

Abstract
Rabbit cells are poorly permissive to HIV-1 infection, but little is known about the nature of this block. Here, we show that the block to infection is mainly at the level of reverse transcription (RT), is independent of the cell receptor used by the virus for entry, cannot be effectively saturated with high doses of virus or virus-like particles, and has a recessive phenotype in human–rabbit heterokaryons. RT complexes (RTCs) extracted from human and rabbit cells have different densities but are both competent for RT in an in vitro endogenous assay. Cell fractionation showed that HIV-1 is trafficked in a different way in human and rabbit cells and that correct intracellular trafficking is linked to efficient RT and high infectivity in vivo. Viral DNA accumulated in rabbit cell nuclei only at a later stage and failed to associate with chromatin, suggesting a further block prior to integration. Our data point to the existence of cellular factors regulating the early stages of intracytoplasmic and possibly intranuclear HIV-1 trafficking.
Keywords: HIV-1, post-entry block, rabbit, recessive, reverse transcription, RTC, trafficking
Lentiviruses have a restricted species tropism. HIV-1 cannot replicate in cells of diverse species, such as rodent, cow, pig, rabbit and non-human primates (1-5). This has impaired the use of these animals as models to study HIV-1 infection and acquired immune-deficiency syndrome (AIDS) but has helped to provide insights into the virus life cycle. Restricted species tropism, however, is not surprising considering that viruses are obligate intracellular parasites and require several host factors to establish infection. Some of the necessary cell factors have been described for different retroviruses, in different cells. In the case of HIV-1, CD4 and the chemokine receptors, CCR5 and CXCR4, act as the cellular receptor and major co-receptors, respectively (6-9). Other host cofactors promote HIV-1 replication but are not absolutely required, for example, cyclophilin A increases HIV-1 infectivity and possibly modulates sensitivity to post-entry restriction factors in human cells (10,11). Interaction with the cell's actin microfilaments stimulates efficient reverse transcription (RT), and microtubules are likely to be used by the RT complex (RTC) to move within the cell (12,13). Importin 7 promotes HIV-1 RTC import into the nucleus, although it may not be necessary in macrophages (14,15). HMGA1 and the barrier to autointegration factor (BAF) bind to viral nucleic acids; BAF is thought to contribute to virus assembly, to prevent self-integration, and to promote correct conformation of HIV-1 pre-integration complex (PIC), and both BAF and HMGA1 stimulate correct retroviral integration in vitro, although HMGA1 does not appear to be necessary for virus infection (16-18). LEGDF/p75 binds to HIV-1 integrase, stimulates in vitro PIC integration activity, and directs PICs to highly transcribed chromatin regions susceptible to LEGDF/p75 regulation (19-21). Once integrated, HIV-1 provirus transcription requires cellular cyclin-dependent kinase 9, recruited to the Long Terminal Repeat LTR by the viral protein Tat (22,23). The unspliced viral mRNA is exported from the nucleus by CRM1, a member of the exportin family, which is recruited by the viral protein Rev (24,25). Finally, assembly and budding from the host cell also requires an entire network of proteins, part of the vacuolar protein sorting pathway (26-30). All these examples emphasize the fact that absence or variation of host factors can determine viral tropism (31,32).
On the contrary, other cellular factors have been shown to inhibit HIV-1 infection. Examples of this are Murr1 (33), APOBEC3G (34-36), INI/hSNF5, PML, and Lv2 (37-39) in human cells and TRIM5α in primate cells (40).
HIV-1 is potently restricted in rabbit cells at a post-entry stage (2,3), however, the nature of this restriction has not been investigated. The block to HIV-1 infection in the rabbit epithelial cell line SIRC (Statens Seruminstitut Rabbit Cornea) is interesting because it occurs at an early stage of infection and it cannot be readily abrogated with high doses of virus or virus-like particles and other retroviruses like SIVmac and murine leukemia virus (MLV) are not restricted (3). In this study, we show that this restriction has a recessive phenotype and is characterized by an aberrant intracellular trafficking of RTCs and a further block before integration. Our results implicate the participation of host cell factors in the correct intracellular trafficking of HIV-1, and they also provide evidence for a link between correct virus trafficking and infectivity.
Results
HIV-1 infection is inefficient in SIRC cells independently of the cell receptor
A post-entry block to HIV-1 infection in rabbit cells has been reported previously (2,3). To assess whether such a block was dependent on the cell receptor used by the virus for entry, we produced viral vectors pseudotyped with vesicular stomatitis virus G envelope VSV-G by transient transfection of 293T cells or collected HIV-1 vector pseudotyped with amphotropic MLV envelope from a stable producer cell line (41). Viral stocks were normalized for RT activity and used to infect SIRC, HeLa, and 293T cells at serial dilutions. As shown in Figure 1, HeLa and 293T cells were up to a few hundred folds more permissive to HIV-1 infection than SIRC cells. The restriction in SIRC cells was partially overcome when more than 650 pg/mL RT of VSV-G-pseudotyped virus was used. However, equivalent values of infection in SIRC and HeLa or 293T cells were never achieved, even with 11 000 pg/mL RT (Figure 1). Infection with HIV-1 pseudotyped with amphotropic MLV envelope was also impaired in SIRC cells compared with both 293T and HeLa cells (Figure 1). To see whether inefficient infection in SIRC cells occurred with different retroviruses, we infected the three cell types with serial dilutions of VSV-G-pseudotyped SIVmac and Moloney MLV (MoMLV) vectors. Titers of SIVmac were approximately fivefold lower in SIRC compared with HeLa and 293T cells at low virus input, but the same percentage of infection was achieved at higher virus input (Figure 1). A modest reduction (twofold to threefold) in infection efficiency was observed in SIRC compared with 293T and HeLa cells with the MoMLV vector, presumably as a consequence of the slower division rate of SIRC cells (Figure 1). Thus the block in SIRC cells was independent of the envelope used and was HIV-1 specific. Similar results were obtained in another fibroblastic rabbit cell line (Supplementary Figure S1). To confirm that a general defect in virus entry was not responsible for the inefficient infection of rabbit cells, we infected HeLa and SIRC cells with the same dose of a HIV-1 vector prepared in the presence or absence of VSV-G (HIV Env-) and total viral RNA measured by quantitative polymerase chain reaction (PCR) after oligo-dT-primed in vitro RT. As shown in Figure 1B, viral RNA levels in HeLa cells were at most twofold higher than those in SIRC cells, and they were reduced by more than 10-fold in both HeLa and SIRC cells infected with HIV (Env-) compared with cells infected with HIV-1 (VSV-G). Taken together, these data indicate that at most a twofold entry defect for HIV-1 in SIRC cells, which does not account for the approximate 80-fold infectivity difference in these conditions (Figure 1C) and is consistent with the fact that the MLV (VSV-G) vector infected HeLa and SIRC cells equally well (Figure 1A).
Figure 1.

HIV-1 is restricted in SIRC cells. (A) HeLa, 293T, and SIRC cells were infected with increasing amounts (RT normalized) of HIV-1 vector pseudotyped with VSV-G (top left panel), HIV-1 vector pseudotyped with amphotropic MLV envelope (top right panel), SIVmac vector pseudotyped with VSV-G (bottom left panel), and NB MLV vector also VSV-G pseudotyped (bottom right panel). All vectors expressed GFP driven from a CMV promoter. The percentage of GFP+ cells (y-axis) was counted by FACS 48 h post-infection and plotted against the amount of input virus. Results of two independent experiments are shown as mean values ± standard error. (B) Viral RNA was extracted from HeLa and SIRC cells 4 h post-infection with the same dose (RT normalized) of HIV-1 (VSV-G) or HIV-1 (Env-) virus, subjected to oligo-dT primed in vitro RT (after DNAse I treatment) and quantified by PCR using GFP primers. With RT, reaction performed in the presence of reverse transcriptase, no RT, control reaction performed in the absence of reverse transcriptase. (C) An aliquot of the cells used for RNA extraction was re-plated and analyzed by FACS 48 h later to quantify infection efficiency.
The block in SIRC cells has a recessive phenotype
To determine whether the block to HIV-1 infection in SIRC cells had a dominant or recessive phenotype, we carried out a fusion assay with 293T and either SIRC or owl monkey (OMK) cells. If, after fusion, the 293T/SIRC heterokaryons were permissive to HIV-1 infection, then it would suggest that there is a factor required by HIV-1, absent in the SIRC cells but provided by the 293T in the syncytia. On the contrary, if the heterokaryons were restrictive to HIV-1 infection, then it would suggest that there is a dominant restriction factor in the SIRC cells that blocks infection. Heterokaryons have previously been used to study SV40 replication and Rous sarcoma virus production (42,43), to investigate the Vif: APOBEC circuit (34) and to study retroviral restriction in OMK and other simian cells (4,44).
Heterokaryons of 293T and OMK cells were also obtained to validate the assay. SIRC and OMK cells were labeled with the amine-reactive fluorescent dye BODIPY 630/650, plated alone or in combination with 293T cells, and 24 h later cells were induced to form syncytia by transfecting a plasmid coding for the highly fusogenic HTLV-1 envelope protein (pV1/HTLV) (4). Twenty-four hours after transfection, cells were infected with VSV-G-pseudotyped HIV-1–green fluorescent protein (HIV-1–GFP) vector, using the amount of virus that had been previously found to infect around 1% of SIRC cells (Figure 1). Cells were analyzed by confocal microscopy (Figure 2A) and by fluorescence-activated cell sorter (FACS) 48 h after infection (Figure 2B). Syncytia were apparent as a population that shifted upward in the side scatter channel (SSC-H) after pV1/HTLV transfection. Syncytia containing BODIPY 630/650 were considered mixed, because cell fusion occurred only when 293T cells were present (not shown). The percentage of FL-1 (GFP) and FL-5 (BODIPY 630/650) positive cells in the gated populations was measured, and representative dot plots are shown in Figure 2B. The number of double positive SIRC/293T heterokaryons was 1.7 times higher than the number of unfused 293T cells and 210 times higher than the number of unfused SIRC cells (Figure 2C). Fusion of 293T with OMK cells did not rescue HIV-1 infectivity compared with unfused OMK cells, consistent with previous reports (4).
