

Abstract
Glucagon-like peptide-1 is a hormone that is encoded in the proglucagon gene. It is mainly produced in enteroendocrine L cells of the gut and is secreted into the blood stream when food containing fat, protein hydrolysate and/or glucose enters the duodenum. Its particular effects on insulin and glucagon secretion have generated a flurry of research activity over the past twenty years culminating in a naturally occurring GLP-1 receptor agonist, exendin-4, now being used to treat type 2 diabetes. GLP-1 engages a specific G-protein coupled receptor that is present in tissues other than the pancreas (brain, kidney, lung, heart, major blood vessels). The most widely studied cell activated by GLP-1 is the insulin-secreting beta cell where its defining action is augmentation of glucose-induced insulin secretion. Upon GLP-1 receptor activation, adenylyl cyclase is activated and cAMP generated, leading, in turn, to cAMP-dependent activation of second messenger pathways, such as the PKA and Epac pathways. As well as short-term effects of enhancing glucose-induced insulin secretion, continuous GLP-1 receptor activation also increases insulin synthesis, and beta cell proliferation and neogenesis. Although these latter effects cannot be currently monitored in humans, there are substantial improvements in glucose tolerance and increases in both first phase and plateau phase insulin secretory responses in type 2 diabetic patients treated with exendin-4. This review we will focus on the effects resulting from GLP-1 receptor activation in islets of Langerhans
Keywords: GLP-1 receptor, exendin-4, insulin synthesis and secretion, exendin (9-39), beta cell, islet of Langerhans, proliferation, differentiation, cAMP, PKA, Epac, PI3 kinase, FoxO1, IRS2, PDX-1
1. Introduction
The incretin effect refers to the augmented insulin secretory response to a glucose load delivered to the gut relative to that achieved by intravenous glucose when the plasma levels of glucose, under both conditions, are comparable. This effect accounts for up to sixty percent of the insulin secretory response following an oral glucose load (Nauck et al., 1986) and is due to the insulinotropic effects of incretin hormones that are released from enteroendocrine cells of the gut. Glucose-dependent insulinotropic peptide (GIP, also referred to as gastric inhibitory polypeptide) and glucagon-like peptide-1 (GLP-1) are the main incretin hormones (Meier et al., 2002; Mojsov et al., 1987) see Table 1 for their amino acid sequences. GLP-1 results from a post-translational cleavage of the product of the glucagon gene by the prohormone convertase PC1/3 (Dhanvantari et al., 2001). The majority of circulating biologically active GLP-1 in man is the GLP-1 (7-36) amide form, with lesser amounts of the bioactive GLP-1 (7-37) form also detectable (Orskov et al., 1994). The actions of GLP-1 have been extensively studied over the last two decades because its acute intravenous infusion or subcutaneous administration lowers blood glucose and increases insulin secretion. Most importantly, it does so in humans suffering from diabetes. Therefore therapeutic strategies based on activating the GLP-1 receptors (GLP-1Rs) on beta (β) cells and enhancing GLP-1’s actions have been developed. One of the major drawbacks to the use of the native peptide in the clinic is its rapid degradation in serum due to the presence of a dipeptidyl peptidase-IV (DPP-IV, also known as CD26) recognition site in the N-terminus (Hansen et al., 1999). This enzyme, present in the blood stream and on cell membranes, cleaves GLP-1 (7-36) peptide to yield the inactive GLP-1 (9-36) form. Therefore, many modifications have been made to GLP-1 to increase its biological half-life and consequently its efficacy in vivo. Exendin-4 (Ex-4, also called exenatide), a GLP-1R agonist is now available for treating type 2 diabetes mellitus (T2DM). This compound is synthesized in the salivary glands of the Heloderma Suspectum or Gila monster lizard, native to Gila county in southern Arizona US. Ex-4 does not possess the DPP-IV recognition site and is a potent insulinotropic agent. Another component of Gila monster saliva, exendin 9-39 (Ex (9-39)) is an antagonist at the GLP-1R and thus has been useful in determining specificity of effects at this receptor in mechanistic studies.
Table 1.
Amino acid sequences for the human gut peptides GLP-1and GIP, and Ex-4 the compound originally isolated from the salivary glands of the heloderma suspectum.
| GLP-1 Numbering | 7 | 11 | 16 | 21 | 26 | 31 | ||
| GLP-1 | HAEGTFTSDVSSYLEGQAAKEFIAWLVKGR | |||||||
| GIP Numbering | 1 | 5 | 10 | 15 | 20 | 25 | 30 | 35 |
| GIP | YAEGTFISDYSIAMDKIHQQDFVNWLLAQKGKKNDWKHNITQ | |||||||
| Ex-4 Numbering | 1 | 5 | 10 | 15 | 20 | 25 | 30 | 35 |
| Ex-4 | HGEGTFTSDLSKQMEEEAVRLFIEWLKNGGPSSGAPPPS | |||||||
Both acute and chronic treatment with GLP-1 and GLP-1R agonists are known to increase insulin secretion and decrease plasma glucose levels in T2DM. Their long-term effects on rodent β cells leading to increased β cell mass through increased β cell proliferation and differentiation in both non-diabetic and diabetic animals have also been extensively studied. However, given the current technical difficulties in assessing human islet mass, the latter properties of the compounds cannot be confirmed in humans.
Many aspects of GLP-1 biology remain unresolved. Here we address a number of those issues including the evidence in the literature for GLP-1 expression in specific cells types of the pancreas, the down-stream signaling of the GLP-1R in those cells and the controversial link between intestinal dumping of food and hypersecretion of GLP-1 resulting in pathologic overgrowth of islet β cells, as a postoperative complication in gastric bypass surgery. Another major issue surrounding the mechanism of action of GLP-1 on β cells is the importance of PKA versus other cAMP signaling pathways, i.e. Epac (GEF). Additionally, and most exciting to investigators in the field, as research on GLP-1’s actions increases, many non-diabetologists are applying their sophisticated techniques to examine the molecular events consequent upon GLP-1R activation in β cells and this has led to many interesting findings that we will cover in this review.
Here we provide a comprehensive review of what is known to date of the molecular events consequent upon GLP-1R activation in the cells of the pancreas.
2. GLP-1R in the pancreas
GLP-1R is a specific seven-transmembrane receptor guanine nucleotide-binding protein (G-protein) coupled receptor (GPCR). It was first cloned from rat pancreatic islets (Thorens, 1992) and later from a human pancreatic insulinoma (Dillon et al., 1993; Thorens et al., 1993) and a gut tumor cell line (Graziano et al., 1993). The rat and human GLP-1Rs exhibit a 95% amino acid homology and are 90% identical (Thorens, 1992; Thorens et al., 1993), differing at 42 amino acid positions (Tibaduiza et al., 2001). The human GLP-1R gene is located on the long arm of chromosome 6p21 (Stoffel et al., 1993). GLP-1R is a 64 kDa protein (Widmann et al., 1995) and although alternate splicing results in two different transcripts for both the rat and the human GLP-1R (Dillon et al., 1993; Thorens, 1992) there has, as yet, been only one functionally distinct GLP-1R described. While various polymorphisms have been associated with the GLP-1R human gene locus (Stoffel et al., 1993), linkage analysis eliminates an association with the majority of T2DM cases, based on the populations studied (Tanizawa et al., 1994; Tokuyama et al., 2004; Yagi et al., 1996; Zhang et al., 1994). One patient diagnosed with T2DM from a Japanese study (Tokuyama et al., 2004) exhibited impairment of insulin secretion, insulin sensitivity and glucose tolerance and had a missense mutation resulting in substitution of threonine 149 with methionine (T149M). The mutated receptor exhibited a reduced affinity in vitro for GLP-1 and Ex-4 (Beinborn et al., 2005).
GPCRs are grouped into four main classes based on sequence similarity, they are classes A, B, C (previously referred to as Class 1, 2 and 3 respectively) and the frizzled family (Foord et al., 2005; NC-IUPHAR). GLP-1R is a member of the Class B family consisting of many classical hormone receptors (Harmar, 2001). Within Class B the receptors for the peptide hormones form a subclass of the glucagon receptor family which also include receptors for glucagon, GLP-2, GIP, growth hormone releasing hormone (GHRH), and secretin (Foord et al., 2005; Harmar, 2004; Mayo et al., 2003). GLP-1, GLP-2 and glucagon are encoded by the same gene and result from post-translational modifications of the proglucagon molecule (Bell, 1986). However, binding of the peptide to its receptor is very specific with no relevant cross-reactivity to receptors for other peptides with the exception of glucagon which binds GLP-1R with 100-1000-fold less affinity than does GLP-1 (Fehmann et al., 1994; Thorens, 1992). Plasma levels of glucagon, in both humans and rodents, do not reach levels where this is likely to be physiologically relevant. All members of the glucagon family of GPCRs are coupled to Gαs subunit with subsequent activation of adenylyl cyclase (AC) and production of cAMP, although some including GLP-1R are capable of signaling through additional G-protein subunits (see section 3.4).
All GPCRs possess seven α-helical transmembrane-domains (TM1–TM7), three extracellular loops (EC1, EC2, EC3), three intracellular loops (IC1, IC2, IC3), an amino terminal extracellular domain and an intracellular carboxyl terminus (Palczewski et al., 2000). The structure of Class B peptide receptors is characterized by an amino-terminus extra-cellular domain of 100-150 amino acids. A number of site directed mutagenesis analyses have been conducted since 1996 on the GLP-1R. Most of these studies were conducted on the rat GLP-1R and Fig. 1 highlights the mutated residues in the various regions of the receptor. Together these studies have formulated a picture of how GLP-1 and Ex-4 bind to this receptor and what regions of GLP-1R are important for agonist recognition. The isolated N-terminus of the rat (Lopez de Maturana et al., 2003; Wilmen et al., 1996; Xiao et al., 2000) and human (Bazarsuren et al., 2002) GLP-1R associate with GLP-1, although with lower affinity than with the native receptor (Bazarsuren et al., 2002; Xiao et al., 2000). Similar to the glucagon receptor and other members of this subfamily, GLP-1R has six cysteine residues in the extracellular region that are highly conserved (Thorens et al., 1993). Disulphide bonds occur between cysteines 46 and 71, 62 and 104, 85 and 126 of human GLP-1R (Bazarsuren et al., 2002). Denaturation of the isolated N-terminal receptor fragment of the rat (Wilmen et al., 1996) or human (Bazarsuren et al., 2002) receptor results in complete loss of affinity for the native peptide. Deletion of portions of the N-terminal of the rat GLP-1R or substitution of amino acids 1-134 with the equivalent sequence of the glucagon receptor eliminates affinity for GLP-1 (Graziano et al., 1996). Graziano and colleagues also demonstrated that a certain peptide specificity is conferred by the 29TVSL32 region as a mutant receptor expressing the equivalent glucagon sequence exhibited a 7-fold decrease in affinity for GLP-1 and an equivalent increase in affinity for glucagon (Graziano et al., 1996). Other residues in the extracellular N-terminal domain that have importance for agonist recognition are highlighted in Fig. 1. Wilmen and colleagues have shown that five out of the six tryptophan residues (W39, W72, W87, W91, W110, W120) on the extracellular domain (W87 is not essential) and in particular, the imidazole ring of W39- are all essential for binding (Wilmen et al., 1996; Wilmen et al., 1997). While Ex-4 interacts primarily with the N-terminal portion of the receptor there is evidence of binding determinants for GLP-1 elsewhere in GLP-1R (Lopez de Maturana et al., 2003): notably residues in the EC1 and TM2 domains are of importance (Xiao et al., 2000). Substitution of the negatively charged aspartate residue at 198 in the TM2 region with the neutral asparagine, does not alter affinity for the receptor indicating that the negative charge is not essential for affinity (Lopez de Maturana and Donnelly, 2002). In contrast, substitution with alanine, at 198 results in a significant reduction in binding to GLP-1 (Lopez de Maturana and Donnelly, 2002; Xiao et al., 2000). However N-terminally truncated exendin-4 (i.e. exendin 9-39) and GLP-1 (i.e. GLP-1 15-36) maintained their affinity for the receptor with the alanine mutation at 198, demonstrating that the aspartate residue is probably important for association of GLP-1R to the N-terminus of GLP-1. Xiao and colleagues show that further charged residues concentrated at the distal TM2/EC1 region (K197, K202, D215 and R227) are also probable binding determinants for GLP-1 (Xiao et al., 2000). Lopez de Maturana performed a further series of double alanine scan mutagenesis studies for the entirety of EC1 (Lopez de Maturana et al., 2004). The majority of the residues in this region did not appear to impact on receptor function with the exception of the substitution of 204MY205 which resulted in an almost 90-fold decrease in GLP-1 binding and a complete absence of biological activity as determined by cAMP production. Further mutagenesis studies on these two residues revealed that the loss of function was due to a loss in hydrophobicity in this region. Another residue in the transmembrane domain important for GLP-1 binding is the positively charged lysine (K288) that is situated in the TM4 region of the rat GLP-1R. Replacing this residue with the neutral alanine or leucine greatly reduces affinity for GLP-1: however substitution with arginine has very little effect on receptor avidity indicating that a positive charge is required at this location for biological function (Al-Sabah and Donnelly, 2003). Finally, as stated above, the T149M mutation in the human GLP-1R is important in the biological activity of GLP-1 exhibiting both a reduced affinity for GLP-1 and a reduced cAMP activation (Beinborn et al., 2005).
Fig 1.
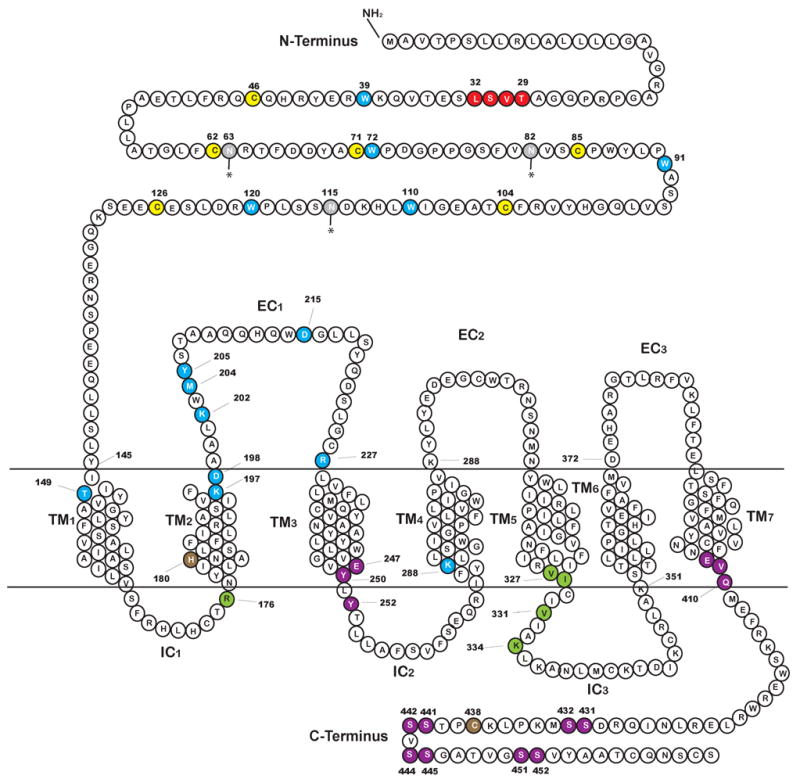
Amino acid sequence of the rat GLP-1R showing the predicted domains, the N-terminal domain, the 7 transmembrane domains (TM1-TM7), the three extracellular domains (EC1, EC2, EC3) and the three intracellular domains (IC1, IC2, IC3). Assignment of these domains is after Thorens (Thorens, 1992). Amino acids that are critical for agonist binding are displayed in blue. The six cysteine residues highly conserved in the Class B receptor family in the N-terminal extracellular region (Thorens et al., 1993) are highlighted in yellow. Amino acids important in binding are shown in blue and are mostly located in the extracellular N-terminal region, in the TM1, TM2, and one in TM4. Glycosylation sites are shown in gray (Goke et al., 1994; Thorens, 1992). Residue H180 is shown is brown as an arginine substitution at this particular point causes both a reduction in affinity for the native ligand and in cAMP production (Heller et al., 1996). Residues known to have a functional importance in binding and/or cAMP activation are highlighted in green and those important in receptor internalization are shown in purple.
Similar to the class A members, the IC3 region of the class B GPCR contains the major determinants required for specific G-protein coupling. A series of papers from the Wheeler laboratory have been instrumental in determining which residues in the IC3 region of the rat receptor are important for G-protein activation. Takhar and colleagues performed a systematic scan in which blocks of 3 or 4 amino acids of the region K334 to K351 in IC3 of GLP-1R (Takhar et al., 1996). Mutation of the 334KLK336 portion produced the most significant reduction in cAMP production while still maintaining affinity for GLP-1 comparable to the wild-type receptor (Takhar et al., 1996). Further specific alanine point mutations of the 334KLK336 region suggested that K334 was principally responsible for the attenuation in cAMP response (Takhar et al., 1996). A following report focusing on point mutations in the N-terminal region of IC3 proximal to the TM5 i.e. the region F321 to L339 revealed the importance of residues V327, I328 and V331 in cAMP stimulation (Mathi et al., 1997). Based on a comparison with a similar region (IC3/TM5 junction) in the M5 muscarinic receptor, Wheeler and colleagues hypothesized that the residues, V 327, I328, V331 and K334 form the hydrophobic face of an α-helical structure and as such would be directly associated with the G-protein. Transfection of two mutated versions of the receptor lacking either the 331VIA333 region of the TM5 domain or the 334KLK336 of the IC3 domain into the insulinoma cell line HIT-T15 showed an absence of GLP-1-induced increases in insulin secretion, cAMP production and Ca2+ channel activation in the β cells expressing the mutated receptor (Salapatek et al., 1999). This pinpointed these regions as being essential for coupling to AC and also highlighted the importance of AC and cAMP in GLP-1 action on the β cell. Some residues in the IC1/TM2 region of the rat GLP-1R have also been found to be of importance in cAMP production. These include H180 (Heller et al., 1996) and R176 (Mathi et al., 1997) although only the latter was associated with an exclusive decrease in cAMP production independent of a loss of affinity of the mutated receptor for GLP-1.
Thorens and co-workers studied internalization of the GLP-1R in a fibroblast cell line transfected with the rat GLP-1R and in the 1056A insulinoma cell line (Widmann et al., 1995). They have shown that GLP-1R is endocytosed via a primarily clathrin coated pit- dependent mechanism and that in the presence of agonist the receptor cycles between the plasma membrane and endosomal compartments. The recognition sequence for the clathrin coated pit is located in the cytoplasmic tail of the receptor and C-terminally truncated mutants exhibit aberrant internalization rates (Vazquez et al., 2005a; Widmann et al., 1997). Widmann and colleagues did not observe internalization of a mutant receptor lacking the last 33 amino acids (Widmann et al., 1997) while Vazquez (Vazquez et al., 2005a) showed a 78% slower internalization of a modified receptor lacking the last 27 amino acids when transfected into fibroblast cell lines. In contrast when the 44 C-terminal amino acids were deleted (GLPR 418R), receptor internalization was only 47% slower with the mutant versus the wild-type GLP-1R, indicating an inhibitory role of the region containing amino acids 419-435 (Vazquez et al., 2005a). Specifically, when the three amino acids located proximal to TM7 (408EVQ410) were replaced with alanine, internalization was found to be much faster. As approximately 40% of the GLPR 418R truncation was internalized when the cells were incubated in hypertonic media (which will disrupt clathrin coated pit-mediated endocytosis) it was postulated that this mutant receptor could be internalized via a faster, uncoated pit pathway (Vazquez et al., 2005a). A recent paper has shown evidence that GLP-1R may also undergo a caveolin-1-dependent trafficking to and from the cell membrane (Syme et al., 2006). The caveolins are a family of scaffolding proteins encoded by three genes (caveolin-1, 2, and 3) that coat caveolae (Cohen et al., 2004). Caveolae are plasmalemmal organelles, with a high lipid content, that, similar to clathrin-coated pits, function as macromolecular vesicular transporters. Syme and co-workers demonstrate using immunoprecipitation that GLP-1R associates with caveolin-1 in the lipid rafts of the cell membrane of MIN6 cells (that endogenously express GLP-1R) and HEK293 cells transfected with a functional green fluorescent protein (GFP)-tagged GLP-1R (Syme et al., 2006). Over expression of a dominant negative form of caveolin-1 (P132L-cav1) inhibited GLP-1 binding and activity in both cell types. Syme and colleagues found a classical caveolin-1 binding motif in the IC2 region (247EGVYLYTLLAFSVF260). They generated two mutated GLP-1Rs using alanine substitution for E247, or Y250 and Y252 simultaneously, E247A and Y250/252A respectively (see Fig 1) that demonstrated both reduced association with caveolin-1 and reduced binding affinity to GLP-1. Similar to Widman and colleagues (Widmann et al., 1995) Syme and co-workers also observed a constitutive cycling of GLP-1R to and from the cell membrane with GFP-tagged GLP-1R being present at the cell membrane and in mobile cytosolic compartments in resting MIN6 cells. Dynamin, a large GTPase, is essential for both clathrin and caveoloae mediated GPCR internalization. Expression of a dominant negative form of dynamin (K44A-dynamin) resulted in a 2.5-fold increase in the amount of GLP-1R at the cell membrane indicating that internalization of GLP-1R was inhibited (Syme et al., 2006). Regulation of GLP-1 internalization is most likely complex, possibly involving both clathrin-coated pit dependent mechanism and a caveolin-dependent mechanism.
Widmann and colleagues have linked internalization of GLP-1R with phosphorylation of three serine doublets located at positions 441/442, 444/445, and 451/452, as removal of these three phosphorylation sites led to a complete suppression of receptor internalization (Widmann et al., 1997). Phosphorylation of these sites also correlates with homologous desensitization of the GLP-1R in cells in vitro (Widmann et al., 1996a; Widmann et al., 1996b; Widmann et al., 1997). Furthermore, the authors demonstrate that heterologous desensitization occurs upon treatment with phorbol 12-myristate 13-acetate (PMA) which results in phosphorylation by protein kinase C (PKC) of 4 serine doublets (431/432, 441/442, 444/445, and 451/452) (Widmann et al., 1996b). Baggio and co-workers examined desensitization in vitro using the INS-1 cell line pretreated for various time intervals with Ex-4 (100 nM for 24 or 72 hr) or with PMA (Baggio et al., 2004b). They followed this with a 1 hr rest period and then re-stimulated with Ex-4 (0.1 nM – 100 nM). They demonstrated a significant downregulation in response at GLP-1R as measured by cAMP production (Baggio et al., 2004b). However, prolonged exposure of both wild type and transgenic mice expressing Ex-4 in a number of tissues (MT-Ex-4) did not adversely affect acute glycemic responses to an intraperitoneal glucose tolerance test (IPGTT) but did in an oral glucose tolerance test (OGTT) (Baggio et al., 2004b). As the authors state this latter observation more likely reflects a desensitization at the level of gastric emptying (GLP-1 is an inhibitor of gastric emptying by its action through vagal afferents; Nauck et al., 1997) as opposed to insulin secretion. Also although insulin content in islets of Ex-4-treated MT-Ex-4 was lower than in islets of wild-type Ex-4-treated mice, levels of transcripts for GLP-1R, PDX-1 and insulin in the pancreata of MT-Ex-4 and wild-type mice receiving treatment were equivalent. Similarly chronic elevation of plasma GLP-1 levels in clinical trials have resulted in effective reduction in blood glucose levels without any loss of potency (see section 9). It therefore must be stated that no physiological significance has been established in vivo for either the homologous or heterologous in vitro desensitization.
There are three N-linked glycosylation sites in the N-terminus extracellular domain (Fig. 1). Inhibition of glycosylation of the GLP-1R in RINm5F cells (Goke et al., 1994) was studied using the antibiotic tunicamycin. Tunicamycin prevents the transfer of the first N-acetylglucosamine residue to dolichol phosphate (Lehle and Tanner, 1976), one of the first intermediates in the synthesis of asparagine-linked glycosylation. Treatment resulted in a concentration dependent reduction in association of the cells with GLP-1 due to a decrease in the number GLP-1 binding sites in the membrane. The reduction in GLP-1R expression at the cell membrane was detected using radiolabeled [125I]GLP-1 and was not a consequence of an inhibition of transcription as mRNA levels in treated cells did not differ (Goke et al., 1994). There was also a reduction in cAMP production and together these results indicate that glycosylation of GLP-1R is necessary for correct insertion into the cell membrane and function. However, the significance of this effect in vivo has not yet been determined.
The GLP-1R is also palmitoylated and replacing C438 with alanine, blocked 3H palmitate incorporation into GLP-1R when transfected into CHO cells (Vazquez et al., 2005b). This substitution also reduced cAMP production 3-fold without loss of receptor processing or redistribution of GLP-1R in the cell (Vazquez et al., 2005b). The loss of receptor function was partially regained by substituting alanine for both serines at positions 431 and 432 (see Fig. 1) and thus palmitoylation of C438 could possibly regulate phosphorylation of these serine residues and could in turn regulate GLP-1R function.
Attempts to characterize the expression pattern of GLP-1R in the pancreas have resulted in numerous and sometimes discordant reports. Earlier experiments indicated a strong expression in the central region of rat islets both by in situ hybridization and immunoreactivity using polyclonal antibodies of GLP-1R (Bullock et al., 1996; Horsch et al., 1997) implying exclusive β cell expression. In contrast, the presence of the receptor on alpha (α), β, and δ cells has been demonstrated by audioradiograph detection of radiolabeled GLP-1 in glucagon, insulin and somatostatin immunoreactive cells in tissue sections from rat pancreata (Heller and Aponte, 1995; Orskov and Poulsen, 1991) suggesting the majority of islet cells express the receptor. The earliest report in 1996 from the Habener laboratory used in situ hybridization on rat tissue sections (Bullock et al., 1996). Similar results were demonstrated a month later by Moens and co-workers who also performed western blot analysis on sorted islet α cells and did not see any GLP-1R protein levels (Moens et al., 1996). Heller, showed in 1997, while in the Habener laboratory, that 20 % of glucagon-positive cells and 76 % of somatostatin-positive cells co-stained for GLP-1R using a polyclonal rabbit antibody (Heller et al., 1997). This is in opposition to a presentation at the American Diabetes Association (ADA) 62nd Annual Scientific Sessions in 2002 in which it was stated that GLP-1R (using a the same rabbit polyclonal antibody to GLP-1R) did not co-localize with glucagon but is only found in the β cells of islets (Romer, 2002). We also did not find the presence of GLP-1R on two α cell lines, INR1-G9 and αTC-1, by immunohistochemistry or western blotting (data not shown). Because GLP-1 in clinical practice actually results in decreased secretion of glucagon we feel it unlikely that any meaningful number of islet α cells express GLP-1R. The physiological effects of GLP-1 on glucagon secretion will be discussed in section 7.
Equally the possible expression of GLP-1R in the ducts is not without controversy. The early reports using in situ hybridization show no expression of the receptor in the ductular network (Bullock et al., 1996) but there are three more recent reports, including one presented at the ADA meeting in 2002, demonstrating a presence in the ducts of the rat, (Romer, 2002; Xu et al., 1999) and human pancreas (Xu et al., 2006). The latter point is important when considering the extra-islet effects of GLP-1 on the putative progenitor cells of the adult pancreas (see section 6). It is probable that the numerous different detection methods used and the systems in which they were applied have resulted in the overall discrepancy in the observation of GLP-1R in the pancreas. A number of new anti-GLP-1R antibodies are now becoming available commercially which will hopefully aid in the clarification of this contentious issue.
3.Second messenger pathways in the β cell activated by GLP-1R
3.1 Stimulation of cAMP production
The GLP-1R is coupled to the Gsα subunit and therefore agonist engagement with the receptor results in activation of AC with consequent production of cAMP (Drucker et al., 1987). At least nine different mammalian membrane-bound isoforms of AC (AC I- AC IX) are known to exist (Hanoune and Defer, 2001). Leech and co-workers performed RT-PCR on extracts from whole rat and human islets showing that AC III, IV,V,VI and VII were present in rat islets and AC V and VI and were found in human islets (Leech et al., 1999). A more recent RT-PCR analysis performed on α and β cells from the islets of Wistar rats clearly shows strong expression of transcripts for AC VI and VIII in β cells and AC II, III, IV, V and VI in α cells (Delmeire et al., 2003). Type VIII AC mRNA expression was also found in RINm5F and INS-1 clonal cell lines (Delmeire et al., 2003). It must be noted that neither Leech and colleagues nor Delmeire and colleagues probed for the presence of type IX AC in islets or individual cells, possibly because AC IX is the only one not activated by forskolin (FSK), a non-specific AC activator that leads to supra-physiological levels of intracellular cAMP in FSK-treated cells (Yan et al., 1998). Type VIII AC is synergistically activated by both Gsα and calcium/calmodulin (Cali et al., 1994) and thus acts as a coincidence detector for glucose and GLP-1 in the β cell. Elevation of glucose concentration (from 1.4 mM to 20 mM) alone did not increase cAMP accumulation in isolated rat primary β cells during a 15 min static incubation (Delmeire et al., 2003). However, the addition of GLP-1 (10 nM) at either low (1.4 mM) or high (20 mM) glucose did and this effect was abrogated by the L-type calcium channel blocker verapamil. In the same study, membranes prepared from RINm5F cells incubated with the G-protein GTPγS.rGsα and calmodulin in the presence of 17 μM [Ca2+] demonstrated a 50 pmol/mg protein−1 min−1 increase above the basal level of cAMP production. This increase was greater than would be the combined effect of either AC stimulant alone, providing evidence of coincidence detection of cAMP and calcium/calmodulin converging at type VIII AC. It must be noted that while Delmeire and colleagues did not observe an increase in intracellular cAMP in response to glucose alone this is most likely due to their cell system which was primary β cells separated by FACS analysis. Isolated β cells are known to produce much lower levels of cAMP than whole islets (Schuit and Pipeleers, 1985) and increased glucose concentrations have been extensively demonstrated to increase cAMP accumulation in whole islets (Grill and Cerasi, 1973; Sharp, 1979).
cAMP is the main mediator of GLP-1 agonist action on acute molecular events in insulin secretion in β cells and overexpression of the GLP-1R in a clonal β cell line leads to increased resting levels of cAMP (Montrose-Rafizadeh et al., 1997a). Although cAMP is a widely adopted second messenger system for many receptors, specificity of response to external stimuli and effect on cell signaling pathways is conferred by regulation of its formation, degradation and spatial regulation by anchoring proteins (Cooper, 2003). Therefore an understanding of the specific regulation of cAMP formation and degradation within β cells is important in examining the downstream effects of this pathway on β cell function. cAMP production is tightly regulated by the balance between the activity of AC and cyclic nucleotide phosphodiesterases (PDEs) that catalyze the hydrolysis of cAMP (Cooper, 2003). There are 11 different families of PDEs representing 21 different gene products (Bender and Beavo, 2006; Conti, 2000). Use of specific pharmacological inhibitors of certain isoforms of PDE has inferred the presence of PDEs 3 and 4 and calcium sensitive PDEs in β cells (Parker et al., 1995; Shafiee-Nick et al., 1995; Sugden and Ashcroft, 1981). Inhibition of PDE3B in particular has demonstrated the firmest evidence for a PDE being implicated in inhibiting insulin secretion. Adenovirus mediated-overexpression of PDE3B in rat islets reduced by 30 % the insulin secretion response to a combination of high glucose (11.1 mM) and GLP-1 (100 nM) over 1 hr when compared to normal islets (Harndahl et al., 2002). In a follow-up study using transgenic mice overexpressing PDE3B under control of the rat insulin 2 promoter, there was a reduced insulin secretion response to intravenous glucose which was both age dependent and increased with the extent of overexpression of PDE3 (Harndahl et al., 2004). In a group of 20-week old mice exhibiting a PDE3B expression 7-fold above basal levels there was a reduction in insulin secretory response (48 % that of wild-type at 1 min peak insulin value) to intravenously administered glucose (1 g/kg) and GLP-1 (1 nmol/kg) (Harndahl et al., 2004). Of note, only males exhibited a reduced response in glucose-induced insulin secretion. The female mice did not exhibit any differences from the wild-type in this regard and hence were not studied in the subsequent GLP-1/glucose induced experiments. Insulin and IGF-1 increase levels of PDE3B in β cells. Treatment of HIT-T15 cells (a hamster clonal β cell line) with IGF-1 (50 nM) roughly halved the insulin secreted in response to GLP-1 (10 nM) in the presence of high levels (12 mM) of glucose (Zhao et al., 1997). This directly correlated to an equivalent reduction in cAMP levels. In conclusion there is ample evidence both in vivo, in isolated islets, and in insulinoma cell lines that PDE3B is major negative regulator of cAMP-mediated GLP-1-induced insulin secretion.
As yet there are no studies examining the involvement of other PDE isoforms in GLP-1-induced insulin secretion. However, Han and colleagues have shown in isolated islets that inhibition of PDE1C but not PDE4 increased glucose-induced insulin secretion in a dose-dependent manner (Han et al., 1999). The combined inhibition of PDE1C, 3 and 4 had as potent an effect on augmentation of insulin secretion by glucose as non-specific inhibition by isobutyl-methylxanthine (IBMX). Interestingly, PDE1C activity was elevated upon glucose stimulation of β cells, pointing to a feedback control of glucose-induced insulin secretion via degradation of cAMP. The authors speculated that it is the increased intracellular calcium from glucose treatment of the islets that is causing activation of the calcium/calmodulin-dependent PDE1C.
A recent paper by Dyachok and colleagues elegantly traced cAMP activation below the cell membrane of INS-1 cells using a ratiometric evanescent wave technique (Dyachok et al., 2006). They demonstrate that there was rapid turnover of cAMP and that cAMP concentrations cycle in response to the application of FSK, glucagon and GLP-1. Glucagon was less efficient than GLP-1, increasing cAMP to a lesser degree and in a smaller proportion of cells. This rapid cycling of cAMP at the cell membrane is most likely conducive to the formation of localized pools of cAMP throughout the β cell and thus could confer the specificity of reaction of GLP-1 versus other G-protein coupled hormones on β cell signaling
cAMP activates further signaling pathways regulating β cell function the two most significant ones being cAMP-dependent protein kinase A (PKA) and the guanine nucleotide exchange factors, both of which are discussed further below.
3.2 Activation of PKA
The rise in cAMP consequent upon G-protein coupled receptor activation results in a significant up-regulation of the activity of PKA, a ubiquitous serine/threonine phosphorylating enzyme (Taylor et al., 1990). The PKA holoenzyme in the inactive state is composed of a regulatory subunit bound non-covalently to two catalytic subunits. There are at least four different regulatory units, type I (RIα, RIβ) and type II (RIIα, RIIβ) which exhibit different affinities for cAMP (Ogreid et al., 1989) and thresholds for activation (Dostmann and Taylor, 1991). Added to these aspects of PKA structure is the different subcellular location of the isozymes owing to their different preferences for the various PKA anchoring proteins (AKAP) which associate with cellular organelles (Skalhegg and Tasken, 2000; Tasken and Aandahl, 2004). This adds to the complexity and diversity of response in different cell types. Type I subunits exhibit a greater affinity for AKAPs that are mainly cytoplasmic and PKA type II is mainly associated with specific cellular structures and organelles (Diviani and Scott, 2001). There are three different catalytic subunits Cα, Cβ, and Cγ. When four molecules of cAMP bind the regulatory subunit dimer (two to each subunit) there is a conformational change in PKA which results in lower affinity for the catalytic subunit and the complex dissociates. The regulatory subunit possesses two cAMP binding sites (known as “A” and “B”) that act cooperatively (Su et al., 1995). It is not clear which isoforms of PKA are present in human β cells: however both PKA type I and II have been isolated from DEAE-cellulose ion-exchange chromatography of rat islets (Sugden et al., 1979). Regulatory unit type RIIα has been detected by western blot in mouse islets (Kashima et al., 2001). Confocal microscopy shows that all three catalytic subunits are present in the mouse insulinoma cell line βTC6, although only the immunofluorescence data for Cα and Cβ were actually presented in the report (Gao et al., 2002).
PKA is a key component in the regulation of insulin secretion by cAMP. It mediates many of the phosphorylation reactions required for secretion by β cells. Inhibition of PKA in isolated islets and insulinoma cell lines diminishes GLP-1- and glucose-mediated insulin secretion (Wang et al., 2001). Thus, basal (nonstimulated) levels of PKA activity are required for optimal glucose-mediated insulin secretion (Eliasson et al., 2003; Kasai et al., 2005a). The PKA anchoring protein inhibitor that blocks association between AKAPs and RII subunit of PKA known as Ht31 peptide (Carr et al., 1991) blocked GLP-1 (1 μM, 3.5-fold increase, GLP-1; 0.7-fold increase in the presence of Ht31)-mediated insulin secretion in both RINm5F and in isolated rat islets (Lester et al., 1997). The study of the complexity of the involvement of AKAP in PKA activation in the context of GLP-1 signaling in the β cell is in its infancy. As well as regulating subcellular concentrations of PKA the anchoring proteins may act as regulators in the activation of PKA and its downstream effectors, and potentially serve to integrate the diverse signaling mechanisms activated by GLP-1. The anchoring protein AKAP18 has been shown to increase cAMP responsive Ca2+ currents when transfected into HEK-293 cells (Fraser et al., 1998). Fraser and colleagues also examined the effects of expressing AKAP18 in the RINm5F insulinoma cell line that does not endogenously express this protein. Transfection of wild-type AKAP18 resulted in a redistribution of the RII subunit of PKA to the cell membrane, while mutant AKAP 18 localized RII to the perinuclear region. RINm5F cells expressing mutant AKAP18 showed a reduced insulin secretory response to GLP-1. RINm5F cells do not endogensously express AKAP18 but these experiments provide some evidence that an anchoring protein may facilitate interaction between PKA and the L-type Ca2+ channel.
Similar pools of PKA associated with various AKAP isoforms may exist at the several points of action downstream of GLP-1R activation in the β cell. Anchoring proteins are also known to integrate and thus coordinate multiple signaling pathways. Lester and coworkers have demonstrated the presence and function in PKA anchoring of the scaffolding protein, IQGAP1, in β cells. PKA was found to co-immunoprecipitate with the calcium/calmodulin binding protein IQGAP1 and the anchoring protein AKAP79 in RINm5F cells (Nauert et al., 2003). Co-localization occurred at the cell membrane and the association was disrupted by Ht31 indicating an indirect association of PKA with IQGAP1 through the anchoring protein. Involvement of IQGAP1 has not yet been explicitly demonstrated for insulin secretion consequent upon GLP-1 mediated activation of PKA. AKAP79 has also been shown to coordinate reversible phosphorylation in a β cell signal transduction complex containing both PKA and the calcium calmodulin phosphatase 3 (also known as calcineurin; Lester et al., 2001). Overexpression of AKAP79 or its human homologue AKAP150 in RIN5mF cells resulted in lower activity of PP-2B, an example of the active regulation by AKAP of its binding partners. PP-2B is known to participate in insulin secretion at a number of different levels in particular in the regulation of insulin transcription (discussed in section 5). To add to the complexity of this whole area of regulation, there is evidence that PDEs are also tethered in the AKAP/PKA complex (Dodge et al., 2001). Scott and colleagues outlined possible regulatory aspects of PKA observed in other cell types that have not yet been explored in the β cell (Alto et al., 2002).
Treatment of βTC6 cells with GLP-1 (100 nM) stimulates translocation of PKA to the nucleus of the cell as determined by confocal microscopy (Gao et al., 2002). In the recent paper (discussed in the previous section) demonstrating cAMP oscillation in β cells it was shown that sustained activation of cAMP by IBMX was necessary to facilitate nuclear translocation of PKA (Dyachok et al., 2006). There are a number of PKA substrates that participate in insulin secretion. These include the IP3 receptor on the endoplasmic reticulum, the GLUT2 glucose transporter and the KATP channel and their regulation in the context of GLP-1-induced insulin secretion are discussed in section 4.
