, in CV-1 cells transfected with the wild-type AR, by D4 (○) or Casodex (●). CAT activity data are normalized against β-gal activity. (B) D4-stimulated CAT activity in CV-1 cells transfected with the T877A mutant AR. (C) Effect of increasing concentrations of D4 on β-gal activity in wild-type AR-transfected CV-1 cells. (D) PSA assay in LNCaP cells. (−), untreated cells (negative control); (+), 50 nM DHT alone (positive control). The ligand was added to the cells at the indicated concentrations together with 50 nM DHT. The films were digitized, and PSA expression data were normalized by α-tubulin content. (Inset) Endogenous AR expression levels in LNCaP cells. The data are mean ± SEM of three independent experiments. ∗, P <0.05. a.u., arbitrary units.
Prostate-Specific Antigen (PSA) Assay and AR Cellular Localization.
The effect of the D4 ligand on the expression of PSA, a marker of endogenous AR receptor activation, was investigated in LNCaP prostate cancer cells (see SI Text). The D4 ligand had a biphasic effect on DHT-stimulated PSA expression (Fig. 2D); an initial increase in expression at 1 μM, followed by a progressive reduction in expression that was >80% at 10 μM. The endogenous AR expression, however, was not modified by ligand treatment (Fig. 2D Inset). As LNCaP cells express both the wild-type AR and the T877A mutant form of the receptor (28), we speculated that the initial increase in PSA expression levels seen in these cells could be caused by activation of the T877A AR mutant by D4 (31). This hypothesis was corroborated in the CAT transactivation assay where the D4 ligand was an agonist of the T887A AR mutant transiently expressed in CV-1 cells (Fig. 2B).
The cellular localization of AR in the presence and absence of ligands was also investigated (Fig. 3) (see SI Text). In the absence of DHT, AR was diffusely distributed in the cytoplasm. Upon treatment with 10 nM DHT for 1 h, AR was completely translocated into the nucleus. When cells were treated with the D4 ligand at 1 μM in the presence and/or absence of 10 nM DHT, AR was also translocated from the cytoplasm to the nucleus, with >90% of the AR translocated to the nucleus. The redistribution of the AR to the nucleus is also seen for the prototypic AR antagonist, bicalutamide (Casodex) (1 μM) (Fig. 3). These observations further demonstrate that the D4 ligand potently interacts with the AR.
Fig. 3.

Nuclear translocation experiments. HeLa cells were transiently transfected with AR. (Left) Translocation of the receptor after treatment was assessed by anti-AR antibody labeling. (Center) The nucleus was identified by DAPI staining. (Right) The overlay of AR labeling with the DAPI staining. The treatments used were as follows: CT, untreated cells; DHT, 10 nM; CDX, Casodex (1 μM); D4, compound D4 (1 μM); or D4 + DHT, compound D4 (1 μM) plus DHT (10 nM). Refer to Fig. 2 for the D4 structure. The data are representative of three independent experiments.
Cross-Docking to a Panel of Nuclear Receptors.