Figure 2.

HIV-1 restriction in SIRC cells has a recessive phenotype. SIRC and OMK cells were labeled with the amine reactive fluorescent dye BODIPY-630/650, fused with 293T cells expressing the highly fusogenic HTLV-1 envelope protein and 24 h later infected with VSV-G-pseudotyped HIV-1–GFP vector. (A) Approximately 24 h post-infection, syncitia were analyzed by confocal microscopy: those containing SIRC or OMK cells (blue labeled) were considered mixed because cell fusion occurred only when 293T cells were present. (B) The same cells were analyzed by FACS and density plots of the forward (FSC-H) and side (SSC-H) scatter measurements are shown. Syncytia appeared as a population that shifted upward in the R2 region of the SSC-H channel after transfection with HTLV-I envelope (bottom panel on the left). This shift was not apparent in mixed 293T and SIRC populations in the absence of HTLV-I envelope (not shown). Cells were analyzed for BODIPY staining, and GFP expression and density plots of the FL-5H channel (BODIPY-630/650) and FL-1H channel (GFP) are shown in the middle panels (for the R1 region) and right panels (R2 region), respectively. SIRC cells were labeled with BODIPY but were poorly infected by the HIV-1 GFP vector (top, middle panel). 293T cells were not labeled with BODIPY but were efficiently infected by the HIV-1 GFP vector (middle, middle panel). Fusion of SIRC cells with 293T cells rescued HIV-1 infection as shown by the increase in the number of double BODIPY/GFP positive cells (bottom, right panel). (C) Histogram showing the percentage of infected cells for each cell type before and after fusion calculated by FACS as shown in panel B. Values represent the average of three independent experiments ± standard deviation. (D) Abrogation assay on HeLa, 293T, OMK, and SIRC cells. Cells were exposed to 11 000 pg/RT of HIV-1–puro vector, infected with a fixed amount of HIV–GFP vector and analyzed by FACS to count the percentage of infected cells. Rescue of infectivity by pre-exposure to HIV-1–Puro is expressed as fold abrogation relative to cells that were not pre-exposed to HIV-1–Puro.
To further test whether the post-entry inhibition of HIV-1 infection was caused by a saturable factor, we performed the abrogation assay as previously described (11) adding a constant amount of HIV–GFP vector (enough to infect 1% of each cell type) in the presence of increasing amounts of an HIV-1 vector carrying the phosphoribosyl transferase gene (HIV–Puro) (approximately from 250 to 11 000 pg/mL RT activity). The principle of this assay is that addition of the first virus (HIV–Puro) saturates restriction factors and increases viral titers of the second virus (HIV–GFP). To validate our assay, we included OMK cells in which a saturable restriction factor has already been characterized (11,45). In the presence of 11 000 pg/mL RT of HIV–Puro, HeLa showed a 3.15-fold abrogation, 293T showed a 17.5-fold abrogation, OMK showed a 100-fold abrogation, and SIRC cells showed a ninefold abrogation (Figure 2D). Thus, HIV-1 infection was similarly reduced in SIRC and OMK cells, but only weakly saturated in SIRC as compared with in OMK cells. Strong saturation of restriction in OMK cells is in agreement with previous studies (3). These results and the relatively weak abrogation indicated that block in SIRC cells had a recessive phenotype, although it remained formally possible that 293T cells provided a factor able to inhibit a dominant restrictor in SIRC cells.
RT is impaired in restricted cells in vivo but not in vitro
The HIV-1, SIVmac, and MLV vectors all expressed the GFP driven by the same cytomegalovirus CMV promoter (46-48), therefore a block to GFP expression in SIRC cells was excluded. Reduced HIV-1 infection levels in SIRC cells could be due to reduced RT or integration or both. To test which step of the viral life cycle was affected in SIRC cells, we used quantitative PCR to measure the amount of early (strong stop), intermediate (GFP), and late (U5/gag) viral DNA at 4, 7, and 16 h post-infection, respectively (Figure 3A and Supplementary Table S1). Intermediate and late RT products were 20- to 30-fold lower in SIRC than HeLa cells at all time points tested. Early RT products were fivefold to 10-fold lower in SIRC than HeLa cells, with the least difference at 7 h post-infection (Figure 3A). All viral stocks (including SIVmac and MoMLV) used for infection were treated with DNAse I and purified through a two-step sucrose cushion. To monitor the effectiveness of the purification procedure and rule out any contamination with plasmid DNA used for virus production, we tested viral stocks by PCR using primers specific for the viral pol and the GFP sequences. No DNA contamination was found in our viral stocks (Figure 3B), demonstrating that we were indeed measuring endogenously reverse transcribed viral DNA by quantitative PCR. Hence, although a 30-fold reduction in viral DNA copy number could not fully account for the potency of the block in SIRC cells, these results clearly showed that HIV-1 RT was defective in rabbit cells.
Figure 3.

HIV-1 RT is defective in SIRC cells. HeLa and SIRC cells were infected with the same dose (RT normalized) of HIV-1–GFP vector pseudotyped with VSV-G envelope, incubated at 4 °C for 2 h to allow virus binding to the cell receptor but not internalization and then incubated at 37 °C for 4, 7, and 16 h. Total DNA was extracted, and the number of viral DNA copies/100 ng total DNA was calculated by real-time PCR using R/U5 (early), GFP (intermediate), or U5/gag (late) primers. Values represent the mean ± standard deviation of triplicate experiments. An aliquot of infected cells was re-plated and analyzed 48 h later by FACS to measure infection efficiency. To control for DNA carry over from transfections performed to generate virus stocks, we subjected eight different virus stocks to 35 cycles PCR using primers specific for pCMVnΔ8.2 gag-pol expression vector (Supplementary Table S1). Serial dilutions of plasmid down to 1 pg were used to monitor the sensitivity of the PCR. Viral stocks were also subjected to PCR with primers specific for the strong stop DNA (+C) and late RT products (− C). MWM, DNA molecular weight markers.
Because RT was impaired, the properties of intracellular RTCs in restricted and non-restricted cells were examined. HeLa and SIRC cells were infected with HIV-1 pseudotyped with VSV-G envelope. Cytoplasmic extracts from acutely infected cells were prepared by Dounce homogenization in hypotonic buffer as previously described (49) and subjected to equilibrium density centrifugation in linear sucrose gradients. Individual fractions were analyzed by PCR to detect viral strong stop DNA, an early product of RT. In HeLa cytoplasmic extracts, the peak of HIV-1 strong stop DNA could be consistently detected in fractions with a density of approximately 1.30 g/mL, in agreement with previous studies (49-51). In SIRC extracts, the peak of strong stop DNA was consistently detected in fractions with a density of 1.20 g/mL (Figure 4B). A weaker strong stop DNA band was also present in fractions with a density of 1.30 g/mL in SIRC cells, and this distribution pattern was observed in another independent experiment, indicating that it was not due to experimental variation (not shown). Moreover, extraction of the nucleic acids from the density gradient fractions with phenol/chloroform followed by ethanol precipitation did not alter the results (not shown), demonstrating that the unusual two-peak distribution in SIRC cells was not due to an inhibitor of the PCR.
Figure 4.

RTCs have a different density in HeLa and SIRC cells but are competent for RT in vitro. Cells were infected with the same dose of HIV-1 vector, incubated 2 h at 4 °C to allow virus binding to the cell receptor and then 4 h at 37 °C. Cytosolic extracts were prepared from infected cells by Dounce homogenization in hypotonic buffer and centrifuged through a 20–70% linear sucrose gradient. Individual fractions were subjected to PCR with primers specific for the strong stop DNA (expected size 140 bp). (A) Density fractions from HeLa cells; (B) density fractions from SIRC cells. The arrow indicates the direction of the gradient (top, low density; bottom, high density). Low molecular weight bands are PCR artifacts. Plasmid DNA was used as a positive control (+C). (C) Regression plot showing the density of each individual fraction measured as described in Materials and Methods. (D) Endogenous RT reaction. The fractions containing the peak of strong stop DNA (fraction 5 for HeLa and fraction 9 for SIRC cells) were subjected to an endogenous RT reaction in the presence (+) or absence (−) of exogenous dNTPs and analyzed by PCR with primers specific for late RT products (HIV SIN2F and HIV FLRC). H, HeLa; S, SIRC; MWM, molecular weight markers. (E) Quantification of endogenous RT activity by qPCR. Samples were normalized for strong stop DNA concentration and subjected to an endogenous reaction as before in the presence or absence of exogenous dNTPs. Products of the reaction were then analyzed by qPCR using primers for late viral DNA products.