3.3 cAMP regulated guanine nucleotide exchange factors (cAMPGEF or Epac)
In islets approximately 40–50% of GLP-1-stimulated insulin secretion is resistant to H89 treatment (Kashima et al., 2001), implying the existence of a second cAMP activated pathway in the GLP-1R signaling cascade. It is now evident that this PKA-independent portion is due to the cAMP-regulated guanine nucleotide exchange factors (cAMPGEFs) cascade, also known as exchange proteins directly associated with cAMP (henceforth referred to as Epac, de Rooij et al., 1998; Kawasaki et al., 1998). These form part of a large family of related non-kinase effectors originally shown to activate the Ras superfamily GEF binding proteins, initially Rap1, but subsequently have been shown to interact with Rab3a (see section 4.6 for further discussion on Rab proteins), which is involved in insulin secretion (Yaekura et al., 2003). There are two variants of GEF that exhibit high specificity for activation by cAMP over other cyclic nucleotides (Rehmann et al., 2003) and they are referred to as Epac1 and Epac2: both of which are found in rat islets and the β cell lines, HIT-T15 and MIN6 (Leech et al., 2000; Ozaki et al., 2000). These isoforms are encoded by distinct genes (de Rooij et al., 1998; Kawasaki et al., 1998). Both isoforms possess GEF (guanine nucleotide exchange factor) binding sites that catalyze the exchange of GTP for GDP on the small G-proteins (Kawasaki et al., 1998). While Epac 1 has one cAMP binding site, Epac 2 has two, and, similar to PKA, these are also referred to as the “A” and “B” binding sites (de Rooij et al., 2000). Unlike PKA, however, these sites do not bind cAMP in a cooperative manner. The A site of Epac 2A (Kd 87 μM) has a much lower affinity than the B site of either Epac (Epac1, Kd 4 μM, and Epac 2B, Kd 1.2 μM). In contrast the Kd for binding of cAMP to PKA is in the range of 0.12–1 μM (Doskeland and Ogreid, 1981; Ekanger et al., 1985). Thus, it is probable that Epac is sensitive to cAMP in a range where PKA is already saturated, which is important when considering the physiological relevance of the pathways in the regulation of insulin secretion and β cell cycle. A novel cAMP analog 8-(4-chloro-phenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate (8CPT-2Me-cAMP) that activates Epac but not PKA (Enserink et al., 2002) has been useful in discerning PKA- versus Epac-dependent pathways. In a study on the protective effect of cAMP activators against palmitate-induced apoptosis in RINm5F cells, protection was conferred by an Epac-dependent mechanism upon stimulation with forskolin but a PKA-dependent component was found at the lower concentrations of cAMP generated by GLP-1 (Kwon et al., 2004b). Anchoring proteins have been shown in muscle cells to bind both PKA and Epac proteins and thus coordinate their regulation (Dodge-Kafka et al., 2005); whether such a scenario exists in β cells is not known.
Epac has also been found to be involved in Ca2+ release from the endoplasmic reticulum and its downstream targets are important in the exocytosis of the insulin secretory vesicles. Both topics are covered below, in section 4, on acute effects of GLP-1 on insulin secretion.
3.4 Calcium/calmodulin pathway
Calcium/calmodulin kinase II (CaM kinase II) is a member of the broad family of ubiquitously expressed Ca2+-dependent kinases. It is a multigene family comprised of four distinct classes, α, β, γ, and δ, encoded by four separate genes (see Braun and Schulman, 1995; Easom, 1999 for review of CaM kinases expressed in the b cell). Upon activation in high Ca2+ levels the enzyme that consists of 8–12 subunits undergoes autophosphorylation and increasing degrees of Ca2+ oscillation results in increasing number of units being autophosphorylated (Easom, 1999). The phosphorylated enzyme has a greater affinity for calmodulin. When stimulation is removed and Ca2+ levels return to basal, the calmodulin eventually dissociates but 20–80% of the autophosphorylated units retain activity in the absence of Ca2+/calmodulin. Resensitization to stimuli requires dephosphorylation by a phosphatase. GLP-1R is known to activate CaM kinase II by increasing intracellular levels of Ca2+ via activation of the L-type voltage-dependent calcium channel (VDCC) and release of Ca2+ from the endoplasmic reticulum (section 4.4).
Calcineurin or protein phosphatase 2B (PP-2B) is a serine/threonine phosphatase (Rusnak and Mertz, 2000) and is unique among other phosphatases of its family (PPI and PP2) in that Ca2+-calmodulin is required for its activation. PP-2B dephosphorylates (on multiples serines) the transcription complex NFAT, exposing its nuclear localization signal (Crabtree, 2001; Rao et al., 1997). The dephosphorylated NFAT complex is maintained in the nucleus as long as Ca2+ concentrations are elevated, thus keeping PP-2B in the activated state (Timmerman et al., 1996). Inhibition of PP-2B by cyclosporin or FK506 (tacrolimus) decreases GLP-1-induced insulin transcription via suppression of binding of NFAT to the insulin promoter region (see section 5.2). Lester and co-workers have shown that β cell substrates such as synapsin 1 undergo rapid and reversible phosphorylation as a consequence of the coordination of both PKA and PP-2B (Lester et al., 2001). They show that both enzymes are co-localized on the anchoring protein AKAP150 in RINm5F cells. When AKAP79, the human homolog of the rat AKAP150, was expressed in the RIN cells, insulin secretion decreased, PP-2B activity was lowered and the cells became insensitive to FK506. Therefore AKAPs probably coordinate reversible phosphorylation events involving PKA and PP-2B in acute insulin secretion and PP-2B activity must be tightly regulated for correct insulin secretion.
3.5 MAPK and PI3 kinase related pathways
GLP-1R is capable of activating the phospholipase C second messenger system (Wheeler et al., 1993). Studies have shown that when stably expressed in Chinese Hamster Ovary (CHO) cells the rat GLP-1R also demonstrates coupling with the G-protein α-subunits, Gq/11α and to a certain extent, Gi1,2α and thus lead to activation of the mitogen-activated protein kinase (MAPK) pathway (Montrose-Rafizadeh et al., 1999). However, direct coupling of GLP-1R to Gq/11α and Gi1,2α has not been demonstrated in a β cell model per se and Montrose-Rafizadeh and colleagues do not rule out the fact that the increased expression levels of GLP-1R in a non-native situation of CHO cells could be responsible for activation of these alternate G-proteins. Recent studies have shown that activation of MAPK-related pathways can occur downstream of GLP-1R-induced cAMP activation. Specifically, the extracellular signal regulated kinases (ERK) 1 and 2 have been shown to be activated in a Ca2+ (Arnette et al., 2003; Gomez et al., 2002) and cAMP dependent manner (Park et al., 2006). ERK1 and ERK2 are the terminal enzymes in a three-kinase cascade, consisting of the Raf kinases that activate the MAP/ERK kinases, MEK1 and MEK2 that, in turn, activate ERK1/2. Activation of ERK1/2 occurs via phosphorylation and translocation to the nucleus and ERK1/2 already present in the nucleus is phosphorylated upon stimulation. Glucose stimulation has been demonstrated to activate both isomers of ERK in β cells (Arnette et al., 2003; Gomez et al., 2002; Khoo and Cobb, 1997). Phosphorylation of ERK1/2 in response to GLP-1 treatment has been demonstrated in several insulinoma cell lines (Arnette et al., 2003; Gomez et al., 2002) and more recently by GLP-1 (Trumper et al., 2005) and Ex-4 (Park et al., 2006) in human islets. The earlier studies in the insulinoma cell lines reported conflicting data indicating either a Raf/Ras-independent mechanism or inconsistent Raf activation due most probably to the different cell types used in the experiments. Through the use of pharmacological inhibitors, these earlier experiments did establish a dependence on Ca2+ influx and release from the endoplasmic reticulum and, in turn, on the Ca2+calmodulin kinases although the direct method of activation of the cascade was not determined. The most direct method of activation of the Raf-MEK-ERK kinase cascade is via small GTPases. These are active when bound to GTP and inactive in the GDP bound complex. In the particular case of the β cell, Rap1 (Trumper et al., 2005), which is activated by Epac, and the active GTP-bound form of Rap1 were found to co-immunoprecipitate with B-Raf. In contrast there was minimal binding of Ras to B-Raf and very little Ras activation of ERK, indicating that the Rap→B-Raf cascade is favored over the Ras→Raf-1 pathway (Trumper et al., 2005).
p38 MAP kinase was also found to be activated by GLP-1 in β cells (Buteau et al., 2001; Kemp and Habener, 2001; Montrose-Rafizadeh et al., 1999) but the exact mechanism by which this occurs is unknown and it is possible that it is regulated by crosstalk from several signaling systems such as the MEK/ERK and the PI3 kinase pathways.
PI3 kinases (phosphoinositide 3-kinases) are implicated in multiple β cell events ranging through growth, survival, metabolism, and channel regulation. There are four classes of PI3 kinases: IA, IB, II and III. Class I enzymes have been studied in β cells (Koyasu, 2003; Stokoe, 2005). The different classes are categorized based on their ability to catalyse phosphorylation of the 3′-OH position of phosphatidylinositol (PtdIns) lipids. Class I PI3 kinases phosphorylate three kinds of phospholipid substrates- phosphatidylinositol (PtdIns), PtdIns(4)P and PtdIns (4,5)P2. Class I enzymes have an approximately 100 kDa catalytic subunit; the α, β, δ, isoforms of p110 in class IA and p110γ in class IB. These interact with a non-catalytic regulatory unit, of which there are five subtypes, for class IA (p85α, p55α, and p50α resulting from alternative splicing of the same gene, and p85β and p55γ encoded by distinct genes) and one for class IB, namely p101. Of these components the catalytic subunit of Class IB p110γ (MacDonald et al., 2004) and the Class IA p110α, 110β subunits along with the generally widely expressed p85α have been identified in β cells (Trumper et al., 2001). The principle difference between Class 1A and 1B is that IB PI3 kinases can be directly activated by free Gβγ subunits released subsequent to activation of a GPCR (Stephens et al., 1997). A study on glucose tolerance in p110γ−/− mice (MacDonald et al., 2004) indicates that this mechanism of PI3 kinase activation is involved in insulin secretion. Loss of this protein causes an insulin secretory defect, rectified by pre-injection in vivo with Ex-4. It also appears from this study that other forms of the enzyme may compensate for the proliferative response attributed to class IB PI3 kinase activation (Buteau et al., 2003). Activation of PI3 kinase by GLP-1 is therefore complex and is regulated by multiple integrated pathways. PI3 kinase activity has been recorded in several β cell types (Buteau et al., 1999; Hui et al., 2003; MacDonald et al., 2003; Rafiq et al., 2000; Trumper et al., 2000). In turn, PI3 kinase recruits the serine threonine kinases PDK1 (phosphoinositide-dependent kinase 1) and PKB (protein kinase B) to the cell membrane where production of phosphatidylinositol-3,4,5-triphosphate (PtdIns[3,4,5]P3) by PI3 kinase will allow phosphorylation and activation of PKB by PDK1 (Lawlor and Alessi, 2001). Many downstream targets of PDK1 are have been found to be phosphorylated following treatment with GLP-1 (10−7 M for 10 min) of MIN6 cells (MacDonald et al., 2003) indicating that the PDK1 system is active in the β cell. All three isoforms of PKB (α, β,γ: also referred to as Akt 1, 2, and 3) are expressed and activated by insulin-like growth factor-1 in the insulinoma cell lines, HIT-15, INS-1 and RINm5F (Holst et al., 1998; Trumper et al., 2001). Phosphorylation of PKB at serine 473 and threonine 308, occurs in response to GLP-1 treatment in INS-1 cells (Trumper et al., 2000). PKB is known to be instrumental for β cell proliferation and survival and will be discussed in sections 6.1 and 6.4 respectively (Bernal-Mizrachi et al., 2001; Tuttle et al., 2001).
PI3 kinase also lies downstream of IRS2 (insulin receptor substrate 2). IRS2 is a substrate of the insulin and IGF1 receptor tyrosine kinases, plays a regulatory role in β cell growth, function and survival (White, 2003). There is some evidence that GLP-1 can directly activate IRS2 by weakly leading to tyrosine phosphorylation of IRS2 and recruitment of p85α was observed in response to GLP-1 (100 nM) following 10 min of treatment of INS-1 cells (Trumper et al., 2000). GLP-1R activation by Ex-4 (10 nM for 4–10 hr) increases IRS2 expression via CREB activation of the IRS2 promoter (Jhala et al., 2003). The IRS2 promoter contains a CRE half site (TGACG) and in chromatin immunoprecipitation assays the IRS2 promoter was amplified from DNA recovered from immunoprecipitates of CREB. Furthermore, activation of IRS2 was inhibited in cell lines and mice expressing a dominant negative form of CREB. Human islets stimulated with Ex-4 (10 nM for 8 hr) prior to activation with IGF1 (10 nM for 10 min) showed increased levels of IRS2 and PKB phosphorylation (Park et al., 2006). Interestingly, while gross protein levels of IRS2 were increased in response to Ex-4 treatment, those of PKB remained unchanged. Treatment with siRNA to IRS2 prior to Ex-4 treatment blocked the ability of Ex-4 to stimulate PKB phosphorylation. Together these results indicate that Ex-4 may promote IRS2 phosphorylation of Serine 473 on PKB. The possibility that IRS2 could stimulate ERK1/2 activation via the mSOS→Raf→Mek1→ERK cascade (Saltiel and Kahn, 2001) was also explored by Park and colleagues in their paper and siRNA to IRS2 was found to have no effect on ERK activation. However, Trumper and colleagues were able to show an association between Rap and PI3 kinase following treatment of human islets by GLP-1 (Trumper et al., 2005). Rap activation of PI3 kinase is dependent on phosphorylation of p85α by tyrosine kinases. An additional GLP-1R-stimulated mechanism for activation of the PI3 kinase pathway by Src tyrosine kinase activation (Buteau et al., 2003), which is known to be a direct effector of Gβγ (Gentili et al., 2006), was found to be an important mechanism in GLP-1-induced β cell proliferation and we discuss this in section 6.1.
A PI3 kinase signaling molecule mammalian target of rapamycin (mTOR) can be activated directly by PKB but there is evidence of a more complicated system of regulation of this molecule in β cells (Kwon et al., 2004a). mTOR is known to phosphorylate and activate a 70 kDa ribosomal protein S6 kinase (S6K1) in response to elevation of cAMP levels by FSK or Ex-4 in β cells of rat islets. This occurs largely as a consequence of increased ATP production, which deactivates the KATP channels, channels that are implicated in directly regulating mTOR activation. The effect of mTOR in general is complex however it is implicated in GLP-1 R mediated increases in β cell proliferation (Kwon et al., 2004a).
4. Acute effects of GLP-1 on glucose sensing and insulin secretion
Glucose-induced insulin secretion
When blood glucose increases postprandially, it equilibrates across the membrane of the β cell through GLUT2 and GLUT1 transporters. It is rapidly phosphorylated to glucose 6-phosphate by glucokinase, which thereafter determines the rate of glycolysis, i.e., acts as the glucose sensor, and pyruvate generation for entry into the tricarboxylic acid (TCA) cycle in mitochondria. Subsequent oxidative metabolism provides the link between the products of glucose metabolism and insulin secretion. The resultant increase in the ATP/ADP ratio in the cytosol causes depolarization of the plasma membrane by closure of the ATP-sensitive K+ channels (KATP channels). This permits opening of voltage-dependent Ca2+ channels accompanied by release of Ca2+ from intracellular stores. This increase in cytosolic Ca2+ ([Ca2+]i) triggers fusion of insulin-containing secretory vesicles to the plasma membrane, and exocytosis of insulin follows rapidly. The process of acute insulin exocytosis is thereby divided into two pathways; 1) the triggering pathway which is the closure of the KATP channels, depolarization and the rise in [Ca2+]i and 2) an amplifying pathway which is an augmentation of Ca2+-induced insulin secretory vesicle exocytosis (Henquin, 2000). Here we will discuss how GLP-1 enhances both of these pathways.
4.2 Glucose sensing
In studies on rodents it has been observed that glucose is rapidly transported into the β cell by the high capacity, low Km glucose transporter GLUT2. In 1996 Thorens and co-workers reported a PKA-dependent phosphorylation of the C-terminal region of GLUT2 consequent upon GLP-1 (10 nM) treatment in single β cells sorted from rodent islets (Thorens et al., 1996). This was abrogated by treatment with H89 and the phosphorylation sites determined from mutation studies in vitro were found to be S489, S501/503 and T510 on the cytoplasmic tail of GLUT2. Surprisingly, this led to a reduction in the rate of glucose uptake. However, two facts must be taken into consideration: 1) the rate of glucose transport in cells is 50–100 times faster than that of phosphorylation, and, 2) glucokinase is the rate-limiting enzyme in the metabolism of glucose. Furthermore as human islets most likely utilize GLUT1 and not GLUT2 in the maintenance of glucose transport into the cell (De Vos et al., 1995). GLUT2-null rodent islets do not display first phase insulin secretion (Thorens et al., 2000). However replacing GLUT2 with GLUT1 corrected this defect in insulin secretion (Thorens et al., 2000). Thus it is likely that in humans glucokinase activity (De Vos et al., 1995) is a more important glucose sensor than the rate of glucose transport into the β cell (Matschinsky, 2002).
GLP-1 not only increases the amount of insulin secreted per cell (Montrose-Rafizadeh et al., 1994) but also sensitizes more β cells to increases in ambient glucose (Holz et al., 1992). This arises from the ability of GLP-1 to modulate the activity of the KATP channels and is discussed in the next section.
4.3 Potassium channels
Of the many potassium channels present in the pancreatic β-cell the ones critical to insulin secretion are the ATP-sensitive K+ channel (KATP channel), the KCa2+ channel and the delayed rectifier channel, Kv (Dukes and Philipson, 1996). The KATP channel and the Kv channel have been found to be under the direct modulation of GLP-1R signaling and they are discussed here.
4.3.1 KATP channels
Pharmacological deactivators of KATP channels have been in clinical use for many years, consequently they have been extensively characterized in terms of their structure, electrophysiology, and their mechanism of action and we refer the reader to these recent reviews (Ashcroft, 2000; Gribble and Reimann, 2003; Matsuo et al., 2005; Nichols, 2006). Briefly, as illustrated in Fig. 2 the KATP channel consists of four pore forming units, Kir6.2 and four SUR1 (140 kDa) regulatory subunits. ATP binds to the cytoplasmic side of Kir6.2 subunit in an Mg2+-dependent manner initiating a conformational change that results in closure of the channel (Gribble et al., 1998). Depolarization and deactivation of the KATP channels are entirely responsible for the first phase of insulin secretion as illustrated by the KATP knockout mice (Miki et al., 2005) and separately by the SUR1 knockout mice (Miki et al., 2005; Shiota et al., 2002). In humans, mutations in either Kir6.2 or KATP result in a severe form of persistent hyperinsulinemic hypoglycemia of infancy (PHHI; Glaser et al., 1994). By contrast, absence of KATP channels in mice results in less severe symptoms where the mice are normoglycemic, displaying glucose intolerance only upon feeding or glucose challenge (Miki et al., 2005; Shiota et al., 2002).
Fig 2.

A schematic drawing outlining the main signaling pathways activated in response to ligand engagement with the GLP-1R and their major downstream effects on acute insulin secretion, insulin synthesis, preservation of β cell function and mass and regulation of proliferation. Pathways are glucose dependent hence the inclusion of glucose metabolism. GLP-1/Ex-4 bind to GLP-1R causing an increase in cAMP (Drucker et al., 1987); this leads to activation of both PKA (Wang et al., 2001) and EPAC (Holz, 2004). Localized low concentrations of cAMP lead to preferential activation of PKA. Higher cell-wide increases of cAMP by the AC stimulator forskolin (FSK) or the phosphodiesterase (PDE) inhibitor IBMX favor the EPAC pathway. cAMP is compartmentalized by PDEs most notably the PDE3B isoform as shown (Harndahl et al., 2004). PKA anchoring proteins (AKAPs) influence the specificity of cAMP response by anchoring the PKA to specific intracellular sites (Lester et al., 1997). Shown here also is the Ca2+/calmodulin binding protein IQGAP1 which co-immunoprecipitates with PKA and AKAP79 (Nauert et al., 2003). cAMP levels are increased as a consequence of ATP activation of AC consequent upon glucose metabolism. Binding of cAMP to the regulatory units of PKA results in release of the catalytic units from PKA and its activation. Sustained oscillatory increases in cAMP by GLP-1R activation lead to translocation of PKA to the nucleus (Dyachok et al., 2006; Gao et al., 2002) where it regulates PDX-1 (Wang et al., 2001) and CREB activation and subsequently insulin transcription (Chepurny et al., 2002; Hay et al., 2005; Kemp and Habener, 2001). Downstream targets of PKA and Epac in acute insulin secretion, include the KATP and Kv channels, the insulin secretory vesicles and the IP3 Ca2+ channels on the endoplasmic reticulum (ER). P38 MAPK, although activated by GLP-1R agonists (Kemp and Habener, 2001; Montrose-Rafizadeh et al., 1999) is not included as the exact mechanism of activation has not been described. Activation of the MEK/ERK pathway is coordinated through both the Epac moieties and the Ca2+/calmodulin kinases (Arnette et al., 2003; Gomez et al., 2002). The effect of PKA on CREB mediated induction of the IRS2 gene is shown, this is a prolonged effect of GLP-1R activation (Jhala et al., 2003). Acutely PI3 kinase is also stimulated by transactivation of the EGF receptor by cSrc-activated betacellulin (BTC; Buteau et al., 2003). Downstream of PI3 kinase are PKB and PKCζ both of which are implicated in β cell proliferation and PKB in prevention of β cell death (Buteau et al., 2001; Wang and Brubaker, 2002). FoxO1 is regulated by phosphorylation by PKB which results in its exclusion from the nucleus thus permitting the nuclear translocation of PDX-1 (Buteau et al., 2006). Finally enhanced ATP production due to increased mobilization of Ca2+ which in turn upregulate mitochondrial dehydrogenases leads to upregulation of mTOR activity and its downstream effector S6K1 (Kwon et al., 2004a). mTOR is implicated in increased β cell mitosis and may also be activated by PKB. GLP-1R activation also leads to stabilization of the insulin transcript by stimulating nucleocytoplasmic translocation of polypyrimidine tract binding protein (PTB) which binds to the U-rich polypyrimidine tract of insulin and insulin secretory vesicle mRNA transcripts thereby stabilizing them (Knoch et al., 2006). Mechanisms that have not been clearly demonstrated are shown by broken arrows.
GLP-1 has been demonstrated to enhance glucose-induced insulin secretion by facilitating closure of KATP channels in what is commonly believed to be a PKA-dependent mechanism (Gromada et al., 1997; Holz et al., 1992; Light et al., 2002). At sub-stimulatory glucose concentrations (5mM) the resting membrane potential of the rat β cell lies between −65 and −53 mV. Electrical activity is initiated at glucose concentrations in the range of 7–8 mM where the membrane potential has reached −50 to −40 mV (Rorsman and Renstrom, 2003). In the β cell this is a characteristic pattern of cyclic oscillations in membrane potential, with superimposed action potentials on the depolarized plateau, followed by repolarized electrically silent intervals. These oscillations between active and silent phases are referred to as bursts. As the glucose concentration is raised further the duration of the active phase of the burst is increased and the repolarized interval between them decreases, until at glucose concentrations above 20 mM the membrane potential is permanently depolarized and the action potentials appear continuous. The application of GLP-1 in the presence of glucose causes a 5–10 mV shift in the membrane potential depending on the glucose concentration (Gromada et al., 1997; Holz et al., 1993; Holz et al., 1992; Light et al., 2002). The reports of the effects of GLP-1 on membrane potential in the various β cell systems are outlined in Table 2. The effect of GLP-1 is not dependent on the time of application and diminishes 5 min after removal of the peptide (Gromada et al., 1997; Holz et al., 1993). As a consequence of GLP-1 increasing the glucose induced membrane depolarization, the interburst membrane potential does not fall back to resting membrane potential, so the cells can begin depolarizing even before they completely recover from inactivation. Therefore, the silent interburst intervals are shorter allowing for greater activation of the Ca2+ channels and increased exocytosis. Pre-incubation with the PKA inhibitor Rp-8-Br-cAMPS (100 μM) for 20 min (Gromada et al., 1997) or H89 (1 μM) for 5 min (Light et al., 2002) abolished GLP-1 induced inhibition of the KATP channels. The mechanism of this glucose dependent action of GLP-1 is now believed to be via phosphorylation of the KATP channel by PKA. Initial experiments indicated that PKA phosphorylation of Kir6.2 (S372) increased channel activity and that phosphorylation of SUR1 (S1571) decreased burst duration and open probability (Beguin et al., 1999). However point mutation analysis has also targeted 1448S as a specific residue on SUR1 that is phosphorylated in response to GLP-1 treatment (Light et al., 2002).
Table 2.
Effects of GLP-1 on membrane potential of the β cell.
| Resting Membrane Potential mV (Glucose Conc. mM) | Stimulatory Concentration of Glucose Applied (mM) | Concentration of GLP-1 Applied (nM) | ΔΨ (mV) depolarization in membrane potential* | System Used | Reference |
|---|---|---|---|---|---|
| −62 (0) | 10 | 10 | 30–35 | Perforated patch; dispersedβ cells from Rat islet of Langerhans | (Holz et al., 1993) |
| −50 (5) | 10 | 10 | 10 | Patch clamp; dispersed β cells from mouse islet of Langerhans | (Gromada et al., 1997) |
| −53 (5) | 5 | 20 | 6.7 | Perforated patch; INS-1 cells | (Light et al., 2002) |
This is the depolarization that is observed in response to GLP-1 application over and above the extent of depolarization in response to glucose alone
It must be noted that while the membrane of the β cell repolarizes within 5 min of the withdrawal of GLP-1 from the patch clamp the effect on exocytosis remains for 10 min after removal. This implies that GLP-1 has some long-term effect on insulin exocytosis distal to that on the KATP channel and increases in [Ca2+]i influx and the nature of this is discussed in section 4.7.
4.3.2 KV channels
While closure of the KATP channel leads to depolarization of the cell membrane re-polarisation is accomplished by the voltage-dependent K+ or Kv channel. Eleven subfamilies of this channel are known to exist in mammals. In a detailed study by Yan and co-workers Kv2.1, Kv3.2, Kv6.2, and Kv9.3 were found on β cells whereas Kv3.1 and Kv6.1 were found on α cells and Kv2.2 on δ cells of human islets (Yan et al., 2004). A summary of expression the various Kv families in insulinoma cells and rat islets has been compiled by MacDonald and Wheeler (MacDonald and Wheeler, 2003). In the resting β cell the Kv channels are closed and they open in response to membrane depolarization following glucose-induced insulin secretion (Roe et al., 1996). In relation to insulin secretion the effects of the Kv2.1 channel has been the one most studied in cellular models as it possibly is the major contributor to the voltage-dependent outward K+ current. Reduction of this channel’s activity by 60–70 %, using a dominant negative form, in rat β cells, resulted in a 60% increase in insulin secretion (MacDonald et al., 2001).
Patch-clamped experiments in rat islets have shown that GLP-1 (10 nM) and Ex-4 (10 nM) can antagonize Kv currents (MacDonald et al., 2002). GLP-1 and Ex-4 treatment induces a 20 mV hyperpolarizing shift in the voltage dependence of steady-state activation of Kv channels. Inclusion of the non-hydrolyzable GTP-analogue GMP-PNP (10 nM) a G-protein activator alone (MacDonald et al., 2002; MacDonald et al., 2003) replicated the effect of Ex-4 whereas the GLP-1R antagonist exendin (9–39) (10-8M; MacDonald et al., 2002) failed to have an impact on the Kv current, indicating a receptor specific effect. This effect is cAMP/PKA-dependent as pre-treatment with the cAMP pathway antagonist Rp-cAMPS (100μM; MacDonald et al., 2002; MacDonald et al., 2003) or the PKA inhibitor H89 (1μM; MacDonald et al., 2003) reduced the effect of GLP-1R agonists on the Kv current. GLP-1R-mediated antagonism of Kv was found not to depend on Epac as inclusion of the Epac activator 8CPT-2Me-cAMP (50 μM) had no effect on the delayed-rectifying current (MacDonald et al., 2003). However treatment with cAMP analog (100 μM) or the constitutively active PKA catalytic subunit (200 units/ml) alone, were both insufficient to replicate the effects of GLP-1R activation. Therefore MacDonald and colleagues concluded that there was an additional signaling pathway activated by GLP-1 required for antagonism of the Kv current. They found this to be the PI3 kinase pathway with subsequent activation of the atypical PKCζ. The activation of PI3 kinase did not occur via direct activation by the G-protein regulated isoform p110γ as Ex-4 was still able to inhibit Kv in p110γ −/− mice. By applying betacellulin or in separate experiments the EGF receptor antagonist AG1428 or the Src kinase inhibitors they were able to demonstrate that this occurred via GLP-1 trans-activation of the EGF receptor (see section 6.1). However, as with PKA, this mechanism was necessary, though not sufficient, to produce a reduction in Kv current, i.e. both pathways synergize to deactivate the Kv channel. Exactly how this occurs still remains to be investigated.
4.4 Elevation of [Ca2+]i
Intracellular Ca2+ [Ca2+]i levels oscillate in response to GLP-1 treatment in INS-1 cells at a frequency that coincides with oscillating intracellular cAMP concentrations (Dyachok et al., 2006). The two signals reinforce one another such that removal of Ca2+ results in loss of signal coherence and cAMP oscillation. Activation either of AC or GLP-1 signaling employs two known methods of increasing Ca2+; firstly by partial activation of the VDCCs thereby causing them to open and allowing influx of calcium and secondly by enhancing calcium-induced Ca2+ release (CICR) from the intracellular stores. Insulin exocytosis is believed to be efficiently coupled to Ca2+ entry through the Ca2+ channel with, as will be outlined in section 4.6, extensive formation of complexes between the channel and the insulin vesicle. However, recently it has been demonstrated that intercellular Ca2+ increases distal to the Ca2+ channel are instrumental in the exocytosis of a subset of highly Ca2+ sensitive pool of insulin secretory vesicles. This subset of insulin secretory vesicles have been clearly defined by two laboratories using both membrane capacitance (Wan et al., 2004; Yang and Gillis, 2004) and carbon-fibre amperometry measurements (Wan et al., 2004; Yang and Gillis, 2004). They are responsive to global rather than localized increases in Ca2+ and are mobilized concurrently with low Ca2+ sensitivity vesicles that are closely associated with voltage-dependent Ca2+ channels. Interestingly Wan and colleagues were able to demonstrate that PKA was able to increase the Ca2+ sensitivity of these vesicles. Under basal conditions as few as ten vesicles are highly Ca2+ sensitive, however Wan and colleagues were able to demonstrate that activation of PKA or PKC by the application of forskolin or PMA respectively in the presence of glucose could increase the numbers of highly Ca2+ sensitive vesicles by up to four-fold. Considering this observation, it is of importance to understand how GLP-1 stimulates release of Ca2+ from the intercellular stores as this would stimulate these highly sensitive Ca2+ vesicles and thus contribute to first phase insulin secretion.
GLP-1 treatment stimulates release of Ca2+ from the endoplasmic reticulum primarily by two mechanisms; one as a result of PKA activation and the second as a result of Epac activation. Both of these are, of course, downstream of cAMP, and CICR fails to occur in the absence of cAMP-elevating agents even when [Ca2+]i levels are high (Kang et al., 2005). The concept that there may be a component of CICR that is not PKA-dependent was first posed by Bode and colleagues who observed the failure of certain specific PKA inhibitors to completely block GLP-1 (10nM; Bode et al., 1999)-and Ex-4 (10nM; Kang et al., 2001)-induced rise in cytosolic free Ca2+. There are two main families of intracellular Ca2+ channels: the inositol 1,4,5 triphosphate receptors (IP3R) and the ryanodine receptors (RyR). Recently it has been suggested that activation of IP3R in response to GLP-1 is PKA-dependent and activation of RyR is a PKA-independent mechanism occurring via Epac 2 (Kang et al., 2003; Tsuboi et al., 2003). There are three distinct mammalian IP3R I, II and III that share considerable sequence homology encoding proteins that are ~300 kDa that exist as tetrameric structures localized in the endoplasmic reticulum (Patel et al., 1999). IP3RI appears to be the most abundant isoform in rodent islets (Lee et al., 1999; Lee and Laychock, 2001). GLP-1 treatment of isolated β cells results in a biphasic response in [Ca2+]i levels; an initial fast transient peak followed by a prolonged effect (Holz et al., 1999). The fast transient increase in [Ca2+]i is inhibited by blocking the L-type VDCC with nimodepine and by pretreatment with ryanodine. The GLP-1-induced fast transient increase in [Ca2+]i also was observed when the membrane potential was clamped at −50 mV but not at −80 mV. This implies that GLP-1 mobilizes the intracellular Ca2+ stores by triggering partial activation of the L-type Ca2+ channel. The RyR is a Ca2+ channel composed of four ~550 kDa ryanodine protomers and four 12 kDa FK506 binding proteins that are the regulatory units FKBP12 or FKB12.6 (Thearle and Brillantes, 2005). There are three distinct genes encoding three ryanodine receptors, types 1, 2, and 3, RyR1, RyR2 and RyR3 respectively and there is some debate over the most prevalent and the most important types in the β cell (reviewed in Bruton et al., 2003). It appears from RNAse protection assays and RT-PCR that RyR2 is the most prevalent form (Islam et al., 1998). Binding of a fluorescent derivative of ryanodine was observed in rodent and human β cells (Holz et al., 1999). The application of the specific Epac activator 8CPT-2Me-cAMP (100μM for 10s) successfully resolved the PKA-independent component as being due to the action of the Epac moieties (Kang et al., 2003). Expression of a dominant negative Epac 2 (Kang et al., 2001; Kang et al., 2003) or use of Epac 2-directed anti-sense oligonucleotides (Kashima et al., 2001) resulted in a loss of insulin secretion and implicated this isoform of the cAMP exchange protein in the activation of CICR. However the role of Epac 1 in this process has not yet been investigated (Holz, 2004). Confirmation that Epac acts predominantly through the RyR was achieved by pre-incubation of INS-1 cells with ryanodine which resulted in blockage of the 8CPT-2Me-cAMP CICR (Kang et al., 2003).
4.5 Metabolic rate
Oxidation of pyruvate by β cell mitochondria is a critical step for the activation of insulin secretion. Increases in mitochondrial concentrations of Ca2+ enhance the metabolic and secretory response of β cells to subsequent challenges with glucose (Wiederkehr and Wollheim, 2006). Calcium activates several matrix dehydrogenases in the TCA cycle including pyruvate dehydrogenase, isocitrate dehydrogenase, and 2-oxoglutarate dehydrogenase.
Using bioluminescence imaging Tsuboi and co-workers have shown that GLP-1-induced CICR increases free intra-mitochondrial concentrations of both Ca2+ and ATP in the MIN6 cell line (Tsuboi et al., 2003). They were able to measure cytosolic and mitochondrial levels of ATP using adenoviruses that expressed mitochondrially targeted luciferase. Similarly the same research group measured mitochondrial Ca2+ concentrations by adenovirus mediated expression of a Ca2+-dependent photoprotein, mitochondrial aequorin (Ainscow and Rutter, 2001). GLP-1 (100 nM) promoted increases in mitochondrial Ca2+ and ATP levels above those seen with glucose alone (Tsuboi et al., 2003). The increases were equivalent either at high (30 mM) or low (3 mM) glucose concentrations but did not occur in the complete absence of glucose. The action was dependent on the mitochondrial metabolism of glucose and not on a stimulation of glycolysis as was confirmed by the addition of oligomycin an inhibitor of mitochondrial F1FoATP synthase. This action was also completely blocked by the addition Rp-cAMP. Forskolin was capable of producing large and more transient increases in mitochondrial Ca2+ that were followed by sustained increases in mitochondrial ATP. This indicated a dependence on intracellular increases in cAMP and the authors then investigated the involvement of Epac2 by transfecting a dominant negative form of this cAMP sensor. The effect of GLP-1 on mitochondrial ATP increases in the presence of glucose under these conditions was halved. This would seem to imply that a second cAMP sensor, namely PKA, and use of H89 also showed a reduction in GLP-1 induced ATP increases. As stated in section 3.5 the increases in ATP are believed to be largely responsible for the activation of the mTOR.
4.6 Exocytosis of insulin secretory vesicles
The process of exocytosis of insulin secretory vesicles has been studied extensively in various β cell cellular systems. We refer the reader to comprehensive reviews on the topic for insulin secretion specifically (Lang, 1999; Rorsman and Renstrom, 2003) and for the general mechanism of exocytosis (Seino and Shibasaki, 2005; Ungermann and Langosch, 2005). However, as GLP-1 exerts effects at various stages of this process we present a brief summary of what is known about the mechanism. As stated above, insulin secretory vesicles must fuse with the plasma membrane in order to discharge their contents. Docking of vesicles is facilitated by a set of SNARE proteins (soluble N-ethylmaleimide sensitive factor attachment protein SNAP receptors) originally described in synaptic vesicle-membrane fusion (Rossetto et al., 1994). SNARE proteins form a superfamily of proteins that consists of 36 members in humans (Jahn and Scheller, 2006). The distinguishing feature of these proteins is a structural motif consisting of an α-helical coiled-coil domain of approximately 60 amino acids, the eponymous SNARE motif (Jahn and Sudhof, 1999). Originally SNARE proteins were subdived into two classes those associated with the transport vesicle (v-SNAREs) and those attached to the membrane of the target compartment or t-SNAREs (Gerst, 1999). More recent classifications reflect the structural differences in the ionic core of the SNARE motif that contains either three highly conserved glutamine (Q) residues and one highly conserved arginine (R) residue (Jahn and Scheller, 2006). Correspondingly the different SNARE motifs are classified into Qa-, Qb-, Qc-, and R-SNARES. The SNARE complex associated with exocytosis of endocrine vesicles is comprised of the v-SNARE proteins synaptobrevin-2/VAMP-2 (vesicle associated membrane protein-2) and syntaxin 1 and the membrane associated v-SNARE SNAP-25 (25 kDa synaptosomal-associated protein). VAMP-2 and syntaxin 1 each contain a single SNARE motif and are classified as belonging to the R and Qa families respectively. The SNAP proteins comprise a small subfamily that contain one Qb-SNARE motif and one Qc-SNARE motif known as the Qbc-SNARES. Together these three SNARE proteins form a very stable complex that is believed to provide the energy required to produce membrane fusion. This complex also facilitates tethering to an adjacent VDCC in the cell membrane and a localized increase in [Ca2+]i consequent upon opening of the VDCC stimulates release of insulin (Rorsman and Renstrom, 2003). SNAP-25 has been observed to undergo phosphorylation by PKC (at Thr138 and Ser187) in PC12 cells (Hepp et al., 2002) and by PKA (at Thr138) in PC12 (Hepp et al., 2002) and adrenal chromaffin cells (Nagy et al., 2004). PKC phosphorylation of Ser187 was also observed in response to glucose and phorbol ester in INS-1 cells (Gonelle-Gispert et al., 2002). There is no correlation between levels of SNAP-25 phosphorylation in large dense-core vesicle exocytosis in PC12 (Hepp et al., 2002) or INS-1 cells (Gonelle-Gispert et al., 2002). Similarly, we demonstrated that tyrosine phosphorylation of SNAP-25 and, correspondingly, GLP-1-induced insulin secretion are attenuated by the tyrosine kinase inhibitor genistein and enhanced by vanadate, a tyrosine phosphatase inhibitor (Zhou and Egan, 1997). Thus, while there is an association between phosphorylation of SNAP-25 and insulin secretion there is as yet no direct evidence of a regulatory role for glucose- or GLP-1- induced phosphorylation of SNAP-25. Further investigation to establish the existence and physiological relevance of PKA phosphorylation of SNAP-25 is required.