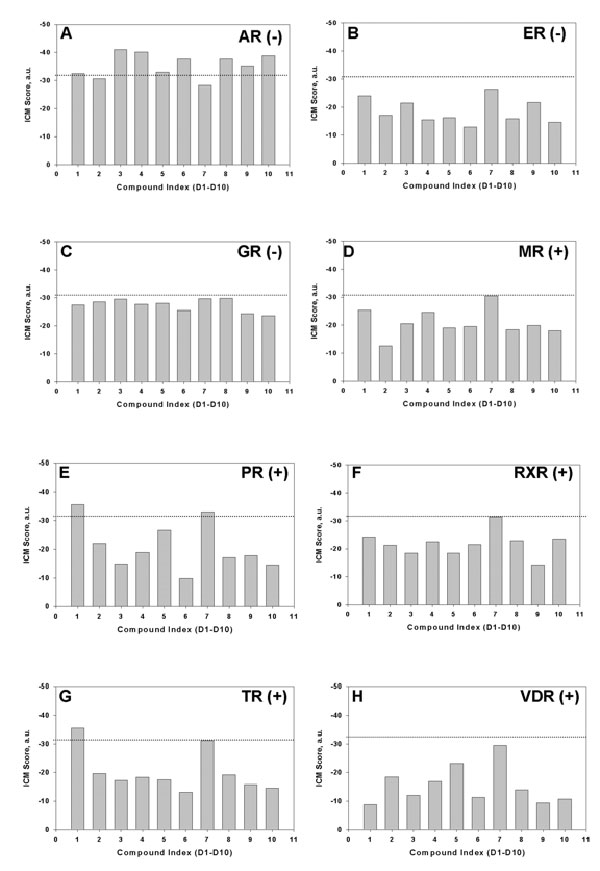
The AR belongs to a large family of nuclear receptors that share high sequence and structure similarity within their LBDs. Previously, we have shown that VLS is able to identify nuclear receptor ligands and predict their promiscuity (32). The D1–D10 FPZ derivatives were systematically docked to the LBDs of a panel of seven nuclear receptors. Our best ligand, D4, scored a below -32.0 docking score [statistically significant ICM docking score cutoff (33)] only in the B5 AR LBD antagonist model, suggesting a low probability of cross-reactivity with other nuclear receptors (SI Fig. 9).
Affinity for Dopaminergic and Serotonergic G Protein-Coupled Receptors (GPCRs).
The phenothiazines, FPZ, APZ, and PCZ, are marketed antipsychotics that interact strongly with serotonergic and dopaminergic GPCRs. To determine whether the D4 ligand maintained affinity for these targets, competitive binding assays at the 5HT2C, 5HT2A, and D2 receptors were performed (see SI Text). In contrast to FPZ, which exhibited high affinity for each of the receptors, the D4 ligand failed to compete for binding at concentrations up to 1 μM (Fig. 4). Thus, the D4 ligand, unlike phenothiazine antipsychotics, binds specifically to AR.
Fig. 4.

Primary target activity of the selected marketed drug derivative. Competitive ligand binding experiments with the primary pharmacological targets of FPZ. (A) Serotonin 5HT2A receptor. (B) Serotonin 5HT2C receptor. (C) Dopamine D2L receptor. The data are mean ± SEM of three independent experiments.
Discussion
Absence of the crystal structure of the AR LBD in an antagonist-bound conformation makes receptor structure-based VLS difficult. The chemical diversity of known AR binders suggests that the AR ligand binding site is capable of substantial induced flexibility to accommodate a binder. Previously, we have generated predictive antagonist-bound models for two nuclear receptors: the thyroid hormone receptor (19) and the retinoic acid receptor α (34). The available structures of antagonist-bound conformations of the homologous nuclear receptors demonstrate that the loop connecting helices H11 and H12 is the most flexible element of their binding site. Therefore, the backbone flexibility of the corresponding AR loop needed to be addressed during modeling.
Computational prediction of the induced fit upon binding of a ligand is a critical unsolved problem (35, 36). Recently, several successful flexible receptor docking protocols were proposed for different systems (37, 38). These protocols are based either on “molding” of the receptor binding pocket around a ligand using global energy optimization (37) or sampling of conformational space of a receptor by normal mode analysis (38). In the current study, the receptor flexibility issue was addressed somewhat differently. The protocol contained two stages: at the first stage alternative backbone conformers were generated, and during the second stage they were further refined by side-chain optimization only. The first stage is an iterative procedure in the course of which each cycle of the receptor backbone and side-chain refinement was followed by ligand docking in the VLS format (see SI Text). Only those receptor conformers that would discriminate between binders (antagonists) and decoy ligands (agonists) were allowed to enter the next refinement cycle. This procedure led to accumulation of structural receptor features, amenable for selective antagonist discrimination. Somewhat surprisingly analysis of the best models revealed that the rmsd difference between them (≈1.0 Å) was primarily caused by differences in the position of side-chains atoms, whereas the position of backbone atoms was essentially equivalent between models.