To test whether HIV-1 RTCs isolated from SIRC cells were defective, we performed an endogenous RT (ERT) assay to compare the in vitro activity of the 1.30 g/mL RTC peak found in HeLa cells with the 1.20 g/mL RTC peak found in SIRC cells. Fractions were incubated with dNTPs at 37 °C for 6–7 h to allow the completion of RT of viral RNA and late RT products detected by PCR (50). In both HeLa and SIRC cells, we observed the synthesis of near full-length HIV-1 DNA (Figure 4D). As a control, the same ERT assay was carried out in the absence of dNTPs, and, as shown in Figure 4D, there was no near full-length HIV-1 DNA in these conditions. To further confirm the activity of the 1.20 g/mL RTC extracted from rabbit cells, we normalized samples for strong stop DNA concentration and examined by quantitative PCR the amount of endogenously synthesized late viral DNA (Figure 4E). RTCs extracted from rabbit cells had the same or even greater relative ability (+dNTPs/−dNTPs) to synthesize late viral DNA products than RTCs extracted from HeLa cells, although SIRC complexes yielded approximately sixfold fewer late RT products than HeLa cell RTCs (Figure 4E).
HIV-1 has a different intracellular distribution in HeLa and SIRC cells
Results so far showed that RT of HIV-1 was impaired in SIRC cells, however, partially purified RTCs could synthesize viral DNA if provided with exogenous dNTPs. Thus, one hypothesis was that the block to infection in SIRC cells could be due to aberrant trafficking of RTCs into subcellular compartments not permissive for viral DNA synthesis. To examine this possibility, we infected HeLa and SIRC cells with the same amount of recombinant VSV-G-pseudotyped HIV, SIV, or MLV vectors. In the case of HIV-1, the amount of virus used resulted in the infection of approximately 2% HeLa cells and ≤0.07% SIRC cells as measured by FACS analysis for the GFP expression 48 h post-infection. Infection was synchronized by the pre-incubation of cells at 4 °C for 2 h to allow virus binding to the receptor but not internalization. Cells were then incubated for 4 h at 37 °C and subjected to sequential lysis in hypotonic, isotonic, high salt, 1% Triton X, and 0.5% SDS-containing buffers (Figure 5A). This kind of procedure causes stepwise disruption of increasingly stronger protein–protein interactions as well as extraction of different intracellular compartments and allows some crude examination of viral trafficking (52).
Figure 5.
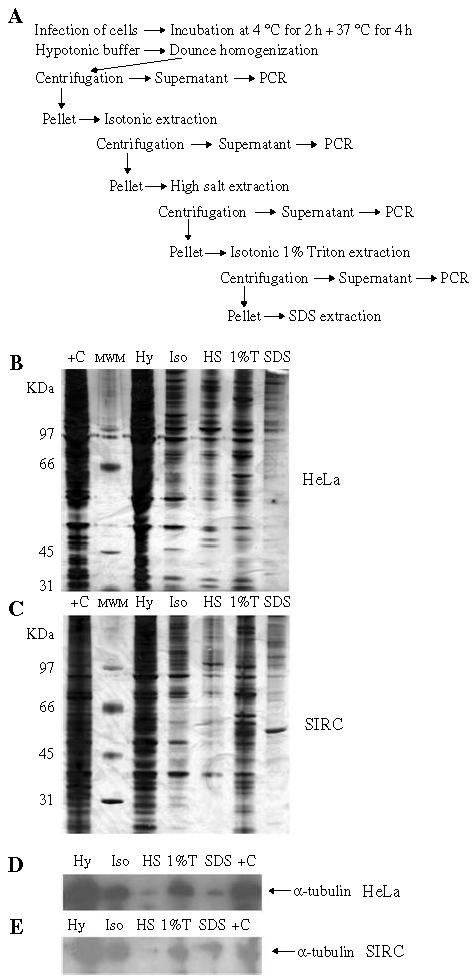
Stepwise fractionation of HeLa and SIRC cells. (A) Schematic representation of the fractionation procedure applied to the cells. (B) Proteins extracted by cell fractionation were concentrated and analyzed by SDS–PAGE and silver stain. Hy, hypotonic fraction; Is, isotonic fraction; HS, high salt fraction; 1% T, 1% Triton-X fraction; SDS, SDS fraction. MWM, molecular weight markers. (C) Total (unfractionated) cell extracts. (D–E) Western blot analysis of the same fractions with an anti-α-tubulin monoclonal antibody.
The effectiveness of the fractionation procedure was monitored by SDS–PAGE and silver staining of the protein content of each individual fraction and by Western blot with a monoclonal antibody against α-tubulin. As shown in Figure 5B,C, silver staining after SDS–PAGE revealed distinct protein patterns in each fraction, and the Western blot showed that similar amounts of tubulin were recovered in the same HeLa and SIRC cells fractions, demonstrating near-equivalent cell lysis (Figure 5D,E). Each fraction was then analyzed for the presence of viral DNA by standard and quantitative PCR (Figure 6). The PCR results confirmed that the overall efficiency of HIV-1 RT at 4 h post-infection was reduced in SIRC cells. Importantly, we found a different intracellular distribution of HIV-1 DNA in HeLa and SIRC cells. HIV-1 RT products appeared mainly in the hypotonic fraction in infected SIRC cells. In HeLa cells, most of HIV-1 reverse transcribed DNA appeared in the isotonic and high salt fractions (Figure 6A). These results were confirmed by quantitative PCR in an independent fractionation experiment (Figure 6B). When the SIVmac vector was used for infection, viral DNA had a similar distribution in both HeLa and SIRC cells, with most DNA being recovered in the hypotonic fraction. Importantly, MLV also had a similar distribution in both HeLa and SIRC cells, and most viral DNA was consistently recovered in the hypotonic fractions (Figure 6B). These results suggested that there was a correlation between virus infectivity and intracellular distribution of viral DNA. Indeed, the SIVmac and MLV vectors infected SIRC cells at higher efficiency than the HIV-1 vector, and most of their DNA was recovered in the same fractions in both cell types. Conversely, most of HIV-1 DNA was recovered in the hypotonic fraction in restricted cells and in the high salt fraction in permissive cells.
Figure 6.

HIV-1 has a different intracellular distribution in HeLa and SIRC cells. Cells were infected with the same dose of HIV-1, SIVmac, or MLV vectors pseudotyped with VSV-G, incubated 2 h at 4 °C and then 4 h at 37 °C and subjected to the stepwise fractionation procedure described in Figure 5A. (A) Total DNA was extracted from each fraction and analyzed by PCR with primers specific for late RT products. H, HeLa cells; S, SIRC cells; MWM, molecular weight markers. (B) The entire procedure was repeated and fractions analyzed by real-time PCR (GFP primers) to measure viral DNA copy number. Bars represent the mean ± standard deviation of triplicate experiments.
To exclude the possibility that we were analyzing the intracellular distribution of the non-restricted virus population only, we carried out the same fractionation procedure in RNAse-free conditions, we treated samples with RNAse-free DNAse to reduce background from endogenously reverse transcribed RTCs, and we used the viral RNA in each fraction as template for first-strand cDNA synthesis. Control reactions were carried out in the absence of RT, and viral cDNA copy number was measured by quantitative PCR (Figure 7). The data revealed two noteworthy aspects. First, in the hypotonic fraction, the amount of viral RNA in SIRC cells was considerably higher than in HeLa cells. This suggested that RTCs were not degraded in SIRC cells, at least at 4 h post-infection. Second, the distribution of viral RNA in both HeLa and SIRC cells mimicked the distribution of endogenously reverse transcribed viral DNA (compare Figures 6B and 7), with the exception of the viral RNA found in the SDS fraction. However, because the bulk of viral RNA was found in the SDS fraction of both HeLa and SIRC cells, it must represent a dead-end pathway unrelated to the restriction phenotype. Control reactions with no RT contained fewer than 100 viral cDNA copies, demonstrating that we were measuring bona fide viral RNA in test samples (Figure 7). These results suggested that distinct virus populations, one restricted and the other not restricted, may not be present in SIRC cells.
Figure 7.
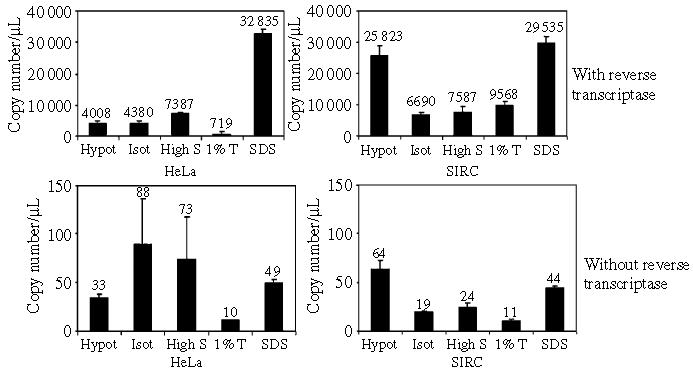
Viral RNA distribution in HeLa and SIRC cells. Cells were infected with the same dose of HIV vector, incubated 2 h at 4 °C and then 4 h at 37 °C. Infected cells were subjected to a stepwise fractionation procedure as described in Figure 5A in RNAse-free conditions. Total RNA was extracted, and samples were treated with RNAse-free DNAse I. After re-purification of the RNA, reverse transcriptase was used to perform first-strand cDNA synthesis. Control reactions were incubated in parallel in the absence of reverse transcriptase. Viral RNA copy number was measured by real-time PCR. Bars represent the mean ± standard deviation of triplicate experiments. Hy, hypotonic buffer; Iso, isotonic buffer; HS, high salt buffer; 1%T, 1% Triton-X buffer; SDS, SDS buffer.