Complex adhesion to the VDCC is aided by participation of certain Rab proteins, a subclass of the Ras superfamily of small G-proteins that have been demonstrated to regulate insulin vesicle exocytosis (Yaekura et al., 2003). Rab proteins cycle between an active GTP bound form associated with vesicles and an inactive GDP bound cytosolic form. In particular, transgenic mice lacking the isoform Rab3A are glucose intolerant and exhibit decreased first phase insulin secretion, consistent with a role for Rab3A in exocytosis (Yaekura et al., 2003). Rab3A has been shown by fractionation of cell organelles from INS-1 cells to be localized with the insulin secretory vesicles. There are four structurally related isoforms of Rab, known as Rab3A, B, C, and D, and while Rab3A is not found in human islets, Rab3B and 3C are (Regazzi et al., 1996). The function of Rab3A in insulin exocytosis is very poorly understood and its importance here is in the context of the complex that it forms with Epac2 and Rim2 (Rab3 interacting molecule Ozaki et al., 2000) linking Rim2 with the insulin secretory vesicle (Shibasaki et al., 2004). While generically many actions of Epac are believed to be Rap1 mediated in the specific case of insulin exocytosis there is evidence that Epac2 interacts directly with this granular protein (Holz, 2004). Rab27A was also recently linked to cAMP modulation of insulin vesicle recruitment to the cell membrane and is discussed in greater detail in section 4.7 (Kasai et al., 2005b).
Rim proteins are a class of multidomain scaffolding proteins encoded by four main genes (RIM1α, 2α, 2β, 2γ, 3γ, 4γ) that regulate exocytosis by direct or indirect interaction with other synaptic proteins (Kaeser and Sudhof, 2005). Transcripts of Rim1 & 2 have been found in pancreatic islets although Rim1 was not found to be expressed in MIN6 cells (Ozaki et al., 2000). A GST tagged form of Rim2 co-immunoprecipitated with Epac2 from MIN6 cells. Kashima and colleagues followed up on this observation and expressed a dominant negative form of Rim2 (lacking certain critical domains) in MIN6 cells to highlight the involvement of this molecule in cAMP-induced insulin secretion (Kashima et al., 2001). Use of this mutant form of Rim2 in MIN6 cells expressing the human preproinsulin gene, inhibited the secretion of human C-peptide in response to the cAMP analog 8-Br-cAMP in 16.7 mM glucose. In conjunction with this, they also used anti-sense oligodeoxynucleotides (ODN) directed at Epac2 to suppress Epac2 expression in mouse islets. While anti-sense ODN treatment alone significantly diminished first and second phase insulin secretion response from islets perifused with 8-Br-cAMP (100 μM) it had no effect on insulin secreted in response to high glucose concentrations. However the effects on first phase and second phase insulin secretion of downregulating Epac2 were only conducted using superphysiological concentrations of glucose (16.7 mM) or 8-Br-cAMP and glucose, no experiments were conducted to examine the effects of GLP-1 perifusion with glucose. So although the authors do invoke these effects for the incretin hormones their relevance has to be questioned given the manner in which the experiments were conducted. The application of 8-Br-cAMP is not the same as treatment with GLP-1 as the former will cause greater global increases in cAMP that could potentially activate Epac in favor of PKA and thus skew the results in favor of an Epac dependent mechanism. Additionally, although the authors do show decreases in static insulin secretion in response to GLP-1 in islets treated with anti-sense to Epac2, this was reported for the 30 min timepoint after GLP-1 treatment. Therefore, no assessment can be made regarding the specific response to GLP-1 in first and second phase insulin secretion in the context of suppressing Epac2. These experiments, while providing evidence that Epac2 is involved in bulk insulin release resulting from incubation of isolated islets with GLP-1, do not yet illustrate the extent of the importance of Epac 2 in GLP-1 action in either early or late phase insulin secretion. Recently Kang and colleagues from the Holz laboratory have demonstrated regulation by both Epac isomers of KATP channel closure through a proposed direct interaction of both Epac isomers with the SUR1 subunit (Kang et al., 2006). Applying the Epac selective cAMP analog 8CPT-2Me-cAMP (100 μM for 30 s) in patch clamp analysis they demonstrated inhibition of whole-cell KATP current of both primary human β cells and INS-1 cells. Transfection of INS-1 cells with a dominant negative form of Epac1 nearly abolished this effect. They also found that myc-epitope tagged forms of both Epac isomers co-immunoprecipitated with full-length FLAG tagged SUR1 in transfected HEK cells which implies direct association with between Epac and the SUR1 subunit. Interestingly, while the specific PKA activator N6-Bnz-cAMP failed to have an effect on the KATP channel the application of either H89 or PKI alone was sufficient to inhibit KATP current.
Epac2 has also been found to be associated with the Ca2+ sensor Piccolo (Shibasaki et al., 2004). Downregulation of Piccolo in MIN6 cells using specific anti-sense ODN treatment results in a decrease in 8-Br-cAMP-induced insulin secretion (Fujimoto et al., 2002). However, as above this has not yet been investigated in the context of GLP-1 induced insulin secretion.
4.7 GLP-1 effects on the readily releasable pool (RRP)
There are believed to be three functionally different pools of insulin secretory vesicles in β cells (Barg et al., 2002; Straub and Sharp, 2004). These are the reserve pool (RP) located deep in the cytoplasm, and two pools located close to the membrane, the readily release pool (RRP) and the immediately releasable pool (IRP). It is estimated that about 5% of insulin vesicles in a cell are actually docked to the membrane and constitute the RRP that undergo exocytosis upon elevation of [Ca2+]i. Of these, approximately 50 vesicles are primed and ready for immediate release; they are referred to as the IRP and are believed to be directly adjacent to the L-type VDCCs. The remaining vesicles within the cytosol further from the membrane comprise the RP of insulin vesicles (Bratanova-Tochkova et al., 2002). Insulin secretion from the β cell in response to a square wave stimulatory increase in glucose concentration from the resting state occurs in two phases both in vitro and in vivo (Cerasi and Luft, 1963; Curry et al., 1968). The first phase is rapid, lasts for about 10–15 min, and reaches a peak within 10 min in the mouse (Barg et al., 2002). In contrast the second phase begins once the IRP has been depleted and recruitment from the RP begins, this plateaus and lasts for the duration of glucose stimulation. The rate-limiting step in the second phase of insulin secretion is the mobilization of vesicles from the RP to the RRP and subsequently to IRP. The kinetics of insulin secretion is slightly different between human and mouse. Termination of the first phase in humans is recognized as a nadir and then insulin secretion gradually increases until it reaches a plateau; thus the rate of transition from RP to IRP changes over this gradual increase.
As pointed out in the discussion on the patch-clamp experiments in section 4.3.1 there is a delayed effect of GLP-1 on insulin exocytosis that remains even after the stimulating effect of the peptide is terminated. This is due to the ability of the peptide to increase the number of insulin secretory vesicles in the RRP. This can be explained in terms of the action of GLP-1 signaling to prolong the activation of the Ca2+ channels. As outlined in section 4.3.1 GLP-1 induces greater depolarization of the β cell membrane thus increasing the number of KATP channels that are closed. Consequently the current undergoes more extensive inactivation before the cells start repolarising. This means that the interburst membrane potential does not fall back to resting membrane potential, so the cells start depolarizing even before they have completely recovered from inactivation (Gromada et al., 1998).
GLP-1 may also potentially mobilize vesicles from the RP via cAMP-dependent activation of Rab27A, a secretory vesicle-associated molecule that has been studied extensively by the Izumi laboratory (Kasai et al., 2005b). Rab27A is one of two isoforms of the Rab27 subfamily - the other being Rab27B. Mice deficient for Rab27A exhibit defects in intracellular migration of melanosomes along the actin filaments (Futter et al., 2004) producing a phenotypic coat color referred to as ashen. The equivalent mutation in humans results in hypopigmentation that co-presents with a severe immune disorder, collectively referred to as Griscelli syndrome 2 (Menasche et al., 2003). Rab27A −/− mice are euglycemic in the fasted state but exhibit glucose intolerance postprandially showing reduced responses in both the first and second phases of insulin secretion (Kasai et al., 2005b). This is a consequence of a reduced number of docked vesicles because of diminished capacity of glucose-stimulated mobilization of vesicles from the RP. Glucose metabolism and ATP production are normal in these mutant mice. The Izumi laboratory have also demonstrated that granuphilin, a specific Rab27A effector molecule showing low affinity for Rab3A in vitro (Yi et al., 2002) forms a complex with the SNARE protein syntaxin 1a thereby facilitating the docking process (Torii et al., 2004). Granuphilin immunostaining shows an aberrant sub-cellular expression pattern in the islets of ashen mice when compared to wildtype controls (Kasai et al., 2005b). Although the ashen mice exhibit normal insulin secretory responses to forskolin this could be due to the massive increase in cAMP produced by this agent that may override the impairment in vesicle mobilization. It is probable, although not yet examined, that the Rab27A/granuphilin complex may also participate in GLP-1 modulation of glucose-induced insulin secretion via the cAMP/Epac pathway.
4.8 PKA-dependent versus PKA-independent effects on insulin exocytosis
We have discussed in various points above the relevant importance of Epac and PKA in GLP-1-modulated insulin exocytosis. However we feel it important to reserve a separate section for a discussion of the literature regarding their relevant contributions to fast exocytosis of insulin secretory vesicles that is directly related to the fast release of Ca2+ from the intracellular Ca2+ stores. There are two basic and opposing theories on this: one is that Epac is solely responsible for the rapid increase in [Ca2+]i and insulin exocytosis and the second, in contrast, emphasizes the importance of residual PKA levels to prime the β cell for rapid insulin release.
Rorsman and colleagues have proposed a mechanism by which a 65 kDa vesicle-associated form of SUR (gSUR) is directly involved in PKA-independent exocytosis (Eliasson et al., 2003; Renstrom et al., 2002). They have based their premise on three main observations: 1) known interactions between SUR1 and Epac2 in a yeast-two-hybrid screen (Ozaki et al., 2000), 2) sulfonylureas are capable of stimulating insulin exocytosis even in β cells from SUR1−/− mice (Eliasson et al., 2003), and, 3) the application of glibenclamide to islets results in vesicle acidification thus aiding insulin exocytosis (Renstrom et al., 2002). The importance of this for GLP-1-mediated exocytosis is that Rorsman and colleagues postulate that this is the main mechanism by which Epac directs fast insulin exocytosis. They believe that the 65kDa protein, via indirect interaction with Epac2, stabilizes the Epac2/Rim2 complex and thus increases association with the ClC-3 chloride channel found on endosomes and vesicles (Jentsch et al., 2002). Acidification of insulin secretory vesicles is an essential part of vesicle release and, as the concentration of H+ increases, the positive charge is offset by a negative charge in order to preserve vesicle stability. The preservation of stimulatory action of sulfonylureas in SUR1−/− β cells led the investigators to postulate that the gSUR was entirely different from that of the KATP SUR1 form.
By contrast Takahashi and colleagues have presented experimental evidence for the importance of PKA in fast glucose-induced exocytosis (Kasai et al., 2002; Takahashi et al., 1999). The model they have developed is a rapid and reversible post-priming step in which ATP acts independently of its effects on [Ca2+] or the KATP channels but requires PKA. Takahashi used amperometry to measure secretion resulting from fusion with the cell membrane combined with a controlled release of Ca2+ using photolysis of a caged calcium compound to initiate Ca2+-dependent secretion from isolated mouse islets. Raising the intracellular ATP levels from 0.1 mM to 3 mM dramatically increased Ca2+-dependent fast insulin exocytosis. This step was not dependent on hydrolysis of ATP as inclusion in the β cell of a hydrolysis-resistant analog of ATP actually potentiated fast exocytosis. Use of a form of ATP that could not support phosphorylation, by contrast, did not exhibit the potentiating effect of ATP. Furthermore, ATP action was dependent on intracellular Mg2+ but was not inhibited by ADP, which is characteristic of phosphorylation reactions. Dependence on cAMP was established using the competitive antagonist of cAMP, Rp-cAMP and on PKA by using H89 (10 μM) both of which blocked the action of ATP on the fast insulin exocytosis. The downstream substrates for the PKA phosphorylation are not known but a potential candidate are the SNAP proteins (Zhou and Egan, 1997). Basal levels of cAMP (and presumably PKA) were sufficient to prime the β cell for this initial burst of vesicle release as FSK, even the presence of high ATP concentrations, did not augment insulin release whereas low concentrations did so. However, as Takahashi and colleagues point out, it is still not possible to rule out that a component of this Ca2+-dependent fast exocytosis could be dependent on another cAMP sensor such as Epac (Kasai et al., 2002).
5. Chronic effects of GLP-1 on insulin synthesis and secretion
Drucker and co-workers initially demonstrated the effect of GLP-1 on increasing insulin mRNA levels in 1987 (Drucker et al., 1987). In 1992 Fehmann and Habener showed that GLP-1 (10 nM) treatment induced the proinsulin gene using a chloramphenicol-acetyltransferase (CAT) reporter gene assay, and it increased insulin mRNA levels and insulin content in the βTC-1 cell line following 24 hr of treatment (Fehmann and Habener, 1992). In 1995 it was shown that prolonged treatment of rat insulinoma cells with GLP-1 (1 or 10 nM for 24 hr) resulted in a 1.5-fold increase in intracellular insulin (Wang et al., 1995). Use of the general transcription inhibitor actinomycin D and the protein synthesis inhibitor cyclohexamide showed that the increase in insulin transcription and consequently insulin translation accounted for the increase in insulin content. However the effect of actinomycin D inhibition did not completely eliminate the GLP-1-induced increases in the levels of insulin transcript. This was the first evidence of an important role for stabilization of the insulin transcript in the GLP-1-mediated increase in intracellular β cell insulin levels, at least in insulinoma cells, during prolonged treatment. By contrast actimomycin D treatment did significantly reduce the effect of GLP-1 upon induction of GLUT1 and hexokinase I genes. Thus it became apparent that the beneficial effects of GLP-1 on insulin secretion arose from the stimulation of transcription in the β cell as well as enhancement of acute insulin secretory responses to glucose. The ability of GLP-1 to induce transcription of the insulin gene was later demonstrated using a luciferase reporter gene assay for the rat insulin I gene in INS-1 cells (Skoglund et al., 2000) where a maximum 2-fold increase in luciferase activity was noted. More recently similar results were also obtained when the luciferase-linked human insulin promoter was transfected into INS-1 cells (Hay et al., 2005).
Here we discuss how GLP-1 treatment increases insulin transcription through stabilization of the insulin transcript and cAMP-dependent and -independent upregulation of the insulin gene. A second mechanism utilized by GLP-1 is the activation of the key β cell transcription factor PDX-1 that binds to the A1, A4/A3 and GG2 regulatory elements of the insulin promoter (see Fig. 3) to stimulate transcription.
Fig 3.
Simplified schema of the human (A) and rat I (B) insulin promoters. The elements known to be regulated downstream of GLP-1R activation are shown in blue. There are four CRE sites in the insulin gene two upstream (CRE1 and CRE2) and two downstream (CRE3 and CRE4) of the transcription start site. With the exception of the first one (CRE1) all were shown to be induced when constructs containing fragments representing the individual sites were transfected into INS-1 cells (Hay et al., 2005). It is probable that the close proximity of the CRE1 site to the A3 element to which PDX-1 binds actively prohibits complex formation at this CRE site. All of the NFAT sites studied in the rat I insulin promoter are responsive to the combination of glucose and GLP-1, although NFAT2 was relatively insensitive to GLP-1 alone (Lawrence et al., 2002)
5.1 Stabilization of the insulin mRNA transcript
The rapidly increased translation of insulin mRNA in response to increasing levels of glucose depends on its 5′ and 3′ untranslated regions (Wicksteed et al., 2001). Polypyrimidine tract binding protein (PTB) binds to the U-rich polypyrimidine tract of mRNAs encoding insulin and insulin secretory vesicle proteins thereby stabilizing them (Knoch et al., 2004; Knoch et al., 2006; Tillmar et al., 2002). Both glucose and GLP-1 stimulate nucleocytoplasmic translocation of PTB1 in INS-1 cells (Knoch et al., 2006). Cytosolic PTB1 quickly upregulates the expression of insulin and the secretory vesicle protein ICA512 a receptor tyrosine protein-phosphatase-like protein associated with insulin secretory vesicles (Knoch et al., 2004). Knoch and colleagues also uncovered a PKA-dependent phosphorylation of PTB1 in INS-1 cells. They confirmed this by showing that inhibition of PKA with H89 (10 μM) as well as inhibition of expression of the α-catalytic subunit and regulatory subunits of PKA by siRNA, resulted in a reduction in phosphorylation at serine 16 (location of the consensus motif for PKA phosphorylation) of PTB1. Selective inhibitors of MEK1/2 and ERK1/2 did not inhibit GLP-1-induced phosphorylation and translocation of PTB1.
5.2 GLP-1 regulation of insulin transcription
The cAMP response element (CRE, TGACGTCA) was initially recognized as an inducible enhancer of genes that can be transcribed in response to elevated cAMP levels (Comb et al., 1986; Montminy et al., 1986). This regulatory element has been characterized as being responsive to a number of basic region leucine zipper transcription (bZIP) factors; however the most studied is the cAMP response element binding (CREB) protein. Phosphorylation of CREB at serine133 induces a conformational change in the CREB molecule permitting it to bind to the co-activator CREB binding protein (CBP; Gonzalez and Montminy, 1989). The resulting complex can regulate transcription of genes containing the palindromic CRE sequence. There are four CRE regions in the human insulin gene (Fig. 3), two sites upstream of the transcription start site (CRE1 and CRE2), one site in the first exon (CRE3) and one site in the first intron (CRE4; Inagaki et al., 1992). Of these CRE2 is the only one conserved between humans and rodents (Inagaki et al., 1992; Philippe and Missotten, 1990). The participation of these CRE sites in GLP-1-induced insulin transcription was studied by transfecting INS-1 cells with constructs containing fragments of the human insulin promoter lacking one or more CRE site, or with constructs having mutations in one or more of the CRE sites, linked to the luciferase reporter gene (Hay et al., 2005). Mutation of all CRE sites except for CRE1 resulted in reduced luciferase activity of the human insulin promoter in response to GLP-1 treatment of the transfected cells. Also interesting is the fact that H89 (10 μM), while completely abolishing the effect of FSK on the luciferase activity representing all four CRE sites, did not completely diminish the effect of GLP-1 at CRE3 and CRE4. This would indicate that CRE3 and CRE4 utilize a cAMP/PKA-independent pathway to modulate insulin transcription at these regulatory elements. Electrophoretic mobility shift assays (EMSA) performed with nuclear extracts from GLP-1 (10 nM)- or FSK (10 μM)- treated INS-1 cells (11.1 mM glucose for 4 hr) incubated with four different oligonucleotides containing one each of the human insulin CRE sites revealed a differential regulation of the CRE sites. In particular a supershift assay using an antibody to PDX-1 was capable of abolishing a major complex formed in the CRE1 reaction mix of nuclear extracts treated with GLP-1. The authors hypothesized therefore that the close proximity of CRE1 to the A3 binding site for PDX-1 could preclude binding of a CRE regulatory complex (Fig. 3).
Most of the work examining elements important in insulin transcription has been performed on the rat insulin I promoter (RIP). The promoter region of the human insulin gene exhibits a 75 % homology with the rat insulin I gene promoter up to bp −240 and then they diverge markedly (Walker et al., 1983). The single RIP (RIP1) CRE site is not palindromic (TGACGTCC) and differs from the canonical CRE site by one C/A nucleotide substitution (Oetjen et al., 1994). In separate studies, the Habener (Kemp and Habener, 2001) and Holz laboratories (Chepurny et al., 2002; Skoglund et al., 2000), using luciferase reporter gene assays in INS-1 cells, found that GLP-1 (10 nM or 100 nM) and Ex-4 (10 nM) induction of rat insulin I gene was not inhibited by H89 (10 μM). In both instances the results obtained are quite clear with no statistical difference between reporter assay results in the presence and absence of H89. Chepurny and colleagues performed further studies to examine this phenomenon and found that simultaneous treatment with the membrane permeable PKA inhibitors 8-Br-Rp-cAMPS (200 μM) or KT 5720 and Ex-4 did not affect transcription. In contrast H89 (1 μM and 10 μM) does have highly significant effects on the induction by FSK (2 μM). Therefore transcriptional regulation of the rat insulin I gene downstream of GLP-1R/cAMP activation would appear to be PKA-independent. However co-transfection with a dominant negative isoform of Epac2 had no effect on Ex-4-mediated activation of RIP1 (Chepurny et al., 2002). Furthermore co-transfection with a dominant negative Gαs did not alter the response of the RIP1 luciferase reporter gene (Kemp and Habener, 2001; Skoglund et al., 2000). In a further dissection of the regulation of the RIP1 by GLP-1 the Holz research team showed that the serine/threonine inhibitor Ro 31-8220 that targets PKC, S6K1, and mitogen- and stress activated protein kinase family of CREB binding proteins blocked the action of Ex-4 on RIP1 (Chepurny et al., 2002). However a role for PKC in the mechanism was eliminated as neither the PKC inhibitor K-252c, downregulation of PKC by pre-treatment with phorbol ester or use of a dominant negative PKCζ had an effect on the action of Ex-4. Inhibition of P38MAPK using SB 203580 was found to lead to an increase in insulin transcription in the presence of either GLP-1 or Ex-4 (Chepurny et al., 2002; Kemp and Habener, 2001). This effect was mediated by the CRE site as deletion of this site reversed the effect of the P38MAPK inhibitor on GLP-1 activation of RIP1.
Further studies by Chepurny and colleagues found that elimination of the A4/A3 site did not result in a reduction in Ex-4-mediated induction of the RIP gene whereas removal of the CRE site did (Chepurny et al., 2002). Therefore the authors called into question the relevance of GLP-1R/PDX-1 mediated regulation of insulin transcription via the A4/A3 site (vide infra). However there are two important aspects to be borne in mind when considering this data. Firstly, PDX-1 also binds to the A1 element of the insulin promoter and Chepurny and colleagues did not examine activity in a rat insulin promoter lacking the A1 element. Secondly, regulation of the rat and human insulin promoters is very different as illustrated by the following example. Co-transfection with dominant negative CREB (A-CREB), a genetically engineered form of CREB that binds to bZIP transcription factors, abolished the effect of GLP-1 (Skoglund et al., 2000) and Ex-4 (Chepurny et al., 2002) at the RIP1. However, co-transfection with a dominant negative form of a related bZIP protein activating transcription factor 2 (ATF-2) did not have an effect on RIP1. This is interesting as ATF-2 was previously demonstrated to mediate human insulin gene transcription via CRE (Ban et al., 2000). Thus a comparison of the studies on the occupancy of the regulatory elements on rat and human insulin promoters in response to GLP-1 treatment should serve a caution to transferring knowledge between the two species. The human insulin promoter is indeed distinct from that of the rodent.
NFAT (nuclear factor of activated T cells) a Ca2+/calmodulin-dependent transcription factor, is activated by dephosphorylation (on multiple serines) by PP-2B, hence is translocated to the nucleus (Rao et al., 1997). PP2-B is a serine/threonine phosphatase (Rusnak and Mertz, 2000) and is unique among other phosphatases of its family (PPI and PP2) in its dependency on Ca2+/calmodulin for its activation. NFAT is expressed in the rat pancreatic β cell (Lawrence et al., 2002) and the dephosphorylated NFAT complex is maintained in the nucleus as long as Ca2+ concentrations are elevated, thus maintaining calcineurin in the activated state (Timmerman et al., 1996). The participation of PP2-B and NFAT in multiple aspects of insulin secretion has been highlighted by the use of the PP2-B inhibitor FK506 (also known as tacrolimus) and cyclosporin A (reviewed in Doyle and Egan, 2003).
Lawrence and colleagues used INS-1 cells to study the effect of PP-2B inhibition and consequently absence of activated NFAT on GLP-1 induction of the insulin gene (Lawrence et al., 2002). After employing a wash out-period of 18 hr in 2 mM glucose they were able to demonstrate a 71-fold increase in rat insulin I reporter gene activity (luciferase reporter gene assay) in response to 100 nM GLP-1 over 6 hr in the presence of 11 mM glucose. This was almost completely inhibited by the addition of FK506 (10 μM) to the medium. Addition of H89 (1–50 μM) in this instance lead to a reduction in the luciferase reporter gene assay (maximal 70% inhibition at 10 μM H89) in INS-1 cells, in contrast to the results obtained by the Holz and Habener laboratories (vide supra). Also interesting was the fact that FSK-mediated activation of the insulin gene was not completely inhibited by PKA inhibitors (maximum 80% inhibition) in the studies by Lawrence and co-workers. This would suggest, as has been found for other GLP-1 actions on the β cell (Kwon et al., 2004b), that PKA is a major regulator of insulin transcription when the levels of cAMP are lower and more localized. There are three putative binding sites [(T/A)GGAAA(A/N)(A/T/C) where N=nucleotide] for NFAT on RIP1 (see Fig. 3). Site directed mutagenesis in each of the individual sites revealed that all three NFAT sequences are important for glucose-dependent GLP-1 induction of the insulin gene (Lawrence et al., 2002). There was a difference in responsiveness with the NFAT1 site being the most responsive to either GLP-1 alone or the combination of glucose and GLP-1, NFAT3 to a lesser extent and finally NFAT2, although responsive to glucose and GLP-1 together, was relatively insensitive to GLP-1 alone. GLP-1 and glucose induction of insulin transcription was abolished by the intracellular calcium chelator BAPTA but only partially inhibited by the L-type VDCC blocker verapamil. These observations would imply that, calcium released from the intracellular stores is important in induction of insulin transcription and is modulated by calcium influx via non-selective ion channels, that GLP-1 is known to activate (Leech and Habener, 1997).
5.3 Regulation of PDX-1
The homeobox transcription factor pancreatic duodenal homeobox-1 (PDX-1, also known as IDX-1, STF1 and IUF1) is essential for pancreatic development and for conserved regulation of insulin transcription. Mice (Jonsson et al., 1994) and humans (Stoffers et al., 1997) completely lacking PDX-1 do not have a pancreas. Mutations in the PDX-1 gene are associated with a form of maturity onset diabetes of the young, MODY4 (Stoffers et al., 1997). PDX-1 expression (Wang et al., 2001), intracellular location (Moede et al., 1999; Rafiq et al., 2000; Rafiq et al., 1998), and DNA binding (Petersen et al., 1998) are known to be responsive to glucose metabolism in the β cell. Indeed PDX-1 is known to regulate acute glucose induction of the insulin gene (Rafiq et al., 1998). Despite earlier reports of PDX-1 knockdown experiments performed in insulinoma cells showing no effect on insulin gene expression (Kajimoto et al., 1997), the central importance for PDX-1 in maintenance of sufficient β cell mass, function, growth and insulin transcription is now clear based on results from both in vivo transgenic models (Kushner et al., 2002; Li et al., 2005c) and from transfection of the human insulin promoter into insulinoma cell lines (Le Lay and Stein, 2006). PDX-1 is also a key effector for the GLP-1R responsive pathways and is critical for the positive effects of GLP-1R agonists on differentiation, proliferation, survival and function of the β cell (Li et al., 2005c). PDX-1 binds to the A1 and A3/A4 elements of the rat and human insulin promoters (Le Lay and Stein, 2006; Ohlsson et al., 1993) (see Fig. 3) via the homeodomain factor TAAT core binding motif. It also binds to the GG2 element of the human promoter which is apparently more critical for the activation of the human insulin gene (Le Lay and Stein, 2006).
We and the Prentki laboratory showed that PDX-1 mRNA and protein levels are increased in response to GLP-1 treatment in rat insulinoma cell lines (Buteau et al., 1999; Wang et al., 1999). In the case of the RIN 1046-38 cells cultured with GLP-1 (10 nM) there was a maximum 1.4-fold increase in PDX-1 mRNA at 3 hr coupled to maximum protein levels at 2–3 hr (Wang et al., 1999). Later we found that chronic treatment with GLP-1R agonists led to upregulation of PDX-1 levels in the endocrine and exocrine pancreas and also to increased nuclear localization of PDX-1 in the β cell (Stoffers et al., 2000). We also found in our experiments in RIN insulinoma cells that GLP-1-induced, but not glucose-induced, nuclear localization of PDX-1 is PKA dependent (Wang et al., 2001). Using a concentration (10 μM) of the PKA inhibitor H89 that we found completely abolished GLP-1-induced insulin secretion in this cell line, (we note this is different from that observed with islets which show a PKA independent component to GLP-1 induced insulin secretion, see section 3.3) we observed a reduction in PDX-1 mRNA and protein levels, nuclear translocation, and, in EMSA assays, a reduction in binding to the A1 element of the rat I insulin promoter in response to simultaneous treatment with GLP-1 and glucose. As the effect of GLP-1 on these parameters of PDX-1 activity in the β cell were not completely abolished it is possible that there may be some dependency of the GLP-1 activation of PDX-1 on PI3 kinase that has been shown to be involved in glucose-induced PDX-1 translocation (Rafiq et al., 2000). Buteau and colleagues did observe a reduction in GLP-1 (10 nM)-induced association of PDX-1 with the rat I insulin promoter when the insulinoma cells were treated with the PI3 kinase inhibitor LY294002 (50μM) (Buteau et al., 1999).
The increases in PDX-1 mRNA and protein levels that we observed in the RIN cells were observed also in normoglycemic mice and in two rodent models of T2DM (Perfetti et al., 2000; Stoffers et al., 2000). Northern blot analysis of total RNA extracted from islets or whole pancreata from 6 and 22 month old Wistar rats treated with GLP-1 (continuous subcutaneous infusion of GLP-1, 1.5 pM/kg·min, for 48 hr) showed that there was a comparable increase in both old and young rats even though the older rats showed a significantly decreased basal level compared to young (Perfetti et al., 2000)
There was a corresponding 4-fold increase in PDX-1 expression in whole pancreatic extracts from old animals treated with GLP-1 as above for 2 days. In contrast GLP-1R−/− mice injected with Ex-4 or treated subcutaneously with GLP-1 did not show an increase in PDX-1 protein levels when compared to wild-type controls (Stoffers et al., 2000). Semi-quantitative analysis of fluorescence intensity of PDX-1 immunoreactivity in C56B16 and diabetic db/db mice revealed an increase in PDX-1 expression not only in the islets but also intense staining in the ductular network.
The mechanism of GLP-1-induced nuclear localization of PDX-1 involves the phosphorylation of a member of the forkhead transcription factors (Fox) of the O subclass, namely FoxO1 (Buteau et al., 2006). FoxO1 is deactivated by phosphorylation (Ser256 in rodents and Ser253 in humans) by the PI3 kinase/PKB pathway a process previously observed in the β cell (Kitamura et al., 2002). In its phosphorylated state FoxO1 is cytoplasmic. FoxO1 and PDX-1 mutually exclude each other from the nucleus of the β cell. Buteau and colleagues recently showed that GLP-1-induced phosphorylation and nuclear exclusion of FoxO1 via transactivation of the EGFR (see section 6.1 for description of this mechanism) in an insulinoma cell line (Buteau et al., 2006). Kawamori and colleagues have found a nuclear export signal on PDX-1 that is activated in response to oxidative stress by the c-Jun NH2-terminal kinase (JNK; Kawamori et al., 2003). Oxidative stress leads to the exclusion of PDX-1 from the nucleus by increasing FoxO1 nuclear expression that is downstream of JNK inactivation of PKB. JNK overexpression reduces the phosphorylation of PKB at serine473 and thereby reduces the extent of FoxO1 phosphorylation (Kawamori et al., 2006). FoxO1 also apparently represses PDX-1 promoter activity by binding to the Foxa2 binding site in the PDX-1 promoter (Kitamura et al., 2002).
6. Regulation of β cell mass
β cell mass is regulated by a balance between β cell proliferation and death. Islet neogenesis is a controversial subject as there is no direct evidence for the existence of a specific pancreatic endocrine stem cell and we reserve discussion of this topic for section 6.2.1. Studies in rodents and humans have and continue to illustrate that the incretin hormones play a central role in the homeostasis of pancreatic β cell mass as well as function and that these two parameters are closely intertwined. Chronic treatment of both normal and diabetic rodents with GLP-1R agonists can result in an increase in β cell mass due to increases in β cell 1) proliferation, 2) neogenesis and/or, 3) decreases in β cell apoptosis. However there are at least two instances in which GLP-1R stimulation alleviated diabetes in rodent models in which there was a decrease in β cell mass. This can be attributed to the improvement in β cell function (Li et al., 2006) and/or a decrease in insulin resistance (Gedulin et al., 2005) therefore less β cell mass is required. Here we review in chronological order the studies in rodents where pancreatic endocrine mass has been modulated by treatment with GLP-1R agonists and studies in cell lines and isolated β cells that outline the mechanism by which this may occur (Fig. 2 & 4). Finally we summarize the data from the rodent studies in which the effects of a GLP-1R agonist were examined in Table 3.
Fig 4.
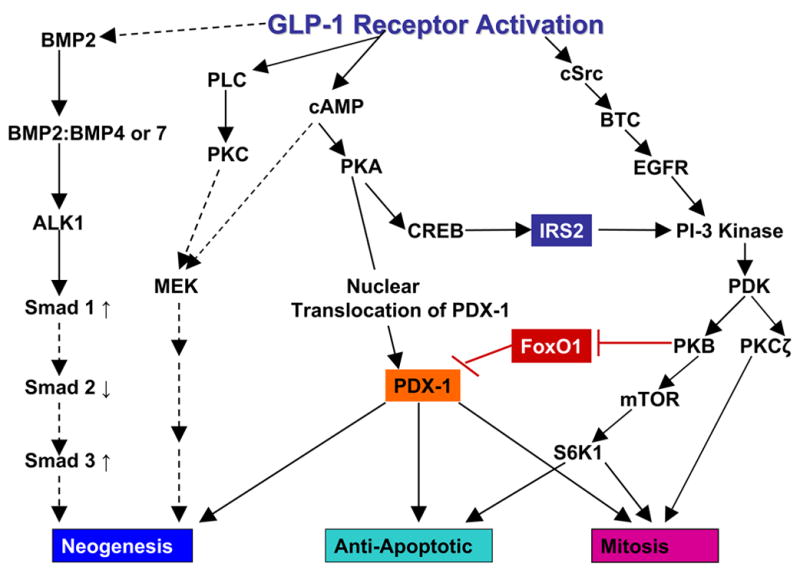
Schema outlining the signaling mechanisms reported to be involved in GLP-1R-induced differentiation/neogenesis of pancreatic precursor cells, proliferation and in the prevention of apoptosis. Dashed lines indicate mechanisms that are either not fully delineated or in the case of the cAMP activation of the MEK pathway are complex and are shown completely in Fig 2. The mechanism shown for involvement of BMP and TGFβ signaling pathways in differentiation is after Gittes and co-workers (Yew et al., 2005; Yew et al., 2004). PKB is shown as inhibiting FoxO1 (by phosphorylation). When FoxO1 is in its active and unphosphorylated state it inhibits the transcription of PDX-1 and its translocation to the nucleus (Kitamura et al., 2002).
Table 3.
Effects of GLP-1R agonists and native GLP-1 in rodent models of diabetes, where changes in β cell mass were assessed.
| Rodent Model | GLP-1R Agonist | Key Findings | Reference |
|---|---|---|---|
| 90–95% Partial pancreatectomy in rats | Ex-4, ip, 1 nmol/kg daily for 10 days starting immediately post surgery | Improved glucose tolerance 25 days post surgery in Ex-4 group
Increase in β cell area in partial pancreatectomy receiving Ex-4 relative to sham controls receiving Ex-4. No increase in β cell BrdU index in partial pancreatectomy receiving Ex-4 relative to surgery alone Conclusion: increase in β cell neogenesis and increased β cell function and insulin content resulted in larger β cell area |
(Xu et al., 1999) |
|
db/db mice
GLP-1R −/− mice |
Ex-4, ip, 1 nmol/kg daily for 2 weeks | Improved glucose tolerance and lower HbA1c
1.76-fold increase in β cell mass Increase in PDX-1 levels in whole pancreatic extracts and increase in number of PDX-1 expressing cells in the ductular network. Conclusion: neogenesis in the ducts initiated by PDX-1 expression, no such effects seen in GLP-1R −/− mice |
(Stoffers et al., 2000) |
| 22 month old glucose intolerant
Wistar rats |
GLP-1, continuous sc infusion, 1.5 pM/kg·min, 5 days | Restoration of glucose tolerance
Increase in PDX-1mRNA and protein levels in whole pancreatic extracts. Initial increase in proliferation of acinar cells at 3 days – gone by 5 days of treatment. Increase in insulin-positive cells in the ducts. Number of insulin-positive cells in any one duct appeared to decrease with the increasing size of the duct. Conclusion: differentiation involved an initial increase in proliferation of exocrine tissue followed by differentiation from the ductular network. |
(Perfetti et al., 2000) |
| STZ Wistar neonate rats |
Ex-4, ip, 3 μg/kg body weight
GLP-1, ip, 400 μg/kg body weight 5 days Once a day Rats observed on day 7 and at 2 months of age. |
Basal hyperglycemia lowered
Glucose tolerance not improved β cell mass increased β cell proliferation not different between STZ saline controls and STZ receiving GLP-1R agonists. Significant increase in number of individual or clusters or insulin-positive cells in ducts Conclusion: pancreatic regeneration due to increased β cell neogenesis with no evidence of increased proliferation or decreased apoptosis |
(Tourrel et al., 2001) |
| db/db mice | Ex-4, ip, 1 nmol/kg daily for 2 weeks | Improved glucose tolerance,delay in the onset of diabetes
1.35-fold increase in β cell mass and 2.3-fold increase in insulin/BrdU positive cells 3.2-fold decrease in β cell apoptosis Number of small islets and single β cells increased Increased protein levels of PKB and ERK1 (but not ERK2) and decrease in Caspase 3 in whole pancreatic extracts Conclusion: PKB and MAPK pathways involved in increasing β cell proliferation and survival following chronic elevation of GLP-1R signaling |
(Wang and Brubaker, 2002) |
| ZDF/GmiTM-fa/fa | GLP-1, continuous sc infusion, 30 pmol/kg·min, 2 days | Improved glucose tolerance
1.6-fold increase in β cell mass 1.4-fold increase in Ki-67/insulin positive cells 3.6-fold decrease in the number of apoptotic β cell nuclei Conclusion: beneficial effects of GLP-1R signaling in ZDF rats are mediated by increased β cell proliferation and decreased apoptosis |
(Farilla et al., 2002) |
|
db/db mice
ob/ob mice |
Liraglutide/NN2211, sc,
100 μg/kg body weight twice a day ob/ob for 2 weeks 200 μg/kg db/db body weight twice a day for 2 weeks |
Non-significant increases in β cell proliferation and β cell mass seen with the 100 μg/kg dose in the ob/ob mice
Significant increase in β cell proliferation (approximately 3-fold) and β cell mass in the 200 μg/kg db/db study Conclusion: Longer pharmacokinetic half-life of NN2211 was reflected in a longer duration of effect than seen with Ex-4 100 μg/kg |
(Rolin et al., 2002) |
| Non-diabetic Sprague Dawley rats | Liraglutide/NN2211, sc, 200 μg/kg body weight, twice a day for 1 or 6 weeks | Fasting blood glucose comparable to vehicle treated controls Sustained lower body weight
Transient increase by 19% in β cell mass after 1 week β cell mass normalized by 6 weeks No change in α cell mass Volume weighted mean islet volume also unchanged Conclusion: In normal rats there is a temporary increase in β cell mass but a sustained decrease in total body weight |
(Bock et al., 2003) |
| 70% Partial pancreatectomy BALB/c mice & GLP-1R −/− mice | Endogenous GLP-1 suppressed by Ex (9-39), sc, 50 pmol/kg·min, for 2 weeks beginning one day prior to surgery | Ex (9-39) did not impair β cell mass regeneration after partial pancreatectomy in BALB/c mice. Regeneration was impaired in the GLP-1R −/− mice | (De Leon et al., 2003) |
| ZDF fa/fa rats
60% Partially pancreatectomized Sprague Dawley rats |
Liraglutide/NN2211, sc, 150 or 30 μg/kg body weight, twice a day for 2 or 6 weeks
Liraglutide/N2211 sc, 150 μg/kg body weight twice a day for 4 days |
6-weeks
Significant reduction in HbA1c and improved glucose tolerance in the high dose group β cell volume increased in both high and low dose groups –only statistically significant for the low dose group Non-significant increases in β cell proliferation with both doses No apparent changes in non-β cell islet mass as measured by a cocktail of antibodies against glucagon, somatostatin, and pancreatic polypeptide No significant increases in β cell volume or proliferation |
(Sturis et al., 2003) |
| Zucker fa/fa 9 weeks old nondiabetic | Ex-4, 3 μg/kg body weight twice a day for 6 weeks | Decreased HbA1c in Ex-4 treated mice and par fed controls
Hyperinsulinemic euglycemic clamp revealed improved insulin sensitivity in Ex-4 treated animals than in the par fed or the controls Animals treated with Ex-4 showed a decrease in β cell mass Conclusion: Pancreatic β cell mass increased hyperbolically with decreasing insulin sensitivity in the controls and par fed animals The decreased β cell mass in Ex-4 treated animals was due increased insulin sensitivity. |
(Gedulin et al., 2005) |
| Sprague Dawley rats
STZ treatment (one injection 65 mg/kg) |
Ex-4 ip 1 nmol/kg daily, begun 1 week following STZ treatment and terminated 10 days later | Improved glucose tolerance in OGTT performed 3 weeks after the start of Ex-4 treatment
2.6-fold increase in β cell mass relative to untreated STZ animals No decrease in apoptosis Conclusion: Pancreatic endocrine regeneration due entirely to neogenesis and proliferation |
(Xu et al., 2006) |
| 4 week old db/db mice | Plasmid encoding GLP-1 fused to mouse IgG1 heavy chain constant region to form a bivalent peptide | Plasmid injected intramuscularly in concert with electroporation at 4 weeks of age and at 6 weeks of age.