The second stage of the protocol involved generation of derivative models (B1-24, F1-24) based on obtained backbone conformers by optimizing side chains only. The optimization protocol was designed to favor models that can discriminate AR antagonists from AR agonists on the basis of the ICM docking score, as the “learning” criteria was biased toward antagonists (SI Figs. 5 and 6).
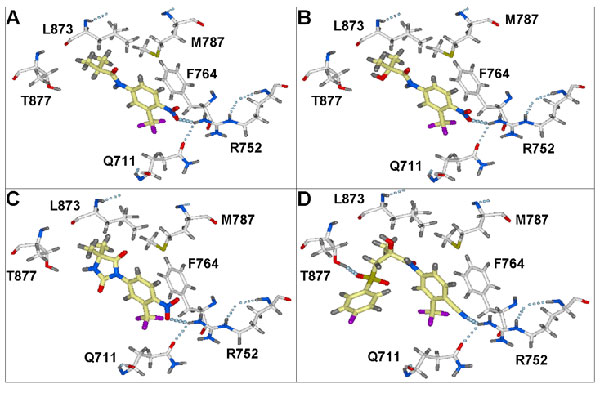
The validation of the best-performing models involved docking of such AR antagonists as flutamide, hydroxyflutamide, nilutamide, and bicalutamide. All of them were found to form a strong hydrogen bond with the R752 residue, located deep in the AR LBD binding pocket (SI Fig. 8). This interaction seems to be essential for proper binding of AR ligands (both agonists and antagonists), anchoring them in the pocket. Indeed, this interaction was observed in the crystal structure of the hAR LBD in complex with the agonist R1881, where 3-keto group of R1881 is involved in hydrogen-bond formation (39). This interaction also appears to be important for certain AR LBD mutants, as it can be observed in the crystal structures of W741L (24) and T877A (23) in complex with bicalutamide and hydroxyflutamide, respectively.
When the database of marketed oral drugs was docked into validated antagonist-biased AR LBD models, members of the phenothiazine family of antipsychotic compounds were among the top predicted binders. Docking experiments with these compounds generated an array of conformations of similar energy, but with rather different orientations for bound drugs. However, a hydrogen bond with the R752 residue was predicted to be frequently formed by either the substituent at position C2 of the phenothiazine system or the hydroxyl of the 1-piperazineethanol group, which is common for these compounds. This observation is in good agreement with predictions on the importance of ligand interaction with the R752 of the receptor (23, 24, 30, 39, 40).
Three of the phenothiazine derivatives (FPZ, APZ, and PCZ; see SI Table 1) exhibited modest binding and antiandrogen activity in vitro. These drugs are routinely used in the clinical treatment of schizophrenia (41). They are thought to principally act by inhibiting the D2 dopamine receptor and the 5HT2 family of serotonin receptors, members of the superfamily of GPCRs (42). During their administration, male patients are reported to experience endocrine side effects, including loss of sexual desire and impotence (43). Among many possible causes of these observed side effects, the weak antiandrogen properties of the compounds identified in the current study could provide a plausible explanation. In fact, the blood concentration achieved during administration of these drugs reaches the micromolar range (www.labcorp.com), and we have demonstrated in vitro AR antagonist activity of FPZ and APZ in the 300 to 1,000 nM ligand concentration range (Fig. 1A).
The antiandrogen activity of these drugs could be attributed to the phenothiazine system, which is topologically similar to the rigid steroid scaffold. The hydrogen bond acceptor group at position C2 mimics groups of a similar nature at the equivalent position of the steroidal ligands (e.g., the 3-keto of the R1881 and DHT). This feature appears to be important for these scaffolds, because perphenazine, which differs from FPZ only by chlorine substitution at C2, did not exhibit any AR-related activity. Thus, it seems plausible that the preferred mode of binding for these compounds involves formation of a hydrogen bond between the C2 substitute of the drug and R752 of the receptor, while the side chain at position N10 could interact with the H11–H12 loop and the H12 helix. This binding mode is similar to that observed for antagonists of other nuclear receptors (30, 44). The antiandrogen properties of several phenothiazine derivatives (SI Table 2) seem to corroborate this assumption. The ability of the D4 ligand to induce translocation of the AR from the cytoplasm into the nucleus also suggests that its antiandrogen activity is mechanistically similar to that of hydroxyflutamide (45) and bicalutamide (Figs. 2A and 3) (45), both of which induce AR translocation from the cytoplasm to the nucleus.