HIV-1 DNA trafficking to the nucleus is delayed in rabbit cells
The reduced ability to reverse transcribe could not fully account for the potency of the HIV-1 infection block in SIRC cells. To investigate whether additional steps were defective in rabbit cells, we carried out a hypotonic lysis and Dounce homogenization of cells infected at different time points followed by the isolation of intact nuclei with a buffer containing 0.4% NP-40 on ice (50). Purified nuclei were then subjected to an additional fractionation step by the Hirt method to separate soluble from chromatin-associated viral DNA (53) (Figure 8A). Individual fractions were analyzed by quantitative PCR for the presence of reverse transcribed viral DNA. Real-time PCR showed that the kinetics of viral DNA association with the nuclei is different in SIRC compared with that in HeLa cells. In HeLa cells, virtually no viral DNA was nucleus associated at 4 h post-infection, however, viral DNA could be found associated with HeLa nuclei (Hirt sup) 7 h post-infection and was chromatin-associated (Hirt pellet) in significant amounts 16 h post-infection (Figure 8B). In SIRC cells, HIV-1 DNA was found mainly in the hypotonic fraction 4 and 7 h post-infection. Viral DNA was associated with the nuclei (Hirt sup) only 16 h post-infection and was not chromatin-associated (Figure 8C). 2LTR circular viral DNA, a form of viral DNA found predominantly in the nuclei (54-56), was detected in both HeLa and SIRC nuclear fractions at 16 h post-infection, albeit at reduced levels in SIRC cells (not shown).
Figure 8.

Association of viral DNA with the nuclei is delayed in SIRC cells. HeLa and SIRC cells were infected with the same dose of HIV-1 vector, incubated 2 h at 4 °C and then 4, 7, and 16 h at 37 °C. (A) Schematic representation of the fractionation procedure designed to separate the cytosol from intact nuclei. Nuclei were then processed into a soluble and chromatin fraction by the Hirt method. DNA was extracted from each fraction and analyzed by real-time PCR (GFP primers) to measure viral DNA copy number. (B) Distribution of viral DNA in HeLa cells. (C) Distribution of viral DNA in SIRC cells. Bars represent the mean ± standard deviation of triplicate experiments. Hypo, hypotonic buffer; NP40, buffer containing 0.4% NP-40.
Overall these data showed that HIV-1 DNA did access the nucleus of SIRC cells, albeit at low efficiency, but could not associate with chromatin suggesting a further restriction to HIV-1 infection, perhaps a block to integration. Indeed, the 20- to 30-fold RT block in SIRC cells could not fully account for the larger difference in viral titers (Figure 3). To test this possibility, we infected HeLa and SIRC cells with the same amount of HIV–GFP vector having a wild-type integrase or the catalytically inactive point mutant integrase D64V to control for background levels of HIV-1 DNA integration and persistence (56-59) and cultured them for 2 weeks. In these conditions, the vast majority of viral DNA detected is integrated, because non-integrated DNA is progressively lost by degradation and dilution. Infected cells were first analyzed by FACS to assess GFP+ cells and then DNA was extracted and analyzed by quantitative PCR to measure provirus copy number. As shown in Figure 9A, the percentage of GFP+ HeLa cells was greatly reduced upon infection with HIV-1 having a defective integrase compared with the normal HIV-1 vector. Consistent with previous results, SIRC cells showed very low levels of infection with both the normal and the integration defective HIV-1 vector. The amount of viral DNA in SIRC cells was substantially reduced compared with HeLa cells and was comparable with the levels measured in cells infected with an integration defective virus (Figure 9A). HeLa cells contained up to 500 times more viral DNA copies than SIRC cells, which fully accounted for the difference in infection efficiency as measured by GFP expression (Figure 9B). Taken together, these data and the data shown in Figure 8 indicated that some step leading to efficient HIV-1 integration might also be defective in SIRC cells.
Figure 9.

HIV-1 does not integrate efficiently in SIRC cells. (A) HeLa and SIRC cells were infected with the same amount of a VSV-G-pseudotyped HIV-1 vector or with a mutant HIV-1–GFP vector having a catalytically inactive point mutant integrase (ΔInt) and passaged for 2 weeks. Cells were analyzed by FACS to detect GFP+ (infected) cells. (B) Total DNA was extracted from the same cells and analyzed by quantitative real-time PCR to measure proviral DNA copy number.
Discussion
A post-entry block to HIV-1 infection in rabbit cells has been previously described but has not been characterized (2,3). We have found that this block is not dependent on the cell receptor used by the virus for entry, appears to be recessive, and is characterized by an aberrant intracellular trafficking of the RTC and by a further impairment before or at integration.
Two lines of evidence support the fact that the block to HIV infection in SIRC cells is recessive. First, saturation experiments by pre-exposing cells to increasing amounts of a virus vector rescued infectivity of the second vector only weakly, in agreement with a previous report (3). This suggested that SIRC cells may not produce a saturable factor able to block HIV-1 infection or that the factor may not be readily available to the incoming virus. Second, upon fusion, heterokaryons of human (293T) and SIRC cells became permissive to HIV-1 infection, whereas heterokaryons of OMK and SIRC cells did not. This also suggested that SIRC cells might not have a dominant restriction factor-like OMK or other primate cells (4,44). In fact, taken together, the results of the abrogation and the fusion assays point to the idea that SIRC cells lack a factor that specifically allows efficient HIV-1 infection. An alternative explanation for our results would be that a factor present in 293T cells could inhibit a restrictor in SIRC cells. A relevant example for this mechanism might be TRIM5γrh, which acts as a dominant negative for TRIM5αrh and rescues HIV-1 infection in rhesus macaque cells (40). Interspecies hetero-multimerization resulting in the inhibition of restriction has also been reported for human and rhesus macaque TRIM5α proteins (60). However, we think that this latter possibility is less likely in light of the potency and the relatively poor saturation of the restriction in SIRC cells and the almost complete rescue obtained upon fusion with human cells. Moreover, the phenotype resulting from TRIM5α consistently showed dominance in human/primate heterokaryons (4,44) and given the inability of 293T cells to rescue HIV-1 infection upon fusion with OMK cells, one would need to postulate a specific interaction of TRIMs belonging to such distant species as Homo sapiens and Oryctolagus cuniculus.
A time-course analysis of viral DNA synthesis in HeLa and SIRC cells showed that RT was inhibited in rabbit cells. The amount of late viral DNA in SIRC cells was 20- to 30-fold lower than that in HeLa cells at 4, 7, and 16 h post-infection. This suggested that RT in SIRC cells was irreversibly blocked rather than delayed. A block to RT could be due to defective RTCs, rapid RTC degradation, and/or aberrant intracellular RTC trafficking. The first possibility was examined by partial RTC purification in linear sucrose gradients. This technique has been used previously to characterize HIV-1 and MLV RTCs from acutely infected cells and is suitable to detect major differences in the biophysical properties of RTCs (51,61-63). Interestingly, most RTCs isolated from acutely infected SIRC cells had a significantly lower density compared with RTCs isolated from HeLa cells. This was not due to experimental variation, because a low RTC density in SIRC cells was found in two independent experiments. Despite different biophysical properties, RTCs isolated from HeLa and SIRC cells were both competent for RT in vitro when provided with exogenous dNTPs. Thus, the block to HIV-1 RT in SIRC cells was unlikely to be caused by an intrinsic defect of intracellular RTCs.
Rapid degradation of the viral genome was also unlikely to explain the poor HIV-1 DNA accumulation in SIRC cells for three reasons. First, the amount of viral DNA measured in SIRC cells was low but did not decrease significantly with time. Second, the amount of HIV-1 RNA in the hypotonic fraction was higher in SIRC than in HeLa cells 4 h post-infection. Third, RTCs recovered from SIRC cells 4 h post-infection were competent for RT if provided with exogenous dNTPs. Nevertheless, on the basis of our data, we cannot exclude that RTC degradation may involve its protein components and may take place at a later stage, and further work is needed to address this issue.
The hypothesis that aberrant RTC trafficking could be linked to reduced levels of HIV-1 RT in SIRC cells was investigated by stepwise cell fractionation in different lysis buffers. This technique has been used to monitor intracellular organelle and protein distribution in different cell types (64-66) and to perform crude examination of intracellular virus trafficking (52). Our cell fractionation procedure revealed that most of HIV-1 DNA is found in isotonic and high salt fractions in HeLa cells and in the hypotonic fraction in SIRC cells. This different distribution is likely to be related to inefficient infection, because it was not observed in the same cell types infected with SIVmac and MLV, two viruses that could infect SIRC cells quite efficiently. Furthermore, most viral RNA was recovered in the hypotonic fraction in SIRC cells, suggesting that HIV-1 RTCs in that fraction were poorly active. The same RTCs were competent for RT if provided with exogenous dNTPs, reinforcing the view that their intracellular location was not conducve to viral DNA synthesis. Although the fractionation procedure is too crude to draw a firm conclusion, one may speculate that in SIRC cells HIV-1 RTCs do not associate with some cytoskeletal component but remain trapped in an easily extractable compartment. Indeed, there is evidence that HIV-1 RTCs associate with cytoskeletal components at some point after cell entry, making them more difficult to extract by low salt buffers (12,13). Another unanticipated result of the cell fractionation was the different intracellular distribution of HIV-1 compared with MLV and SIVmac DNA in the same cell type. All vectors were pseudotyped with VSV-G, thus in this case different viral trafficking was likely to be determined by viral proteins other than env. It is unclear at present why HIV-1 appears to require an intracellular trafficking distinct from the one required by SIVmac and MLV for efficient infection.