Fasting blood glucose levels were lower and fasting insulin levels were higher relative to controls 12 weeks after the first injection. Conclusion: Administration of GLP-1 via this method did have long lasting anti-diabetogenic effect on these animals. |
(Kumar et al., 2006) |
6.1 β cell proliferation
GLP-1 (10 nM) was shown to induce an increase in DNA synthesis as measured by tritiated thymidine incorporation in the INS832/13 insulinoma cell line and in rat pancreatic islets, following a 24 hr incubation (Buteau et al., 1999). 3[H]uptake in the INS-1 cells was concurrent with an increase in PI3 kinase activity and was blocked by the PI3 kinase inhibitors wortmannin and LY 294002. Subsequently, Buteau and colleagues demonstrated that the atypical isoform zeta of protein kinase C (PKCζ) a downstream effector of PI3 kinase, is rapidly (within 5 min) translocated to the nucleus of INS-1 cells in response to GLP-1 treatment (Buteau et al., 2001). Inhibition of p38 MAPK with SB203580 and use of a PKCζ pseudosubstrate but not classical PKC (α, β, and γ) pseudosubstrates significantly decreased the effect of GLP-1 on INS-1 cell proliferation. Adenoviral-mediated overexpression of wild type PKCζ led to a small but significant increase in β cell proliferation but expression of the kinase dead dominant negative form decreased the proliferative response by 60%. Furthermore, in a third publication from the Prentki laboratory it was shown that GLP-1 activation of PI3 kinase could be inhibited by PP1, a c-Src inhibitor as well as AG1478, an EGFR inhibitor (Buteau et al., 2003). They hypothesized that c-Src stimulated tyrosine phosphorylation of EGFR by cleavage and ectodomain shedding of the membrane anchored betacellulin (BTC), an EGFR ligand. Using fluorescence activated cell sorting (FACS) they showed that cell surface levels of BTC were decreased following GLP-1 treatment of INS-1 cells. They also demonstrated that a metalloproteinase inhibitor GM6001 and an anti-BTC neutralizing antibody both suppressed the GLP-1 proliferative effect. The authors concluded that GLP-1 increased INS-1 cell proliferation by transactivation of the EGFR with subsequent activation of PI3 kinase (Fig. 2 and 4). This is followed by activation and translocation to the nucleus of PKCζ. How exactly PKCζ exerts its downstream effects on β cell proliferation is not known but in other cell types it is known to modulate the phosphorylation state and consequently the activity of PKB (Shizukuda and Buttrick, 2002). Thus, as discussed above in section 5.3, PKB will deactivate FoxO1 that will, in turn, facilitate acute nuclear translocation of existing PDX-1 and the synthesis of further PDX-1 by displacing this inhibitor of PDX-1 transcription from the promoter region of the homeodomain transcription factor. Five to six week old IRS2−/− mice treated with Ex-4 failed to demonstrate increases in BrdU incorporation into β cells (Park et al., 2006). Mice with a β cell specific inactivation of PDX-1 also do not display a proliferative response to Ex-4 treatment (Li et al., 2005c). Together all of these reports would suggest the proposed mechanism for proliferation that is shown in Fig. 3 where IRS2 leads to activation of PI3 kinase that in turn will regulate PDX-1 via deactivation of FoxO1.
The first evidence that GLP-1R agonists could act as mitogenic factors for β cells in vivo came with treatment of rats that had undergone a partial pancreatectomy, with Ex-4 (Xu et al., 1999). Following a 90–95 % pancreatectomy, Ex-4 (1 nmol/kg body weight) was administered to one group of animals for 10 days, another group received saline injections. They also had two sham operated groups that received either saline or Ex-4. Partially pancreatectomized animals that received treatment showed improved tolerance to an oral glucose challenge, a lower HbA1c and a greater β cell mass than those that underwent surgery and received saline. Proliferation was quantified by the extent of incorporation of the thymidine analog, bromodeoxyuridine (BrdU) into DNA synthesizing cells. An increase in β cell BrdU labeling index was not observed in pancreatectomized rats that received Ex-4 relative to pancreatectomized animals that received saline injections. However there was an increase in β cell proliferation in the pancreata of sham operated animals that received the peptide relative to those receiving saline alone. The β cells had not hypertrophied as the mean cross-sectional area of the pancreatectomized group receiving Ex-4 was not significantly elevated from the sham operated or sham operated that received Ex-4. Insulin content in the whole pancreas was greater in both groups of Ex-4-treated animals. Considerable regeneration occurs in the 90% partial pancreatectomy model that was enhanced by treatment with Ex-4. The results would imply that neogenesis or differentiation was the major contributor to formation of new β cells (see section 6.2.2). However, the improved insulin granulation, synthesis and β cell function were also major contributors to the overall improvement in glucose tolerance. What is also interesting is that the number of glucagon-positive cells also increased in the sham operated animals that received Ex-4 although the BrdU labeling index of α cells was not determined. This would imply that GLP-1R agonists are capable of stimulating regrowth of α cells either directly or indirectly through a paracrine mechanism.
A model of spontaneous T2DM and a model that has been very well studied by people in the diabetes field is the Zucker diabetic fatty rat (ZDF/GmiTM-fa/fa). These rats have a defect in their leptin receptor and therefore overeat, become obese, hence are insulin resistant and at about 10 weeks of age they develop diabetes. The lesion in the endocrine pancreas is comparable to T2DM and is characterized by an inadequate β cell mass to meet requirement for insulin caused by increased rate of β cell apoptosis and a decrease in β cell proliferation. Farilla and colleagues treated these rats with a continuous 2-day infusion of GLP-1 (30 pmol/kg·min) via an Alzet microosmotic pump implanted in the interscapular region (Farilla et al., 2002). They measured proliferation of the β cells by co-staining for Ki-67 (labels cells in all phases of growth) and insulin and found a 1.6-fold increase in labeling index of β cells relative to untreated animals. Histopathology analysis of the staining revealed qualitative differences in location and distribution of proliferating cells in the pancreata of the control and treated groups. In the control animals the Ki-67 positive cells were individual cells randomly distributed throughout the endocrine, exocrine and ductal compartments of the pancreas. In the pancreata of the GLP-1-treated animals there were two distinct patterns of Ki-67 immunostaining; a) individual cells alone that co-stained for insulin and Ki-67 located within the islets as well as individual insulin- and Ki-67-positive cells throughout the pancreatic tissue, and, b) aggregates of islet-like but insulin-negative proliferating cells of about 10 cells in size were observed in the exocrine pancreas and not in untreated controls. At about the same time the Brubaker laboratory (Wang and Brubaker, 2002) also observed an increase in β cell area (1.35-fold) and proliferation (2.3-fold) in mice exhibiting a defect in the leptin receptor following treatment with Ex-4 (1 nmol/kg, ip, daily for 2 weeks).
A recent review has discussed many of the important cell cycle proteins in the G1/S phase transition, the checkpoint in cell cycle progression critical for postnatal β cell growth hence maintenance of β cell mass (Cozar-Castellano et al., 2006). Two of the three Cyclin D proteins, namely Cyclin D1 and D2, are expressed in β cells and the absence of Cyclin D2 results eventually in overt diabetes in Cyclin D2−/− mice (Georgia and Bhushan, 2004; Kushner et al., 2005). The cyclins coordinate with the cyclin-dependent kinases (cdks) to phosphorylate pRB, a member of the retinoblastoma family of proteins, thus releasing them from the E2F transcription factors (Cozar-Castellano et al., 2006). Members of E2F family of transcription factors are the effectors that control the G1/S transition and in particular transgenic mice lacking E2F1 display defective insulin secretion in response to a glucose challenge due to inadequate β cell mass and a disregulation in PDX-1 (Fajas et al., 2004). In two separate recent publications GLP-1R activation has been shown to regulate Cyclin D expression in models of β cell growth (Friedrichsen et al., 2006; Kim et al., 2006). Surprisingly both reports demonstrated that GLP-1 or Ex-4 treatment in the INS-1 insulinoma cell line caused significant increases in Cyclin D1 mRNA expression but had little effect on Cyclin D2 expression. Friedrichsen and colleagues investigated GLP-1-induced proliferation in monolayers of freshly isolated neonatal rat islets as well as INS-1 cells. Using pharmacological inhibition they showed that this process was PKA-, PI3 kinase- and MEK/ERK-dependent (Friedrichsen et al., 2006). They examined Cyclin D1 expression in GLP-1 (100 nM) treated INS-1 cells at 6 and 12 hr and found it to be increased 100 and 37% respectively above basal levels using qRT-PCR analysis. They transfected the cell line with a luciferase linked Cyclin D1 promoter and found GLP-1 (100 nM for 24 hr) activation of transcription of Cyclin D1 to be PKA-, PI3 kinase-, and MEK/ERK-dependent. Kim and co-workers examined protein levels of Cyclin D1 in response to Ex-4 treatment (10 nM for up to 6 hr) and demonstrated a PKA dependency but no inhibition of Cyclin D1 expression by ERK inhibition or increase in response to the inclusion of exogenous ERK during the Ex-4 treatment (Kim et al., 2006). This might indicate alternate pathways of activation by the two GLP-1R agonists; however activation by both peptides would need to be examined in the same system using identical detection techniques for Cyclin D1 gene expression at both the transcriptional and translational level to confirm. Kim and co-workers found that a CRE binding site on the Cyclin D1 promoter was induced by treatment with Ex-4 (Kim et al., 2006). They investigated this further using EMSA and ChIP analysis and provided convincing evidence for increased association of phosphoCREB in response to Ex-4 treatment. We have also observed an increase in Cyclin D1 protein expression in response to Ex-4 treatment of the RIN insulinoma cell line and our preliminary data indicates that the Notch system is also involved in regulation of this protein by GLP-1R activation (Doyle et al., 2006).
Preliminary data presented by Rankin and colleagues from the Kushner laboratory in which 14 and 20 month old, partially pancreatectomized (50%) mice were exposed chronically to BrdU by addition of the compound to their drinking water, showed that Ex-4 did not cause a significant increase in β cell proliferation (Rankin et al., 2006). The Kushner laboratory had earlier used this labeling technique to show that β cell proliferation was very low in one year old mice (Teta et al., 2005). Given the slow proliferation rate of β cells this would have to be considered to be a very accurate method of analysis. This is probably why they did observe increased proliferation in response to Ex-4 treatment in the younger pancreatectomized mice, in contrast to the earlier experiment with rats from Bonner-Weir’s laboratory. However it remains to be determined if the absence of response in the older animals is a strain or species dependent effect as increases in β cell proliferation have clearly been demonstrated in glucose intolerant rats as outlined above. Table 3 outlines the studies on rodent models in which the impact of GLP-1R agonist treatment on β cell mass has been assessed. While the ability of chronic GLP-1R stimulation to promote new formation of β cells (next section) and to prevent apoptosis (section 6.4) is not always consistent there is little debate over its effects on proliferation in young mice.
6.2 GLP-1 and β cell neogenesis
6.2.1 Islet neogenesis: the current hypotheses
To discuss this second possible regenerative pathway initiated by chronic GLP-1R activation it is necessary to give some background on islet neogenesis in the adult pancreas and the current state of the field. The de novo formation of islet cells in the adult pancreas and in particular β cell neogenesis is a controversial process with evidence indicating that it can occur, especially in the rodent and some recent evidence from transgenic mice showing otherwise.
In 1993 Bonner-Weir and colleagues performed a 90% partial pancreatectomy on rats and documented by immunohistochemistry, the presence of insulin-positive cells from the ductular network in the remnant of the pancreas (Bonner-Weir et al., 1993). This led Bonner-Weir to develop a model of a facultative pancreatic stem cell, not necessarily a ductal cell per se, but one that resides in the ductular network and is responsive to metabolic demands. That there are no specific markers for this cell has remained the crux of the debate since then. Many adult in vivo models have been used to demonstrate the potential for the ductular network to give rise to new β cells, including interferon gamma (IFN-γ) overexpression, plastic wrapping of the pancreatic duct (in order to induce mild pancreatitis) and administration of gastrin and EGF (Bonner-Weir and Weir, 2005; Trucco, 2005). In 2004 Dor and co-workers reported work using a partial pancreatectomy on transgenic mice in which insulin producing cells were indelibly labeled using a Cre/Lox system (Dor et al., 2004). Insulin-positive cells arising directly from the putative stem cell (or any cell that was not previously producing insulin) in the duct would not be labeled. However, no β cells (i.e. insulin-containing cells) were observed that did not also contain the label, pointing to mitosis of existing β cells as the sole method of endocrine pancreatic regeneration in the mouse. However as Dor performed only a 70% partial pancreatectomy it is questionable if the extent of the insult on the pancreas was sufficient to induce the putative pancreatic stem cell to differentiate. Bulter and co-workers have investigated β cell mass in pancreatic tissue from autopsies of lean and obese patients in normoglycemic and glucose intolerant states and patients diagnosed with T2DM (Butler et al., 2003). Rates of β cell apoptosis were increased in T2DM patients (3-fold in obese and 10-fold in lean cases) compared to normoglycemic individuals with no compensatory increase in proliferation rates, which was similar in all groups. The extent of islet neogenesis as measured by the appearance of insulin-positive cells in the ductular network was equivalent in the normoglycemic and diabetic individuals. Butler and co-workers have also demonstrated evidence of new β cell formation in pancreatic biopsies from patients that had been diagnosed with type 1 diabetes (T1DM) many years prior to the biopsy (Meier et al., 2005). Indeed, they found no correlation between duration of the diabetes and extent of insulin-positive cells. They observed occasional insulin-positive cells in the ducts as well as T lymphocyte and macrophage infiltrates and periductal fibrosis, consistent with on-going destruction of β cells as they appear from ducts. Thus they hypothesized that regeneration of insulin-positive cells from cells residing in ducts was a continuous process in T1DM. Unfortunately this was not clinically relevant as the new β cells were susceptible to autoimmune destruction. These observations raise the issue of the relevance of the observations by Dor and co-workers in the mouse pancreas to the human context, given the differences in mouse and human islet architecture, and milieu (Bouwens and Pipeleers, 1998), together with the greater overall heterogeneity in the endocrine cell patterning of the human islet (Brissova et al., 2005).
In 2000 Peck and colleagues showed that it was possible to cultivate and expand a cell with neuroendocrine-type morphology from the mouse pancreas that could then give rise to islet-like clusters expressing insulin and glucagon (Ramiya et al., 2000). When implanted into the subcapsular region of the kidney of diabetic female NOD/Uf mice, (although they had slightly elevated blood glucose levels relative to controls) they survived insulin-free for up to 55 days. Due to the method of isolation (whole pancreas digestion with sucrose fractionation to isolate an islet rich population) it is impossible to identify the origin of this specific cell type. Whether in the native pancreas they reside in the islet or the exocrine tissue will be unknown until a specific marker has been identified. Since this work was published a number of groups have repeated and confirmed that a such a precursor cell exists in both rodent (Ta et al., 2006) and human (Seeberger et al., 2006; Todorov et al., 2006) pancreata. One study would seem to corroborate the work by Dor and colleagues as it showed that human insulin-positive cells undergo an epithelial to mesenchymal transition when kept in culture (Gershengorn et al., 2004). These cells are isolated in a manner similar to that of Peck and co-workers and also undergo long-term culture, followed by a re-differentiation procedure to produce insulin-, C-peptide- and glucagon-positive clusters. As yet no extensive studies have been conducted to define the ability of these cells to reverse diabetes in an animal model. Also, whether β cells can undergo such a dedifferentiation process in vivo is highly questionable; the transition that Gershengorn and co-workers observe is most likely unique to the in vitro conditions. Of particular interest is the recent paper from Levine’s laboratory (Hao et al., 2006) in which they isolated and heritably marked a fairly homogeneous population of CK19 cells (a marker for pancreatic ductal cells) from digested human pancreata with a lentiviral vector expressing EGFP or ddsRed. Using antibiotic resistance to selectively culture the labeled cells they expanded the CK19-positive pool of cells and then implanted them under the kidney capsule of mice and left them for 3 months. Not much insulin immunoreactivity was observed in implants in normoglycemic mice. This apparently was not further improved by treatment of the recipient mice with Ex-4. However 10% of the labeled cells were insulin-positive when the mice received a graft also containing clusters of immortalized human fetal cells. This is the first convincing evidence using lineage tracing to demonstrate that insulin-positive cells could arise from the duct cells in the pancreas. In section 6.2.3 we discuss the potential of GLP-1R agonists to differentiate such precursor cells in vitro.
6.2.2 GLP-1R agonist effects in vivo on β cell differentiation
Determining the effects of GLP-1R on differentiation is assessed indirectly by a) investigating for the presence of insulin-positive cells in the ductular network and/or b) comparing increases in β cell mass with rates of proliferation to see if they can be entirely responsible for increases in the mass. Xu and colleagues showed that there was an increase in β cell area following the combination of partial pancreatectomy and Ex-4 treatment (as discussed in section 6.1) in rats but did not observe an increase in β cell proliferation relative to pancreatectomized animals that did not receive Ex-4 treatment (Xu et al., 1999). It is possible that β cell growth in the pancreas of the pancreatectomized animals was already at its maximum and further treatment with Ex-4 could not improve upon this. To find an explanation for the increase in β cell mass in the Ex-4 treated rats Xu and colleagues looked for endocrine hormone expression in the ducts of the injured pancreas. The number of extra-islet insulin and glucagon positive cells in the ductular network in the sham operated animals that received Ex-4 was assessed as being greater at 7-days post the operation than in the untreated sham operated animals. Therefore they concluded that neogenesis of islets from the ductal network was a major contribution to expansion of pancreatic endocrine mass following Ex-4 treatment.
In 22 month old Wistar rats treated for 5 days with a subcutaneous infusion of GLP-1 we observed an increased β cell area (Perfetti et al., 2000). We also observed an obvious increase in clusters of insulin-positive cells in the pancreatic ductular network of the treated rats. The size of the insulin-positive cluster was inversely proportional to the size of the ducts in which they were located. We also found a significant increase in β cell area and in total PDX-1 isolated from the whole mouse pancreas of treated versus controls. Levels of PDX-1 are known to increase during pancreatic regeneration and it also seems to be essential for GLP-1R mediated differentiation of endocrine precursors (see next section and Table 3). Tourrel and colleagues actually performed a quantitative analysis of the number of insulin-positive clusters in ducts in an animal model of diabetes that received GLP-1R agonist treatment (Tourrel et al., 2001). They treated newborn Wistar rats with STZ (100 μg/g body weight). Then they immediately began treatment for 5 days with an sc injection of GLP-1 (400 μg/kg body weight) or Ex-4 (3 μg/kg body weight) and subsequently examined β cell neogenesis in the rat pancreata at day 7 of the experiment. Both the number of isolated β cells in the ductular epithelium and the number of β cell clusters (2–10 cells in size) proximal to ducts, was increased significantly in the STZ mice that received the GLP-1R agonists. Similar to recent observations by Xu and colleagues (Xu et al., 2006) in STZ treated Sprague Dawley rats Tourrel and co-workers did not observe any effect of GLP-1 or Ex-4 on apoptosis.
6.2.3 In vitro determination of the mechanism of β cell differentiation by GLP-1R agonists
In vitro studies have been conducted on acinar and ductal cell lines to assess the ability of GLP-1R agonists to stimulate endocrine hormone production in these cell types. These also provide an easier model to study the underlying mechanism in neogenesis as stimulated by GLP-1 treatment. We have examined the effect of chronic GLP-1 R activation in both a rat acinar cell line, AR42J cells (Zhou et al., 1999b) and a human ductal cell line, Capan-1 cells (Zhou et al., 2002).
When AR42J cells were treated for 3 days with GLP-1 (10 nM) or Ex-4 (1 nM) approximately 20% were insulin-positive and 50% were glucagon-positive with 20% exhibiting co-staining for both hormones. This conversion was blocked by the addition of the MEK inhibitor PD98059 and partially hindered by the addition of H89. However cultivation in FSK (10 μM) for 3 days did not result in the same degree of insulin expression (2%) as did GLP-1 or Ex-4. This would imply a differentiation mechanism that is co-dependent on the MEK/ERK and PKA pathways. It is possible that since the Epac moieties can, in combination with the Ca2+ calmodulin kinases, activate the MEK/ERK pathways, that they may also be implicated. However the inability of FSK to cause differentiation to the same degree as GLP-1 or Ex-4 implies the involvement of pathways other than those activated by cAMP. Simultaneous treatment with the PKC inhibitor 1-O-hexadecyl-2-O-methyl-rac-glycerol and Ex-4 completely blocked differentiation. This PKC inhibitor is a diether analogue of diacylglycerol and therefore it should inhibit only the classical and novel isoforms of PKC and not affect the atypical form of PKC, PKCζ that has been implicated in GLP-1R-mediated proliferation (section 6.1). However the precise involvement of PKC, and which isoforms are involved in differentiation of the AR42J cells has not yet been investigated. AR42J cells were maintained in media supplemented with fetal bovine serum (FBS, 10%) that certainly contains some insulin. However we confirmed, 1) expression of insulin in the cells by RT-PCR, 2) demonstrated glucose (10 mM)-responsive insulin release (0.65 versus 0.05 pg insulin/μg protein, treated versus control), and, also, 3) found that protein levels of PDX-1 were increased by immunoblotting. Thus uptake of insulin from the medium was not a major contribution to the insulin-positivity observed in the immunostaining assay (Rajagopal et al., 2003).
The Gittes laboratory (Yew et al., 2004) have expanded on these observations and performed a dose-response curve for Ex-4 (1 pM – 100 nM) conversion by quantifying insulin II, pdx-1, and IAPP mRNA levels (Yew and colleagues supplemented their media with 20% FBS). They found that 5 pM of Ex-4 was the most effective dose over a 3 day treatment period. In contrast to our results they did find a few untreated cells expressing insulin. Surprisingly, levels of Foxa2 (HNF3β) a known transcription factor regulator of pdx-1 (Wu et al., 1997), were suppressed in response to Ex-4 treatment, indicating that it is not necessary for expression of pdx-1 in this context (vide infra). The combination of activin and betacellulin has also been shown to induce differentiation of the AR42J cells (Mashima et al., 1996). Activin binds to and activates the transforming growth factor-β (TGFβ) receptors (Fleisch et al., 2006). There are three mammalian forms of TGFβ, 1, 2, and 3, that can potentially bind three ubiquitously expressed TGFβ receptors, TβR I, II and III. TβRI and TβRII are serine-threonine kinases. Almost all cells secrete latent TGFβ and therefore activation of TGFβ acts as the checkpoint for regulation. Following activation of TGFβ the TβRI and TβRII receptor dimers form a hetero-tetrameric complex, and subsequently TβRI phosphorylates receptor-regulated Smad proteins 2 and 3 which mediate activin signaling. Gittes and colleagues found evidence of synergy between the TGFβ and GLP-1R pathways in Ex-4-induced differentiation of this cell line. Ex-4 treatment increased Smad 3 but decreased Smad 2 mRNA and protein levels in the AR42J cell line. Levels of Smad 4, a binding partner of both Smads 2 and 3, remained unchanged. Morpholine ring anti-sense directed against Smad 2 significantly blocked Ex-4-induced increases in insulin II, pdx-1, pax4, and pax 6, mRNA. This inhibition of Smad 2 expression prevented the Ex-4 induced increases in Smad 3 mRNA in this cell line. Use of anti-sense against Smad 3 during Ex-4 treatment dramatically increased insulin and Pax6 levels but decreased the amount of pdx-1 and IAPP transcripts. Finally simultaneous use of anti-sense against both Smad 2 and 3 was equivalent to inhibition of Smad 2 alone. Based on these knockdown experiments Yew and co-workers concluded that Smad 2 is essential for early commitment to an endocrine fate and that Smad 3 is instrumental in advancing the differentiation of the cells (Fig 4). Next the authors investigated AR42J cells for expression of activin ligands and TGFβ isoforms and found very little expression of inhibin A and B (activin monomers). However TGFβ1 and TGFβ2 were expressed and TGFβ1 was upregulated following Ex-4 treatment. Use of a TGFβ pan-neutralising antibody completely blocked Ex-4 differentiation of the AR42J cells decreasing levels of Smad 2 and Smad 3 mRNA. This indicates the ability of Ex-4 to decrease Smad 2 levels is probably independent of TGFβ signaling. Antisense against the TβR type I form, Alk 5, which is upregulated following Ex-4 treatment inhibited differentiation. Finally, simultaneous treatment with exogenous TGFβ1 (10 ng/ml) increased insulin mRNA levels 10- to 15- fold over Ex-4 induced insulin-transcription alone. Gittes and colleagues have also demonstrated this synergy between the GLP-1R and TGFβ pathways in the developing pancreas using mouse e11.5 pancreas treated with exogenous TGFβ1 and Ex-4 (Tei et al., 2005). When administered together there was a 4.5-fold increase in insulin-positive differentiation, greater than that seen with either compound alone. Of note Ex-4 (100 pM for 6 days) alone, caused only a slight increase in the numbers of cells immunoreactive for insulin but no increase in those positive for glucagon.
In a follow-up study the Gittes laboratory found evidence that GLP-1R-induced differentiation of AR42J cells involves activation of the bone morphogenetic protein (BMP) signaling pathway first, followed by the TGFβ isoform signaling mechanism (Fig 4; Yew et al., 2005). BMPs are cytokines that also form part of the TGFβ superfamily (Zhang and Li, 2005). Yew and co-workers found endogenous expression of transcripts for the BMP Smads 1 and 8, the BMP-2 ligand and activin receptor-like kinase-1 (ALK-1, which activates Smads 1 and 5) were increased and Smad 5 decreased, following Ex-4 treatment of the AR42J cell line. When the cells were also exposed to BMP ligand inhibitors there was a suppression of the increase in both the BMP Smads 1 and 8, as well as Smad 3 in the TGFβ pathway observed in response to Ex-4 treatment. Inhibition of the BMP pathway did not prevent the Ex-4-induced decrease in Smad 2 and 5 transcript levels. Morpholino anti-sense against ALK-1 blocked Ex-4 induced expression of insulin II, pdx-1, and pax 4 seemingly by blocking the increase in Smad 3 mRNA levels. As in the case with BMP-ligand inhibition Smad 2 levels were unaffected. Use of anti-sense against the ALK-1 activator BMP-2 also did not effect Smad 2 or 5 expression but did reduce insulin II, pdx-1 and pax-4 mRNA levels. Based on these results Gittes proposed a possible mechanism (see Fig 4) by which BMP protein ALK-1, was activated by BMP-2 heterodimer formation with either BMP4 or 7. ALK-1 in turn upregulates Smad1, which leads to a suppression of Smad2 and commitment to the endocrine hormone differentiation pathway. Upregulation of Smad3 was required for the progression to a more mature phenotype.
The importance of PDX-1 in GLP-1R mediated differentiation of ductal cells has been demonstrated by us and investigators in the Perfetti laboratory. Using the human ductal cell line Capan-1 we demonstrated that Ex-4 treatment (0.1 nM for up to 5 days) increased the percentage of hormone-positive cells from 8% (in medium supplemented with 10% serum) to 40% (Zhou et al., 2002). Ex-4 treatment increased both the total level, as measured in immunoblots of whole cell extracts, as well as nuclear levels of PDX-1, following incubation for 72 hrs. Overexpression of PDX-1 increased the number of insulin-positive cells to 80% of the total population. Correspondingly use of anti-sense against pdx-1 completely blocked the effect of Ex-4. Using EMSA we observed that there was a 12-fold increase in association of Foxa2 with the PDX-1 promoter region (oligonucleotide equivalent to −2109 to −2088) in nuclear extracts from cells treated with Ex-4 for 24 hr. However we did not determine whether Foxa2 was absolutely essential for upregulation of PDX-1 and differentiation of this cell line, a question that arises given the results presented by Yew and colleagues in the differentiation of the AR42J cells (vide supra). Perfetti and colleagues compared GLP-1-mediated differentiation of ARIP cells, a rat ductal cell line expressing PDX-1, with a human ductal cell line PANC1 that does not express PDX-1 (Hui et al., 2001). They found that the PANC1 cells were unaltered by GLP-1 treatment alone but upon transfection of PDX-1 were capable of expressing both insulin and glucagon positivity.
Finally, with the interest in generating new sources of β cells there have been several papers published examining the effects of GLP-1 or Ex-4 on precursor cells isolated from a number of different mammalian pancreata and these studies are summarized in Table 4. The main observation of all these papers is that GLP-1R-mediated differentiation requires cells that are progressively more mature. There are several papers included in this list that suggest that PDX-1 expression is a prerequisite for GLP-1R signaling to cause differentiation.
Table 4.
In vitro cell systems used to study the differentiation properties of GLP-1R agonists in various cell types.
| AR42J
Rat adenocarcinoma cell line |
GLP-1 (10 nM)
Ex-4 (1 nM) |
3 day treatment resulted in insulin, glucagon and PP positive cells, co-staining for insulin and glucagon observed, glucose responsive insulin secretion seen at 10 mM glucose, PKC, MEK and PKA dependent process. | (Zhou et al., 1999b) |
| ARIP
Rat ductal cell line PANC-1 Human ductal cell line |
GLP-1 (10 nM) | PDX-1 essential for the conversion of the cells as PANC-1 cells lacking PDX-1 did not exhibit insulin expression | (Hui et al., 2001) |
| βlox5
Immortalized cell line derived from purified population of human islets |
Ex-4 (10 nM) | Transfection of PDX-1, spherical cell-cell contact and GLP-1R stimulation required for optimal insulin expression | (de la Tour et al., 2001) |
| Immortalized pancreas cells from the H-2kb-tsA58 transgenic mouse | GLP-1 NN2211 derivative (250 nM) | Expression of insulin and Ngn3 prior to treatment with GLP-1, GLP-1 did not augment insulin expression, authors concluded that cells were too immature, see (Koizumi et al., 2005) | (Klein et al., 2001) |
| AR42J
Rat adenocarcinoma cell line |
GLP-1 (10 nM) | Increase in insulin, PDX-1, NeuroD/β2 and Nkx6.1 | (Zhu et al., 2002) |
| Capan-1 Human ductal cell line | Ex-4 (0.1 nM) | Insulin, glucagon and PDX-1 expression, cAMP increased, MEK dependent | (Zhou et al., 2002) |
| Expanded human islet cells of neuroendocrine phenotype | Ex-4 (10 nM) | Increase in insulin expression decrease in nestin express | (Abraham et al., 2002) |
| Fetal Porcine islet-like clusters | GLP-1 (100 nM) | Increase in PDX-1 expression, glucose sensitivity, no increase in the size of the clusters | (Hardikar et al., 2002) |
| AR42J | Ex-4 (5 pM) | Differentiation for 3 days produced insulin II, pdx-1, IAPP and Pax4 positive cells TGFβ and BMP dependent process | (Yew et al., 2005; Yew et al., 2004) |
| Sca-1+/c-Kit+ sorted cells from adult mouse salivary glands | GLP-1 (20 nM) | Spherical cell-to-cell contact required for GLP-1-mediated differentiation, insulin and glucagon co-expression. | (Hisatomi et al., 2004) |
| Immortalized pancreas cells from the H-2kb-tsA58 transgenic mouse | GLP-1 (20 nM) | Adenovirus mediated expression of PDX-1 required before GLP-1 treatment is effective | (Koizumi et al., 2005) |
| Monkey Embryonic Stem Cells | Ex-4 (1 nM) | C-peptide, PDX-1 and NeuroD expressed in treated cells | (Lester et al., 2004) |
| Expanded clonal population of mouse pancreatic neuroendocrine- | Ex-4 (10 nM) | Used in the culture medium along with other pancreatic hormone differentiation factors, 130 bFGF, EGF and Activin-A | (Seaberg et al., 2004) |
6.3 Potential GLP-1R effects after gastric bypass surgery
New observations related to gut factors and the control of β cell mass have recently been made in patients following bariatric surgery, implicating endogenous GLP-1 as a possible pathogenic factor. Service and colleagues reported that six patients after Roux-en-Y gastric bypass surgery had postprandial hypoglycemia and neuroglycopenia with elevated insulin levels that led surgeons to perform partial pancreatectomies for control of symptoms (Service et al., 2005). Therefore pancreatic specimens were available for histological evaluation and the patients were diagnosed as having nesidioblastosis and enlarged islets. The authors interpreted their findings as possibly resulting from large amount of trophic (humoral) factors i.e. GLP-1, being released as a result of the dumping of food into the lower small bowel leading to islet overgrowth. However, the control pancreata used for histological comparison and from whom islet sizes were reported to be smaller were from obese subjects that were not nearly as heavy as the subjects who had undergone surgery (BMIs of 34 versus 50). The staining appeared to have been done at a different time and/or with different methodology or under different fixative conditions than the staining of the patients that had bariatric surgery. It is quite possible that the conditions of the pancreata from the bariatric-treated patients resulted from the preceding obesity and were not post-surgical events. None of the six were reported to have diabetes prior to their bypass and so must have had robust insulin secretory capacity. The six patients had a median loss of 44 % of their pre-operative weight (they are roughly half the weight they were before surgery): this is a massive weight loss and could mean that the β cell mass/islet secretory capacity had not yet re-set itself from its previous capacity.
The Service report was followed a few months later by a similar short communication from Patti and colleagues, who reported on three Roux-en-Y gastric bypass patients who had severe postprandial hypoglycemia, again with hyperinsulinemia, on whom surgeons performed partial pancreatectomies, in one case a drastic 80% pancreatectomy (Patti et al., 2005). Their pathological examination led them to conclude that islet hyperplasia was present in all three patients and they postulated that high GLP-1 plasma levels post-surgery were causative. However, the data from Patti and co-workers is open to interpretation. Their reasons for claiming there was islet hyperplasia present were based on the following: 1) that islets appeared close to ducts (suggesting that islets were being induced to form from ducts), 2) occasional insulin-positive cells were present in ducts, and 3) β cell area in one patient was considered to be high (3.3 % of total pancreatic area). However, the observation of islets in close proximity to ducts is a perfectly normal anatomical finding in humans (Bouwens and Pipeleers, 1998; Watanabe et al., 1999), insulin-positive cells are a frequent occurrence in human pancreatic ducts and 3.3 % is in fact within the normal range for total pancreatic islet area (Butler et al., 2003). Indeed, if islet hyperplasia were pathologic, one would expect fasting hypoglycemia to be the most problematic clinical finding and not postprandial hypoglycemia. The patient who underwent 80 % pancreatectomy and one of the patients who underwent distal pancreatectomy, had little or no relief from their symptoms. This is contradictory to increased β cell mass being the cause of the hypoglycemia and leads to the conclusion that surgery should probably not have been performed. Another interpretation of the data from Patti and colleagues, as well from Service and colleagues, is that the substantial weight loss due to the bypass surgery causes marked reductions in insulin resistance, occurring in the setting of β cell hypertrophy as a result of the preceding obesity. None of the three patients were reported to have suffered from diabetes prior to their gastric bypass and therefore, as with the Service report, they must be presumed to have had robust hypertrophic and hyperactive islets leading up to the bypass. It would not be unrealistic to expect that it may take months to years for the full homeostatic mechanism controlling insulin secretion/insulin sensitivity to re-set itself after profound weight loss. It must be remembered that it took years for the patients to reach their levels of obesity and so re-setting their homeostatic mechanisms is not likely to be quickly accomplished. Two recent publications comparing postprandial levels of the gut peptides PYY and GLP-1 in humans before and after Roux-en-Y gastric bypass surgery have demonstrated significant increases in both hormones (le Roux et al., 2006; Morinigo et al., 2006). In light of the results produced by Patti and co-workers, any type of pancreatectomy should be avoided in patients who have postprandial hypoglycemia following gastric bypass surgery.
Butler and colleagues carried out a histological evaluation of the same six pancreata as in the Service report and concluded that there was no evidence of increased β cell formation (either islet neogenesis or β cell replication) or decreased β cell loss in the patients after gastric bypass surgery. Their only positive finding was that in patients with post-gastric bypass surgery hypoglycemia, β cell nuclear diameter was greater than that of BMI-matched control subjects but appeared appropriate for the BMI of the patients before surgery (Meier et al., 2006a). It therefore now appears more certain that in some patients there is failure of insulin secretory mechanisms (and possibly β cell mass) to reset to the decreased requirement for insulin secretion as a consequence of massive and precipitous weight loss after gastric bypass surgery.
6.4 β cell toxicity and death: protective effects of GLP-1R agonists
GLP-1-induced protection against the deleterious effects of the diabetic milieu, (i.e. increased cytokine-, glucose- and lipo-toxicity) shown in both T1- and T2DM is an aspect of the preservation of β cell mass observed in rodent models of both major forms of diabetes. GLP-1R activation has been demonstrated both in human islets in vitro and in rodent models in vivo to reduce β cell apoptosis. The rate of β cell apoptosis is very low (Scaglia et al., 1997) and therefore models of injury to the endocrine pancreas have to be used to measure potential protective effects of GLP-1R agonists against the demise of the β cell. The first published model was the inbred Zucker diabetic fatty rat (ZDF/GmiTM-fa/fa, previously described in section 6.1) to which GLP-1 was infused over 2 days (Farilla et al., 2002). Perfetti and colleagues used Alzet microosmotic pumps implanted in the interscapsular region of the ZDF rats to slowly infuse GLP-1 (30 pmol/kg·min) for 2 days. Following this period the pumps were removed and then the rats were left for 4 days. Six days after the beginning of the infusion an intraperitoneal glucose tolerance test (IPGTT) was performed (Farilla et al., 2002). The rats were euthanized on day 7 and their pancreata excised. Perfetti and colleagues assessed the degree of terminal deoxynucleotidyl transferase-mediated dUTP-digoxigenin nick end labeling (TUNEL) as a measure the number of apoptotic cells in the islets of untreated controls versus in those of the ZDF rats receiving the incretin hormone. In the control group they observed several aggregates of apoptotic cells in the exocrine parenchyma and in close proximity to the islets and fragmented nuclear apoptotic cells within the islets themselves. In the GLP-1-treated animals there was no clustering of apoptotic cells and the islets were virtually free of apoptotic cells. Correspondingly there was a reduction in immunostaining for the cysteine-aspartic-acid-protease involved in initiation of apoptosis, i.e. caspase-3, in the GLP-1-treated rats. In summary, GLP-1 produced a 3.6-fold drop in the total number of apoptotic β cells, a 1.4-fold increase in the number of Ki-67 positive or dividing β cells, and a 1.6-fold expansion of β cell mass relative to controls. This was reflected in an improvement of the parameters of the IPGTT. Drucker and colleagues used the low dose streptozotocin (STZ) model of β cell destruction in mice to examine the effects of Ex-4 on β cell apoptosis (Li et al., 2003). In a detailed set of experiments, STZ either alone, or combination with Ex-4, was administered to GLP-1R−/− mice, and wildtype-, CD1 and C57BL/6 mice. Co-administration of Ex-4 to the wildtype mice reduced morning fed blood glucose levels and glycemic excursions following an oral glucose tolerance test. The STZ/GLP-1R−/− mice displayed increased fasting blood glucose relative both to wildtype receiving STZ alone or combination with Ex-4. This indicates that endogenous GLP-1 has a protective effect as the wildtype mice exhibited higher levels of endogenous GLP-1 production. Indeed increased levels of apoptosis were shown in the islets of the GLP-1R−/− mice relative to the wildtype STZ treated and control mice. Ex-4 (100 nM) also induced a cytoprotective effect in monolayers of β cells derived from mouse islets, incubated with the combination of cytokines interleukin 1 β (IL-1β, 1 ng/ml) and tumour necrosis factor α (TNF-α, 5 ng/ml). Both of these cytokines are released by T cells and macrophages during an autoimmune response and are instrumental in the destruction of β cells (Cnop et al., 2005).