The D4 biological data are in good agreement with its predicted binding mode in the B5 AR LBD conformer (SI Fig. 9). The phenomenon of AR antagonism is believed to arise from the inability of the AR LBD in antagonist conformation to interact with LXXLL leucine-rich and aromatic-rich FXXLF motifs of nuclear steroid receptor coactivators (SRCs) (20, 21). These motifs interact with the AR LBD through a hydrophobic interface formed by helices 3, 4, 5, and 12 of the receptor (SI Fig. 9). Binding of an antagonist ligand is believed to disrupt this interface. It can be seen from SI Fig. 9 that D4 binding is predicted to dislodge the H12 helix by pushing the H11–H12 flexible loop through its naphthalene substituent. The H12 helix then occupies the binding site of the SRC peptide.
The presented data demonstrate that the D4 ligand, derived from the phenothiazine-marketed drug scaffold, interacts specifically with the AR and behaves as an antiandrogen. This was further confirmed by resistance of androgen-independent PC3 prostate cancer cells to D4 treatment (data not shown) and in silico cross-docking experiments with homologous nuclear receptors (see SI Fig. 10). However, repurposing of drugs requires not only improvement of activity at “off targets” but abolition of activity at the original, primary drug targets. Competitive binding experiments, involving the classic pharmacological targets of FPZ (serotonin and dopamine GPCRs), demonstrated that the D4 ligand has markedly reduced affinity for these receptors (Fig. 4). These data confirm that modification of phenothiazine drug scaffolds can lead to repurposing toward novel therapeutic targets.
Conclusions
In the present work, we have successfully demonstrated application of computational biology to the identification of antiandrogen scaffolds. Compounds derived from phenothiazines, currently marketed as antipsychotics, possessed submicromolar antiandrogen activity and are promising leads for further optimization. The work also suggests that, where validated models are available, virtual screening of drug candidates may provide a mechanism for better understanding of the potential side-effect profile of drugs. The methodology developed can be applied to the discovery of antiandrogen lead candidates against metastatic mutant forms of the AR.
Materials and Methods
Database Preparation.
Three databases were constructed and used to evaluate and optimize the models of the AR LBD. Database 1 contained 24 AR antagonists identified from literature (flutamide, hydroxyflutamide, nilutamide and derivatives, bicalutamide and derivatives, and isoxazolone derivatives) (46–50). Database 2, contained 88 known agonists and antagonists specific for androgen, estrogen, progesterone, and other homologous nuclear receptors (32) kindly provided by M. Totrov (Molsoft, La Jolla, CA). Database 3 contained 5,000 compounds of similar molecular weight, randomly selected from the ChemBridge drug-like compound database.
Model Preparation and Optimization.