The magnitude of the RT defect (20- to 30-fold) could not fully account for the block to HIV-1 infection in SIRC cells as determined by FACS analysis (100- to 300-fold). Provirus silencing was considered very unlikely because inefficient infection was observed with HIV-1 (data not shown), SIVmac, and MLV vectors expressing GFP from the same CMV promoter. Cell fractionation experiments showed that HIV-1 DNA association with the nuclei was delayed in SIRC compared with HeLa cells. Moreover, nuclear-associated DNA was found in both a soluble and a chromatin-associated form in HeLa cells but only in the soluble form in SIRC cells. This suggested that at least some HIV-1 DNA could access SIRC nuclei, as confirmed by the presence of 2LTR circular forms, but could not integrate efficiently. Accordingly, proviral DNA copy number was reduced in SIRC cells compared with HeLa cells in long-term cultures to an extent that matched infection levels measured by FACS. These data point to a further block after nuclear entry but before integration in rabbit cells. This integration block could be due to a defective PIC, to aberrant intranuclear trafficking or to the inability of a rabbit cellular factor to interact with HIV-1 PICs. We are currently trying to define PIC function by sensitive in vitro integration assays (17,67-70).
It will be important to identify both the cellular and the viral determinants of this unusual block to HIV-1 infection to understand the factors that influence intracellular virus trafficking in human cells. Moreover, the infection of rabbit cells overexpressing CD4 and CCR5 with high-titer HIV-1 has been reported, although the kinetic of virus spread was not analyzed (71). Thus, a detailed understanding of the nature of this block may provide an important contribution to the development of rabbit as a small animal model for HIV-1 replication (72,73).
Materials and Methods
Cells
HeLa, 293T, and SIRC cells were grown in DMEM supplemented with heat-inactivated 10% fetal calf serum (FCS) (Gibco/BRL, Invitrogen, Paisley, UK) in 5% CO2/95% air at 37 °C. SIRC cells were genotyped by PCR with a set of primers specific for the Oryctolagus cuniculus genome (clone 16796957A1, NCBI accession number AC158743) and the identity of the PCR fragment confirmed by sequencing. OMK cells were also grown in 5% CO2/95% air at 37 °C in minimal essential Eagle's medium supplemented with 10% FCS, non-essential amino acids, penicillin 100 U/mL, streptomycin 100 μg/mL (Gibco/BRL), and l-glutamine.
Vector production
HIV-1 viral vectors were prepared by the transfection of subconfluent 293T cells on a 10-cm dish diameter with a mixture of 18-μL Fugene-6 (Roche Molecular Biochemicals, Basel, Switzerland) in 200-μL Optimem media (Gibco/BRL) and 1 μg pMD.G (vesicular stomatitis virus envelope protein or VSV-G expression vector), 1.2 μg of pCMVΔ8.2 (HIV-1 gag-pol expression vector) (46) or p64V8.2 for integrase mutant virus (58,59) and 1.5 μg of the retroviral vector. Three different vectors were used: CSGW, a self-inactivating HIV-1 retroviral expression vector encoding enhanced GFP (eGFP), CSPW encoding the phosphoribosyl transferase gene (conferring puromycin resistance), and pHR′ a non–self-inactivating vector encoding eGFP (46,74). To make SIVmac vectors, the same protocol was followed using 1 μg of pSIV3+ (SIVmac gag-pol expression vector) and 1.5 μg of both SIVeGFP and pMD.G vectors (75,76). To make MLV vectors, 293T cells were transfected with 1 μg of gag-pol expression vectors pC1G3 N or phCMV-intron (77), 1.2 μg of pMDG, and 1.5 μg of the eGFP encoding vector pCNCG (Oxford Biomedica, Oxford, UK). Media was changed to fresh DMEM plus 10% FCS the next day, and supernatant was collected 48, 72, and 96 h post-transfection. The supernatant was filtered through a 0.45-μm filter and pH was adjusted with HEPES pH 7.4 (10 mm final). Viral stocks were incubated in the presence of DNAse I (70 U/mL) with 5 mm MgCl2 for 1 h at 37 °C and purified through a 25–45% sucrose gradient by centrifugation at 68 726 × g at 4 °C for 2 h in a Sorvall Discovery Surespin Rotor. Stable HIV-1 vector (pseudotyped with MLV amphotropic envelope) producer cells ISTAR and 100R26 were kindly provided by Yasuhiro Ikeda (41). Supernatant from these cells was harvested, filter sterilized through a 0.45-μm filter, and kept frozen in aliquots at −70 °C.
Viral titer determination and standardization
RT activity of HIV–GFP, HIV–Puro, ISTAR, and 100R26 viruses was measured by the Lenti-RT™ Activity Assay (Cavidi Tech, Uppsala, Sweden) following the manufacturer's instructions. RT values were used to standardize infection in different cell types. Viral titers of HIV-1, SIV, and MLV vectors expressing eGFP were measured on HeLa, 293T, SIRC, and OMK cells by FACS. Briefly, 2 × 105 cells/well were plated in 6-well trays and infected by addition of 10-fold serial dilutions of viral stocks (10–0.0001 μL/well) in the presence of 8 μg/mL polybrene. Viral titers were determined 48 h post-infection by FACS.
Abrogation assay
Abrogation assays were performed as described (78) by infecting 2 × 105 cells/well with 10-fold serial dilutions of HIV-1–Puro virus together with a constant amount of HIV–GFP virus (enough to infect 1% of the respective cell type). Cells were analyzed by FACS 48 h post-infection.
Fusion assay
SIRC and OMK cells were washed with PBS and incubated with BODIPY 630/650 (Molecular Probes and Invitrogen) diluted 1:2000 in Optimem (4 mL total volume) for 50 min at 37 °C in the dark. Cells were washed twice with PBS and plated in 6-well trays on their own or in combination with 293T cells. Eight hours after plating, cells were transfected with 1.5 μg of pV1/HTLV (4) using Fugene-6. Thirty-six hours post-transfection, cells were infected with an amount of virus sufficient to infect 1% of SIRC or 0.5% OMK cells. Forty-eight hours post-infection, cells were analyzed by FACS. Cells were first analyzed according to their size and granularity in a forward scatter (FSC-H) and SSC-H density plots. A population of cells with increased size appeared only when 293T were present and pV1/HTLV was transfected. This population was gated and analyzed in a dot plot for GFP expression (FL-1) and BODIPY 630/650 signal (FL-5). Double positive cells were considered as infected syncytia containing SIRC or OMK cells fused with 293T cells.
Cell fractionation
About 5 × 106 SIRC or HeLa cells were plated in T-175 flasks. Twenty-four hours later, each flask was infected with an HIV-1 or SIVmac vector with a multiplicity of infection (MOI) of 0.3 in the presence of 8 μg/mL polybrene. Samples were incubated for 2 h at 4 °C to allow virus binding but not internalization and then for 4 h at 37 °C. Cells were then trypsinized and washed in PBS. All subsequent manipulations were carried out at 4 °C. The pellet containing the infected cells was resuspended in five volumes of hypotonic buffer [10 mM HEPES (pH 7.9), 1.5 mM MgCl2, 10 mM KCl, 5 mM dithiothreitol (DTT), 20 μg aprotinin/mL, 20 μg leupeptin/mL] and centrifuged for 5 min at 1100 ×g in an Eppendorf microcentrifuge. Supernatant was kept in a separate tube, and the pellet was resuspended in five volumes of hypotonic buffer and incubated for 10 min on ice. Cells were homogenized with 10–15 strokes in a Dounce homogenizer. Disruption of cell membrane but integrity of nuclei was monitored by Trypan blue staining. Samples were centrifuged at 3300 × g for 15 min. The supernatant was clarified by centrifugation at 7500 × g for 20 min. The pellet was resuspended in isotonic buffer [10 mM Tris HCl (pH 7.4), 160 mM KCl, 5 mM MgCl2, 1 mM DTT, 20 μg of aprotinin/mL, 20 μg of leupeptin/mL] and centrifuged at 4 °C at 3300 × g for 15 min. The supernatant was collected, and the pellet was resuspended in high salt buffer [0.5 m KCl, 10 mm TRIS HCl (pH 7.4), 5 mm MgCl2] + 1 mm DTT + 20 μg/mL aprotinin + 20 μg/mL leupeptin and centrifuged as before. The supernatant from this step was collected, and the pellet was resuspended in a 1% Triton-X buffer [0.16 m KCl, 10 mm TRIS HCl (pH 7.4), 5 mm MgCl2, 1% Triton] + 1 mm DTT + 20 μg/mL aprotinin + 20 μg/mL leupeptin and centrifuged at 4 °C at 7500 × g for 10 min. The supernatant from this step was collected, and the pellet was resuspended in SDS buffer [20 mm Tris HCl (pH 8.1), 0.4% SDS, 10 mm EDTA]. Aliquots of all samples were digested with proteinase K (10 μg/mL) in SDS buffer [20 mm Tris HCl (pH 8.1), 0.4% SDS, 10 mm EDTA] at 55 °C for 16–18 h followed by phenol–chloroform extraction and ethanol precipitation of nucleic acids.
Purification of nuclei
To purify intact nuclei, we lysed cells by Dounce homogenization in hypotonic buffer as described. The resulting pellet was gently resuspended on ice in 0.4% NP-40 (IGEPAL CA-630) (SIGMA, St. Louis, MO, USA) in isotonic buffer [10 mm Tris (pH 7.4), 160 mm KCl, 5 mm MgCl2] + 1 mm DTT + 20 μg/mL aprotinin + 20 μg/mL leupeptin and quickly centrifuged at 700 × g for 5 min at 4 °C. The supernatant was collected and the pellet washed once in isotonic buffer + 1 mm DTT + 20 μg/mL aprotinin + 20 μg/mL leupeptin. The integrity of nuclei was checked at high magnification by light microscopy in the presence of trypan blue. The remaining sample was further fractionated into a soluble and insoluble part by the Hirt method (53), digested by proteinase K and nucleic acids purified by phenol–chloroform extraction and ethanol precipitation.