Perfetti and colleagues examined the mechanism of reactive oxygen species (ROS)-induced apoptosis in vitro in the MIN6 cell line (Hui et al., 2003). The extent of hydrogen-peroxide (50 μM for 30 min) induced apoptosis was reduced by prior GLP-1 (10 nM for 16 hr) administration as demonstrated by 1) reduced number of fragmented/damaged nuclei (from 60 % to 20 %, measured by Hoechst staining), 2) reduction in DNA fragmentation, 3) reduced cleavage by caspase 3 of the proenzyme form of the repair enzyme, poly-(ADP-ribose)-polymerase (PARP) and 4) increased levels of the mitochondrial membrane stabilizers and anti-apoptotic Bcl-2 family of proteins. Specificity to the GLP-1R was indicated as some of the experiments were performed using the GLP-1R antagonist exendin (9-39) (100 nM) and it was found to have no effect on reducing DNA fragmentation whereas Ex-4 had such an effect. Mechanistically, Hui and colleagues found that the PI3 kinase and cAMP pathways were both instrumental in GLP-1-mediated preservation of cell viability in the presence of ROS. The pharmacological inhibitors of PI3 kinase and cAMP, LY294002 (50 μM) and Rp-cAMP (50 μM) respectively, were found to significantly reduce the effect of GLP-1, whereas the MAPK inhibitor, PD098059 (50 μM) did not do so. Similar results were recently obtained using the long acting GLP-1R agonist liraglutide that has a modification so as to eliminate proteolysis by DPP-IV (Bregenholt et al., 2005). In a later study from the Perfetti group GLP-1 (10 nM) was shown to reduce caspase 3 and increase Bcl-2 expression in human islets cultured over 3–5 days (Farilla et al., 2003). This was also accompanied by an increase in immunoreactivity in the individual β cells for insulin, insulin mRNA levels and in glucose stimulated insulin secretion.
Buteau and colleagues examined the ability of GLP-1 to protect human islets against glucolipotoxicity (Buteau et al., 2004). Dispersed human islet cells plated on poly-ornithine treated glass coverslips and cultivated for a period of 24 hr in the presence of 25 mM glucose and/or palmitate (0.4 mM) were completely protected against the gluco- or lipotoxicity, respectively, by simultaneous incubation with GLP-1 (10 nM) as assessed by Hoechst and TUNEL staining. In the same study Buteau and co-workers then explored the mechanism of protection by using INS832/13 cells. In particular they examined the contribution of the PI3 Kinase/PKB pathway. PKB has been shown to exhibit protective effects against FFA-induced apoptosis and to be a key survival gene for β cells (Tuttle et al., 2001; Wrede et al., 2002). Expression of a dominant negative form of PKB in INS832/13 cells completely reversed the protective effect of GLP-1 against both glucose and palmitate induced apoptosis whereas overexpression of a constitutively active form of PKB completely eliminated the toxic effects of both treatments both in the presence and absence of GLP-1 (Buteau et al., 2004). A downstream target of PKB is the nuclear factor-κB (NF-κB)/Rel family of transcription factors that have been shown to regulate anti-apoptotic proteins including Bcl-2 (Mattson, 2005). A pharmacological inhibitor of NF-κB, BAY 11-7082 ((E)-3-[4-methylphenylsulfonyl]-2-propenenitrile, 20 μM) increased both the basal and combined, glucose and palmitate-induced apoptosis (Buteau et al., 2004). The inhibitor also blocked the action of GLP-1 in protecting the insulinoma cells. EMSA assays showed that GLP-1 increased NF-κB DNA binding activity in the clonal β cells by about 80 % over a 2 hr incubation period, a value that remained unchanged in the presence of high glucose and/or palmitate. The significance of these observations on NF-κB binding activity is not clear as the exact role of NF-κB in the β cell is not completely delineated at present and may vary during development compared to the situation in adult islets. NF-κB is activated by phosphorylation and subsequent degradation of its inhibitor κB (IκB). Once phosphorylated NF-κB enters the nucleus and acts as a transcription factor regulating the transcription of genes associated with both pro- and anti-apoptotic processes depending on the cell context (Mattson, 2005). This activation step has been demonstrated to occur in MIN6 cells in response to Ca2+ influx (Bernal-Mizrachi et al., 2001). Attenuation of NF-kB activation in β cells by overexpression of IκBα under control of the PDX-1 promoter leads to glucose intolerance and a downregulation of GLUT2, uncoupling protein 2 and the vesicle protein Rab3c (Norlin et al., 2005). Therefore it seems that NF-κB plays a positive role in insulin secretion; however whether it mediates or prevents cytokine-induced β cell death is subject to debate and further investigation (Baker et al., 2001; Chang et al., 2003; Heimberg et al., 2001; Park et al., 2003). It is known that nutrients and cytokines employ two different pathways to initiate apoptosis (Cnop et al., 2005). Glucose and free fatty acids (FFAs) trigger apoptosis by causing ER stress, which is NF-κB independent. Cytokines initiate β cell death via an NF-κB dependent mechanism that results in caspase 3 activation, raising the question of the relevance of the observations of Buteau and co-workers. Of course it should always be remembered that the findings in insulinoma cells may not reflect what occurs in primary β cells and results obtained using insulinoma cells in turn will vary from cell line to cell line.
The Brubaker laboratory has also studied the involvement of PKB in GLP-1R-mediated protection from cytokine induced apoptosis and necrosis in INS-1E cells (Li et al., 2005a; Wang et al., 2004). Regulation of PKB activity is achieved by two regulatory phosphorylation sites one in the activation loop within the kinase domain Thr 308 and Ser 473 in the C-terminal of the regulatory domain (Li et al., 2005a). Phosphorylation at both sites is required for complete activation. Brubaker and colleagues first demonstrated that PKB was rapidly (5 min) phosphorylated at Ser 473 (2.7-fold above basal) in response to GLP-1 (10 nM) treatment, a process reversed by treatment with the PI3 kinase inhibitor wortmannin (Wang et al., 2004). Then they examined the dependency of anti-apoptotic properties of Ex-4 (10 nM for 18 hr) on PKB activation in this insulinoma cell line (Li et al., 2005a). They used adenovirus-mediated expression of a constitutively active PKBα or a kinase dead PKBα and monitored, 1) apoptosis as assessed by Hoechst 33342 expression 2) caspase 3 activation, 3) degree of necrosis as determined by iNOS levels, and 4) total glycogen synthase kinase β (GSK3β) levels, when the cells were treated with a mixture of cytokines, IL-1β, TNF-α and interferon-γ (10–50 ng/ml) in the absence or presence of Ex-4 (Li et al., 2005a). In all of the assays it was demonstrated that the presence of the active form of PKB enhanced the pro-survival properties of Ex-4 whereas this function of GLP-1R activation was lost in cells expressing the kinase dead form of PKB. Park and colleagues (Park et al., 2006) also found that IGF1 (10 nM for 1–10 min) increases in phosphorylated PKB (Ser 473) were increased about 2-fold in human islets in response to treatment with Ex-4 (5 nM for 8 hr) although total expression levels remained unchanged. This has further implications for β cell survival dependent on downstream signaling to IRS2 and is discussed below.
Perfetti and co-workers expressed the GLP-1 fragment of the proglucagon gene under control of the rat insulin II promoter in MIN6 cells (D’Amico et al., 2005) and showed that this conferred protection against a cocktail of immunosuppressive reagents (sirolimus 25 ng/ml, mycophenolate 17.5 μg/ml and FK506 75 ng/ml) a commonly used regimen in organ transplantation. As discussed above in section 5.2, FK506 inhibits insulin synthesis but it has also been shown to cause reversible toxic effects to pancreatic islets. In biopsies taken from pancreata in 20 simultaneous kidney-pancreas transplant recipients on immunosuppressive regimens including FK506 and cyclosporin A, islet cell toxicity was observed in the form of cytoplasmic swelling, vacuolization and loss of secretory vesicles (Drachenberg et al., 1999). In the MIN6 cells D’Amico and colleagues (D’Amico et al., 2005) did observe increased necrosis and apoptosis, caspase 3 levels and levels of the proapoptotic markers PARP and Smac/Diablo in response to the cocktail of immunosuppressive drugs that they administered. The cells expressing GLP-1 exhibited high levels of Bcl-2 and were resistant to the effects of the immunosuppressive drugs. To distinguish between the ability of GLP-1 to increase insulin content and secretion and effects on β cell survival they treated cells simultaneously with diazoxide to inhibit insulin secretion. They found that, while diazoxide treatment of MIN6 cells reduced insulin secretion considerably, it did not significantly diminish the resistance of the GLP-1-transfected cells to the immunosuppressive drugs.
Glucocorticoids are another class of drugs known to affect insulin secretion and to induce hyperglycemia which can lead to steroid-induced diabetes. Previously it was not known whether these drugs could actually cause β cell death. A recent paper by Ranta and co-workers showed that dexamethasone (0.1 μM) administration to INS-1 cells resulted in increased apoptosis as measured by 1) TUNEL assay, 2) increased caspase 3 activity, 3) PARP cleavage and 4) decreased Bcl-2 expression (Ranta et al., 2006). As in the models described already Ex-4 (10 nM) protected against dexamethasone-induced death. In agreement with the concept of a cAMP component to the mechanism of protection forskolin (10 μM) was also found to inhibit the apoptotic effects of dexamethasone. The authors used H89 (10 μM) and KT5720 (5 μM) to pharmacologically inhibit PKA in order to show that Ex-4 protected against cell death in a PKA-dependent manner. The specific Epac activator 8CPT-Me-cAMP (50 μM) did not mimic the effects of Ex-4. This implied that the cAMP/PKA pathway is important for the anti-apoptotic pathway stimulated by GLP-1R activation. As discussed earlier in section 3.3 under conditions of low and localized subcellular increases in cAMP as in those stimulated by GLP-1R activation of AC the preferred pathway appears to be via PKA activation rather than through Epac2. Kwon and co-workers have demonstrated that this is the case for the anti-apoptotic properties of GLP-1 in which they also found a PKA-dependent component of the mechanism in the prevention of FFA-induced apoptosis in RINm5F cells (Kwon et al., 2004b).
The importance of cAMP/CREB mediated survival of β cells downstream of GLP-1R and forskolin activation of AC was established in vivo by the White and Montminy laboratories using a transgenic mouse expressing a dominant negative form of CREB (referred to as A-CREB) specifically in the β cell (Jhala et al., 2003). Initially they demonstrated in vitro using MIN6 cells expressing A-CREB that GLP-1R activation led to transcriptional upregulation of IRS2 in a CREB-dependent fashion. They found that the IRS2 promoter contains a CREB half-site (TGACG) and they were successfully able to recover the IRS2 promoter in genomic DNA following a chromatin immunoprecipitation assay using an anti-CREB antibody. Furthermore in vivo functional significance for cAMP/CREB in β cell survival was established in the A-CREB transgenic mice which demonstrated reduced β cell area and increased staining for caspase 3 and 6 exclusive to the β cell compartment of the islet.
IRS2 −/− mice, while developing normally, display increased insulin resistance and decreased compensatory β cell hyperplasia, increasing apoptosis and therefore become diabetic (Kubota et al., 2000; Withers et al., 1998). The age of onset of diabetes in the transgenic mice is strain-dependent, with C57B6 mice on the SBA background developed by the Kadowaki laboratory (Kubota et al., 2000) showing a later age of onset (10 weeks) than those developed on the 129/Sv background used by the White laboratory (6–8 weeks; Withers et al., 1998). There are also strain-dependent differences in PDX-1 mRNA levels, and β cell area with the C57B6/SBA mice showing less severe reductions in both parameters possibly explaining their reduced propensity to diabetes at an earlier age. PDX-1 was shown by the White group to be significantly ablated in the IRS2−/− mice (Kushner et al., 2002) leading them to hypothesize that this was the central lesion in the reduced β cell mass observed in their mice. When PDX-1 expression was increased in the IRS2 −/− mice developed on the C57B6/129/Sv background by crossing them with mice expressing multiple copies of PDX-1 under control of the PDX-1 promoter their, β cell area was restored and the severity of their diabetes was reduced (Kushner et al., 2002).
As stated above in section 5.3 when IRS2 is activated FoxO1 will be deactivated by phosphorylation by PI3 kinase and thus will no longer inhibit PDX-1 transcription or nuclear translocation. The involvement of IRS2 in Ex-4-mediated protection against apoptosis was illustrated in vivo by the White group. When IRS2−/− mice were treated with Ex-4 (via osmotic pump 300 pmol/kg of body weight per day or 150 pmol ip injection every 12 hr) Ex-4 did not prevent the progressive β cell loss that occurs in this phenotype (Park et al., 2006). It would seem from the experiments in these mice that increased GLP-1R-mediated β cell survival is dependent on an intact IRS2 signaling cascade. Transgenic mice heterozygous for PDX-1 expression exhibit a greater degree of β cell apoptosis and caspase activity (Johnson et al., 2003). When mice with a β cell specific defect in PDX-1 were treated with Ex-4 there was no decrease in apoptotic nuclei compared with wild-type littermates (Li et al., 2005c). Therefore both IRS2 and PDX-1 appear to be important for β cell survival and for the ability of GLP-1R agonists to protect against apoptosis and β cell death in general.
Finally a recent paper (Chen et al., 2006) presents evidence that Ex-4 downregulates thioredoxin-interacting protein (TXNIP) an apparent pro-apoptotic factor in the β cell (Minn et al., 2005). INS-1 cells treated with Ex-4 (100 nM for 24 h) exhibited a reduction in TXNIP mRNA with a concomitant decrease in caspase-3 and Bax transcript levels (Chen et al., 2006). Correspondingly viral-mediated overexpression of TXNIP in INS-1 cells reduced the capacity of Ex-4 to protect against hydrogen peroxide induced apoptosis. Finally islets isolated from C3H/HeJ mice treated daily for one week with Ex-4 (24 nmol/kg) exhibited significantly lower amounts of TXNIP transcript by RT-PCR when compared to saline treated controls.
6.5 GLP-1 effects on β cell lipolysis
Increased plasma levels of free fatty acids (FFAs) are a risk factor for T2DM leading to formation of triglycerides in adipocytes (Kashyap et al., 2003). Increased intracellular levels of FFAs in the long-term are detrimental to β cell function (Zhou et al., 1998). In contrast, in low levels of glucose and FFA, fat storage is an important source of energy for β cells (Malaisse et al., 1985). GLP-1 has been shown to stimulate fatty acid synthesis from triglyceride stores in both clonal β cell lines and in rat islets. Yaney and colleagues showed that acute treatment of the HIT cell line with GLP-1 (10 nM for 1 hr) increased formation of FFA via a PKA-dependent activation of hormone sensitive lipase which led to the rapid breakdown of internal stores of triglycerides (Yaney et al., 2001). Treatment with the acyl synthetase (acyl CoA) inhibitor Triacsin C (which will inhibit FFA conversion to long chain (LC)-acyl CoA) increased the efflux of FFA in response to GLP-1. Treatment with the lipase inhibitor, orlistat inhibited the effect of forskolin on insulin secretion thereby leading the authors to hypothesize that LC-acyl CoA plays an influential role in cAMP mediated augmentation of insulin secretion. They propose that LC-CoA accumulate in the cytosol due to increased malonyl CoA production. Malonyl CoA will inhibit carnitine palmitoyl transferase thus inhibiting mitochondrial uptake and oxidation of LC-CoA. Corkey and colleagues had previously shown evidence for the ability of LC-CoA to regulate various steps in insulin secretion ranging from ion channel activation, calcium flux, to regulation of PKC (Deeney et al., 1992; Larsson et al., 1996; Yaney et al., 2000).
In contrast to the Corkey laboratory, Winzell and Ahrén did not find any evidence of acute effects of GLP-1 (100 nM for 1 hr) on palmitate oxidation or islet lipolysis in isolated normal mouse islets (Winzell and Ahrén, 2004). However islets isolated from animals fed a high fat diet and treated with Ex-4 over a period of 16 days did show increased palmitate oxidation and islet lipolysis relative to those on a high fat diet not receiving Ex-4 treatment (Winzell and Ahrén, 2004). Bulotta and colleagues showed that GLP-1R-mediated stimulation of de novo fatty acid palmitate synthesis in a rat and a human pancreatic ductal cell line was an important aspect of the differentiation of these cells into endocrine hormone cells (Bulotta et al., 2003). Nauck and co-workers recently studied the effect of administering GLP-1 (1.2 pmol kg−1 min−1) to healthy fasted male human volunteers over a 390 min period starting 30 min prior to eating a solid meal on postprandial triglyceride plasma levels (Meier et al., 2006b). They found that triglyceride levels were significantly increased in the control placebo group relative to the group receiving GLP-1. Thus long-term treatment with GLP-1 may prevent the formation of triglycerides in β cells and thereby reduce β cell toxicity in the diabetic state, which would be an added bonus to the use of GLP-1R in the treatment of diabetes.
6.6 Effects of GLP-1R activation during pancreatic development and in the neonatal pancreas
Pancreatic endocrine cell development and islet formation in rodents has undergone intensive investigation recently. One of the prime motivating factors is the potential application of ontogenic factors and specific pancreatic progenitor cell markers to the differentiation of islet cells precursors to treat diabetes. Transgenic mouse models applying both cell- and time- specific expression of lineage markers and/or repression or overexpression of various pancreatic transcription factors and regulators thereof, have led to a temporal model for transcription factor expression in the various pancreatic cell lineages (Wilson et al., 2003). In the rodent there are two phases of islet cell expansion in utero. The first occurring at mouse embryonic day 9 consists almost entirely of glucagon-positive cells (Pictet et al., 1972). The second wave of expansion of endocrine cells is noted at day 15 (Han et al., 1986). There is evidence of considerable restructuring of the endocrine pancreas through islet cell apoptosis in neo-nates (Trudeau et al., 2000). The GLP-1R transcript has been detected in rat fetal islets taken at embryonic day 21 and in neonate suckling rats (Garcia-Flores et al., 2001). In addition GLP-1 itself is produced in adult α cells that express low levels of PC1/3 convertase resulting in the processing of proglucagon to GLP-1 and GLP-2. Similarly PC1/3 has been found in the first wave of glucagon-positive cells in the endocrine pancreas indicating the possibility of GLP-1 production in these cells (Wilson et al., 2002).. The number of glucagon/PC1/3 positive cells decreased with embryonic age of the pancreas. PC1/3 is expressed in all islet cells and acts in concert with PC2 in the β cell to cleave pro-insulin into insulin however it is the virtually exclusive presence of PC1/3 that is responsible for the conversion of proglucagon into GLP-1, GLP-2 and glicenten in the L cells of the gut (Scopsi et al., 1995). It is not certain (as nearly all antibodies raised against GLP-1 and available for use at the time of Wilson and colleagues’ publication were also immunoreactive for proglucagon) whether GLP-1 is present but the presence of the enzyme responsible for conversion implies the possibility that GLP-1 is present and could therefore exert an effect on endocrine development in the pancreas.
Incubation of rat islets from the 21 day old fetuses in normoglycemic glucose concentration (5.5 mM) results in an increase in mRNA levels of GLP-1R relative to the levels seen with low (2.8 mM) or high concentrations of glucose (>20mM; Garcia-Flores et al., 2001). A similar phenomenon has been observed in adult islets (Abrahamsen and Nishimura, 1995). Fetal islets were more effective than adult islets at utilizing and oxidizing glucose but were less sensitive to glucose induced insulin secretion (Garcia-Flores et al., 2001). GLP-1 treatment (100 nM) of the 21-day old fetal islets was able to significantly enhance insulin response to both, low (1.67 mM), normoglycemic (5.5 mM) and supraphysiologic (16.7 mM), concentrations of glucose. However the rat fetal islets secrete significantly less insulin in response to glucose alone than do the adult islets in the same conditions. As GLP-1 is presumed to be present in the embryonic pancreas the lack of robust response was considered by Garcia-Flores and colleagues to be due to the immaturity of the glucose sensing or insulin secretory machinery of the fetal β cells. Considering that exposure of the pancreas to gut-derived GLP-1 will occur upon the first ingestion of food it is probable that GLP-1 could play a major part in sensitizing the neonatal islets to glucose when first ingested. It would be very interesting to know absolute levels of GLP-1 present in the pancreas and/or embryo per se and to compare them with levels, post-partum, and upon first weaning to gauge the relative importance of GLP-1 at these particular timepoints in islet development. As discussed in section 6.2.3 treatment of mouse embryonic pancreas e11.5 with Ex-4 (100pM for 6 days) results in increased numbers of insulin positive cells (Tei et al., 2005).
A role for GLP-1 in neonatal regenerative responses to insult has been established using streptozotocin (STZ, 70 mg/kg) treatment of 4 day old rats that were then monitored for a total of 40 days (Thyssen et al., 2006). This is a well-characterized model showing a 60 % reduction in the number of insulin-immunoreactive cells. The insertion of microosmotic pumps slowly releasing GLP-1 (9-39) (an apparent antagonist at GLP-1R; Montrose-Rafizadeh et al., 1997b) over a 2 week period (terminating on day 19) allowed the authors to define the contribution of this incretin to the regenerative process. Some interesting observations arose out of this study. Firstly there was an increase in circulating levels of GLP-1 on day 8 of the experiment due to an increase in the number of pancreatic glucagon positive cells (over days 8 to 14) obviously capable of processing and secreting GLP-1. Four days following treatment with STZ an increased number of insulin positive cells adjacent to pancreatic ducts was observed. Untreated controls showed a substantial increase in islet volume-weighted mean islet volume between days 4 and 8 but a decline between days 8 and 20 due most probably to remodeling by apoptosis. Animals receiving the STZ treatment were euglycemic and displayed normal levels of insulin mRNA by the end of the observation period. However the addition of the GLP-1R antagonist Ex (9-39) retarded the recovery of the endocrine pancreas from STZ treatment exhibiting intolerance to glucose. Similarly adult rats in a low dose STZ protocol also exhibited higher levels of circulating GLP-1 and intraislet GLP-1 immunoreactivity (Nie et al., 2000).
Postnatal administration of Ex-4 has been shown to alleviate diabetes linked with uteroplacental insufficiency and fetal growth retardation in a rodent model of same (Stoffers et al., 2003). Simmons and colleagues previously used a rat model of intra-uterine growth retardation by ligation of the bilateral uterine artery in late gestation period of the rat (Simmons et al., 2001). This results in the development of diabetes at 15–26 weeks of age due to a progressive decline in β cell mass with accompanying insulin secretory defects. Treatment of the intrauterine growth retarded (IUGR) rats with Ex-4 (1 nmol·kg body wt−1·day−1) on postnatal days 1–6 prevented the development of diabetes seen in the IUGR rats at 8 months of age. The IUGR rats treated with Ex-4 remained healthy and normoglycemic up to 18 months of age by which time all of the IUGR rats had expired (Stoffers et al., 2003). This was due to the ability of Ex-4 to preserve the pancreatic β cell mass that declined in the IUGR rats beginning at 7 weeks of age. This decrease in islet cell mass is not due to apoptosis but to a reduced β cell proliferation rate and reduced β cell differentiation. Treatment with Ex-4 normalized the proliferation rate in the IUGR rats. In humans there is a statistical correlation between poor fetal growth and the development of T2DM later in life (de Rooij et al., 2006; Hales et al., 1991; Ravelli et al., 1976). Therefore knowledge of the potential of Ex-4 to counteract this problem in rodents could be a precedent for the treatment of human beings. It is interesting to note that ex vivo perfusions of the human placenta with Ex-4 indicated negligible transfer of the peptide across the placenta; thus maternal use of the peptide during gestation would seem not to result in the fetus coming into contact with modulating concentrations of the insulinotrope (Hiles et al., 2003).
7. Effects on glucagon secretion, are they direct or indirect?
As already explained above (section 2) there is controversy about the presence of GLP-1Rs on α cells and if present, they are on but a few cells. The functional assays examining effects of GLP-1 on α cells vary. Heller and Aponte performed dose response analysis of GLP-1 treatment of whole islets and did not see any increase in glucagon secretion (Heller and Aponte, 1995). Moens and colleagues also failed to elicit cAMP production in rat α cells in response to 1 nM GLP-1 (Moens et al., 1996): but it should be remembered that it can be difficult to measure intracellular cAMP in small numbers of primary cells. Likewise when Franklin and co-workers measured glucagon secretion from fluorescence activated cell sorted (FACS) individual rat α cells GLP-1 was shown to have no effect on pyruvate-stimulated glucagon secretion (Franklin et al., 2005). They also stated that no transcripts for GLP-1R were found in the cells. However capacitance measurements and glucagon secretion experiments in response to GLP-1 performed by Ding and co-workers showed a potentiation of glucagon secretion evoked by voltage-clamp depolarizations indicating a functional GLP-1R on FACS sorted rat α cells (Ding et al., 1997). In glucagonoma cells (INR1-G9) that have been transfected with human GLP-1R, GLP-1 stimulation leads to glucagon secretion and increases in intracellular cAMP not inhibition (Dillon et al., 2005). The wild-type INR1-G9 cells do not contain transcripts for GLP-1R (Dillon et al., 2005 ;Fig 5). Matsumura and colleagues (Matsumura et al., 1992) have shown that GLP-1 decreased cAMP and suppressed glucagon secretion in INR1-G9 cells. However, given that we and the Dillon laboratory do not observe the expression of GLP-1R in this cell line, the mechanism for this is difficult to comprehend.
Fig 5.
INRI G9 cells were transfected with the rat GLP-1R. (A), (B), RT-PCR and western blot of INR1 G9 cells demonstrate the absence of GLP-1R gene and protein in native cells but its presence in transfected cells. CHO and RIN cells serve as negative and positive controls, respectively. (C), Intracellular cAMP levels in transfected cells in response to GLP-1(10 nM). (D), GLP-1-mediated glucagon (30 min) secretion into the medium of transfected cells shows no glucose-dependency.
GLP-1 infusions suppress glucagon secretion i.e. are glucagonostatic, in both healthy subjects as well as subjects with T1DM and T2DM (Gutniak et al., 1992). The suppression of glucagon secretion in vivo, therefore, especially in humans where δ cells are present throughout islets, is likely due to increased intra-islet release of somatostatin and/or insulin by GLP-1 (Fehmann et al., 1995). Increased somatostatin secretion in response to GLP-1 has been shown in both perfused rat pancreas (Schmid et al., 1990) and isolated islets (Fehmann et al., 1995). GIP, in contrast, actually increases glucagon secretion in healthy subjects under normoglycemic conditions (Meier et al., 2003) while having no effect in T2DM (Creutzfeldt and Nauck, 1992). The inference is that GIP is not glucagonotropic under hyperglycemic conditions. No one has yet looked for the presence of GIP receptors on α cells, but it is likely that they are present, accounting for the increased glucagon secretion seen under normoglycemic conditions. If the GIPR is present on α cells then GIP infusions would be expected to increase cAMP levels and induce glucagon secretion. The experiments described above illustrate the diversity of action apparent between GIP and GLP-1 on the pancreas. Our assessment of the literature to date is that GLP-1R is not expressed on α cells in vivo. Expressing GLP-1R in α cells in vitro causes glucagon secretion and increases in intracellular cAMP levels in response to GLP-1. Therefore any effects of GLP-1 on glucagon secretion in vivo are likely due to secondary effects, consequent to β and/or δ cell activation.
8. Effects on exocrine and ductal pancreatic cells
8.1 Exocrine pancreatic secretion
Fehmann and colleagues were the first to study the effect of GLP-1 on pancreatic acinar secretions (Fehmann et al., 1990). They examined the synergistic action of GLP-1 (10 pM) and cholecystokinin-8 (CCK-8, 1 nM–1 pM) on isolated rat pancreatic acini and found that GLP-1 had no impact on CCK-induced amylase secretion. Eng and colleagues performed a dose response curve and found that both Ex-4 and GLP-1 could increase cAMP levels in dispersed guinea pig acini but did not actually increase amylase release independent of treatment with CCK-8 (Eng et al., 1992; Raufman et al., 1992). The observed increases in cAMP seen with both peptides in the guinea pig acini were inhibited by Ex (9-39) indicating that the GLP-1R did indeed mediate the effects (Eng et al., 1992; Raufman et al., 1992). In contrast to the results of Fehmann and colleagues, Eng did see an increase in amylase secretion when either GLP-1 or Ex-4 were applied simultaneously with CCK-8. Eng and colleagues explain the differences as resulting from the low concentration of GLP-1 used by Fehmann and an apparent greater sensitivity of the guinea pig to GLP-1. Eng and co-workers state that they did not observe a very strong synergistic action of GLP-1 and CCK-8 in rat pancreatic acini. Also at 10 pM of GLP-1 Eng did not observe an effect on CCK-8 induced amylase release, but the potentiating action of GLP-1 was observed at 1 nM or greater in either guinea pig or rat pancreatic acini.
We have used the AR42J cell line derived from a rat pancreatic tumor to examine the effect of GLP-1 on exocrine cells (Zhou et al., 1999a). We confirmed the presence by RT-PCR of the GLP-1R on these cells and examined cAMP stimulation, [Ca2+]i, and amylase release. Treatment for 10 min with GLP-1 (10 nM) or Ex-4 (0.1 nM) caused a 1.5-fold and a 3-fold increase in intracellular cAMP levels, respectively. These results are compatible in magnitude with those observed by Eng and colleagues. We found in contrast to earlier results from the Eng group performed in guinea pigs (Malhotra et al., 1992) that GLP-1R stimulation did increase [Ca2+]i levels. Exposure of the AR42J cells to GLP-1 of which 1 nM achieved maximum amplitudes, elicited [Ca2+]i responses in approximately 50% of the cells. These responses occurred at a slower rate and showed smaller amplitudes than were observed with CCK treatment. We also found that there was no effect of GLP-1 on CCK-induced amylase secretion. We further examined the mechanism and observed that while CCK-8 produced extensive tyrosine phosphorylation of several cellular proteins, GLP-1 did not. Genestein blocked CCK-induced phosphorylation events and amylase secretion, and vanadate increased amylase secretion. This would imply that tyrosine phosphorylation is required for amylase release in rat acinar cells and that GLP-1 does not stimulate this pathway sufficiently to elicit amylase secretion. Therefore we conclude that guinea pig pancreatic acinar physiology is substantially different from that of the rat.
8.2 Pancreatic exocrine and ductal cell growth
GLP-1R activation either in vitro in ductal or acinar cell lines or in vivo in rodents causes an initial burst of proliferation followed by cell cycle arrest leading to differentiation of a large fraction of these cells into pancreatic hormone expressing cells. Specifically treatment of AR42J cells with GLP-1 (10 nM) for 24 hr resulted in 80 % of the cells exhibiting uptake of BrdU versus 12% on day 3 of treatment (Zhou et al., 1999b). In contrast following three days of treatment 60 % of the control cells were still proliferating. At this point almost 70 % of the treated cell population exhibited hormone immunoreactivity. Using proliferating cell nuclear antigen (PCNA) as a marker of exocrine proliferation in 6 and 22 month old wistar rats treated with GLP-1 (described in section 5.3) we observed an obvious (16.6 versus 6.2% of acinar cells, treated versus control) increase in acinar cell growth following 48 hr of treatment that dissipated by the fifth day of treatment at which point there was a 1.4-fold increase in β cell mass (Perfetti et al., 2000).
Bulotta and colleagues in the Perfetti laboratory quantified increases in cell number and cell cycle distribution in a rat pancreatic ductal cell line, ARIP, treated with GLP-1 (10 nM for 12 hr, 24 hr or 48 hr) following induction of cell cycle arrest (Bulotta et al., 2002). Unlike the observations in the acinar cells no initial increase in cell proliferation was observed as measured by the number of cells in cultures treated with GLP-1 (17 % lower than in untreated controls). This was accompanied by an increase in the number of cells in G0-G1 phase and reduction of those in the G2-M and S phases. They demonstrated that this was due to decrease in G1 cyclin-dependent-kinase inhibitors p27Kip1 and p21Cip1 following 24 hr of treatment. Immunostaining and semi-quantitative RT-PCR analysis of the treated and control populations of cells showed that at this stage of cultivation the cells in medium with GLP-1 were expressing insulin. What is interesting about this study is that the induction of insulin gene expression and loss of the ductal marker CK20 correlated very well. CK20 immunostaining was absent from the GLP-1 treated cells at 48 hr when levels of insulin mRNA were shown to be at their highest. This indicates a well orchestrated GLP-1 induced conversion of the progenitor cells reminiscent of the plasticity of these cells and their ability to respond to metabolic demand, in the milieu of the pancreas.
Should chronic activation of GLP-1R in pancreatic ductal cells increase proliferation of the cells as our studies in the AR42J cells and in the Wistar rat would seem to suggest then the potential for GLP-1 agonists to induce pancreatic adenocarcinoma arises (Hezel et al., 2006). This was addressed by Koehler and Drucker who recently reported a comprehensive study examining the proliferative effects of Ex-4 on human pancreatic cancer cell lines both in vitro and following transplantation in nude mice (Koehler and Drucker, 2006). They found the presence of GLP-1R in three human pancreatic ductal adenocarcinoma (CAPAN-1, CFPAC-1, and PL 45) and two carcinoma cell lines (PANC-1 and Hs 766T) but not in HPAC cells. When treated with Ex-4 (5 or 50 nM for up to 5 days) there was no increase in proliferation above that seen in the presence of 10% FBS. The CF-PAC-1 and PL 45 cell lines were the only ones studied that exhibited increases in intra-cellular cAMP and PI3 kinase activity respectively in response to Ex-4 treatment. When these two cells lines were implanted subcutaneously into nude mice that were subsequently treated with Ex-4 (24 nmol/kg) for four weeks there was no increase in the weight of the tumors. Neither was there an increase in BrdU incorporation in the implants. Therefore the Drucker group concluded that Ex-4 did not activate proliferation in these cell lines. It must be noted that although the conditions for the assay were similar in both cases Koehler and colleagues did not observe an increase in cAMP in CAPAN-1 cells in response to Ex-4 treatment although this has been reported (Fig. 4; Zhou et al., 2002). However upon closer examination of the data presented by Koehler and colleagues (Fig 2B; Koehler and Drucker, 2006) there is an elevation of cAMP observed at concentrations of Ex-4 at 10 nM and above when sampled at 10 min. Finally Koehler and colleagues also did not observe a protective effect of Ex-4 against drug-induced apoptosis in any of the cell lines.
9. GLP-1R−/− Mice
GLP-1R−/− mice display abnormally high blood glucose levels after an intraperitoneal glucose challenge demonstrating that GLP-1 is important for clearance of the glucose load, irrespective of the site of glucose entry into the circulation (Scrocchi et al., 1996). As anticipated from the known actions of GLP-1, they also exhibit mild fasting hyperglycemia and glucose intolerance after oral glucose that is associated with reduced glucose-stimulated insulin secretion. Despite evidence that pharmacological GLP-1 levels potently inhibit short-term food intake, GLP-1R−/− mice have normal body weight and food intake. Moreover, GLP-1R signaling is not required for maintenance of glucose competence in pancreatic β cells as glucose-induced insulin release is normal in islets isolated from GLP-1R −/− mice (Flamez et al., 1998) but the islets demonstrate abnormalities in basal and glucose-stimulated cytosolic Ca2+ (Flamez et al., 1999). In line with normal glucose-induced insulin secretion from isolated islets, fasted animals have no significant changes in fasting insulin mRNA and content in their pancreata (Scrocchi et al., 1998). There are non-significant reductions in insulin mRNA and pancreatic insulin content in the fed state. Although pharmacological levels of GLP-1 inhibit glucagon secretion, GLP-1R −/− mice have normal fasting and postprandial levels of glucagon and display normal whole-body glucose utilization (Scrocchi et al., 1998).
Lack of GLP-1R signalling is partially compensated for by GIP as both GIP secretion and GIP action are up-regulated in GLP-1R −/− mice (Pederson et al., 1998). This probably explains the mild phenotype of these animals. Interestingly, despite evidence implicating GLP-1R signaling as an important pathway for regulating β-cell proliferation and survival β cell mass is reported to be normal in GLP-1R −/− mice (Li et al., 2003). However, GLP-1R−/− mice exhibit morphological abnormalities: the pancreata display less of the large sized islets and the distribution of β cells is no longer solely in the periphery of islets (Ling et al., 2001).
10. GLP-1R agonists and GLP-1 analogs in the treatment of diabetes
10.1 GLP-1 as an insulinotropic agent
In 1986 and 1987 GLP-1 was shown to have insulinotropic properties in rodents (Holst et al., 1986; Mojsov et al., 1987) and there was little if any doubt about its potency from these early experiments. The first human experiments were performed by Bloom and co-workers in 1987 (Kreymann et al., 1987). They showed that infusing GLP-1 intravenously so as to reach plasma concentrations in the apparently physiological range lead to increased glucose-dependent insulin secretion. This glucose-dependency was reminiscent of what was known at that time about GIP, the other incretin. It also only stimulates insulin secretion when plasma glucose concentrations were in the rising phase of the curve (Elahi et al., 1984)
10.2 Native GLP-1 and treatment of diabetes
There was a hiatus of a few years following the discovery that GLP-1 is insulinotropic before the peptide was administered to diabetic subjects. This arose from the assertion that it would not be insulinotropic in that disease, the disappointing results with infusing GIP in diabetic subjects remaining uppermost in the minds of clinical researchers. GIP, even when given at very high concentrations did not increase insulin secretion or lower blood glucose in diabetic patients. However, it was soon obvious that GLP-1 did not mimic the pharmacologic profile of action of GIP. In 1992, exogenous administration of GLP-1 as a continuous intravenous infusion at a dose rate of 0.75 pmol/kg/min was anti-diabetogenic in both type 1 and type 2 diabetic subjects (Gutniak et al., 1992). The interpretation of the data was that GLP-1 lowered post-prandial glucose levels in type 1 subjects because it delayed gastric emptying. It lowered fasting and post-prandial glucose levels in type 2 diabetic patients because it increased insulin secretion and decreased glucagon secretion, as well as decreasing gastric emptying. Further studies showed that the effects in humans were consistent (Elahi et al., 1994; Nathan et al., 1992; Nauck et al., 1993a; Nauck et al., 1993b). Furthermore, GLP-1 was capable of lowering blood glucose even in patients with long-standing and severe T2DM and even in patients who no longer responded to sulfonylureas.
These exciting findings stimulated clinical researchers to explore the potential use of the peptide in the treatment of T2DM. It immediately became clear that simple subcutaneous injections gave but a weak and transient effect in insulin secretion and plasma glucose levels (Nauck et al., 1996): the reason being the cleavage of the histidine and alanine within 1–2 minutes from the N-terminus of GLP-1 by DPP-1V; the remaining fragment does not activate the GLP-1R (Hansen et al., 1999). Nonetheless, 7 days of bolus subcutaneous administration of GLP-1 before breakfast, lunch and dinner significantly improved post-prandial glucose levels and decreased plasma lipid levels (Juntti-Berggren et al., 1996). In another study, overnight intravenous GLP-1 lowered fasting and post-prandial plasma glucose levels to near-normal levels in subjects with T2DM (Rachman et al., 1997). It even reduced fasting and postprandial glucose levels after buccal absorption of a tablet containing 119 nmol of GLP-1 (Gutniak et al., 1997). Of particular importance were two studies of the effects of 6 weeks and 3 months continuous subcutaneous infusion of GLP-1 via MiniMed pumps in subjects with T2DM (Meneilly et al., 2003; Zander et al., 2002). In the first study GLP-1 at a dose of 4.9 pmol/kg.min caused a reduction in fasting glucose (by approximately 80 mg/dl) and hemoglobin A1c (by 1.3%) (Zander et al., 2002). Body weight was slightly decreased, fasting free fatty acids were also reduced, and the treatment was well tolerated with few adverse events. Fasting plasma glucagon levels, however, were not lower. In the second study of elderly patients (mean age 72 years), some of whom had diabetes for up to 13 years, half the patients had their oral diabetic agents withheld and were given GLP-1 infusion of up to 3.2 pmol/kg.min for 3 months (Meneilly et al., 2003). The other half continued with their usual diabetic medications. At the beginning and end of the study, the patients were subjected to a glucose clamp in order to study any improvements in insulin secretion. The GLP-1 infusion clearly restored first phase insulin secretion and improved plateau phase secretion. Hemoglobin A1c levels were equally maintained in both groups of patients. Body weights did not change in either group, and no adverse event occurred. Once again, however, plasma glucagon levels were not significantly lower in the subjects that received GLP-1 infusion. Additionally, insulin pulse mass and pulsatile insulin secretion were significantly increased by the chronic subcutaneous infusion of GLP-1. Approximate entropy, a measure of irregularity of insulin release, was also greatly improved by GLP-1 treatment (Meneilly et al., 2005).