An initial approximation of the antagonist-bound conformation of the AR LBD was derived by combining comparative protein modeling with global energy optimization. Because the crystal structure of the AR LBD in antagonist conformation is still not available, a segmented approach to assembly of the AR LBD model was used. Briefly, the AR LBD structure for residues 669–885 was modeled by using the crystal structure of the glucocorticoid receptor in an antagonist (RU-486) bound conformation [Protein Data Bank entry 1nhz (30)] as template. To model the conformation of the loop, connecting helices H11 and H12, and the H12 helix (residues 886–910), a different approach was used. First, weighted harmonic restraints were imposed between Ca of residues 892–910 (H12) of AR LBD and Ca of equivalent residues of H12 (residues 535–547) of the crystal structure of the estrogen receptor in antagonist-bound conformation [Protein Data Bank entry 1err (51)]. Then, ϕ, ψ, and χ angles of the residues comprising the AR LBD flexible loop (residues 887–891) were freed, and the energy of the system was globally optimized in internal coordinate space by using the biased probability Monte Carlo (BPMC) procedure (52, 53) within the ICM program (Molsoft). This initial crude model was used to dock a small set of known nuclear receptor agonists and antagonists, composed of testosterone, DHT, RU-486, flutamide, hydroxyflutamide, nilutamide, and bicalutamide, using the ligand-docking procedure within the ICM program. Each receptor–ligand complex was then refined by the BPMC procedure applied to the flexible loop backbone and receptor side chains in the vicinity of the ligand, while allowing the ligand to move. The set of ligands was then redocked to each of the refined receptor conformations. The resulting receptor–ligand complexes were manually inspected, and receptor conformations that achieved the best docking score separation between antagonists and agonists were chosen. The selected conformations were then used to generate a new set of refined receptor–ligand complexes, and the procedure was repeated iteratively (≈15 iterations) to achieve a reproducible agonist/antagonist separation. This procedure resulted in the generation of two receptor conformations favoring antagonist molecules with respect to the ICM docking score. These models were designated as B-model (bicalutamide ligand preferring) and F-model (flutamide ligand preferring) and selected for further optimization and refinement.
The optimization of selected receptor conformations is described in SI Fig. 5. Briefly, the database containing published structures of 24 AR antagonists and their derivatives was docked independently into each of the models, generating 48 receptor–ligand complexes. This database was docked at least three times, and the lowest-energy, ligand-bound conformations were retained. Then, for each receptor–ligand complex, conformations of the receptor side chains in the vicinity of the ligand were optimized by global energy minimization with biased probability Monte Carlo in internal coordinate space (52, 53). Enrichment factors for identification of target-specific compounds were determined as described (32), with certain modifications. Briefly, docking score thresholds for the retention of 1% and 10% of the 5,000 compounds in the trial database (50 and 500, respectively), were determined after docking of database 3 three times to each of the 48 refined models. These threshold values were subsequently applied to the focused library of 88 known nuclear hormone receptor agonists/antagonists after docking to each of the refined models. To evaluate selectivity of individual AR conformers toward AR antagonist ligands, the enrichment factor corresponding to the 1% docking score cutoff was computed by counting focused library hits only for AR antagonist ligands within this threshold. The enrichment factor for the 10% docking score threshold was computed by counting all of the nuclear receptor-focused library hits within this cutoff, thus providing an estimate of the ligand cross-reactivity potential of a particular AR conformer. The two models with the highest 1% docking score cutoff enrichment factors were selected for use in VLS experiments with the marketed oral drugs database (10).
Supplementary Material
Acknowledgments
We thank Dr. Maxim Totrov and Colin Smith for their help in setting up and optimizing the ICM docking procedures and Dr. Siva Kolluri (Oregon State University, Corvallis, OR) for valuable suggestions about in vitro experiments. This work was supported in part by Department of Defense Postdoctoral Fellowship Award W81XWH-04-1-0181 (to A.V.C.), a Swiss National Science Foundation postdoctoral fellowship award (to W.H.B.), and National Health and Medical Research Council of Australia Project Grant 400133. A.C. is a Senior Research Fellow and P.M.S. is a Principal Research Fellow of the National Health and Medical Research Council of Australia.
Abbreviations
- AR
androgen receptor
- HTS
high-throughput screening
- VLS
virtual ligand screening
- LBD
ligand binding domain
- DHT
dihydrotestosterone
- CAT
chloramphenicol acetyl transferase
- PSA
prostate-specific antigen
- FPZ
fluphenazine
- APZ
acetophenazine
- PCZ
periciazine
- GPCR
G protein-coupled receptor.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0609752104/DC1.