Long-term infection
1.5 106 SIRC or HeLa cells were plated in T-75 flasks. Twenty-four hours later, cells were infected with an HIV-1 vector, an HIV-1 integrase-mutant vector, or an NB-MLV vector at an MOI of 0.3 in the presence of 8 μg/mL polybrene. Forty-eight hours later an aliquot of infected cells was analyzed by FACS. After 2 weeks, cells were trypsinized and resuspended in PBS. Some of the cells were analyzed by FACS, and total DNA was extracted from the rest with the Qiamp® DNA Minikit (Qiagen, Hilden, Germany) and subjected to real-time PCR analysis (see Real Time PCR section below).
RT-PCR
5 × 106 SIRC or HeLa cells were plated in T-175 flasks. Twenty-four hours later, cells were infected with an HIV-1 vector with an MOI of 0.3 in the presence of 8 μg/mL polybrene. Samples were incubated for 2 h at 4 °C to allow virus binding but not internalization and then for 4 h at 37 °C. Cells were then trypsinized and washed in PBS. All subsequent manipulations were carried out at 4 °C and at RNase-free conditions. The same cell fractionation procedure was followed as described above, and the nucleic acids were ethanol precipitated, resuspended in RNase-free water, and digested with 10 U/mL RNAse-free DNase I at 37 °C for 1 h. For first-strand cDNA synthesis, RNA was mixed with Oligo dT20 probe, biotin-labeled (Roche Molecular Biochemicals) (200 pmol), 1 mm dNTPs mix in 10 μL total volume following the manufacturer's instructions. Samples were incubated at 65 °C for 5 min, placed on ice for 1 min, and then mixed with 10 mm MgCl2, 2 mm DTT, 40 units of RNAseOUT and 200 units of Super Script III Reverse Transcriptase (Invitrogen) in its own buffer. Samples were incubated at 50 °C for 50 min followed by 1 min at 85 °C and treated with RNAse H at 37 °C for 20 min. The reaction products were then used as template for PCR. Control reactions were performed in the absence of Super Script III reverse transcriptase.
RTC density assay
Cytosolic extracts were prepared by Dounce homogenization in hypotonic buffer as described above from 5 × 106 acutely infected HeLa, SIRC, or OMK cells, and RTCs were purified onto 20–70% continuous linear sucrose gradients as previously described (16,50). Gradients were fractionated by puncturing the bottom of the tube and collecting 12 fractions. Fractions were kept frozen at −70 °C.
Endogenous RT assay
Reactions were carried out in 60 μL of buffer [10 mm Tris HCl (pH 8.1), 15 mm NaCl, 6 mm MgCl2, 1 mm DTT, 2 mm each dNTP]. Fifteen microliters from the density equilibrium fractions was added to the buffer and incubated for ≥6 h at 37 °C. The products of RT were detected by PCR using 5 μL of the endogenous reaction as template (16). As a control, the same mix was prepared for each sample but without dNTPs. PCR products were separated onto a 7.5% polyacrylamide gel and stained with SYBR-Gold Nucleic Acid Gel Stain (Molecular Probes) and analyzed by the phosphorimager STORM 860 (Molecular Dynamics and Amersham Biosciences, Little Chalfont, UK).
Real-time PCR
TaqMan quantitative PCR was performed as described (3, 79, 80, and Supplementary Table S1). For the amplification of intermediate and late products of RT (GFP and late primers), 0.3 pmol of each primer (for primer sequences and probes please refer to Supplementary Table S1), 0.15 pmol of the probe and 12.5 μL of Quantitect Probe Master Mix (Qiagen) and 208 ng of carrier DNA were used in a final volume of 25 μL. For amplification of early products of RT (RU5 primers), 200 nm of primers and probe were used with 25 μL of Quantitect Probe Master Mix (Qiagen) and 208 ng of carrier DNA were used in a final volume of 50 μL. Carrier DNA was prepared with salmon sperm DNA at a concentration of 0.1 mg/mL. The standards were prepared with CNCG or pHR′ plasmids in 10-fold dilutions from 105 to 107 copies/μL. About 100–500 ng of template DNA were added per reaction.
PCR amplification
All PCR were performed in a final volume of 50 μL containing 1 × PCR buffer (Promega, Madison, WI, USA), 100 μm of each dNTP, 2.5 mm MgCl2, 5 U of Taq polymerase (Promega), and 30 pmol of each primer. For primer sequences refer to Supplementary Table S1. Cycle parameters were as follows: 94 °C for 3 min the first cycle; 94 °C for 1 min, 55 °C for 30 s, and 68 °C for 1 min for 25–30 cycles; followed by one final extension cycle at 68 °C for 10 min. PCR products were resolved on a 1.5% agarose gel and visualized by ethidium bromide.
SDS–PAGE and Western blot
For protein analyses, 300 μL of each fraction from the stepwise fractionation experiment were first diluted in 1.2 ml of ice-cold 50 mM sodium phosphate buffer (pH 7.4) in the presence of 10% (v/v) trichloroacetic acid. Fractions were incubated at −20 °C for 16 h and centrifuged for 30 min at 4 °C at maximum speed in a minicentrifuge. Pellets were washed once in a solution of ice-cold 80% acetone in distilled H2O and resuspended in 20 μL 2× SDS loading buffer [0.5 M Tris HCl (pH 6.8), 1% SDS, 10% glycerol, 0.1% bromophenol blue, 1 mM EDTA, 10 mM DTT, 20 μg of aprotinin/mL, 2 μg of leupeptin hemisulfate/mL, 10 μg of phenylmethylsulfonyl fluoride] and the pH was adjusted to approximately 7.0 by the addition of 1 μL of 1.5 m Tris HCl (pH 8.8). Samples were resolved onto a 6–12.5% SDS–PAGE and either stained with Silver Stain Plus (Bio-Rad, Hercules, CA, USA) following the manufacturer's instructions or transferred to a polyvinylidene difluoride (PVDF) membrane (Bio-Rad). Membranes were incubated with MCA77G rat MAb against α-tubulin (Serotec, Oxford, UK) for 1 h at room temperature after blocking with 10% non-fat milk (Tesco, London, UK). A horseradish peroxidase-conjugated secondary antibody was used diluted 1:3000 (anti-rat IgG; Dako, Glostrup, Denmark). Enhanced chemiluminescence was used to develop the blots (Amersham, Little Chalfont, UK). Autoradiography films were exposed for different periods of time to ensure linearity of the signal.
Supplementary Material
Figure S1. HeLa, 293T, SIRC, and the rabbit embryonic skin fibroblastic cell line EREp (ATCC reference number CRL 6498) were infected with increasing amounts (RT normalized) of HIV-1 (top panel), SIVmac (middle panel), and NB MLV (bottom panel) vector pseudotyped with VSV-G. All vectors expressed GFP driven from a CMV promoter. The percentage of GFP+ cells (y-axis) was counted by FACS 48 h post-infection and plotted against the amount of input virus. Results of two independent experiments are shown as mean values ± standard error.
Table S1. Sequences and positions of primers and probes used for real-time qPCR and for standard PCR are indicated.
These materials are available as part of the online article from http://www.blackwell-synergy.com
Acknowledgments
We thank Laura Ylinen, Zuzana Keckesova, Robin Weiss, Mary Collins, and Paul Kellam for helpful discussions and advice and Greg Towers for critically reading the manuscript. We are grateful to Paul Bieniasz, Theodora Hatziioannou, and Greg Towers for the generous gift of reagents. TC is supported by the CONACYT, Mexico, and AF is a Wellcome Trust University Fellow.