Collectively, these studies using native GLP-1 show the potential of this agent to treat subjects with T2DM. They also show that the GLP-1R was not downregulated even by long-term stimulation with its native ligand and provide ‘proof-of-concept’ for the pharmaceutical industry to develop GLP-1R-based therapy. Continuous GLP-1 infusion is not a practical way of lowering blood glucose levels. There are therefore two obvious options remaining: use GLP-1 analogs or GLP-1R agonists that are resistant to DPP-1V activity and/or inhibit the enzymatic activity of DPP-1V.
10.3 GLP-1R agonists and DPP-1V inhibitors
One such agonist, Ex-4 (exenatide), is now a treatment for T2DM. As stated in the introduction this is a 39 amino acid peptide isolated from the salivary gland of the Gila monster lizard and it has 50 % amino acid homology to GLP-1 (see Table 1). It is not lizard GLP-1 (the lizard also synthesizes GLP-1; Chen and Drucker, 1997) and is encoded by a distinct Ex-4 gene. Ex-4 is not a substrate for DPP-IV because it contains histidine-glycine at its N-terminus and has a half-life of 4–5 hrs due to renal elimination. It binds and activates the GLP-1R with greater potency than native GLP-1 (Montrose-Rafizadeh et al., 1997b) and it has long-term antihyperglycemic actions in diabetic mice (Greig et al., 1999). In acute experiments it was found to be insulinotropic and glucagonostatic in both non-diabetic and type 2 diabetic subjects (Egan et al., 2002). Additionally, similar to native GLP-1, it restores first phase insulin secretion in type 2 diabetic subjects in response to glucose (Fehse et al., 2005). Furthermore, in subjects with T2DM, twice-daily subcutaneous injections of Ex-4 (daily dose 12 to 96 pmol/kg) for 1 month reduced post-prandial glucose levels and stimulated insulin secretion, leading to a reduction in HbA1c from 9.1 % to 8.3 % (Egan et al., 2003). Ex-4 is a potent inhibitor of gastric emptying (Egan et al., 2003) and it causes a progressive decline in weight in subjects that have been followed out to 82 weeks (Blonde et al., 2006; Buse et al., 2004; DeFronzo et al., 2005; Kendall et al., 2005). Attempts to develop DPP-1V-resistant GLP-1 analogs are well under way but none are yet approved by regulatory agencies for long-term use (see Table 5).
Table 5.
GLP-1R agonists currently in clinical use or in development for clinical use. Information is current from the websites of the companies developing the drugs at the time of writing.
| Company | Name | Compound | Indication | Stage of Development | Date |
|---|---|---|---|---|---|
| Amylin/Eli Lilly | Byetta | Ex-4 | Adjunctive therapy with metformin for type 2 diabetes | Marketed | 29th April 2005 FDA approval |
| Amylin/Alkermes/Eli Lilly | Exenatide LAR | Ex-4 long acting peptide for weekly SC injection | T2DM | Phase III | 24th March 2006 |
| Amylin | AC2592 | Continuous infusion of GLP-1 | Congestive heart failure | Phase II | Late 2004 |
| Conju Chem | PC-DAC™:Ex-4 | Exendin-4 | T2DM; HbA1c 6.5–11% | Phase I/II Clinical Trials | 25th January 2006 |
| Roche/Ipsen | BIM 51077 | GLP-1 Analogue | T2DM | Phase I Clinical Trials | 21st October 2003 |
| Novo Nordisk | Liraglutide /NN2211 | Modified GLP-1 compound (Knudsen et al., 2000) | T2DM | Phase II Clinical Trials | On-going |
| Zealand Pharma/Sanofi-Aventis | ZP10 | Modified GLP-1 compound for SC injection | T2DM | Phase II Clinical Trials completed | March 2005 |
| Theratechnologies | TH0318 | Modified GLP-1 compound for SC injection | T2DM | Phase I Clinical Trials completed | 23rd March 2005 |
| Transition Therapeutics/Novo Nordisk | GLP1-I.N.T™. | GLP-1 in combination with gastrin | Islet regeneration | Pre-clinical | 16th June 2005 |
| Human Genome Sciences/GlaxoSmithKline | GSK716155, Albugon | GLP-1 –derivatised with albumin (Baggio et al., 2004a) | T2DM | Phase I Clinical Trials | 10th January 2006 |
Oral DPP-1V inhibitors are being developed. Human data for vildagliptin (Ahren et al., 2005) has been submitted under New Drug Applications for review by the FDA. Sitagliptin (Bergman et al., 2006; Herman et al., 2005) was approved for marketing in October 2006. Saxagliptin (Augeri et al., 2005) and denagliptin are in phase III and phase II clinical trials respectively. As DPP-1V is involved in activating and inactivating many peptides (Augustyns et al., 1999), besides both GLP-1 and GIP, its long-term effects in humans are hard to fully predict.
11. Summary
GLP-1R activation has many beneficial effects on acute insulin secretion and the maintenance of correct β cell glucose sensing, transcriptional synthesis, proliferation and survival. This is most likely due to the activation and integration of multiple pathways consequent upon engagement of GLP-1 agonists with the receptor. Therefore for clinicians the use of GLP-1R agonists would seem to be the perfect treatment for chronic β cell failure in T2DM. However there is as yet only one GLP-1R agonist, exenatide, on the market.
GLP-1 increases the amount of insulin secreted by each β cell in response to glucose, and in addition, it increases the number of glucose responsive β cells: both effects are a consequence of its ability to enhance cAMP production. Agents such as forskolin also have this ability but it has become apparent that the regionalized nature of the increases induced by GLP-1 confers specificity and efficacy for the signaling mechanisms that modulate the machinery of insulin secretion. A major question for biochemists is precisely which of the pathways activated are the critical ones for GLP-1 to exert its specific effects on the β cell. Some effects require integration of multiple pathways and others are highly dependent on one major pathway in particular. This is particularly important for pharmacological harnessing of activators of these pathways if modulation of only one or a few aspects of β cell biology is desired. It should be apparent from our review that there is still some debate in the literature on this issue. Therefore what seems to be emerging from the analysis of the literature reviewed here is that 1) a high-codependency of multiple pathways to enhance β cell function, and 2) the spatial and temporal patterning of cAMP production in the β cell, are two important aspects of GLP-1 regulation of β cell function.
The relative contributions of the effect of GLP-1R activation on proliferation, differentiation and apoptosis to either preserving or increasing β cell mass are not known. Inability to accurately measure apoptosis will also hinder the acquisition of a complete picture. The effect of GLP-1 on β cell function also impacts on its contribution to β cell mass as improved function requires less β cell mass to respond to demand for insulin. All of the studies so far performed have used a variety of rodent models of diabetes and generally have examined only one or two aspects of GLP-1R regulation of β cell mass and function. Therefore it is difficult to quantify the exact contributions of the individual effects of chronic GLP-1 treatment on the dynamics of β cell mass. Also the extent to which GLP-1 stimulates these cell cycle mechanisms in the remaining four hormonal cell types within the islet is not known. Of course as pointed out in the Introduction as we cannot measure β cell mass in humans it is impossible at present to gauge the contribution of this aspect of β cell regulation to the improvement in diabetes at present.
Acknowledgments
MED would like to thank Ammon B. Peck. JME would like to thank Byeung-Jun Kim and Olga D. Carlson for figure 5 data and Tina Roberson for editorial assistance. The authors were supported by the Intramural Research program of the NIH, National Institute on Aging and have no conflict of interest in the writing of this review.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abraham EJ, Leech CA, Lin JC, Zulewski H, Habener JF. Insulinotropic hormone glucagon-like peptide-1 differentiation of human pancreatic islet-derived progenitor cells into insulin-producing cells. Endocrinology. 2002;143:3152–3161. doi: 10.1210/endo.143.8.8973. [DOI] [PubMed] [Google Scholar]
- Abrahamsen N, Nishimura E. Regulation of glucagon and glucagon-like peptide-1 receptor messenger ribonucleic acid expression in cultured rat pancreatic islets by glucose, cyclic adenosine 3′,5′-monophosphate, and glucocorticoids. Endocrinology. 1995;136:1572–1578. doi: 10.1210/endo.136.4.7534705. [DOI] [PubMed] [Google Scholar]
- Ahren B, Pacini G, Foley JE, Schweizer A. Improved meal-related beta-cell function and insulin sensitivity by the dipeptidyl peptidase-IV inhibitor vildagliptin in metformin-treated patients with type 2 diabetes over 1 year. Diabetes Care. 2005;28:1936–1940. doi: 10.2337/diacare.28.8.1936. [DOI] [PubMed] [Google Scholar]
- Ainscow EK, Rutter GA. Mitochondrial priming modifies Ca2+ oscillations and insulin secretion in pancreatic islets. Biochem J. 2001;353:175–180. doi: 10.1042/0264-6021:3530175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Sabah S, Donnelly D. The positive charge at Lys-288 of the glucagon-like peptide-1 (GLP-1) receptor is important for binding the N-terminus of peptide agonists. FEBS Lett. 2003;553:342–346. doi: 10.1016/s0014-5793(03)01043-3. [DOI] [PubMed] [Google Scholar]
- Alto N, Carlisle Michel JJ, Dodge KL, Langeberg LK, Scott JD. Intracellular targeting of protein kinases and phosphatases. Diabetes. 2002;51(Suppl 3):S385–388. doi: 10.2337/diabetes.51.2007.s385. [DOI] [PubMed] [Google Scholar]
- Arnette D, Gibson TB, Lawrence MC, January B, Khoo S, McGlynn K, Vanderbilt CA, Cobb MH. Regulation of ERK1 and ERK2 by glucose and peptide hormones in pancreatic beta cells. J Biol Chem. 2003;278:32517–32525. doi: 10.1074/jbc.M301174200. [DOI] [PubMed] [Google Scholar]
- Ashcroft SJ. The beta-cell K(ATP) channel. J Membr Biol. 2000;176:187–206. doi: 10.1007/s00232001095. [DOI] [PubMed] [Google Scholar]
- Augeri DJ, Robl JA, Betebenner DA, Magnin DR, Khanna A, Robertson JG, Wang A, Simpkins LM, Taunk P, Huang Q, Han SP, Abboa-Offei B, Cap M, Xin L, Tao L, Tozzo E, Welzel GE, Egan DM, Marcinkeviciene J, Chang SY, Biller SA, Kirby MS, Parker RA, Hamann LG. Discovery and preclinical profile of Saxagliptin (BMS-477118): a highly potent, long-acting, orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J Med Chem. 2005;48:5025–5037. doi: 10.1021/jm050261p. [DOI] [PubMed] [Google Scholar]
- Augustyns K, Bal G, Thonus G, Belyaev A, Zhang XM, Bollaert W, Lambeir AM, Durinx C, Goossens F, Haemers A. The unique properties of dipeptidyl-peptidase IV (DPP IV / CD26) and the therapeutic potential of DPP IV inhibitors. Curr Med Chem. 1999;6:311–327. [PubMed] [Google Scholar]
- Baggio LL, Huang Q, Brown TJ, Drucker DJ. A recombinant human glucagon-like peptide (GLP)-1-albumin protein (albugon) mimics peptidergic activation of GLP-1 receptor-dependent pathways coupled with satiety, gastrointestinal motility, and glucose homeostasis. Diabetes. 2004a;53:2492–2500. doi: 10.2337/diabetes.53.9.2492. [DOI] [PubMed] [Google Scholar]
- Baggio LL, Kim JG, Drucker DJ. Chronic exposure to GLP-1R agonists promotes homologous GLP-1 receptor desensitization in vitro but does not attenuate GLP-1R-dependent glucose homeostasis in vivo. Diabetes. 2004b;53(Suppl 3):S205–214. doi: 10.2337/diabetes.53.suppl_3.s205. [DOI] [PubMed] [Google Scholar]
- Baker MS, Chen X, Cao XC, Kaufman DB. Expression of a dominant negative inhibitor of NF-kappaB protects MIN6 beta-cells from cytokine-induced apoptosis. J Surg Res. 2001;97:117–122. doi: 10.1006/jsre.2001.6121. [DOI] [PubMed] [Google Scholar]
- Ban N, Yamada Y, Someya Y, Ihara Y, Adachi T, Kubota A, Watanabe R, Kuroe A, Inada A, Miyawaki K, Sunaga Y, Shen ZP, Iwakura T, Tsukiyama K, Toyokuni S, Tsuda K, Seino Y. Activating transcription factor-2 is a positive regulator in CaM kinase IV-induced human insulin gene expression. Diabetes. 2000;49:1142–1148. doi: 10.2337/diabetes.49.7.1142. [DOI] [PubMed] [Google Scholar]
- Barg S, Eliasson L, Renstrom E, Rorsman P. A subset of 50 secretory granules in close contact with L-type Ca2+ channels accounts for first-phase insulin secretion in mouse beta-cells. Diabetes. 2002;51(Suppl 1):S74–82. doi: 10.2337/diabetes.51.2007.s74. [DOI] [PubMed] [Google Scholar]
- Bazarsuren A, Grauschopf U, Wozny M, Reusch D, Hoffmann E, Schaefer W, Panzner S, Rudolph R. In vitro folding, functional characterization, and disulfide pattern of the extracellular domain of human GLP-1 receptor. Biophys Chem. 2002;96:305–318. doi: 10.1016/s0301-4622(02)00023-6. [DOI] [PubMed] [Google Scholar]
- Beguin P, Nagashima K, Nishimura M, Gonoi T, Seino S. PKA-mediated phosphorylation of the human K(ATP) channel: separate roles of Kir6.2 and SUR1 subunit phosphorylation. Embo J. 1999;18:4722–4732. doi: 10.1093/emboj/18.17.4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beinborn M, Worrall CI, McBride EW, Kopin AS. A human glucagon-like peptide-1 receptor polymorphism results in reduced agonist responsiveness. Regul Pept. 2005;130:1–6. doi: 10.1016/j.regpep.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Bell GI. The glucagon superfamily: precursor structure and gene organization. Peptides. 1986;7(Suppl 1):27–36. doi: 10.1016/0196-9781(86)90160-9. [DOI] [PubMed] [Google Scholar]
- Bender AT, Beavo JA. Cyclic n–cleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev. 2006;58:488–520. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- Bergman AJ, Stevens C, Zhou Y, Yi B, Laethem M, De Smet M, Snyder K, Hilliard D, Tanaka W, Zeng W, Tanen M, Wang AQ, Chen L, Winchell G, Davies MJ, Ramael S, Wagner JA, Herman GA. Pharmacokinetic and pharmacodynamic properties of multiple oral doses of sitagliptin, a dipeptidyl peptidase-IV inhibitor: a double-blind, randomized, placebo-controlled study in healthy male volunteers. Clin Ther. 2006;28:55–72. doi: 10.1016/j.clinthera.2006.01.015. [DOI] [PubMed] [Google Scholar]
- Bernal-Mizrachi E, Wen W, Stahlhut S, Welling CM, Permutt MA. Islet beta cell expression of constitutively active Akt1/PKB alpha induces striking hypertrophy, hyperplasia, and hyperinsulinemia. J Clin Invest. 2001;108:1631–1638. doi: 10.1172/JCI13785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blonde L, Klein EJ, Han J, Zhang B, Mac SM, Poon TH, Taylor KL, Trautmann ME, Kim DD, Kendall DM. Interim analysis of the effects of exenatide treatment on A1C, weight and cardiovascular risk factors over 82 weeks in 314 overweight patients with type 2 diabetes. Diabetes Obes Metab. 2006;8:436–447. doi: 10.1111/j.1463-1326.2006.00602.x. [DOI] [PubMed] [Google Scholar]
- Bock T, Pakkenberg B, Buschard K. The endocrine pancreas in non-diabetic rats after short-term and long-term treatment with the long-acting GLP-1 derivative NN2211. Apmis. 2003;111:1117–1124. doi: 10.1111/j.1600-0463.2003.apm1111207.x. [DOI] [PubMed] [Google Scholar]
- Bode HP, Moormann B, Dabew R, Goke B. Glucagon-like peptide 1 elevates cytosolic calcium in pancreatic beta-cells independently of protein kinase A. Endocrinology. 1999;140:3919–3927. doi: 10.1210/endo.140.9.6947. [DOI] [PubMed] [Google Scholar]
- Bonner-Weir S, Baxter LA, Schuppin GT, Smith FE. A second pathway for regeneration of adult exocrine and endocrine pancreas. A possible recapitulation of embryonic development. Diabetes. 1993;42:1715–1720. doi: 10.2337/diab.42.12.1715. [DOI] [PubMed] [Google Scholar]
- Bonner-Weir S, Weir GC. New sources of pancreatic beta-cells. Nat Biotechnol. 2005;23:857–861. doi: 10.1038/nbt1115. [DOI] [PubMed] [Google Scholar]
- Bouwens L, Pipeleers DG. Extra-insular beta cells associated with ductules are frequent in adult human pancreas. Diabetologia. 1998;41:629–633. doi: 10.1007/s001250050960. [DOI] [PubMed] [Google Scholar]
- Bratanova-Tochkova TK, Cheng H, Daniel S, Gunawardana S, Liu YJ, Mulvaney-Musa J, Schermerhorn T, Straub SG, Yajima H, Sharp GW. Triggering and augmentation mechanisms, granule pools, and biphasic insulin secretion. Diabetes. 2002;51(Suppl 1):S83–90. doi: 10.2337/diabetes.51.2007.s83. [DOI] [PubMed] [Google Scholar]
- Braun AP, Schulman H. The multifunctional calcium/calmodulin-dependent protein kinase: from form to function. Annu Rev Physiol. 1995;57:417–445. doi: 10.1146/annurev.ph.57.030195.002221. [DOI] [PubMed] [Google Scholar]
- Bregenholt S, Moldrup A, Blume N, Karlsen AE, Nissen Friedrichsen B, Tornhave D, Knudsen LB, Petersen JS. The long-acting glucagon-like peptide-1 analogue, liraglutide, inhibits beta-cell apoptosis in vitro. Biochem Biophys Res Commun. 2005;330:577–584. doi: 10.1016/j.bbrc.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Brissova M, Fowler MJ, Nicholson WE, Chu A, Hirshberg B, Harlan DM, Powers AC. Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. J Histochem Cytochem. 2005;53:1087–1097. doi: 10.1369/jhc.5C6684.2005. [DOI] [PubMed] [Google Scholar]
- Bruton JD, Lemmens R, Shi CL, Persson-Sjogren S, Westerblad H, Ahmed M, Pyne NJ, Frame M, Furman BL, Islam MS. Ryanodine receptors of pancreatic beta-cells mediate a distinct context-dependent signal for insulin secretion. Faseb J. 2003;17:301–303. doi: 10.1096/fj.02-0481fje. [DOI] [PubMed] [Google Scholar]
- Bullock BP, Heller RS, Habener JF. Tissue distribution of messenger ribonucleic acid encoding the rat glucagon-like peptide-1 receptor. Endocrinology. 1996;137:2968–2978. doi: 10.1210/endo.137.7.8770921. [DOI] [PubMed] [Google Scholar]
- Bulotta A, Hui H, Anastasi E, Bertolotto C, Boros LG, Di Mario U, Perfetti R. Cultured pancreatic ductal cells undergo cell cycle re-distribution and beta-cell-like differentiation in response to glucagon-like peptide-1. J Mol Endocrinol. 2002;29:347–360. doi: 10.1677/jme.0.0290347. [DOI] [PubMed] [Google Scholar]
- Bulotta A, Perfetti R, Hui H, Boros LG. GLP-1 stimulates glucose-derived de novo fatty acid synthesis and chain elongation during cell differentiation and insulin release. J Lipid Res. 2003;44:1559–1565. doi: 10.1194/jlr.M300093-JLR200. [DOI] [PubMed] [Google Scholar]
- Buse JB, Henry RR, Han J, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in sulfonylurea-treated patients with type 2 diabetes. Diabetes Care. 2004;27:2628–2635. doi: 10.2337/diacare.27.11.2628. [DOI] [PubMed] [Google Scholar]
- Buteau J, El-Assaad W, Rhodes CJ, Rosenberg L, Joly E, Prentki M. Glucagon-like peptide-1 prevents beta cell glucolipotoxicity. Diabetologia. 2004;47:806–815. doi: 10.1007/s00125-004-1379-6. [DOI] [PubMed] [Google Scholar]
- Buteau J, Foisy S, Joly E, Prentki M. Glucagon-like peptide 1 induces pancreatic beta-cell proliferation via transactivation of the epidermal growth factor receptor. Diabetes. 2003;52:124–132. doi: 10.2337/diabetes.52.1.124. [DOI] [PubMed] [Google Scholar]
- Buteau J, Foisy S, Rhodes CJ, Carpenter L, Biden TJ, Prentki M. Protein kinase Czeta activation mediates glucagon-like peptide-1-induced pancreatic beta-cell proliferation. Diabetes. 2001;50:2237–2243. doi: 10.2337/diabetes.50.10.2237. [DOI] [PubMed] [Google Scholar]
- Buteau J, Roduit R, Susini S, Prentki M. Glucagon-like peptide-1 promotes DNA synthesis, activates phosphatidylinositol 3-kinase and increases transcription factor pancreatic and duodenal homeobox gene 1 (PDX-1) DNA binding activity in beta (INS-1)-cells. Diabetologia. 1999;42:856–864. doi: 10.1007/s001250051238. [DOI] [PubMed] [Google Scholar]
- Buteau J, Spatz ML, Accili D. Transcription Factor FoxO1 Mediates Glucagon-Like Peptide-1 Effects on Pancreatic {beta}-Cell Mass. Diabetes. 2006;55:1190–1196. doi: 10.2337/db05-0825. [DOI] [PubMed] [Google Scholar]
- Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- Cali JJ, Zwaagstra JC, Mons N, Cooper DM, Krupinski J. Type VIII adenylyl cyclase. A Ca2+/calmodulin-stimulated enzyme expressed in discrete regions of rat brain. J Biol Chem. 1994;269:12190–12195. [PubMed] [Google Scholar]
- Carr DW, Stofko-Hahn RE, Fraser ID, Bishop SM, Acott TS, Brennan RG, Scott JD. Interaction of the regulatory subunit (RII) of cAMP-dependent protein kinase with RII-anchoring proteins occurs through an amphipathic helix binding motif. J Biol Chem. 1991;266:14188–14192. [PubMed] [Google Scholar]
- Cerasi E, Luft R. Plasma-Insulin Response to Sustained Hyperglycemia Induced by Glucose Infusion in Human Subjects. Lancet. 1963;41:1359–1361. doi: 10.1016/s0140-6736(63)90740-2. [DOI] [PubMed] [Google Scholar]
- Chang I, Kim S, Kim JY, Cho N, Kim YH, Kim HS, Lee MK, Kim KW, Lee MS. Nuclear factor kappaB protects pancreatic beta-cells from tumor necrosis factor-alpha-mediated apoptosis. Diabetes. 2003;52:1169–1175. doi: 10.2337/diabetes.52.5.1169. [DOI] [PubMed] [Google Scholar]
- Chen J, Couto FM, Minn AH, Shalev A. Exenatide inhibits beta-cell apoptosis by decreasing thioredoxin-interacting protein. Biochem Biophys Res Commun. 2006;346:1067–1074. doi: 10.1016/j.bbrc.2006.06.027. [DOI] [PubMed] [Google Scholar]
- Chen YE, Drucker DJ. Tissue-specific expression of unique mRNAs that encode proglucagon-derived peptides or exendin 4 in the lizard. J Biol Chem. 1997;272:4108–4115. doi: 10.1074/jbc.272.7.4108. [DOI] [PubMed] [Google Scholar]
- Chepurny OG, Hussain MA, Holz GG. Exendin-4 as a stimulator of rat insulin I gene promoter activity via bZIP/CRE interactions sensitive to serine/threonine protein kinase inhibitor Ro 31-8220. Endocrinology. 2002;143:2303–2313. doi: 10.1210/endo.143.6.8870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cnop M, Welsh N, Jonas JC, Jorns A, Lenzen S, Eizirik DL. Mechanisms of Pancreatic {beta}-Cell Death in Type 1 and Type 2 Diabetes: Many Differences, Few Similarities. Diabetes. 2005;54(Suppl 2):S97–S107. doi: 10.2337/diabetes.54.suppl_2.s97. [DOI] [PubMed] [Google Scholar]
- Cohen AW, Hnasko R, Schubert W, Lisanti MP. Role of caveolae and caveolins in health and disease. Physiol Rev. 2004;84:1341–1379. doi: 10.1152/physrev.00046.2003. [DOI] [PubMed] [Google Scholar]
- Comb M, Birnberg NC, Seasholtz A, Herbert E, Goodman HM. A cyclic AMP- and phorbol ester-inducible DNA element. Nature. 1986;323:353–356. doi: 10.1038/323353a0. [DOI] [PubMed] [Google Scholar]
- Conti M. Phosphodiesterases and cyclic nucleotide signaling in endocrine cells. Mol Endocrinol. 2000;14:1317–1327. doi: 10.1210/mend.14.9.0534. [DOI] [PubMed] [Google Scholar]
- Cooper DM. Regulation and organization of adenylyl cyclases and cAMP. Biochem J. 2003;375:517–529. doi: 10.1042/BJ20031061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozar-Castellano I, Fiaschi-Taesch N, Bigatel TA, Takane KK, Garcia-Ocana A, Vasavada R, Stewart AF. Molecular Control of Cell Cycle Progression in the Pancreatic {beta}-Cell. Endocr Rev. 2006;27:356–370. doi: 10.1210/er.2006-0004. [DOI] [PubMed] [Google Scholar]
- Crabtree GR. Calcium, calcineurin, and the control of transcription. J Biol Chem. 2001;276:2313–2316. doi: 10.1074/jbc.R000024200. [DOI] [PubMed] [Google Scholar]
- Creutzfeldt W, Nauck M. Gut hormones and diabetes mellitus. Diabetes Metab Rev. 1992;8:149–177. doi: 10.1002/dmr.5610080206. [DOI] [PubMed] [Google Scholar]
- Curry DL, Bennett LL, Grodsky GM. Dynamics of insulin secretion by the perfused rat pancreas. Endocrinology. 1968;83:572–584. doi: 10.1210/endo-83-3-572. [DOI] [PubMed] [Google Scholar]
- D’Amico E, Hui H, Khoury N, Di Mario U, Perfetti R. Pancreatic beta-cells expressing GLP-1 are resistant to the toxic effects of immunosuppressive drugs. J Mol Endocrinol. 2005;34:377–390. doi: 10.1677/jme.1.01655. [DOI] [PubMed] [Google Scholar]
- de la Tour D, Halvorsen T, Demeterco C, Tyrberg B, Itkin-Ansari P, Loy M, Yoo SJ, Hao E, Bossie S, Levine F. Beta-cell differentiation from a human pancreatic cell line in vitro and in vivo. Mol Endocrinol. 2001;15:476–483. doi: 10.1210/mend.15.3.0604. [DOI] [PubMed] [Google Scholar]
- De Leon DD, Deng S, Madani R, Ahima RS, Drucker DJ, Stoffers DA. Role of endogenous glucagon-like peptide-1 in islet regeneration after partial pancreatectomy. Diabetes. 2003;52:365–371. doi: 10.2337/diabetes.52.2.365. [DOI] [PubMed] [Google Scholar]
- de Rooij J, Rehmann H, van Triest M, Cool RH, Wittinghofer A, Bos JL. Mechanism of regulation of the Epac family of cAMP-dependent RapGEFs. J Biol Chem. 2000;275:20829–20836. doi: 10.1074/jbc.M001113200. [DOI] [PubMed] [Google Scholar]
- de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- de Rooij SR, Painter RC, Roseboom TJ, Phillips DI, Osmond C, Barker DJ, Tanck MW, Michels RP, Bossuyt PM, Bleker OP. Glucose tolerance at age 58 and the decline of glucose tolerance in comparison with age 50 in people prenatally exposed to the Dutch famine. Diabetologia. 2006;49:637–643. doi: 10.1007/s00125-005-0136-9. [DOI] [PubMed] [Google Scholar]
- De Vos A, Heimberg H, Quartier E, Huypens P, Bouwens L, Pipeleers D, Schuit F. Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression. J Clin Invest. 1995;96:2489–2495. doi: 10.1172/JCI118308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeney JT, Tornheim K, Korchak HM, Prentki M, Corkey BE. Acyl-CoA esters modulate intracellular Ca2+ handling by permeabilized clonal pancreatic beta-cells. J Biol Chem. 1992;267:19840–19845. [PubMed] [Google Scholar]
- DeFronzo RA, Ratner RE, Han J, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care. 2005;28:1092–1100. doi: 10.2337/diacare.28.5.1092. [DOI] [PubMed] [Google Scholar]
- Delmeire D, Flamez D, Hinke SA, Cali JJ, Pipeleers D, Schuit F. Type VIII adenylyl cyclase in rat beta cells: coincidence signal detector/generator for glucose and GLP-1. Diabetologia. 2003;46:1383–1393. doi: 10.1007/s00125-003-1203-8. [DOI] [PubMed] [Google Scholar]
- Dhanvantari S, Izzo A, Jansen E, Brubaker PL. Coregulation of glucagon-like peptide-1 synthesis with proglucagon and prohormone convertase 1 gene expression in enteroendocrine GLUTag cells. Endocrinology. 2001;142:37–42. doi: 10.1210/endo.142.1.7870. [DOI] [PubMed] [Google Scholar]
- Dillon JS, Lu M, Bowen S, Homan LL. The recombinant rat glucagon-like peptide-1 receptor, expressed in an alpha-cell line, is coupled to adenylyl cyclase activation and intracellular calcium release. Exp Clin Endocrinol Diabetes. 2005;113:182–189. doi: 10.1055/s-2005-837526. [DOI] [PubMed] [Google Scholar]
- Dillon JS, Tanizawa Y, Wheeler MB, Leng XH, Ligon BB, Rabin DU, Yoo-Warren H, Permutt MA, Boyd AE., 3rd Cloning and functional expression of the human glucagon-like peptide-1 (GLP-1) receptor. Endocrinology. 1993;133:1907–1910. doi: 10.1210/endo.133.4.8404634. [DOI] [PubMed] [Google Scholar]
- Ding WG, Renstrom E, Rorsman P, Buschard K, Gromada J. Glucagon-like peptide I and glucose-dependent insulinotropic polypeptide stimulate Ca2+-induced secretion in rat alpha-cells by a protein kinase A-mediated mechanism. Diabetes. 1997;46:792–800. doi: 10.2337/diab.46.5.792. [DOI] [PubMed] [Google Scholar]
- Diviani D, Scott JD. AKAP signaling complexes at the cytoskeleton. J Cell Sci. 2001;114:1431–1437. doi: 10.1242/jcs.114.8.1431. [DOI] [PubMed] [Google Scholar]
- Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, Langeberg LK, Kapiloff MS, Scott JD. The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature. 2005;437:574–578. doi: 10.1038/nature03966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge KL, Khouangsathiene S, Kapiloff MS, Mouton R, Hill EV, Houslay MD, Langeberg LK, Scott JD. mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP signaling module. Embo J. 2001;20:1921–1930. doi: 10.1093/emboj/20.8.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429:41–46. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- Doskeland SO, Ogreid D. Binding proteins for cyclic AMP in mammalian tissues. Int J Biochem. 1981;13:1–19. doi: 10.1016/0020-711x(81)90131-2. [DOI] [PubMed] [Google Scholar]
- Dostmann WR, Taylor SS. Identifying the molecular switches that determine whether (Rp)-cAMPS functions as an antagonist or an agonist in the activation of cAMP-dependent protein kinase I. Biochemistry. 1991;30:8710–8716. doi: 10.1021/bi00099a032. [DOI] [PubMed] [Google Scholar]
- Doyle ME, Egan JM. Pharmacological agents that directly modulate insulin secretion. Pharmacol Rev. 2003;55:105–131. doi: 10.1124/pr.55.1.7. [DOI] [PubMed] [Google Scholar]
- Doyle ME, Kim BJ, Carlson OD, Jo GJ, Zhou J, Egan JM. Notch Mediates Insulinotropic Signaling in β-Cells. Diabetes. 2006;55:A361. [Google Scholar]
- Drachenberg CB, Klassen DK, Weir MR, Wiland A, Fink JC, Bartlett ST, Cangro CB, Blahut S, Papadimitriou JC. Islet cell damage associated with tacrolimus and cyclosporine: morphological features in pancreas allograft biopsies and clinical correlation. Transplantation. 1999;68:396–402. doi: 10.1097/00007890-199908150-00012. [DOI] [PubMed] [Google Scholar]
- Drucker DJ, Philippe J, Mojsov S, Chick WL, Habener JF. Glucagon-like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line. Proc Natl Acad Sci U S A. 1987;84:3434–3438. doi: 10.1073/pnas.84.10.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukes ID, Philipson LH. K+ channels: generating excitement in pancreatic beta-cells. Diabetes. 1996;45:845–853. doi: 10.2337/diab.45.7.845. [DOI] [PubMed] [Google Scholar]
- Dyachok O, Isakov Y, Sagetorp J, Tengholm A. Oscillations of cyclic AMP in hormone-stimulated insulin-secreting beta-cells. Nature. 2006;439:349–352. doi: 10.1038/nature04410. [DOI] [PubMed] [Google Scholar]
- Easom RA. CaM kinase II: a protein kinase with extraordinary talents germane to insulin exocytosis. Diabetes. 1999;48:675–684. doi: 10.2337/diabetes.48.4.675. [DOI] [PubMed] [Google Scholar]
- Egan JM, Clocquet AR, Elahi D. The insulinotropic effect of acute exendin-4 administered to humans: comparison of nondiabetic state to type 2 diabetes. J Clin Endocrinol Metab. 2002;87:1282–1290. doi: 10.1210/jcem.87.3.8337. [DOI] [PubMed] [Google Scholar]
- Egan JM, Meneilly GS, Elahi D. Effects of 1-mo bolus subcutaneous administration of exendin-4 in type 2 diabetes. Am J Physiol Endocrinol Metab. 2003;284:E1072–1079. doi: 10.1152/ajpendo.00315.2002. [DOI] [PubMed] [Google Scholar]
- Ekanger R, Sand TE, Ogreid D, Christoffersen T, Doskeland SO. The separate estimation of cAMP intracellularly bound to the regulatory subunits of protein kinase I and II in glucagon-stimulated rat hepatocytes. J Biol Chem. 1985;260:3393–3401. [PubMed] [Google Scholar]
- Elahi D, Andersen DK, Muller DC, Tobin JD, Brown JC, Andres R. The enteric enhancement of glucose-stimulated insulin release. The role of GIP in aging, obesity, and non-insulin-dependent diabetes mellitus. Diabetes. 1984;33:950–957. doi: 10.2337/diab.33.10.950. [DOI] [PubMed] [Google Scholar]
- Elahi D, McAloon-Dyke M, Fukagawa NK, Meneilly GS, Sclater AL, Minaker KL, Habener JF, Andersen DK. The insulinotropic actions of glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (7-37) in normal and diabetic subjects. Regul Pept. 1994;51:63–74. doi: 10.1016/0167-0115(94)90136-8. [DOI] [PubMed] [Google Scholar]
- Eliasson L, Ma X, Renstrom E, Barg S, Berggren PO, Galvanovskis J, Gromada J, Jing X, Lundquist I, Salehi A, Sewing S, Rorsman P. SUR1 regulates PKA-independent cAMP-induced granule priming in mouse pancreatic B-cells. J Gen Physiol. 2003;121:181–197. doi: 10.1085/jgp.20028707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng J, Kleinman WA, Singh L, Singh G, Raufman JP. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J Biol Chem. 1992;267:7402–7405. [PubMed] [Google Scholar]
- Enserink JM, Christensen AE, de Rooij J, van Triest M, Schwede F, Genieser HG, Doskeland SO, Blank JL, Bos JL. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat Cell Biol. 2002;4:901–906. doi: 10.1038/ncb874. [DOI] [PubMed] [Google Scholar]
- Fajas L, Annicotte JS, Miard S, Sarruf D, Watanabe M, Auwerx J. Impaired pancreatic growth, beta cell mass, and beta cell function in E2F1 (−/− )mice. J Clin Invest. 2004;113:1288–1295. doi: 10.1172/JCI18555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farilla L, Bulotta A, Hirshberg B, Li Calzi S, Khoury N, Noushmehr H, Bertolotto C, Di Mario U, Harlan DM, Perfetti R. Glucagon-like peptide 1 inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets. Endocrinology. 2003;144:5149–5158. doi: 10.1210/en.2003-0323. [DOI] [PubMed] [Google Scholar]
- Farilla L, Hui H, Bertolotto C, Kang E, Bulotta A, Di Mario U, Perfetti R. Glucagon-like peptide-1 promotes islet cell growth and inhibits apoptosis in Zucker diabetic rats. Endocrinology. 2002;143:4397–4408. doi: 10.1210/en.2002-220405. [DOI] [PubMed] [Google Scholar]
- Fehmann HC, Goke B, Weber V, Goke R, Trautmann ME, Richter G, Arnold R. Interaction of glucagon-like peptide-1 (7-36)amide and cholecystokinin-8 in the endocrine and exocrine rat pancreas. Pancreas. 1990;5:361–365. doi: 10.1097/00006676-199005000-00019. [DOI] [PubMed] [Google Scholar]
- Fehmann HC, Habener JF. Insulinotropic hormone glucagon-like peptide-I(7-37) stimulation of proinsulin gene expression and proinsulin biosynthesis in insulinoma beta TC-1 cells. Endocrinology. 1992;130:159–166. doi: 10.1210/endo.130.1.1309325. [DOI] [PubMed] [Google Scholar]
- Fehmann HC, Hering BJ, Wolf MJ, Brandhorst H, Brandhorst D, Bretzel RG, Federlin K, Goke B. The effects of glucagon-like peptide-I (GLP-I) on hormone secretion from isolated human pancreatic islets. Pancreas. 1995;11:196–200. doi: 10.1097/00006676-199508000-00014. [DOI] [PubMed] [Google Scholar]
- Fehmann HC, Jiang J, Schweinfurth J, Dorsch K, Wheeler MB, Boyd AE, 3rd, Goke B. Ligand-specificity of the rat GLP-I receptor recombinantly expressed in Chinese hamster ovary (CHO-) cells. Z Gastroenterol. 1994;32:203–207. [PubMed] [Google Scholar]
- Fehse F, Trautmann M, Holst JJ, Halseth AE, Nanayakkara N, Nielsen LL, Fineman MS, Kim DD, Nauck MA. Exenatide augments first- and second-phase insulin secretion in response to intravenous glucose in subjects with type 2 diabetes. J Clin Endocrinol Metab. 2005;90:5991–5997. doi: 10.1210/jc.2005-1093. [DOI] [PubMed] [Google Scholar]