References
- 1.Raju TN. Lancet. 2000;356:346–352. doi: 10.1016/s0140-6736(05)73635-7. [DOI] [PubMed] [Google Scholar]
- 2.Song Y, Connor DT, Sercel AD, Sorenson RJ, Doubleday R, Unangst PC, Roth BD, Beylin VG, Gilbertsen RB, Chan K, et al. J Med Chem. 1999;42:1161–1169. doi: 10.1021/jm980570y. [DOI] [PubMed] [Google Scholar]
- 3.Youngman MA, McNally JJ, Lovenberg TW, Reitz AB, Willard NM, Nepomuceno DH, Wilson SJ, Crooke JJ, Rosenthal D, Vaidya AH, Dax SL. J Med Chem. 2000;43:346–350. doi: 10.1021/jm990468g. [DOI] [PubMed] [Google Scholar]
- 4.Gilligan PJ, Robertson DW, Zaczek R. J Med Chem. 2000;43:1641–1660. doi: 10.1021/jm990590f. [DOI] [PubMed] [Google Scholar]
- 5.Liu K, Xu L, Szalkowski D, Li Z, Ding V, Kwei G, Huskey S, Moller DE, Heck JV, Zhang BB, Jones AB. J Med Chem. 2000;43:3487–3494. doi: 10.1021/jm000285q. [DOI] [PubMed] [Google Scholar]
- 6.Doman TN, McGovern SL, Witherbee BJ, Kasten TP, Kurumbail R, Stallings WC, Connolly DT, Shoichet BK. J Med Chem. 2002;45:2213–2221. doi: 10.1021/jm010548w. [DOI] [PubMed] [Google Scholar]
- 7.Jenck F, Wichmann J, Dautzenberg FM, Moreau JL, Ouagazzal AM, Martin JR, Lundstrom K, Cesura AM, Poli SM, Roever S, et al. Proc Natl Acad Sci USA. 2000;97:4938–4943. doi: 10.1073/pnas.090514397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baxter A, Bennion C, Bent J, Boden K, Brough S, Cooper A, Kinchin E, Kindon N, McInally T, Mortimore M, et al. Bioorg Med Chem Lett. 2003;13:2625–2628. doi: 10.1016/s0960-894x(03)00561-4. [DOI] [PubMed] [Google Scholar]
- 9.Shoichet BK. Nature. 2004;432:862–865. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vieth M, Siegel MG, Higgs RE, Watson IA, Robertson DH, Savin KA, Durst GL, Hipskind PA. J Med Chem. 2004;47:224–232. doi: 10.1021/jm030267j. [DOI] [PubMed] [Google Scholar]
- 11.Evans BE, Rittle KE, Bock MG, DiPardo RM, Freidinger RM, Whitter WL, Lundell GF, Veber DF, Anderson PS, Chang RS, et al. J Med Chem. 1988;31:2235–2246. doi: 10.1021/jm00120a002. [DOI] [PubMed] [Google Scholar]
- 12.Buxton IL, Cheek OD, Eckman D, Westfall DP, Sanders KM, Keef KD. Circ Res. 1993;72:387–395. doi: 10.1161/01.res.72.2.387. [DOI] [PubMed] [Google Scholar]
- 13.Hoyer D. In: Peripheral Actions of 5-HT. Fozard JR, editor. Oxford: Oxford Univ Press; 1989. pp. 72–99. [Google Scholar]
- 14.Bymaster FP, Nelson DL, DeLapp NW, Falcone JF, Eckols K, Truex LL, Foreman MM, Lucaites VL, Calligaro DO. Schizophr Res. 1999;37:107–122. doi: 10.1016/s0920-9964(98)00146-7. [DOI] [PubMed] [Google Scholar]
- 15.Stemp G, Evans JM. In: Medicinal Chemistry: The Role of Organic Chemistry in Drug Research. Ganellin CR, Roberts SM, editors. London: Academic; 1993. pp. 141–162. [Google Scholar]
- 16.Baures PW, Oza VB, Peterson SA, Kelly JW. Bioorg Med Chem. 1999;7:1339–1347. doi: 10.1016/s0968-0896(99)00066-8. [DOI] [PubMed] [Google Scholar]