References
- 1.McKnight A, Clapham PR, Weiss R. HIV-2 and SIV infection of nonprimate cell lines expressing human CD4+ restrictions to replication at distinct stages. Virology. 1994;201:8–18. doi: 10.1006/viro.1994.1260. [DOI] [PubMed] [Google Scholar]
- 2.Hofmann W, Schubert D, LaBonte J, Munson L, Gibson S, Scammell J, Ferrigno P, Sodroski J. Species-specific, postentry barriers to primate immunodeficiency virus infection. J Virol. 1999;73:10020–10028. doi: 10.1128/jvi.73.12.10020-10028.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Besnier C, Takeuchi Y, Towers G. Restriction of lentivirus in monkeys. Proc Natl Acad Sci USA. 2002;99:11920–11925. doi: 10.1073/pnas.172384599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cowan S, Hatziioannou T, Cunningham T, Muesing MA, Gottlinger HG, Bieniasz PD. Cellular inhibitors with Fv1-like activity restrict human and simian immunodeficiency virus tropism. Proc Natl Acad Sci USA. 2002;99:11914–11919. doi: 10.1073/pnas.162299499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Song B, Javanbakht H, Perron M, Park DH, Stremlau M, Sodroski J. Retrovirus restriction by TRIM5alpha variants from old world and new world primates. J Virol. 2005;79:3930–3937. doi: 10.1128/JVI.79.7.3930-3937.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dalgleish AG, Beverley PC, Clapham PR, Crawford DH, Greaves MF, Weiss RA. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature. 1984;312:763–767. doi: 10.1038/312763a0. [DOI] [PubMed] [Google Scholar]
- 7.Klatzmann D, Champagne E, Chamaret S, Gruest J, Guetard D, Hercend T, Gluckman JC, Montagnier L. T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nature. 1984;312:767–768. doi: 10.1038/312767a0. [DOI] [PubMed] [Google Scholar]
- 8.Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272:872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- 9.Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA. CC CKR5: a RANTES, MIP-1 alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science. 1996;272:1955–1958. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- 10.Luban J, Bossolt KL, Franke EK, Kalpana GV, Goff SP. Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell. 1993;73:1067–1078. doi: 10.1016/0092-8674(93)90637-6. [DOI] [PubMed] [Google Scholar]
- 11.Towers GJ, Hatziioannou T, Cowan S, Goff SP, Luban J, Bieniasz PD. Cyclophilin A modulates the sensitivity of HIV-1 to host restriction factors. Nat Med. 2003;9:1138–1143. doi: 10.1038/nm910. [DOI] [PubMed] [Google Scholar]
- 12.Bukrinskaya A, Brichacek B, Mann A, Stevenson M. Establishment of a functional human immunodeficiency virus type 1 (HIV-1) reverse transcription complex involves the cytoskeleton. J Exp Med. 1998;188:2113–2125. doi: 10.1084/jem.188.11.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McDonald D, Vodicka MA, Lucero G, Svitkina TM, Borisy GG, Emerman M, Hope TJ. Visualization of the intracellular behavior of HIV in living cells. J Cell Biol. 2002;159:441–452. doi: 10.1083/jcb.200203150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fassati A, Görlich D, Harrison I, Zaytseva L, Mingot JM. Nuclear import of HIV-1 intracellular reverse transcription complex is mediated by importin 7. EMBO J. 2003;22:3675–3685. doi: 10.1093/emboj/cdg357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zielske SP, Stevenson M. Importin 7 may be dispensable for human immunodeficiency virus type 1 and simian immunodeficiency virus infection of primary macrophages. J Virol. 2005;79:11541–11546. doi: 10.1128/JVI.79.17.11541-11546.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen H, Engelman A. The barrier-to-autointegration protein is a host factor for HIV type 1 integration. Proc Natl Acad Sci USA. 1998;95:15270–15274. doi: 10.1073/pnas.95.26.15270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farnet CM, Bushman FD. HIV-1 cDNA integration: requirement of HMG I (Y) protein for function of preintegration complexes in vitro. Cell. 1997;88:483–492. doi: 10.1016/s0092-8674(00)81888-7. [DOI] [PubMed] [Google Scholar]
- 18.Beitzel B, Bushman F. Construction and analysis of cells lacking the HMGA gene family. Nucleic Acids Res. 2003;31:5025–5032. doi: 10.1093/nar/gkg684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ciuffi A, Llano M, Poeschla E, Hoffmann C, Leipzig J, Shinn P, Ecker JR, Bushman F. A role for LEDGF/p75 in targeting HIV DNA integration. Nat Med. 2005;11:1287–1289. doi: 10.1038/nm1329. [DOI] [PubMed] [Google Scholar]
- 20.Van Maele B, Busschots K, Vandekerckhove L, Christ F, Debyser Z. Cellular co-factors of HIV-1 integration. Trends Biochem Sci. 2006;31:98–105. doi: 10.1016/j.tibs.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 21.Cherepanov P, Sun ZY, Rahman S, Maertens G, Wagner G, Engelman A. Solution structure of the HIV-1 integrase-binding domain in LEDGF/p75. Nat Struct Mol Biol. 2005;12:526–532. doi: 10.1038/nsmb937. [DOI] [PubMed] [Google Scholar]
- 22.Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell. 1998;92:451–462. doi: 10.1016/s0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- 23.Bieniasz PD, Grdina TA, Bogerd HP, Cullen BR. Recruitment of cyclin T1/P-TEFb to an HIV type 1 long terminal repeats promoter proximal RNA target is both necessary and sufficient for full activation of transcription. Proc Natl Acad Sci USA. 1999;96:7791–7796. doi: 10.1073/pnas.96.14.7791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malim MH, Hauber J, Le SY, Maizel JV, Cullen BR. The HIV-1 rev trans-activator acts through a structured target sequence to activate nuclear export of unspliced viral mRNA. Nature. 1989;338:254–257. doi: 10.1038/338254a0. [DOI] [PubMed] [Google Scholar]
- 25.Yi R, Bogerd HP, Cullen BR. Recruitment of the Crm1 nuclear export factor is sufficient to induce cytoplasmic expression of incompletely spliced human immunodeficiency virus mRNAs. J Virol. 2002;76:2036–2042. doi: 10.1128/jvi.76.5.2036-2042.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Cote M, Rich RL, Myszka DG, Sundquist WI. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell. 2001;107:55–65. doi: 10.1016/s0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- 27.Martin-Serrano J, Zang T, Bieniasz PD. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat Med. 2001;7:1313–1319. doi: 10.1038/nm1201-1313. [DOI] [PubMed] [Google Scholar]
- 28.Strack B, Calistri A, Craig S, Popova E, Gottlinger HG. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell. 2003;114:689–699. doi: 10.1016/s0092-8674(03)00653-6. [DOI] [PubMed] [Google Scholar]
- 29.Von Schwedler UK, Stuchell MM, Müller B, Ward DM, Chung HY, Morita E, Wang H, Davis T, He GP, Cimbora DM, Scott A, Kräusslich HG, Kaplan J, Morham SG, Sundquist WI. The protein network of HIV budding. Cell. 2003;114:701–713. doi: 10.1016/s0092-8674(03)00714-1. [DOI] [PubMed] [Google Scholar]
- 30.Martin-Serrano J, Eastman SW, Chung W, Bieniasz PD. HECT ubiquitin ligases link viral and cellular PPXY motifs to the vacuolar protein-sorting pathway. J Cell Biol. 2005;168:89–101. doi: 10.1083/jcb.200408155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bieniasz PD, Cullen BR. Multiple blocks to human immunodeficiency virus type 1 replication in rodent cells. J Virol. 2000;74:9868–9877. doi: 10.1128/jvi.74.21.9868-9877.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yap MW, Nisole S, Stoye JP. A single amino acid change in the SPRY domain of human Trim5 leads to HIV-1 restriction. Curr Biol. 2005;15:73–78. doi: 10.1016/j.cub.2004.12.042. [DOI] [PubMed] [Google Scholar]
- 33.Ganesh L, Burstein E, Guha-Niyogi A, Louder MK, Mascola JR, Klomp LW, Wijmenga C, Duckett CS, Nabel GJ. The gene product Murr1 restricts HIV-1 in resting CD4+ lymphocytes. Nature. 2003;426:853–857. doi: 10.1038/nature02171. [DOI] [PubMed] [Google Scholar]
- 34.Simon JHM, Gaddis NC, Fouchier RA, Malim MH. Evidence for a newly discovered cellular anti-HIV-1 phenotype. Nat Med. 1998;4:1397–1400. doi: 10.1038/3987. [DOI] [PubMed] [Google Scholar]
- 35.Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 36.Sheehy AM, Gaddis NC, Malim MH. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med. 2003;9:1404–1407. doi: 10.1038/nm945. [DOI] [PubMed] [Google Scholar]
- 37.Yung E, Sorin M, Wang EJ, Perumal S, Ott D, Kalpana GV. Specificity of interaction of INI1/hSNF5 with retroviral integrases and its functional significance. J Virol. 2004;78:2222–2231. doi: 10.1128/JVI.78.5.2222-2231.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turelli P, Doucas V, Craig E, Mangeat B, Klages N, Evans R, Kalpana G, Trono D. Cytoplasmic recruitment of INI1 and PML of incoming HIV preintegration complexes: interference with early steps of viral replication. Mol Cell. 2001;7:1245–1254. doi: 10.1016/s1097-2765(01)00255-6. [DOI] [PubMed] [Google Scholar]
- 39.Marchant D, Neil SJ, Aubin K, Schmitz C, McKnight A. An envelope-determined, pH-independent endocytic route of viral entry determines the susceptibility of human immunodeficiency virus type 1 (HIV-1) and HIV-2 to Lv2 restriction. J Virol. 2005;79:9410–9418. doi: 10.1128/JVI.79.15.9410-9418.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. The cytoplasmic body component TRIM5α restricts HIV-1 infection in Old World monkeys. Nature. 2004;427:848–853. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- 41.Ikeda Y, Takeuchi Y, Martin F, Cosset FL, Mitrophanous K, Collins M. Continuous high-titer HIV-1 vector production. Nat Biotechnol. 2003;21:569–572. doi: 10.1038/nbt815. [DOI] [PubMed] [Google Scholar]
- 42.Svoboda J, Dourmashkin R. Rescue of Rous sarcoma virus from virogenic mammalian cells associated with chicken cells and treated with Sendai virus. J Gen Virol. 1969;4:523–529. doi: 10.1099/0022-1317-4-4-523. [DOI] [PubMed] [Google Scholar]