- Flamez D, Gilon P, Moens K, Van Breusegem A, Delmeire D, Scrocchi LA, Henquin JC, Drucker DJ, Schuit F. Altered cAMP and Ca2+ signaling in mouse pancreatic islets with glucagon-like peptide-1 receptor null phenotype. Diabetes. 1999;48:1979–1986. doi: 10.2337/diabetes.48.10.1979. [DOI] [PubMed] [Google Scholar]
- Flamez D, Van Breusegem A, Scrocchi LA, Quartier E, Pipeleers D, Drucker DJ, Schuit F. Mouse pancreatic beta-cells exhibit preserved glucose competence after disruption of the glucagon-like peptide-1 receptor gene. Diabetes. 1998;47:646–652. doi: 10.2337/diabetes.47.4.646. [DOI] [PubMed] [Google Scholar]
- Fleisch MC, Maxwell CA, Barcellos-Hoff MH. The pleiotropic roles of transforming growth factor beta in homeostasis and carcinogenesis of endocrine organs. Endocr Relat Cancer. 2006;13:379–400. doi: 10.1677/erc.1.01112. [DOI] [PubMed] [Google Scholar]
- Foord SM, Bonner TI, Neubig RR, Rosser EM, Pin JP, Davenport AP, Spedding M, Harmar AJ. International Union of Pharmacology. XLVI. G protein-coupled receptor list. Pharmacol Rev. 2005;57:279–288. doi: 10.1124/pr.57.2.5. [DOI] [PubMed] [Google Scholar]
- Franklin I, Gromada J, Gjinovci A, Theander S, Wollheim CB. Beta-cell secretory products activate alpha-cell ATP-dependent potassium channels to inhibit glucagon release. Diabetes. 2005;54:1808–1815. doi: 10.2337/diabetes.54.6.1808. [DOI] [PubMed] [Google Scholar]
- Fraser ID, Tavalin SJ, Lester LB, Langeberg LK, Westphal AM, Dean RA, Marrion NV, Scott JD. A novel lipid-anchored A-kinase Anchoring Protein facilitates cAMP-responsive membrane events. Embo J. 1998;17:2261–2272. doi: 10.1093/emboj/17.8.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrichsen BN, Neubauer N, Lee YC, Gram VK, Blume N, Petersen JS, Nielsen JH, Moldrup A. Stimulation of pancreatic beta-cell replication by incretins involves transcriptional induction of cyclin D1 via multiple signalling pathways. J Endocrinol. 2006;188:481–492. doi: 10.1677/joe.1.06160. [DOI] [PubMed] [Google Scholar]
- Fujimoto K, Shibasaki T, Yokoi N, Kashima Y, Matsumoto M, Sasaki T, Tajima N, Iwanaga T, Seino S. Piccolo, a Ca2+ sensor in pancreatic beta-cells. Involvement of cAMP-GEFII.Rim2.Piccolo complex in cAMP-dependent exocytosis. J Biol Chem. 2002;277:50497–50502. doi: 10.1074/jbc.M210146200. [DOI] [PubMed] [Google Scholar]
- Futter CE, Ramalho JS, Jaissle GB, Seeliger MW, Seabra MC. The role of Rab27a in the regulation of melanosome distribution within retinal pigment epithelial cells. Mol Biol Cell. 2004;15:2264–2275. doi: 10.1091/mbc.E03-10-0772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Young RA, Trucco MM, Greene SR, Hewlett EL, Matschinsky FM, Wolf BA. Protein kinase A translocation and insulin secretion in pancreatic beta-cells: studies with adenylate cyclase toxin from Bordetella pertussis. Biochem J. 2002;368:397–404. doi: 10.1042/BJ20020999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Flores M, Zueco JA, Alvarez E, Blazquez E. Expression of glucagon-like peptide-1 (GLP-1) receptor and the effect of GLP-1-(7-36) amide on insulin release by pancreatic islets during rat ontogenic development. Eur J Biochem. 2001;268:514–520. doi: 10.1046/j.1432-1327.2001.01865.x. [DOI] [PubMed] [Google Scholar]
- Gedulin BR, Nikoulina SE, Smith PA, Gedulin G, Nielsen LL, Baron AD, Parkes DG, Young AA. Exenatide (exendin-4) improves insulin sensitivity and {beta}-cell mass in insulin-resistant obese fa/fa Zucker rats independent of glycemia and body weight. Endocrinology. 2005;146:2069–2076. doi: 10.1210/en.2004-1349. [DOI] [PubMed] [Google Scholar]
- Gentili C, Boland R, Russo de Boland A. Implication of Gbetagamma proteins and c-SRC tyrosine kinase in parathyroid hormone-induced signal transduction in rat enterocytes. J Endocrinol. 2006;188:69–78. doi: 10.1677/joe.1.06397. [DOI] [PubMed] [Google Scholar]
- Georgia S, Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin Invest. 2004;114:963–968. doi: 10.1172/JCI22098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershengorn MC, Hardikar AA, Wei C, Geras-Raaka E, Marcus-Samuels B, Raaka BM. Epithelial-to-mesenchymal transition generates proliferative human islet precursor cells. Science. 2004;306:2261–2264. doi: 10.1126/science.1101968. [DOI] [PubMed] [Google Scholar]
- Gerst JE. SNAREs and SNARE regulators in membrane fusion and exocytosis. Cell Mol Life Sci. 1999;55:707–734. doi: 10.1007/s000180050328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser B, Chiu KC, Anker R, Nestorowicz A, Landau H, Ben-Bassat H, Shlomai Z, Kaiser N, Thornton PS, Stanley CA, et al. Familial hyperinsulinism maps to chromosome 11p14–15.1, 30 cM centromeric to the insulin gene. Nat Genet. 1994;7:185–188. doi: 10.1038/ng0694-185. [DOI] [PubMed] [Google Scholar]
- Goke R, Just R, Lankat-Buttgereit B, Goke B. Glycosylation of the GLP-1 receptor is a prerequisite for regular receptor function. Peptides. 1994;15:675–681. doi: 10.1016/0196-9781(94)90095-7. [DOI] [PubMed] [Google Scholar]
- Gomez E, Pritchard C, Herbert TP. cAMP-dependent protein kinase and Ca2+ influx through L-type voltage-gated calcium channels mediate Raf-independent activation of extracellular regulated kinase in response to glucagon-like peptide-1 in pancreatic beta-cells. J Biol Chem. 2002;277:48146–48151. doi: 10.1074/jbc.M209165200. [DOI] [PubMed] [Google Scholar]
- Gonelle-Gispert C, Costa M, Takahashi M, Sadoul K, Halban P. Phosphorylation of SNAP-25 on serine-187 is induced by secretagogues in insulin-secreting cells, but is not correlated with insulin secretion. Biochem J. 2002;368:223–232. doi: 10.1042/BJ20020896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–680. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- Graziano MP, Hey PJ, Borkowski D, Chicchi GG, Strader CD. Cloning and functional expression of a human glucagon-like peptide-1 receptor. Biochem Biophys Res Commun. 1993;196:141–146. doi: 10.1006/bbrc.1993.2226. [DOI] [PubMed] [Google Scholar]
- Graziano MP, Hey PJ, Strader CD. The amino terminal domain of the glucagon-like peptide-1 receptor is a critical determinant of subtype specificity. Receptors Channels. 1996;4:9–17. [PubMed] [Google Scholar]
- Greig NH, Holloway HW, De Ore KA, Jani D, Wang Y, Zhou J, Garant MJ, Egan JM. Once daily injection of exendin-4 to diabetic mice achieves long-term beneficial effects on blood glucose concentrations. Diabetologia. 1999;42:45–50. doi: 10.1007/s001250051111. [DOI] [PubMed] [Google Scholar]
- Gribble FM, Reimann F. Sulphonylurea action revisited: the post-cloning era. Diabetologia. 2003;46:875–891. doi: 10.1007/s00125-003-1143-3. [DOI] [PubMed] [Google Scholar]
- Gribble FM, Tucker SJ, Haug T, Ashcroft FM. MgATP activates the beta cell KATP channel by interaction with its SUR1 subunit. Proc Natl Acad Sci U S A. 1998;95:7185–7190. doi: 10.1073/pnas.95.12.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grill V, Cerasi E. Activation by glucose of adenyl cyclase in pancreatic islets of the rat. FEBS Lett. 1973;33:311–314. doi: 10.1016/0014-5793(73)80218-2. [DOI] [PubMed] [Google Scholar]
- Gromada J, Bokvist K, Ding WG, Holst JJ, Nielsen JH, Rorsman P. Glucagon-like peptide 1 (7-36) amide stimulates exocytosis in human pancreatic beta-cells by both proximal and distal regulatory steps in stimulus-secretion coupling. Diabetes. 1998;47:57–65. doi: 10.2337/diab.47.1.57. [DOI] [PubMed] [Google Scholar]
- Gromada J, Ding WG, Barg S, Renstrom E, Rorsman P. Multisite regulation of insulin secretion by cAMP-increasing agonists: evidence that glucagon-like peptide 1 and glucagon act via distinct receptors. Pflugers Arch. 1997;434:515–524. doi: 10.1007/s004240050431. [DOI] [PubMed] [Google Scholar]
- Gutniak M, Orskov C, Holst JJ, Ahren B, Efendic S. Antidiabetogenic effect of glucagon-like peptide-1 (7-36)amide in normal subjects and patients with diabetes mellitus. N Engl J Med. 1992;326:1316–1322. doi: 10.1056/NEJM199205143262003. [DOI] [PubMed] [Google Scholar]
- Gutniak MK, Larsson H, Sanders SW, Juneskans O, Holst JJ, Ahren B. GLP-1 tablet in type 2 diabetes in fasting and postprandial conditions. Diabetes Care. 1997;20:1874–1879. doi: 10.2337/diacare.20.12.1874. [DOI] [PubMed] [Google Scholar]
- Hales CN, Barker DJ, Clark PM, Cox LJ, Fall C, Osmond C, Winter PD. Fetal and infant growth and impaired glucose tolerance at age 64. Bmj. 1991;303:1019–1022. doi: 10.1136/bmj.303.6809.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JH, Rall L, Rutter WJ. Selective expression of rat pancreatic genes during embryonic development. Proc Natl Acad Sci U S A. 1986;83:110–114. doi: 10.1073/pnas.83.1.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han P, Werber J, Surana M, Fleischer N, Michaeli T. The calcium/calmodulin-dependent phosphodiesterase PDE1C down-regulates glucose-induced insulin secretion. J Biol Chem. 1999;274:22337–22344. doi: 10.1074/jbc.274.32.22337. [DOI] [PubMed] [Google Scholar]
- Hanoune J, Defer N. Regulation and role of adenylyl cyclase isoforms. Annu Rev Pharmacol Toxicol. 2001;41:145–174. doi: 10.1146/annurev.pharmtox.41.1.145. [DOI] [PubMed] [Google Scholar]
- Hansen L, Deacon CF, Orskov C, Holst JJ. Glucagon-like peptide-1-(7-36)amide is transformed to glucagon-like peptide-1-(9-36)amide by dipeptidyl peptidase IV in the capillaries supplying the L cells of the porcine intestine. Endocrinology. 1999;140:5356–5363. doi: 10.1210/endo.140.11.7143. [DOI] [PubMed] [Google Scholar]
- Hao E, Tyrberg B, Itkin-Ansari P, Lakey JR, Geron I, Monosov EZ, Barcova M, Mercola M, Levine F. Beta-cell differentiation from nonendocrine epithelial cells of the adult human pancreas. Nat Med. 2006;12:310–316. doi: 10.1038/nm1367. [DOI] [PubMed] [Google Scholar]
- Hardikar AA, Wang XY, Williams LJ, Kwok J, Wong R, Yao M, Tuch BE. Functional maturation of fetal porcine beta-cells by glucagon-like peptide 1 and cholecystokinin. Endocrinology. 2002;143:3505–3514. doi: 10.1210/en.2001-211344. [DOI] [PubMed] [Google Scholar]
- Harmar AJ. Family-B G-protein-coupled receptors. Genome Biol. 2001;2:REVIEWS3013. doi: 10.1186/gb-2001-2-12-reviews3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmar AJ. Clinical endocrinology and metabolism. Receptors for gut peptides. Best Pract Res Clin Endocrinol Metab. 2004;18:463–475. doi: 10.1016/j.beem.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Harndahl L, Jing XJ, Ivarsson R, Degerman E, Ahren B, Manganiello VC, Renstrom E, Holst LS. Important role of phosphodiesterase 3B for the stimulatory action of cAMP on pancreatic beta-cell exocytosis and release of insulin. J Biol Chem. 2002;277:37446–37455. doi: 10.1074/jbc.M205401200. [DOI] [PubMed] [Google Scholar]
- Harndahl L, Wierup N, Enerback S, Mulder H, Manganiello VC, Sundler F, Degerman E, Ahren B, Holst LS. Beta-cell-targeted overexpression of phosphodiesterase 3B in mice causes impaired insulin secretion, glucose intolerance, and deranged islet morphology. J Biol Chem. 2004;279:15214–15222. doi: 10.1074/jbc.M308952200. [DOI] [PubMed] [Google Scholar]
- Hay CW, Sinclair EM, Bermano G, Durward E, Tadayyon M, Docherty K. Glucagon-like peptide-1 stimulates human insulin promoter activity in part through cAMP-responsive elements that lie upstream and downstream of the transcription start site. J Endocrinol. 2005;186:353–365. doi: 10.1677/joe.1.06205. [DOI] [PubMed] [Google Scholar]
- Heimberg H, Heremans Y, Jobin C, Leemans R, Cardozo AK, Darville M, Eizirik DL. Inhibition of cytokine-induced NF-kappaB activation by adenovirus-mediated expression of a NF-kappaB super-repressor prevents beta-cell apoptosis. Diabetes. 2001;50:2219–2224. doi: 10.2337/diabetes.50.10.2219. [DOI] [PubMed] [Google Scholar]
- Heller RS, Aponte GW. Intra-islet regulation of hormone secretion by glucagon-like peptide-1-(7--36) amide. Am J Physiol. 1995;269:G852–860. doi: 10.1152/ajpgi.1995.269.6.G852. [DOI] [PubMed] [Google Scholar]
- Heller RS, Kieffer TJ, Habener JF. Point mutations in the first and third intracellular loops of the glucagon-like peptide-1 receptor alter intracellular signaling. Biochem Biophys Res Commun. 1996;223:624–632. doi: 10.1006/bbrc.1996.0945. [DOI] [PubMed] [Google Scholar]
- Heller RS, Kieffer TJ, Habener JF. Insulinotropic glucagon-like peptide I receptor expression in glucagon-producing alpha-cells of the rat endocrine pancreas. Diabetes. 1997;46:785–791. doi: 10.2337/diab.46.5.785. [DOI] [PubMed] [Google Scholar]
- Henquin JC. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes. 2000;49:1751–1760. doi: 10.2337/diabetes.49.11.1751. [DOI] [PubMed] [Google Scholar]
- Hepp R, Cabaniols JP, Roche PA. Differential phosphorylation of SNAP-25 in vivo by protein kinase C and protein kinase A. FEBS Lett. 2002;532:52–56. doi: 10.1016/s0014-5793(02)03629-3. [DOI] [PubMed] [Google Scholar]
- Herman GA, Stevens C, Van Dyck K, Bergman A, Yi B, De Smet M, Snyder K, Hilliard D, Tanen M, Tanaka W, Wang AQ, Zeng W, Musson D, Winchell G, Davies MJ, Ramael S, Gottesdiener KM, Wagner JA. Pharmacokinetics and pharmacodynamics of sitagliptin, an inhibitor of dipeptidyl peptidase IV, in healthy subjects: results from two randomized, double-blind, placebo-controlled studies with single oral doses. Clin Pharmacol Ther. 2005;78:675–688. doi: 10.1016/j.clpt.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006;20:1218–1249. doi: 10.1101/gad.1415606. [DOI] [PubMed] [Google Scholar]
- Hiles RA, Bawdon RE, Petrella EM. Ex vivo human placental transfer of the peptides pramlintide and exenatide (synthetic exendin-4) Hum Exp Toxicol. 2003;22:623–628. doi: 10.1191/0960327103ht402oa. [DOI] [PubMed] [Google Scholar]
- Hisatomi Y, Okumura K, Nakamura K, Matsumoto S, Satoh A, Nagano K, Yamamoto T, Endo F. Flow cytometric isolation of endodermal progenitors from mouse salivary gland differentiate into hepatic and pancreatic lineages. Hepatology. 2004;39:667–675. doi: 10.1002/hep.20063. [DOI] [PubMed] [Google Scholar]
- Holst JJ, Orskov C, Schwartz TW, Buhl T, Baldissera F. Pro glucagon 78–105, a potent insulinotropic from the lower small intestine. Diabetologia. 1986;29:549A. [Google Scholar]
- Holst LS, Mulder H, Manganiello V, Sundler F, Ahren B, Holm C, Degerman E. Protein kinase B is expressed in pancreatic beta cells and activated upon stimulation with insulin-like growth factor I. Biochem Biophys Res Commun. 1998;250:181–186. doi: 10.1006/bbrc.1998.9166. [DOI] [PubMed] [Google Scholar]
- Holz GG. Epac: A new cAMP-binding protein in support of glucagon-like peptide-1 receptor-mediated signal transduction in the pancreatic beta-cell. Diabetes. 2004;53:5–13. doi: 10.2337/diabetes.53.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz GG, Leech CA, Heller RS, Castonguay M, Habener JF. cAMP-dependent mobilization of intracellular Ca2+ stores by activation of ryanodine receptors in pancreatic beta-cells. A Ca2+ signaling system stimulated by the insulinotropic hormone glucagon-like peptide-1-(7-37) J Biol Chem. 1999;274:14147–14156. doi: 10.1074/jbc.274.20.14147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz GGt, Kuhtreiber WM, Habener JF. Pancreatic beta-cells are rendered glucose-competent by the insulinotropic hormone glucagon-like peptide-1(7-37) Nature. 1993;361:362–365. doi: 10.1038/361362a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz GHt, Kuhtreiber WM, Habener JF. Induction of glucose competence in pancreatic beta cells by glucagon-like peptide-1(7-37) Trans Assoc Am Physicians. 1992;105:260–267. [PubMed] [Google Scholar]
- Horsch D, Goke R, Eissele R, Michel B, Goke B. Reciprocal cellular distribution of glucagon-like peptide-1 (GLP-1) immunoreactivity and GLP-1 receptor mRNA in pancreatic islets of rat. Pancreas. 1997;14:290–294. doi: 10.1097/00006676-199704000-00012. [DOI] [PubMed] [Google Scholar]
- Hui H, Nourparvar A, Zhao X, Perfetti R. Glucagon-like peptide-1 inhibits apoptosis of insulin-secreting cells via a cyclic 5′-adenosine monophosphate-dependent protein kinase A- and a phosphatidylinositol 3-kinase-dependent pathway. Endocrinology. 2003;144:1444–1455. doi: 10.1210/en.2002-220897. [DOI] [PubMed] [Google Scholar]
- Hui H, Wright C, Perfetti R. Glucagon-like peptide 1 induces differentiation of islet duodenal homeobox-1-positive pancreatic ductal cells into insulin-secreting cells. Diabetes. 2001;50:785–796. doi: 10.2337/diabetes.50.4.785. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Maekawa T, Sudo T, Ishii S, Seino Y, Imura H. c-Jun represses the human insulin promoter activity that depends on multiple cAMP response elements. Proc Natl Acad Sci U S A. 1992;89:1045–1049. doi: 10.1073/pnas.89.3.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam MS, Leibiger I, Leibiger B, Rossi D, Sorrentino V, Ekstrom TJ, Westerblad H, Andrade FH, Berggren PO. In situ activation of the type 2 ryanodine receptor in pancreatic beta cells requires cAMP-dependent phosphorylation. Proc Natl Acad Sci U S A. 1998;95:6145–6150. doi: 10.1073/pnas.95.11.6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, Scheller RH. SNAREs--engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- Jahn R, Sudhof TC. Membrane fusion and exocytosis. Annu Rev Biochem. 1999;68:863–911. doi: 10.1146/annurev.biochem.68.1.863. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ, Stein V, Weinreich F, Zdebik AA. Molecular structure and physiological function of chloride channels. Physiol Rev. 2002;82:503–568. doi: 10.1152/physrev.00029.2001. [DOI] [PubMed] [Google Scholar]
- Jhala US, Canettieri G, Screaton RA, Kulkarni RN, Krajewski S, Reed J, Walker J, Lin X, White M, Montminy M. cAMP promotes pancreatic beta-cell survival via CREB-mediated induction of IRS2. Genes Dev. 2003;17:1575–1580. doi: 10.1101/gad.1097103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JD, Ahmed NT, Luciani DS, Han Z, Tran H, Fujita J, Misler S, Edlund H, Polonsky KS. Increased islet apoptosis in Pdx1+/− mice. J Clin Invest. 2003;111:1147–1160. doi: 10.1172/JCI16537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature. 1994;371:606–609. doi: 10.1038/371606a0. [DOI] [PubMed] [Google Scholar]
- Juntti-Berggren L, Pigon J, Karpe F, Hamsten A, Gutniak M, Vignati L, Efendic S. The antidiabetogenic effect of GLP-1 is maintained during a 7-day treatment period and improves diabetic dyslipoproteinemia in NIDDM patients. Diabetes Care. 1996;19:1200–1206. doi: 10.2337/diacare.19.11.1200. [DOI] [PubMed] [Google Scholar]
- Kaeser PS, Sudhof TC. RIM function in short- and long-term synaptic plasticity. Biochem Soc Trans. 2005;33:1345–1349. doi: 10.1042/BST0331345. [DOI] [PubMed] [Google Scholar]
- Kajimoto Y, Watada H, Matsuoka T, Kaneto H, Fujitani Y, Miyazaki J, Yamasaki Y. Suppression of transcription factor PDX-1/IPF1/STF-1/IDX-1 causes no decrease in insulin mRNA in MIN6 cells. J Clin Invest. 1997;100:1840–1846. doi: 10.1172/JCI119712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang G, Chepurny OG, Holz GG. cAMP-regulated guanine nucleotide exchange factor II (Epac2) mediates Ca2+-induced Ca2+ release in INS-1 pancreatic beta-cells. J Physiol. 2001;536:375–385. doi: 10.1111/j.1469-7793.2001.0375c.xd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang G, Chepurny OG, Malester B, Rindler MJ, Rehmann H, Bos JL, Schwede F, Coetzee WA, Holz GG. cAMP Sensor Epac As A Determinant Of ATP-Sensitive Potassium Channel Activity In Human Pancreatic Beta Cells And Rat INS-1 Cells. J Physiol. 2006;573:595–609. doi: 10.1113/jphysiol.2006.107391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang G, Chepurny OG, Rindler MJ, Collis L, Chepurny Z, Li WH, Harbeck M, Roe MW, Holz GG. A cAMP and Ca2+ coincidence detector in support of Ca2+-induced Ca2+ release in mouse pancreatic beta cells. J Physiol. 2005;566:173–188. doi: 10.1113/jphysiol.2005.087510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang G, Joseph JW, Chepurny OG, Monaco M, Wheeler MB, Bos JL, Schwede F, Genieser HG, Holz GG. Epac-selective cAMP analog 8-pCPT-2′-O-Me-cAMP as a stimulus for Ca2+-induced Ca2+ release and exocytosis in pancreatic beta-cells. J Biol Chem. 2003;278:8279–8285. doi: 10.1074/jbc.M211682200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H, Hatakeyama H, Kishimoto T, Liu TT, Nemoto T, Takahashi N. A new quantitative (two-photon extracellular polar-tracer imaging-based quantification (TEPIQ)) analysis for diameters of exocytic vesicles and its application to mouse pancreatic islets. J Physiol. 2005a;568:891–903. doi: 10.1113/jphysiol.2005.093047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H, Suzuki T, Liu TT, Kishimoto T, Takahashi N. Fast and cAMP-sensitive mode of Ca(2+)-dependent exocytosis in pancreatic beta-cells. Diabetes. 2002;51(Suppl 1):S19–24. doi: 10.2337/diabetes.51.2007.s19. [DOI] [PubMed] [Google Scholar]
- Kasai K, Ohara-Imaizumi M, Takahashi N, Mizutani S, Zhao S, Kikuta T, Kasai H, Nagamatsu S, Gomi H, Izumi T. Rab27a mediates the tight docking of insulin granules onto the plasma membrane during glucose stimulation. J Clin Invest. 2005b;115:388–396. doi: 10.1172/JCI22955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashima Y, Miki T, Shibasaki T, Ozaki N, Miyazaki M, Yano H, Seino S. Critical role of cAMP-GEFII--Rim2 complex in incretin-potentiated insulin secretion. J Biol Chem. 2001;276:46046–46053. doi: 10.1074/jbc.M108378200. [DOI] [PubMed] [Google Scholar]
- Kashyap S, Belfort R, Gastaldelli A, Pratipanawatr T, Berria R, Pratipanawatr W, Bajaj M, Mandarino L, DeFronzo R, Cusi K. A sustained increase in plasma free fatty acids impairs insulin secretion in nondiabetic subjects genetically predisposed to develop type 2 diabetes. Diabetes. 2003;52:2461–2474. doi: 10.2337/diabetes.52.10.2461. [DOI] [PubMed] [Google Scholar]
- Kawamori D, Kajimoto Y, Kaneto H, Umayahara Y, Fujitani Y, Miyatsuka T, Watada H, Leibiger IB, Yamasaki Y, Hori M. Oxidative stress induces nucleo-cytoplasmic translocation of pancreatic transcription factor PDX-1 through activation of c-Jun NH(2)-terminal kinase. Diabetes. 2003;52:2896–2904. doi: 10.2337/diabetes.52.12.2896. [DOI] [PubMed] [Google Scholar]
- Kawamori D, Kaneto H, Nakatani Y, Matsuoka TA, Matsuhisa M, Hori M, Yamasaki Y. The forkhead transcription factor Foxo1 bridges the JNK pathway and the transcription factor PDX-1 through its intracellular translocation. J Biol Chem. 2006;281:1091–1098. doi: 10.1074/jbc.M508510200. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM. A family of cAMP-binding proteins that directly activate Rap1. Science. 1998;282:2275–2279. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- Kemp DM, Habener JF. Insulinotropic hormone glucagon-like peptide 1 (GLP-1) activation of insulin gene promoter inhibited by p38 mitogen-activated protein kinase. Endocrinology. 2001;142:1179–1187. doi: 10.1210/endo.142.3.8026. [DOI] [PubMed] [Google Scholar]
- Kendall DM, Riddle MC, Rosenstock J, Zhuang D, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in patients with type 2 diabetes treated with metformin and a sulfonylurea. Diabetes Care. 2005;28:1083–1091. doi: 10.2337/diacare.28.5.1083. [DOI] [PubMed] [Google Scholar]
- Khoo S, Cobb MH. Activation of mitogen-activating protein kinase by glucose is not required for insulin secretion. Proc Natl Acad Sci U S A. 1997;94:5599–5604. doi: 10.1073/pnas.94.11.5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Kang JH, Park YG, Ryu GR, Ko SH, Jeong IK, Koh KH, Rhie DJ, Yoon SH, Hahn SJ, Kim MS, Jo YH. Exendin-4 induction of cyclin D1 expression in INS-1 beta-cells: involvement of cAMP-responsive element. J Endocrinol. 2006;188:623–633. doi: 10.1677/joe.1.06480. [DOI] [PubMed] [Google Scholar]
- Kitamura T, Nakae J, Kitamura Y, Kido Y, Biggs WH, 3rd, Wright CV, White MF, Arden KC, Accili D. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J Clin Invest. 2002;110:1839–1847. doi: 10.1172/JCI200216857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein T, Frandsen U, Heller RS, Serup P. IMPAN cells: a pancreatic model for differentiation into endocrine cells. Arch Biochem Biophys. 2001;395:259–263. doi: 10.1006/abbi.2001.2579. [DOI] [PubMed] [Google Scholar]
- Knoch KP, Bergert H, Borgonovo B, Saeger HD, Altkruger A, Verkade P, Solimena M. Polypyrimidine tract-binding protein promotes insulin secretory granule biogenesis. Nat Cell Biol. 2004;6:207–214. doi: 10.1038/ncb1099. [DOI] [PubMed] [Google Scholar]
- Knoch KP, Meisterfeld R, Kersting S, Bergert H, Altkruger A, Wegbrod C, Jager M, Saeger HD, Solimena M. cAMP-dependent phosphorylation of PTB1 promotes the expression of insulin secretory granule proteins in beta cells. Cell Metab. 2006;3:123–134. doi: 10.1016/j.cmet.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Knudsen LB, Nielsen PF, Huusfeldt PO, Johansen NL, Madsen K, Pedersen FZ, Thogersen H, Wilken M, Agerso H. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J Med Chem. 2000;43:1664–1669. doi: 10.1021/jm9909645. [DOI] [PubMed] [Google Scholar]
- Koehler JA, Drucker DJ. Activation of glucagon-like peptide-1 receptor signaling does not modify the growth or apoptosis of human pancreatic cancer cells. Diabetes. 2006;55:1369–1379. doi: 10.2337/db05-1145. [DOI] [PubMed] [Google Scholar]
- Koizumi M, Doi R, Fujimoto K, Ito D, Toyoda E, Mori T, Kami K, Kawaguchi Y, Gittes GK, Imamura M. Pancreatic epithelial cells can be converted into insulin-producing cells by GLP-1 in conjunction with virus-mediated gene transfer of pdx-1. Surgery. 2005;138:125–133. doi: 10.1016/j.surg.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Koyasu S. The role of PI3K in immune cells. Nat Immunol. 2003;4:313–319. doi: 10.1038/ni0403-313. [DOI] [PubMed] [Google Scholar]
- Kreymann B, Williams G, Ghatei MA, Bloom SR. Glucagon-like peptide-1 7-36: a physiological incretin in man. Lancet. 1987;2:1300–1304. doi: 10.1016/s0140-6736(87)91194-9. [DOI] [PubMed] [Google Scholar]
- Kubota N, Tobe K, Terauchi Y, Eto K, Yamauchi T, Suzuki R, Tsubamoto Y, Komeda K, Nakano R, Miki H, Satoh S, Sekihara H, Sciacchitano S, Lesniak M, Aizawa S, Nagai R, Kimura S, Akanuma Y, Taylor SI, Kadowaki T. Disruption of insulin receptor substrate 2 causes type 2 diabetes because of liver insulin resistance and lack of compensatory beta-cell hyperplasia. Diabetes. 2000;49:1880–1889. doi: 10.2337/diabetes.49.11.1880. [DOI] [PubMed] [Google Scholar]
- Kumar M, Hunag Y, Glinka Y, Prud’homme GJ, Wang Q. Gene therapy of diabetes using a novel GLP-1/IgG1-Fc fusion construct normalizes glucose levels in db/db mice. Gene Ther. 2006 doi: 10.1038/sj.gt.3302836. [DOI] [PubMed] [Google Scholar]
- Kushner JA, Ciemerych MA, Sicinska E, Wartschow LM, Teta M, Long SY, Sicinski P, White MF. Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Mol Cell Biol. 2005;25:3752–3762. doi: 10.1128/MCB.25.9.3752-3762.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushner JA, Ye J, Schubert M, Burks DJ, Dow MA, Flint CL, Dutta S, Wright CV, Montminy MR, White MF. Pdx1 restores beta cell function in Irs2 knockout mice. J Clin Invest. 2002;109:1193–1201. doi: 10.1172/JCI14439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon G, Marshall CA, Pappan KL, Remedi MS, McDaniel ML. Signaling elements involved in the metabolic regulation of mTOR by nutrients, incretins, and growth factors in islets. Diabetes. 2004a;53(Suppl 3):S225–232. doi: 10.2337/diabetes.53.suppl_3.s225. [DOI] [PubMed] [Google Scholar]
- Kwon G, Pappan KL, Marshall CA, Schaffer JE, McDaniel ML. cAMP Dose-dependently prevents palmitate-induced apoptosis by both protein kinase A- and cAMP-guanine nucleotide exchange factor-dependent pathways in beta-cells. J Biol Chem. 2004b;279:8938–8945. doi: 10.1074/jbc.M310330200. [DOI] [PubMed] [Google Scholar]
- Lang J. Molecular mechanisms and regulation of insulin exocytosis as a paradigm of endocrine secretion. Eur J Biochem. 1999;259:3–17. doi: 10.1046/j.1432-1327.1999.00043.x. [DOI] [PubMed] [Google Scholar]
- Larsson O, Deeney JT, Branstrom R, Berggren PO, Corkey BE. Activation of the ATP-sensitive K+ channel by long chain acyl-CoA. A role in modulation of pancreatic beta-cell glucose sensitivity. J Biol Chem. 1996;271:10623–10626. doi: 10.1074/jbc.271.18.10623. [DOI] [PubMed] [Google Scholar]
- Lawlor MA, Alessi DR. PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci. 2001;114:2903–2910. doi: 10.1242/jcs.114.16.2903. [DOI] [PubMed] [Google Scholar]
- Lawrence MC, Bhatt HS, Easom RA. NFAT regulates insulin gene promoter activity in response to synergistic pathways induced by glucose and glucagon-like peptide-1. Diabetes. 2002;51:691–698. doi: 10.2337/diabetes.51.3.691. [DOI] [PubMed] [Google Scholar]
- Le Lay J, Stein R. Involvement of PDX-1 in activation of human insulin gene transcription. J Endocrinol. 2006;188:287–294. doi: 10.1677/joe.1.06510. [DOI] [PubMed] [Google Scholar]
- le Roux CW, Aylwin SJ, Batterham RL, Borg CM, Coyle F, Prasad V, Shurey S, Ghatei MA, Patel AG, Bloom SR. Gut hormone profiles following bariatric surgery favor an anorectic state, facilitate weight loss, and improve metabolic parameters. Ann Surg. 2006;243:108–114. doi: 10.1097/01.sla.0000183349.16877.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B, Jonas JC, Weir GC, Laychock SG. Glucose regulates expression of inositol 1,4,5-trisphosphate receptor isoforms in isolated rat pancreatic islets. Endocrinology. 1999;140:2173–2182. doi: 10.1210/endo.140.5.6738. [DOI] [PubMed] [Google Scholar]
- Lee B, Laychock SG. Inositol 1,4,5-trisphosphate receptor isoform expression in mouse pancreatic islets: effects of carbachol. Biochem Pharmacol. 2001;61:327–336. doi: 10.1016/s0006-2952(00)00559-1. [DOI] [PubMed] [Google Scholar]
- Leech CA, Castonguay MA, Habener JF. Expression of adenylyl cyclase subtypes in pancreatic beta-cells. Biochem Biophys Res Commun. 1999;254:703–706. doi: 10.1006/bbrc.1998.9906. [DOI] [PubMed] [Google Scholar]
- Leech CA, Habener JF. Insulinotropic glucagon-like peptide-1-mediated activation of non-selective cation currents in insulinoma cells is mimicked by maitotoxin. J Biol Chem. 1997;272:17987–17993. doi: 10.1074/jbc.272.29.17987. [DOI] [PubMed] [Google Scholar]
- Leech CA, Holz GG, Chepurny O, Habener JF. Expression of cAMP-regulated guanine nucleotide exchange factors in pancreatic beta-cells. Biochem Biophys Res Commun. 2000;278:44–47. doi: 10.1006/bbrc.2000.3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehle L, Tanner W. The specific site of tunicamycin inhibition in the formation of dolichol-bound N-acetylglucosamine derivatives. FEBS Lett. 1976;72:167–170. doi: 10.1016/0014-5793(76)80922-2. [DOI] [PubMed] [Google Scholar]
- Lester LB, Faux MC, Nauert JB, Scott JD. Targeted protein kinase A and PP-2B regulate insulin secretion through reversible phosphorylation. Endocrinology. 2001;142:1218–1227. doi: 10.1210/endo.142.3.8023. [DOI] [PubMed] [Google Scholar]
- Lester LB, Kuo HC, Andrews L, Nauert B, Wolf DP. Directed differentiation of rhesus monkey ES cells into pancreatic cell phenotypes. Reprod Biol Endocrinol. 2004;2:42. doi: 10.1186/1477-7827-2-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester LB, Langeberg LK, Scott JD. Anchoring of protein kinase A facilitates hormone-mediated insulin secretion. Proc Natl Acad Sci U S A. 1997;94:14942–14947. doi: 10.1073/pnas.94.26.14942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, El-Kholy W, Rhodes CJ, Brubaker PL. Glucagon-like peptide-1 protects beta cells from cytokine-induced apoptosis and necrosis: role of protein kinase B. Diabetologia. 2005a;48:1339–1349. doi: 10.1007/s00125-005-1787-2. [DOI] [PubMed] [Google Scholar]
- Li LX, MacDonald PE, Ahn DS, Oudit GY, Backx PH, Brubaker PL. Role of phosphatidylinositol 3-kinasegamma in the beta-cell: interactions with glucagon-like peptide-1. Endocrinology. 2006;147:3318–3325. doi: 10.1210/en.2006-0155. [DOI] [PubMed] [Google Scholar]
- Li WC, Horb ME, Tosh D, Slack JM. In vitro transdifferentiation of hepatoma cells into functional pancreatic cells. Mech Dev. 2005b;122:835–847. doi: 10.1016/j.mod.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Li Y, Cao X, Li LX, Brubaker PL, Edlund H, Drucker DJ. beta-Cell Pdx1 expression is essential for the glucoregulatory, proliferative, and cytoprotective actions of glucagon-like peptide-1. Diabetes. 2005c;54:482–491. doi: 10.2337/diabetes.54.2.482. [DOI] [PubMed] [Google Scholar]
- Li Y, Hansotia T, Yusta B, Ris F, Halban PA, Drucker DJ. Glucagon-like peptide-1 receptor signaling modulates beta cell apoptosis. J Biol Chem. 2003;278:471–478. doi: 10.1074/jbc.M209423200. [DOI] [PubMed] [Google Scholar]
- Light PE, Manning Fox JE, Riedel MJ, Wheeler MB. Glucagon-like peptide-1 inhibits pancreatic ATP-sensitive potassium channels via a protein kinase A- and ADP-dependent mechanism. Mol Endocrinol. 2002;16:2135–2144. doi: 10.1210/me.2002-0084. [DOI] [PubMed] [Google Scholar]
- Ling Z, Wu D, Zambre Y, Flamez D, Drucker DJ, Pipeleers DG, Schuit FC. Glucagon-like peptide 1 receptor signaling influences topography of islet cells in mice. Virchows Arch. 2001;438:382–387. doi: 10.1007/s004280000374. [DOI] [PubMed] [Google Scholar]
- Lopez de Maturana R, Donnelly D. The glucagon-like peptide-1 receptor binding site for the N-terminus of GLP-1 requires polarity at Asp198 rather than negative charge. FEBS Lett. 2002;530:244–248. doi: 10.1016/s0014-5793(02)03492-0. [DOI] [PubMed] [Google Scholar]
- Lopez de Maturana R, Treece-Birch J, Abidi F, Findlay JB, Donnelly D. Met-204 and Tyr-205 are together important for binding GLP-1 receptor agonists but not their N-terminally truncated analogues. Protein Pept Lett. 2004;11:15–22. doi: 10.2174/0929866043478491. [DOI] [PubMed] [Google Scholar]
- Lopez de Maturana R, Willshaw A, Kuntzsch A, Rudolph R, Donnelly D. The isolated N-terminal domain of the glucagon-like peptide-1 (GLP-1) receptor binds exendin peptides with much higher affinity than GLP-1. J Biol Chem. 2003;278:10195–10200. doi: 10.1074/jbc.M212147200. [DOI] [PubMed] [Google Scholar]
- MacDonald PE, Ha XF, Wang J, Smukler SR, Sun AM, Gaisano HY, Salapatek AM, Backx PH, Wheeler MB. Members of the Kv1 and Kv2 voltage-dependent K(+) channel families regulate insulin secretion. Mol Endocrinol. 2001;15:1423–1435. doi: 10.1210/mend.15.8.0685. [DOI] [PubMed] [Google Scholar]