- 17.Schapira M, Raaka BM, Samuels HH, Abagyan R. Proc Natl Acad Sci USA. 2000;97:1008–1013. doi: 10.1073/pnas.97.3.1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cavasotto CN, Liu G, James SY, Hobbs PD, Peterson VJ, Bhattacharya AA, Kolluri SK, Zhang XK, Leid M, Abagyan R, et al. J Med Chem. 2004;47:4360–4372. doi: 10.1021/jm030651g. [DOI] [PubMed] [Google Scholar]
- 19.Schapira M, Raaka BM, Das S, Fan L, Totrov M, Zhou Z, Wilson SR, Abagyan R, Samuels HH. Proc Natl Acad Sci USA. 2003;100:7354–7359. doi: 10.1073/pnas.1131854100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hur E, Pfaff SJ, Payne ES, Gron H, Buehrer BM, Fletterick RJ. PLoS Biol. 2004;2:e274. doi: 10.1371/journal.pbio.0020274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Estebanez-Perpina E, Moore JM, Mar E, Delgado-Rodrigues E, Nguyen P, Baxter JD, Buehrer BM, Webb P, Fletterick RJ, Guy RK. J Biol Chem. 2005;280:8060–8068. doi: 10.1074/jbc.M407046200. [DOI] [PubMed] [Google Scholar]
- 22.Matias PM, Carrondo MA, Coelho R, Thomaz M, Zhao XY, Wegg A, Crusius K, Egner U, Donner P. J Med Chem. 2002;45:1439–1446. doi: 10.1021/jm011072j. [DOI] [PubMed] [Google Scholar]
- 23.Bohl CE, Miller DD, Chen J, Bell CE, Dalton JT. J Biol Chem. 2005;280:37747–37754. doi: 10.1074/jbc.M507464200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bohl CE, Gao W, Miller DD, Bell CE, Dalton JT. Proc Natl Acad Sci USA. 2005;102:6201–6206. doi: 10.1073/pnas.0500381102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.An J, Totrov M, Abagyan R. Mol Cell Proteomics. 2005;4:752–761. doi: 10.1074/mcp.M400159-MCP200. [DOI] [PubMed] [Google Scholar]
- 26.Sack JS, Kish KF, Wang C, Attar RM, Kiefer SE, An Y, Wu GY, Scheffler JE, Salvati ME, Krystek SR, Jr, et al. Proc Natl Acad Sci USA. 2001;98:4904–4909. doi: 10.1073/pnas.081565498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Veldscholte J, Ris-Stalpers C, Kuiper GG, Jenster G, Berrevoets C, Claassen E, van Rooij HC, Trapman J, Brinkmann AO, Mulder E. Biochem Biophys Res Commun. 1990;173:534–540. doi: 10.1016/s0006-291x(05)80067-1. [DOI] [PubMed] [Google Scholar]
- 28.Zhao XY, Malloy PJ, Krishnan AV, Swami S, Navone NM, Peehl DM, Feldman D. Nat Med. 2000;6:703–706. doi: 10.1038/76287. [DOI] [PubMed] [Google Scholar]
- 29.Krishnan AV, Zhao XY, Swami S, Brive L, Peehl DM, Ely KR, Feldman D. Endocrinology. 2002;143:1889–1900. doi: 10.1210/endo.143.5.8778. [DOI] [PubMed] [Google Scholar]
- 30.Kauppi B, Jakob C, Farnegardh M, Yang J, Ahola H, Alarcon M, Calles K, Engstrom O, Harlan J, Muchmore S, et al. J Biol Chem. 2003;278:22748–22754. doi: 10.1074/jbc.M212711200. [DOI] [PubMed] [Google Scholar]
- 31.Wetherill YB, Petre CE, Monk KR, Puga A, Knudsen KE. Mol Cancer Ther. 2002;1:515–524. [PubMed] [Google Scholar]
- 32.Schapira M, Abagyan R, Totrov M. J Med Chem. 2003;46:3045–3059. doi: 10.1021/jm0300173. [DOI] [PubMed] [Google Scholar]