- 43.Watkins JF, Dulbecco R. Production of SV40 virus in heterokaryons of transformed and susceptible cells. Proc Natl Acad Sci USA. 1967;58:1396–1403. doi: 10.1073/pnas.58.4.1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Münk C, Brandt SM, Lucero G, Landau NR. A dominant block to HIV-1 replication at reverse transcription in simian cells. Proc Natl Acad Sci USA. 2002;99:13843–13848. doi: 10.1073/pnas.212400099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sayah DM, Sokolskaja E, Berthoux L, Luban J. Cyclophilin A retro-transposition into TRIM5 explains owl monkey resistance to HIV-1. Nature. 2004;430:569–573. doi: 10.1038/nature02777. [DOI] [PubMed] [Google Scholar]
- 46.Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 47.Mangeot PE, Negre D, Dubois B, Winter AJ, Leissner P, Mehtali M, Kaiserlian D, Cosset FL, Darlix JL. Development of minimal lentivirus vectors derived from simian immunodeficiency virus (SIVmac251) and their use for gene transfer into human dendritic cells. J Virol. 2000;74:8307–8315. doi: 10.1128/jvi.74.18.8307-8315.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Soneoka Y, Cannon PM, Ramsdale EE, Griffiths JC, Romano G, Kingsman SM, Kingsman AJ. A transient three-plasmid expression system for the production of high titer retroviral vectors. Nucleic Acids Res. 1995;23:628–633. doi: 10.1093/nar/23.4.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nermut MV, Fassati A. Structural analysis of purified human immunodeficiency virus type 1 intracellular reverse transcription complexes. J Virol. 2003;73:8196–8206. doi: 10.1128/JVI.77.15.8196-8206.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fassati A, Goff SP. Characterization of intracellular reverse transcription complexes of human immunodeficiency virus type 1. J Virol. 2001;75:3626–3635. doi: 10.1128/JVI.75.8.3626-3635.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heinzinger NK, Bukinsky MI, Haggerty SA, Ragland AM, Kewalramani V, Lee MA, Gendelman HE, Ratner L, Stevenson M, Emerman M. The Vpr protein of human immunodeficiency virus type 1 influences nuclear localization of viral nucleic acids in nondividing host cells. Proc Natl Acad Sci USA. 1994;91:7311–7315. doi: 10.1073/pnas.91.15.7311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gao G, Goff SP. Somatic cell mutants resistant to retrovirus replication: intracellular blocks during the early stages of infection. Mol Biol Cell. 1999;10:1705–1717. doi: 10.1091/mbc.10.6.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hirt B. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol. 1967;26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- 54.Coffin JM, Hughes SH, Varmus HE. Retroviruses. New York, USA: Cold Spring Harbor Laboratory Press; 1997. [PubMed] [Google Scholar]
- 55.Fritsch E, Temin HM. Formation and structure of infectious DNA of spleen necrosis virus. J Virol. 1977;21:119–130. doi: 10.1128/jvi.21.1.119-130.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li L, Olvera JM, Yoder KE, Mitchell RS, Butler SL, Lieber M, Martin SL, Bushman FD. Role of the non-homologous DNA end joining pathway in the early steps of retroviral infection. EMBO J. 2001;20:3272–3281. doi: 10.1093/emboj/20.12.3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Butler SL, Johnson EP, Bushman FD. Human immunodeficiency virus cDNA metabolism: notable stability of two-long terminal repeat circles. J Virol. 2002;76:3739–3747. doi: 10.1128/JVI.76.8.3739-3747.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leavitt AD, Shiue L, Varmus HE. Site-directed mutagenesis of HIV-1 integrase demonstrates differential effects on integrase functions in vitro. J Biol Chem. 1993;268:2113–2119. [PubMed] [Google Scholar]
- 59.Leavitt AD, Robles G, Alesandro N, Varmus HE. Human immunodeficiency virus type 1 integrase mutants retain in vitro integrase activity yet fail to integrate viral DNA efficiently during infection. J Virol. 1996;70:721–728. doi: 10.1128/jvi.70.2.721-728.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Berthoux L, Sebastian S, Sayah DM, Luban J. Disruption of human TRIM5alpha antiviral activity by nonhuman primate orthologues. J Virol. 2005;79:7883–7888. doi: 10.1128/JVI.79.12.7883-7888.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bowerman B, Brown PO, Bishop JM, Varmus HE. A nucleoprotein complex mediates the integration of retroviral DNA. Genes Dev. 1989;3:469–478. doi: 10.1101/gad.3.4.469. [DOI] [PubMed] [Google Scholar]
- 62.Fassati A, Goff SP. Characterization of intracellular reverse transcription complexes of Moloney murine leukemia virus. J Virol. 1999;73:8919–8925. doi: 10.1128/jvi.73.11.8919-8925.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yuan B, Fassati A, Yueh A, Goff SP. Characterization of Moloney murine leukemia virus p12 mutants blocked during early events of infection. J Virol. 2002;76:10801–10810. doi: 10.1128/JVI.76.21.10801-10810.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lim MJ, Chiang ET, Hechtman HB, Shepro D. Inflammation-induced subcellular redistribution of VE-cadherin, actin, and gamma-catenin in cultured human lung microvessel endothelial cells. Microvasc Res. 2001;62:366–382. doi: 10.1006/mvre.2001.2355. [DOI] [PubMed] [Google Scholar]
- 65.Ramsby ML, Makowski GS, Khairallah EA. Differential detergent fractionation of isolated hepatocytes: biochemical, immunochemical and two-dimensional gel electrophoresis characterization of cytoskeletal and noncytoskeletal compartments. Electrophoresis. 1994;15:265–277. doi: 10.1002/elps.1150150146. [DOI] [PubMed] [Google Scholar]
- 66.Wang Q, Patton WF, Hechtman HB, Shepro D. A novel anti-inflammatory peptide inhibits endothelial cell cytoskeletal rearrangement, nitric oxide synthase translocation, and paracellular permeability increases. J Cell Physiol. 1997;172:171–182. doi: 10.1002/(SICI)1097-4652(199708)172:2<171::AID-JCP4>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 67.Lu R, Vandegraaff N, Cherepanov P, Engelman A. Lys-34, dispensable for integrase catalysis, is required for preintegration complex function and human immunodeficiency virus type 1 replication. J Virol. 2005;79:12584–12591. doi: 10.1128/JVI.79.19.12584-12591.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen H, Engelman A. Characterization of a replication-defective human immunodeficiency virus type 1 att site mutant that is blocked after the 3′ processing step of retroviral integration. J Virol. 2000;74:8188–8193. doi: 10.1128/jvi.74.17.8188-8193.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hansen MS, Smith GJR, Kafri T, Molteni V, Siegel JS, Bushman FD. Integration complexes derived from HIV vectors for rapid assays in vitro. Nat Biotechnol. 1999;17:578–582. doi: 10.1038/9886. [DOI] [PubMed] [Google Scholar]
- 70.Brooun A, Richman DD, Kornbluth RS. HIV-1 preintegration complexes preferentially integrate into longer target DNA molecules in solution as detected by a sensitive, polymerase chain reaction-based integration assay. J Biol Chem. 2001;276:46946–46952. doi: 10.1074/jbc.M108000200. [DOI] [PubMed] [Google Scholar]
- 71.Speck RF, Penn ML, Wimmer J, Esser U, Hague BF, Kindt TJ, Atchison RE, Goldsmith MA. Rabbit cells expressing human CD4 and human CCR5 are highly permissive for human immunodeficiency virus type 1 infection. J Virol. 1998;72:5728–5734. doi: 10.1128/jvi.72.7.5728-5734.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Filice G, Cereda PM, Varnier OE. Infection of rabbits with human immunodeficiency virus. Nature. 1988;335:366–369. doi: 10.1038/335366a0. [DOI] [PubMed] [Google Scholar]
- 73.Kulaga H, Folks T, Rutledge R, Truckenmiller ME, Gugel E, Kindt TJ. Infection of rabbits with human immunodeficiency virus 1. A small animal model for acquired immunodeficiency syndrome. J Exp Med. 1989;169:321–326. doi: 10.1084/jem.169.1.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Demaison C, Parsley K, Brouns G, Scherr M, Battmer K, Kinnon C, Grez M, Thrasher AJ. High-level transduction and gene expression in hematopoietic repopulating cells using a human immunodeficiency virus type 1-based lentiviral vector containing an internal spleen focus forming virus promoter. Hum Gene Ther. 2002;13:803–813. doi: 10.1089/10430340252898984. [DOI] [PubMed] [Google Scholar]
- 75.Nègre D, Mangeot PE, Duisit G, Blanchard S, Vidalain PO, Leissner P, Winter AJ, Rabourdin-Combe C, Mehtali M, Moullier P, Darlix JL, Cosset FL. Characterization of novel safe lentiviral vectors derived from simian immunodeficiency virus (SIVmac251) that efficiently transduce mature human dendritic cells. Gene Ther. 2000;7:1613–1623. doi: 10.1038/sj.gt.3301292. [DOI] [PubMed] [Google Scholar]
- 76.Nègre D, Duisit G, Mangeot PE, Moullier P, Darlix JL, Cosset FL. Lentiviral vectors derived from simian immunodeficiency virus. Curr Top Microbiol Immunol. 2002;261:53–74. doi: 10.1007/978-3-642-56114-6_3. [DOI] [PubMed] [Google Scholar]
- 77.Bock M, Bishop KN, Towers G, Stoye JP. Use of a transient assay for studying the genetic determinants of Fv1 restriction. J Virol. 2000;77:13403–13406. doi: 10.1128/jvi.74.16.7422-7430.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Towers G, Collins M, Takeuchi Y. Abrogation of Ref1 retrovirus restriction in human cells. J Virol. 2002;76:2548–2550. doi: 10.1128/jvi.76.5.2548-2550.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Butler SL, Hansen MST, Bushman FD. A quantitative assay for HIV DNA integration in vivo. Nat Med. 2001;7:631–634. doi: 10.1038/87979. [DOI] [PubMed] [Google Scholar]
- 80.Julias JG, Ferris AL, Boyer PL, Hughes SH. Replication of phenotypically mixed human immunodeficiency virus type 1 virions containing catalytically active and catalytically inactive reverse transcriptase. J Virol. 2001;75:6537–6546. doi: 10.1128/JVI.75.14.6537-6546.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]