- 33.Totrov M, Abagyan R. In: Istrail S, Pevzner P, Waterman M, editors. Proceedings of the Third Annual International Conference on Computational Molecular Biology; New York: Association for Computing Machinery; 1999. pp. 312–320. [Google Scholar]
- 34.Schapira M, Raaka BM, Samuels HH, Abagyan R. BMC Struct Biol J. 2001;1:1. doi: 10.1186/1472-6807-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carlson HA, Masukawa KM, Rubins K, Bushman FD, Jorgensen WL, Lins RD, Briggs JM, McCammon JA. J Med Chem. 2000;43:2100–2114. doi: 10.1021/jm990322h. [DOI] [PubMed] [Google Scholar]
- 36.Carlson HA, McCammon JA. Mol Pharmacol. 2000;57:213–218. [PubMed] [Google Scholar]
- 37.Cavasotto CN, Abagyan RA. J Mol Biol. 2004;337:209–225. doi: 10.1016/j.jmb.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 38.Cavasotto CN, Kovacs JA, Abagyan RA. J Am Chem Soc. 2005;127:9632–9640. doi: 10.1021/ja042260c. [DOI] [PubMed] [Google Scholar]
- 39.Matias PM, Donner P, Coelho R, Thomaz M, Peixoto C, Macedo S, Otto N, Joschko S, Scholz P, Wegg A, et al. J Biol Chem. 2000;275:26164–26171. doi: 10.1074/jbc.M004571200. [DOI] [PubMed] [Google Scholar]
- 40.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 41.Turner M, Eerdekens E, Jacko M, Eerdekens M. Int Clin Psychopharmacol. 2004;19:241–249. doi: 10.1097/01.yic.0000133500.92025.20. [DOI] [PubMed] [Google Scholar]
- 42.Cai G, Gurdal H, Smith C, Wang HY, Friedman E. Mol Pharmacol. 1999;56:989–996. doi: 10.1124/mol.56.5.989. [DOI] [PubMed] [Google Scholar]
- 43.Shirai M, Yamanaka M, Shiina H, Igawa M, Fujime M, Lue TF, Dahiya R. Int J Impot Res. 2003;15:391–396. doi: 10.1038/sj.ijir.3901050. [DOI] [PubMed] [Google Scholar]
- 44.Renaud JP, Rochel N, Ruff M, Vivat V, Chambon P, Gronemeyer H, Moras D. Nature. 1995;378:681–689. doi: 10.1038/378681a0. [DOI] [PubMed] [Google Scholar]
- 45.Farla P, Hersmus R, Trapman J, Houtsmuller AB. J Cell Sci. 2005;118:4187–4198. doi: 10.1242/jcs.02546. [DOI] [PubMed] [Google Scholar]
- 46.Van Dort ME, Robins DM, Wayburn B. J Med Chem. 2000;43:3344–3347. doi: 10.1021/jm000163y. [DOI] [PubMed] [Google Scholar]
- 47.Van Dort ME, Jung YW. Bioorg Med Chem Lett. 2001;11:1045–1047. doi: 10.1016/s0960-894x(01)00146-9. [DOI] [PubMed] [Google Scholar]
- 48.Ishioka T, Kubo A, Koiso Y, Nagasawa K, Itai A, Hashimoto Y. Bioorg Med Chem. 2002;10:1555–1566. doi: 10.1016/s0968-0896(01)00421-7. [DOI] [PubMed] [Google Scholar]
- 49.Cogan PS, Koch TH. J Med Chem. 2003;46:5258–5270. doi: 10.1021/jm0303305. [DOI] [PubMed] [Google Scholar]
- 50.Yin D, He Y, Perera MA, Hong SS, Marhefka C, Stourman N, Kirkovsky L, Miller DD, Dalton JT. Mol Pharmacol. 2003;63:211–223. doi: 10.1124/mol.63.1.211. [DOI] [PubMed] [Google Scholar]
- 51.Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 52.Abagyan R, Totrov M. J Mol Biol. 1994;235:983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- 53.Abagyan R, Totrov M. J Comp Chem. 1999;151:402–421. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}