- MacDonald PE, Joseph JW, Yau D, Diao J, Asghar Z, Dai F, Oudit GY, Patel MM, Backx PH, Wheeler MB. Impaired glucose-stimulated insulin secretion, enhanced intraperitoneal insulin tolerance, and increased beta-cell mass in mice lacking the p110gamma isoform of phosphoinositide 3-kinase. Endocrinology. 2004;145:4078–4083. doi: 10.1210/en.2004-0028. [DOI] [PubMed] [Google Scholar]
- MacDonald PE, Salapatek AM, Wheeler MB. Glucagon-like peptide-1 receptor activation antagonizes voltage-dependent repolarizing K(+) currents in beta-cells: a possible glucose-dependent insulinotropic mechanism. Diabetes. 2002;51(Suppl 3):S443–447. doi: 10.2337/diabetes.51.2007.s443. [DOI] [PubMed] [Google Scholar]
- MacDonald PE, Wang X, Xia F, El-kholy W, Targonsky ED, Tsushima RG, Wheeler MB. Antagonism of rat beta-cell voltage-dependent K+ currents by exendin 4 requires dual activation of the cAMP/protein kinase A and phosphatidylinositol 3-kinase signaling pathways. J Biol Chem. 2003;278:52446–52453. doi: 10.1074/jbc.M307612200. [DOI] [PubMed] [Google Scholar]
- MacDonald PE, Wheeler MB. Voltage-dependent K(+) channels in pancreatic beta cells: role, regulation and potential as therapeutic targets. Diabetologia. 2003;46:1046–1062. doi: 10.1007/s00125-003-1159-8. [DOI] [PubMed] [Google Scholar]
- Malaisse WJ, Malaisse-Lagae F, Sener A, Hellerstrom C. Participation of endogenous fatty acids in the secretory activity of the pancreatic B-cell. Biochem J. 1985;227:995–1002. doi: 10.1042/bj2270995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra R, Singh L, Eng J, Raufman JP. Exendin-4, a new peptide from Heloderma suspectum venom, potentiates cholecystokinin-induced amylase release from rat pancreatic acini. Regul Pept. 1992;41:149–156. doi: 10.1016/0167-0115(92)90044-u. [DOI] [PubMed] [Google Scholar]
- Mashima H, Ohnishi H, Wakabayashi K, Mine T, Miyagawa J, Hanafusa T, Seno M, Yamada H, Kojima I. Betacellulin and activin A coordinately convert amylase-secreting pancreatic AR42J cells into insulin-secreting cells. J Clin Invest. 1996;97:1647–1654. doi: 10.1172/JCI118591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathi SK, Chan Y, Li X, Wheeler MB. Scanning of the glucagon-like peptide-1 receptor localizes G protein-activating determinants primarily to the N terminus of the third intracellular loop. Mol Endocrinol. 1997;11:424–432. doi: 10.1210/mend.11.4.9913. [DOI] [PubMed] [Google Scholar]
- Matschinsky FM. Regulation of pancreatic beta-cell glucokinase: from basics to therapeutics. Diabetes. 2002;51(Suppl 3):S394–404. doi: 10.2337/diabetes.51.2007.s394. [DOI] [PubMed] [Google Scholar]
- Matsumura T, Itoh H, Watanabe N, Oda Y, Tanaka M, Namba M, Kono N, Matsuyama T, Komatsu R, Matsuzawa Y. Glucagonlike peptide-1(7-36)amide suppresses glucagon secretion and decreases cyclic AMP concentration in cultured In-R1-G9 cells. Biochem Biophys Res Commun. 1992;186:503–508. doi: 10.1016/s0006-291x(05)80836-8. [DOI] [PubMed] [Google Scholar]
- Matsuo M, Kimura Y, Ueda K. KATP channel interaction with adenine nucleotides. J Mol Cell Cardiol. 2005;38:907–916. doi: 10.1016/j.yjmcc.2004.11.021. [DOI] [PubMed] [Google Scholar]
- Mattson MP. NF-kappaB in the survival and plasticity of neurons. Neurochem Res. 2005;30:883–893. doi: 10.1007/s11064-005-6961-x. [DOI] [PubMed] [Google Scholar]
- Mayo KE, Miller LJ, Bataille D, Dalle S, Goke B, Thorens B, Drucker DJ. International Union of Pharmacology. XXXV. The glucagon receptor family. Pharmacol Rev. 2003;55:167–194. doi: 10.1124/pr.55.1.6. [DOI] [PubMed] [Google Scholar]
- Meier JJ, Bhushan A, Butler AE, Rizza RA, Butler PC. Sustained beta cell apoptosis in patients with long-standing type 1 diabetes: indirect evidence for islet regeneration? Diabetologia. 2005;48:2221–2228. doi: 10.1007/s00125-005-1949-2. [DOI] [PubMed] [Google Scholar]
- Meier JJ, Butler AE, Galasso R, Butler PC. Hyperinsulinemic Hypoglycemia After Gastric Bypass Surgery Is Not Accompanied by Islet Hyperplasia or Increased {beta}-Cell Turnover. Diabetes Care. 2006a;29:1554–1559. doi: 10.2337/dc06-0392. [DOI] [PubMed] [Google Scholar]
- Meier JJ, Gallwitz B, Siepmann N, Holst JJ, Deacon CF, Schmidt WE, Nauck MA. Gastric inhibitory polypeptide (GIP) dose-dependently stimulates glucagon secretion in healthy human subjects at euglycaemia. Diabetologia. 2003;46:798–801. doi: 10.1007/s00125-003-1103-y. [DOI] [PubMed] [Google Scholar]
- Meier JJ, Gethmann A, Gotze O, Gallwitz B, Holst JJ, Schmidt WE, Nauck MA. Glucagon-like peptide 1 abolishes the postprandial rise in triglyceride concentrations and lowers levels of non-esterified fatty acids in humans. Diabetologia. 2006b;49:452–458. doi: 10.1007/s00125-005-0126-y. [DOI] [PubMed] [Google Scholar]
- Meier JJ, Nauck MA, Schmidt WE, Gallwitz B. Gastric inhibitory polypeptide: the neglected incretin revisited. Regul Pept. 2002;107:1–13. doi: 10.1016/s0167-0115(02)00039-3. [DOI] [PubMed] [Google Scholar]
- Menasche G, Ho CH, Sanal O, Feldmann J, Tezcan I, Ersoy F, Houdusse A, Fischer A, de Saint Basile G. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1) J Clin Invest. 2003;112:450–456. doi: 10.1172/JCI18264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meneilly GS, Greig N, Tildesley H, Habener JF, Egan JM, Elahi D. Effects of 3 months of continuous subcutaneous administration of glucagon-like peptide 1 in elderly patients with type 2 diabetes. Diabetes Care. 2003;26:2835–2841. doi: 10.2337/diacare.26.10.2835. [DOI] [PubMed] [Google Scholar]
- Meneilly GS, Veldhuis JD, Elahi D. Deconvolution analysis of rapid insulin pulses before and after six weeks of continuous subcutaneous administration of glucagon-like peptide-1 in elderly patients with type 2 diabetes. J Clin Endocrinol Metab. 2005;90:6251–6256. doi: 10.1210/jc.2004-2100. [DOI] [PubMed] [Google Scholar]
- Miki T, Minami K, Shinozaki H, Matsumura K, Saraya A, Ikeda H, Yamada Y, Holst JJ, Seino S. Distinct effects of glucose-dependent insulinotropic polypeptide and glucagon-like peptide-1 on insulin secretion and gut motility. Diabetes. 2005;54:1056–1063. doi: 10.2337/diabetes.54.4.1056. [DOI] [PubMed] [Google Scholar]
- Minn AH, Hafele C, Shalev A. Thioredoxin-interacting protein is stimulated by glucose through a carbohydrate response element and induces beta-cell apoptosis. Endocrinology. 2005;146:2397–2405. doi: 10.1210/en.2004-1378. [DOI] [PubMed] [Google Scholar]
- Moede T, Leibiger B, Pour HG, Berggren P, Leibiger IB. Identification of a nuclear localization signal, RRMKWKK, in the homeodomain transcription factor PDX-1. FEBS Lett. 1999;461:229–234. doi: 10.1016/s0014-5793(99)01446-5. [DOI] [PubMed] [Google Scholar]
- Moens K, Heimberg H, Flamez D, Huypens P, Quartier E, Ling Z, Pipeleers D, Gremlich S, Thorens B, Schuit F. Expression and functional activity of glucagon, glucagon-like peptide I, and glucose-dependent insulinotropic peptide receptors in rat pancreatic islet cells. Diabetes. 1996;45:257–261. doi: 10.2337/diab.45.2.257. [DOI] [PubMed] [Google Scholar]
- Mojsov S, Weir GC, Habener JF. Insulinotropin: glucagon-like peptide I (7-37) co-encoded in the glucagon gene is a potent stimulator of insulin release in the perfused rat pancreas. J Clin Invest. 1987;79:616–619. doi: 10.1172/JCI112855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montminy MR, Sevarino KA, Wagner JA, Mandel G, Goodman RH. Identification of a cyclic-AMP-responsive element within the rat somatostatin gene. Proc Natl Acad Sci U S A. 1986;83:6682–6686. doi: 10.1073/pnas.83.18.6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montrose-Rafizadeh C, Avdonin P, Garant MJ, Rodgers BD, Kole S, Yang H, Levine MA, Schwindinger W, Bernier M. Pancreatic glucagon-like peptide-1 receptor couples to multiple G proteins and activates mitogen-activated protein kinase pathways in Chinese hamster ovary cells. Endocrinology. 1999;140:1132–1140. doi: 10.1210/endo.140.3.6550. [DOI] [PubMed] [Google Scholar]
- Montrose-Rafizadeh C, Egan JM, Roth J. Incretin hormones regulate glucose-dependent insulin secretion in RIN 1046-38 cells: mechanisms of action. Endocrinology. 1994;135:589–594. doi: 10.1210/endo.135.2.8033807. [DOI] [PubMed] [Google Scholar]
- Montrose-Rafizadeh C, Wang Y, Janczewski AM, Henderson TE, Egan JM. Overexpression of glucagon-like peptide-1 receptor in an insulin-secreting cell line enhances glucose responsiveness. Mol Cell Endocrinol. 1997a;130:109–117. doi: 10.1016/s0303-7207(97)00079-8. [DOI] [PubMed] [Google Scholar]
- Montrose-Rafizadeh C, Yang H, Rodgers BD, Beday A, Pritchette LA, Eng J. High potency antagonists of the pancreatic glucagon-like peptide-1 receptor. J Biol Chem. 1997b;272:21201–21206. doi: 10.1074/jbc.272.34.21201. [DOI] [PubMed] [Google Scholar]
- Morinigo R, Moize V, Musri M, Lacy AM, Navarro S, Marin JL, Delgado S, Casamitjana R, Vidal J. Glucagon-like peptide-1, peptide YY, hunger, and satiety after gastric bypass surgery in morbidly obese subjects. J Clin Endocrinol Metab. 2006;91:1735–1740. doi: 10.1210/jc.2005-0904. [DOI] [PubMed] [Google Scholar]
- Nagy G, Reim K, Matti U, Brose N, Binz T, Rettig J, Neher E, Sorensen JB. Regulation of releasable vesicle pool sizes by protein kinase A-dependent phosphorylation of SNAP-25. Neuron. 2004;41:417–429. doi: 10.1016/s0896-6273(04)00038-8. [DOI] [PubMed] [Google Scholar]
- Nathan DM, Schreiber E, Fogel H, Mojsov S, Habener JF. Insulinotropic action of glucagonlike peptide-I-(7-37) in diabetic and nondiabetic subjects. Diabetes Care. 1992;15:270–276. doi: 10.2337/diacare.15.2.270. [DOI] [PubMed] [Google Scholar]
- Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest. 1993a;91:301–307. doi: 10.1172/JCI116186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauck MA, Homberger E, Siegel EG, Allen RC, Eaton RP, Ebert R, Creutzfeldt W. Incretin effects of increasing glucose loads in man calculated from venous insulin and C-peptide responses. J Clin Endocrinol Metab. 1986;63:492–498. doi: 10.1210/jcem-63-2-492. [DOI] [PubMed] [Google Scholar]
- Nauck MA, Kleine N, Orskov C, Holst JJ, Willms B, Creutzfeldt W. Normalization of fasting hyperglycaemia by exogenous glucagon-like peptide 1 (7-36 amide) in type 2 (non-insulin-dependent) diabetic patients. Diabetologia. 1993b;36:741–744. doi: 10.1007/BF00401145. [DOI] [PubMed] [Google Scholar]
- Nauck MA, Niedereichholz U, Ettler R, Holst JJ, Orskov C, Ritzel R, Schmiegel WH. Glucagon-like peptide 1 inhibition of gastric emptying outweighs its insulinotropic effects in healthy humans. Am J Physiol. 1997;273:E981–988. doi: 10.1152/ajpendo.1997.273.5.E981. [DOI] [PubMed] [Google Scholar]
- Nauck MA, Wollschlager D, Werner J, Holst JJ, Orskov C, Creutzfeldt W, Willms B. Effects of subcutaneous glucagon-like peptide 1 (GLP-1 [7-36 amide]) in patients with NIDDM. Diabetologia. 1996;39:1546–1553. doi: 10.1007/s001250050613. [DOI] [PubMed] [Google Scholar]
- Nauert JB, Rigas JD, Lester LB. Identification of an IQGAP1/AKAP79 complex in beta-cells. J Cell Biochem. 2003;90:97–108. doi: 10.1002/jcb.10604. [DOI] [PubMed] [Google Scholar]
- NC-IUPHAR: Internal Union of Pharmacology (IUPHAR) Receptor Database
- Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440:470–476. doi: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- Nie Y, Nakashima M, Brubaker PL, Li QL, Perfetti R, Jansen E, Zambre Y, Pipeleers D, Friedman TC. Regulation of pancreatic PC1 and PC2 associated with increased glucagon-like peptide 1 in diabetic rats. J Clin Invest. 2000;105:955–965. doi: 10.1172/JCI7456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norlin S, Ahlgren U, Edlund H. Nuclear factor-{kappa}B activity in {beta}-cells is required for glucose-stimulated insulin secretion. Diabetes. 2005;54:125–132. doi: 10.2337/diabetes.54.1.125. [DOI] [PubMed] [Google Scholar]
- Oetjen E, Diedrich T, Eggers A, Eckert B, Knepel W. Distinct properties of the cAMP-responsive element of the rat insulin I gene. J Biol Chem. 1994;269:27036–27044. [PubMed] [Google Scholar]
- Ogreid D, Ekanger R, Suva RH, Miller JP, Doskeland SO. Comparison of the two classes of binding sites (A and B) of type I and type II cyclic-AMP-dependent protein kinases by using cyclic nucleotide analogs. Eur J Biochem. 1989;181:19–31. doi: 10.1111/j.1432-1033.1989.tb14689.x. [DOI] [PubMed] [Google Scholar]
- Ohlsson H, Karlsson K, Edlund T. IPF1, a homeodomain-containing transactivator of the insulin gene. Embo J. 1993;12:4251–4259. doi: 10.1002/j.1460-2075.1993.tb06109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orskov C, Poulsen SS. Glucagonlike peptide-I-(7-36)-amide receptors only in islets of Langerhans. Autoradiographic survey of extracerebral tissues in rats. Diabetes. 1991;40:1292–1296. doi: 10.2337/diab.40.10.1292. [DOI] [PubMed] [Google Scholar]
- Orskov C, Rabenhoj L, Wettergren A, Kofod H, Holst JJ. Tissue and plasma concentrations of amidated and glycine-extended glucagon-like peptide I in humans. Diabetes. 1994;43:535–539. doi: 10.2337/diab.43.4.535. [DOI] [PubMed] [Google Scholar]
- Ozaki N, Shibasaki T, Kashima Y, Miki T, Takahashi K, Ueno H, Sunaga Y, Yano H, Matsuura Y, Iwanaga T, Takai Y, Seino S. cAMP-GEFII is a direct target of cAMP in regulated exocytosis. Nat Cell Biol. 2000;2:805–811. doi: 10.1038/35041046. [DOI] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Park H, Ahn Y, Park CK, Chung HY, Park Y. Interleukin-6 protects MIN6 beta cells from cytokine-induced apoptosis. Ann N Y Acad Sci. 2003;1005:242–249. doi: 10.1196/annals.1288.036. [DOI] [PubMed] [Google Scholar]
- Park S, Dong X, Fisher TL, Dunn S, Omer AK, Weir G, White MF. Exendin-4 uses Irs2 signaling to mediate pancreatic Beta cell growth and function. J Biol Chem. 2006;281:1159–1168. doi: 10.1074/jbc.M508307200. [DOI] [PubMed] [Google Scholar]
- Parker JC, VanVolkenburg MA, Ketchum RJ, Brayman KL, Andrews KM. Cyclic AMP phosphodiesterases of human and rat islets of Langerhans: contributions of types III and IV to the modulation of insulin secretion. Biochem Biophys Res Commun. 1995;217:916–923. doi: 10.1006/bbrc.1995.2858. [DOI] [PubMed] [Google Scholar]
- Patel S, Joseph SK, Thomas AP. Molecular properties of inositol 1,4,5-trisphosphate receptors. Cell Calcium. 1999;25:247–264. doi: 10.1054/ceca.1999.0021. [DOI] [PubMed] [Google Scholar]
- Patti ME, McMahon G, Mun EC, Bitton A, Holst JJ, Goldsmith J, Hanto DW, Callery M, Arky R, Nose V, Bonner-Weir S, Goldfine AB. Severe hypoglycaemia post-gastric bypass requiring partial pancreatectomy: evidence for inappropriate insulin secretion and pancreatic islet hyperplasia. Diabetologia. 2005;48:2236–2240. doi: 10.1007/s00125-005-1933-x. [DOI] [PubMed] [Google Scholar]
- Pederson RA, Satkunarajah M, McIntosh CH, Scrocchi LA, Flamez D, Schuit F, Drucker DJ, Wheeler MB. Enhanced glucose-dependent insulinotropic polypeptide secretion and insulinotropic action in glucagon-like peptide 1 receptor −/− mice. Diabetes. 1998;47:1046–1052. doi: 10.2337/diabetes.47.7.1046. [DOI] [PubMed] [Google Scholar]
- Perfetti R, Zhou J, Doyle ME, Egan JM. Glucagon-like peptide-1 induces cell proliferation and pancreatic-duodenum homeobox-1 expression and increases endocrine cell mass in the pancreas of old, glucose-intolerant rats. Endocrinology. 2000;141:4600–4605. doi: 10.1210/endo.141.12.7806. [DOI] [PubMed] [Google Scholar]
- Petersen HV, Peshavaria M, Pedersen AA, Philippe J, Stein R, Madsen OD, Serup P. Glucose stimulates the activation domain potential of the PDX-1 homeodomain transcription factor. FEBS Lett. 1998;431:362–366. doi: 10.1016/s0014-5793(98)00776-5. [DOI] [PubMed] [Google Scholar]
- Philippe J, Missotten M. Functional characterization of a cAMP-responsive element of the rat insulin I gene. J Biol Chem. 1990;265:1465–1469. [PubMed] [Google Scholar]
- Pictet RL, Clark WR, Williams RH, Rutter WJ. An ultrastructural analysis of the developing embryonic pancreas. Dev Biol. 1972;29:436–467. doi: 10.1016/0012-1606(72)90083-8. [DOI] [PubMed] [Google Scholar]
- Rachman J, Barrow BA, Levy JC, Turner RC. Near-normalisation of diurnal glucose concentrations by continuous administration of glucagon-like peptide-1 (GLP-1) in subjects with NIDDM. Diabetologia. 1997;40:205–211. doi: 10.1007/s001250050664. [DOI] [PubMed] [Google Scholar]
- Rafiq I, da Silva Xavier G, Hooper S, Rutter GA. Glucose-stimulated preproinsulin gene expression and nuclear trans-location of pancreatic duodenum homeobox-1 require activation of phosphatidylinositol 3-kinase but not p38 MAPK/SAPK2. J Biol Chem. 2000;275:15977–15984. doi: 10.1074/jbc.275.21.15977. [DOI] [PubMed] [Google Scholar]
- Rafiq I, Kennedy HJ, Rutter GA. Glucose-dependent translocation of insulin promoter factor-1 (IPF-1) between the nuclear periphery and the nucleoplasm of single MIN6 beta-cells. J Biol Chem. 1998;273:23241–23247. doi: 10.1074/jbc.273.36.23241. [DOI] [PubMed] [Google Scholar]
- Rajagopal J, Anderson WJ, Kume S, Martinez OI, Melton DA. Insulin staining of ES cell progeny from insulin uptake. Science. 2003;299:363. doi: 10.1126/science.1077838. [DOI] [PubMed] [Google Scholar]
- Ramiya VK, Maraist M, Arfors KE, Schatz DA, Peck AB, Cornelius JG. Reversal of insulin-dependent diabetes using islets generated in vitro from pancreatic stem cells. Nat Med. 2000;6:278–282. doi: 10.1038/73128. [DOI] [PubMed] [Google Scholar]
- Rankin MM, Zajac AL, Teta M, Long SY, Kushner JA. Beta Cell Regeneration Capacity Acutely Declines with Advanced Age. Diabetes. 2006;55:A78. [Google Scholar]
- Ranta F, Avram D, Berchtold S, Dufer M, Drews G, Lang F, Ullrich S. Dexamethasone induces cell death in insulin-secreting cells, an effect reversed by exendin-4. Diabetes. 2006;55:1380–1390. doi: 10.2337/db05-1220. [DOI] [PubMed] [Google Scholar]
- Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- Raufman JP, Singh L, Singh G, Eng J. Truncated glucagon-like peptide-1 interacts with exendin receptors on dispersed acini from guinea pig pancreas. Identification of a mammalian analogue of the reptilian peptide exendin-4. J Biol Chem. 1992;267:21432–21437. [PubMed] [Google Scholar]
- Ravelli GP, Stein ZA, Susser MW. Obesity in young men after famine exposure in utero and early infancy. N Engl J Med. 1976;295:349–353. doi: 10.1056/NEJM197608122950701. [DOI] [PubMed] [Google Scholar]
- Regazzi R, Ravazzola M, Iezzi M, Lang J, Zahraoui A, Andereggen E, Morel P, Takai Y, Wollheim CB. Expression, localization and functional role of small GTPases of the Rab3 family in insulin-secreting cells. J Cell Sci. 1996;109(Pt 9):2265–2273. doi: 10.1242/jcs.109.9.2265. [DOI] [PubMed] [Google Scholar]
- Rehmann H, Schwede F, Doskeland SO, Wittinghofer A, Bos JL. Ligand-mediated activation of the cAMP-responsive guanine nucleotide exchange factor Epac. J Biol Chem. 2003;278:38548–38556. doi: 10.1074/jbc.M306292200. [DOI] [PubMed] [Google Scholar]
- Renstrom E, Barg S, Thevenod F, Rorsman P. Sulfonylurea-mediated stimulation of insulin exocytosis via an ATP-sensitive K+ channel-independent action. Diabetes. 2002;51(Suppl 1):S33–36. doi: 10.2337/diabetes.51.2007.s33. [DOI] [PubMed] [Google Scholar]
- Roe MW, Worley JF, 3rd, Mittal AA, Kuznetsov A, DasGupta S, Mertz RJ, Witherspoon SM, 3rd, Blair N, Lancaster ME, McIntyre MS, Shehee WR, Dukes ID, Philipson LH. Expression and function of pancreatic beta-cell delayed rectifier K+ channels. Role in stimulus-secretion coupling. J Biol Chem. 1996;271:32241–32246. doi: 10.1074/jbc.271.50.32241. [DOI] [PubMed] [Google Scholar]
- Rolin B, Larsen MO, Gotfredsen CF, Deacon CF, Carr RD, Wilken M, Knudsen LB. The long-acting GLP-1 derivative NN2211 ameliorates glycemia and increases beta-cell mass in diabetic mice. Am J Physiol Endocrinol Metab. 2002;283:E745–752. doi: 10.1152/ajpendo.00030.2002. [DOI] [PubMed] [Google Scholar]
- Romer JKP, Heller RS, Knudsen LB, Tornehave D. In vivo expression of the GLP-1 receptor is restricted to insulin producing beta-cells in normal rat, mouse, and human intact pancreatic islets. Diabetes. 2002:1403. [Google Scholar]
- Rorsman P, Renstrom E. Insulin granule dynamics in pancreatic beta cells. Diabetologia. 2003;46:1029–1045. doi: 10.1007/s00125-003-1153-1. [DOI] [PubMed] [Google Scholar]
- Rossetto O, Schiavo G, Montecucco C, Poulain B, Deloye F, Lozzi L, Shone CC. SNARE motif and neurotoxins. Nature. 1994;372:415–416. doi: 10.1038/372415a0. [DOI] [PubMed] [Google Scholar]
- Rusnak F, Mertz P. Calcineurin: form and function. Physiol Rev. 2000;80:1483–1521. doi: 10.1152/physrev.2000.80.4.1483. [DOI] [PubMed] [Google Scholar]
- Salapatek AM, MacDonald PE, Gaisano HY, Wheeler MB. Mutations to the third cytoplasmic domain of the glucagon-like peptide 1 (GLP-1) receptor can functionally uncouple GLP-1-stimulated insulin secretion in HIT-T15 cells. Mol Endocrinol. 1999;13:1305–1317. doi: 10.1210/mend.13.8.0321. [DOI] [PubMed] [Google Scholar]
- Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- Scaglia L, Cahill CJ, Finegood DT, Bonner-Weir S. Apoptosis participates in the remodeling of the endocrine pancreas in the neonatal rat. Endocrinology. 1997;138:1736–1741. doi: 10.1210/endo.138.4.5069. [DOI] [PubMed] [Google Scholar]
- Schmid R, Schusdziarra V, Aulehner R, Weigert N, Classen M. Comparison of GLP-1 (7-36amide) and GIP on release of somatostatin-like immunoreactivity and insulin from the isolated rat pancreas. Z Gastroenterol. 1990;28:280–284. [PubMed] [Google Scholar]
- Schuit FC, Pipeleers DG. Regulation of adenosine 3′,5′-monophosphate levels in the pancreatic B cell. Endocrinology. 1985;117:834–840. doi: 10.1210/endo-117-3-834. [DOI] [PubMed] [Google Scholar]
- Scopsi L, Gullo M, Rilke F, Martin S, Steiner DF. Proprotein convertases (PC1/PC3 and PC2) in normal and neoplastic human tissues: their use as markers of neuroendocrine differentiation. J Clin Endocrinol Metab. 1995;80:294–301. doi: 10.1210/jcem.80.1.7829629. [DOI] [PubMed] [Google Scholar]
- Scrocchi LA, Brown TJ, MaClusky N, Brubaker PL, Auerbach AB, Joyner AL, Drucker DJ. Glucose intolerance but normal satiety in mice with a null mutation in the glucagon-like peptide 1 receptor gene. Nat Med. 1996;2:1254–1258. doi: 10.1038/nm1196-1254. [DOI] [PubMed] [Google Scholar]
- Scrocchi LA, Marshall BA, Cook SM, Brubaker PL, Drucker DJ. Identification of glucagon-like peptide 1 (GLP-1) actions essential for glucose homeostasis in mice with disruption of GLP-1 receptor signaling. Diabetes. 1998;47:632–639. doi: 10.2337/diabetes.47.4.632. [DOI] [PubMed] [Google Scholar]
- Seaberg RM, Smukler SR, Kieffer TJ, Enikolopov G, Asghar Z, Wheeler MB, Korbutt G, van der Kooy D. Clonal identification of multipotent precursors from adult mouse pancreas that generate neural and pancreatic lineages. Nat Biotechnol. 2004;22:1115–1124. doi: 10.1038/nbt1004. [DOI] [PubMed] [Google Scholar]
- Seeberger KL, Dufour JM, Shapiro AM, Lakey JR, Rajotte RV, Korbutt GS. Expansion of mesenchymal stem cells from human pancreatic ductal epithelium. Lab Invest. 2006;86:141–153. doi: 10.1038/labinvest.3700377. [DOI] [PubMed] [Google Scholar]
- Seino S, Shibasaki T. PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol Rev. 2005;85:1303–1342. doi: 10.1152/physrev.00001.2005. [DOI] [PubMed] [Google Scholar]
- Service GJ, Thompson GB, Service FJ, Andrews JC, Collazo-Clavell ML, Lloyd RV. Hyperinsulinemic hypoglycemia with nesidioblastosis after gastric-bypass surgery. N Engl J Med. 2005;353:249–254. doi: 10.1056/NEJMoa043690. [DOI] [PubMed] [Google Scholar]
- Shafiee-Nick R, Pyne NJ, Furman BL. Effects of type-selective phosphodiesterase inhibitors on glucose-induced insulin secretion and islet phosphodiesterase activity. Br J Pharmacol. 1995;115:1486–1492. doi: 10.1111/j.1476-5381.1995.tb16641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp GW. The adenylate cyclase-cyclic AMP system in islets of Langerhans and its role in the control of insulin release. Diabetologia. 1979;16:287–296. doi: 10.1007/BF01223617. [DOI] [PubMed] [Google Scholar]
- Shibasaki T, Sunaga Y, Fujimoto K, Kashima Y, Seino S. Interaction of ATP sensor, cAMP sensor, Ca2+ sensor, and voltage-dependent Ca2+ channel in insulin granule exocytosis. J Biol Chem. 2004;279:7956–7961. doi: 10.1074/jbc.M309068200. [DOI] [PubMed] [Google Scholar]
- Shiota C, Larsson O, Shelton KD, Shiota M, Efanov AM, Hoy M, Lindner J, Kooptiwut S, Juntti-Berggren L, Gromada J, Berggren PO, Magnuson MA. Sulfonylurea receptor type 1 knock-out mice have intact feeding-stimulated insulin secretion despite marked impairment in their response to glucose. J Biol Chem. 2002;277:37176–37183. doi: 10.1074/jbc.M206757200. [DOI] [PubMed] [Google Scholar]
- Shizukuda Y, Buttrick PM. Protein kinase C-zeta modulates thromboxane A(2)-mediated apoptosis in adult ventricular myocytes via Akt. Am J Physiol Heart Circ Physiol. 2002;282:H320–327. doi: 10.1152/ajpheart.2002.282.1.H320. [DOI] [PubMed] [Google Scholar]
- Simmons RA, Templeton LJ, Gertz SJ. Intrauterine growth retardation leads to the development of type 2 diabetes in the rat. Diabetes. 2001;50:2279–2286. doi: 10.2337/diabetes.50.10.2279. [DOI] [PubMed] [Google Scholar]
- Skalhegg BS, Tasken K. Specificity in the cAMP/PKA signaling pathway. Differential expression,regulation, and subcellular localization of subunits of PKA. Front Biosci. 2000;5:D678–693. doi: 10.2741/skalhegg. [DOI] [PubMed] [Google Scholar]
- Skoglund G, Hussain MA, Holz GG. Glucagon-like peptide 1 stimulates insulin gene promoter activity by protein kinase A-independent activation of the rat insulin I gene cAMP response element. Diabetes. 2000;49:1156–1164. doi: 10.2337/diabetes.49.7.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens LR, Eguinoa A, Erdjument-Bromage H, Lui M, Cooke F, Coadwell J, Smrcka AS, Thelen M, Cadwallader K, Tempst P, Hawkins PT. The G beta gamma sensitivity of a PI3K is dependent upon a tightly associated adaptor, p101. Cell. 1997;89:105–114. doi: 10.1016/s0092-8674(00)80187-7. [DOI] [PubMed] [Google Scholar]
- Stoffel M, Espinosa R, 3rd, Le Beau MM, Bell GI. Human glucagon-like peptide-1 receptor gene. Localization to chromosome band 6p21 by fluorescence in situ hybridization and linkage of a highly polymorphic simple tandem repeat DNA polymorphism to other markers on chromosome 6. Diabetes. 1993;42:1215–1218. doi: 10.2337/diab.42.8.1215. [DOI] [PubMed] [Google Scholar]
- Stoffers DA, Desai BM, DeLeon DD, Simmons RA. Neonatal exendin-4 prevents the development of diabetes in the intrauterine growth retarded rat. Diabetes. 2003;52:734–740. doi: 10.2337/diabetes.52.3.734. [DOI] [PubMed] [Google Scholar]
- Stoffers DA, Kieffer TJ, Hussain MA, Drucker DJ, Bonner-Weir S, Habener JF, Egan JM. Insulinotropic glucagon-like peptide 1 agonists stimulate expression of homeodomain protein IDX-1 and increase islet size in mouse pancreas. Diabetes. 2000;49:741–748. doi: 10.2337/diabetes.49.5.741. [DOI] [PubMed] [Google Scholar]
- Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet. 1997;15:106–110. doi: 10.1038/ng0197-106. [DOI] [PubMed] [Google Scholar]
- Stokoe D. The phosphoinositide 3-kinase pathway and cancer. Expert Rev Mol Med. 2005;7:1–22. doi: 10.1017/S1462399405009361. [DOI] [PubMed] [Google Scholar]
- Straub SG, Sharp GW. Hypothesis: one rate-limiting step controls the magnitude of both phases of glucose-stimulated insulin secretion. Am J Physiol Cell Physiol. 2004;287:C565–571. doi: 10.1152/ajpcell.00079.2004. [DOI] [PubMed] [Google Scholar]
- Sturis J, Gotfredsen CF, Romer J, Rolin B, Ribel U, Brand CL, Wilken M, Wassermann K, Deacon CF, Carr RD, Knudsen LB. GLP-1 derivative liraglutide in rats with beta-cell deficiencies: influence of metabolic state on beta-cell mass dynamics. Br J Pharmacol. 2003;140:123–132. doi: 10.1038/sj.bjp.0705397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Dostmann WR, Herberg FW, Durick K, Xuong NH, Ten Eyck L, Taylor SS, Varughese KI. Regulatory subunit of protein kinase A: structure of deletion mutant with cAMP binding domains. Science. 1995;269:807–813. doi: 10.1126/science.7638597. [DOI] [PubMed] [Google Scholar]
- Sugden MC, Ashcroft SJ. Cyclic nucleotide phosphodiesterase of rat pancreatic islets. Effects of Ca2+, calmodulin and trifluoperazine. Biochem J. 1981;197:459–464. doi: 10.1042/bj1970459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugden MC, Ashcroft SJ, Sugden PH. Protein kinase activities in rat pancreatic islets of Langerhans. Biochem J. 1979;180:219–229. doi: 10.1042/bj1800219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syme CA, Zhang L, Bisello A. Caveolin-1 Regulates Cellular Trafficking and Function of the Glucagon-Like Peptide 1 Receptor. Mol Endocrinol. 2006 doi: 10.1210/me.2006-0178. [DOI] [PubMed] [Google Scholar]
- Ta M, Choi Y, Atouf F, Park CH, Lumelsky N. The defined combination of growth factors controls generation of long-term-replicating islet progenitor-like cells from cultures of adult mouse pancreas. Stem Cells. 2006;24:1738–1749. doi: 10.1634/stemcells.2005-0367. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Kadowaki T, Yazaki Y, Ellis-Davies GC, Miyashita Y, Kasai H. Post-priming actions of ATP on Ca2+-dependent exocytosis in pancreatic beta cells. Proc Natl Acad Sci U S A. 1999;96:760–765. doi: 10.1073/pnas.96.2.760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takhar S, Gyomorey S, Su RC, Mathi SK, Li X, Wheeler MB. The third cytoplasmic domain of the GLP-1[7-36 amide] receptor is required for coupling to the adenylyl cyclase system. Endocrinology. 1996;137:2175–2178. doi: 10.1210/endo.137.5.8612565. [DOI] [PubMed] [Google Scholar]
- Tanizawa Y, Riggs AC, Elbein SC, Whelan A, Donis-Keller H, Permutt MA. Human glucagon-like peptide-1 receptor gene in NIDDM. Identification and use of simple sequence repeat polymorphisms in genetic analysis. Diabetes. 1994;43:752–757. doi: 10.2337/diab.43.6.752. [DOI] [PubMed] [Google Scholar]
- Tasken K, Aandahl EM. Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol Rev. 2004;84:137–167. doi: 10.1152/physrev.00021.2003. [DOI] [PubMed] [Google Scholar]
- Taylor SS, Buechler JA, Yonemoto W. cAMP-dependent protein kinase: framework for a diverse family of regulatory enzymes. Annu Rev Biochem. 1990;59:971–1005. doi: 10.1146/annurev.bi.59.070190.004543. [DOI] [PubMed] [Google Scholar]
- Tei E, Mehta S, Tulachan SS, Yew H, Hembree M, Preuett B, Snyder CL, Yamataka A, Miyano T, Gittes GK. Synergistic endocrine induction by GLP-1 and TGF-beta in the developing pancreas. Pancreas. 2005;31:138–141. doi: 10.1097/01.mpa.0000172566.70619.58. [DOI] [PubMed] [Google Scholar]
- Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of beta-cells in aged adult mice. Diabetes. 2005;54:2557–2567. doi: 10.2337/diabetes.54.9.2557. [DOI] [PubMed] [Google Scholar]
- Thearle M, Brillantes AM. Unique characteristics of the geriatric diabetic population and the role for therapeutic strategies that enhance glucagon-like peptide-1 activity. Curr Opin Clin Nutr Metab Care. 2005;8:9–16. doi: 10.1097/00075197-200501000-00003. [DOI] [PubMed] [Google Scholar]
- Thorens B. Expression cloning of the pancreatic beta cell receptor for the gluco-incretin hormone glucagon-like peptide 1. Proc Natl Acad Sci U S A. 1992;89:8641–8645. doi: 10.1073/pnas.89.18.8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorens B, Deriaz N, Bosco D, DeVos A, Pipeleers D, Schuit F, Meda P, Porret A. Protein kinase A-dependent phosphorylation of GLUT2 in pancreatic beta cells. J Biol Chem. 1996;271:8075–8081. doi: 10.1074/jbc.271.14.8075. [DOI] [PubMed] [Google Scholar]
- Thorens B, Guillam MT, Beermann F, Burcelin R, Jaquet M. Transgenic reexpression of GLUT1 or GLUT2 in pancreatic beta cells rescues GLUT2-null mice from early death and restores normal glucose-stimulated insulin secretion. J Biol Chem. 2000;275:23751–23758. doi: 10.1074/jbc.M002908200. [DOI] [PubMed] [Google Scholar]
- Thorens B, Porret A, Buhler L, Deng SP, Morel P, Widmann C. Cloning and functional expression of the human islet GLP-1 receptor. Demonstration that exendin-4 is an agonist and exendin-(9-39) an antagonist of the receptor. Diabetes. 1993;42:1678–1682. doi: 10.2337/diab.42.11.1678. [DOI] [PubMed] [Google Scholar]
- Thyssen S, Arany E, Hill DJ. Ontogeny of Regeneration of {beta}-Cells in the Neonatal Rat after Treatment with Streptozotocin. Endocrinology. 2006;147:2346–2356. doi: 10.1210/en.2005-0396. [DOI] [PubMed] [Google Scholar]
- Tibaduiza EC, Chen C, Beinborn M. A small molecule ligand of the glucagon-like peptide 1 receptor targets its amino-terminal hormone binding domain. J Biol Chem. 2001;276:37787–37793. doi: 10.1074/jbc.M106692200. [DOI] [PubMed] [Google Scholar]
- Tillmar L, Carlsson C, Welsh N. Control of insulin mRNA stability in rat pancreatic islets. Regulatory role of a 3′-untranslated region pyrimidine-rich sequence. J Biol Chem. 2002;277:1099–1106. doi: 10.1074/jbc.M108340200. [DOI] [PubMed] [Google Scholar]
- Timmerman LA, Clipstone NA, Ho SN, Northrop JP, Crabtree GR. Rapid shuttling of NF-AT in discrimination of Ca2+ signals and immunosuppression. Nature. 1996;383:837–840. doi: 10.1038/383837a0. [DOI] [PubMed] [Google Scholar]
- Todorov I, Omori K, Pascual M, Rawson J, Nair I, Valiente L, Vuong T, Matsuda T, Orr C, Ferreri K, Smith CV, Kandeel F, Mullen Y. Generation of human islets through expansion and differentiation of non-islet pancreatic cells discarded (pancreatic discard) after islet isolation. Pancreas. 2006;32:130–138. doi: 10.1097/01.mpa.0000202945.78331.93. [DOI] [PubMed] [Google Scholar]
- Tokuyama Y, Matsui K, Egashira T, Nozaki O, Ishizuka T, Kanatsuka A. Five missense mutations in glucagon-like peptide 1 receptor gene in Japanese population. Diabetes Res Clin Pract. 2004;66:63–69. doi: 10.1016/j.diabres.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Torii S, Takeuchi T, Nagamatsu S, Izumi T. Rab27 effector granuphilin promotes the plasma membrane targeting of insulin granules via interaction with syntaxin 1a. J Biol Chem. 2004;279:22532–22538. doi: 10.1074/jbc.M400600200. [DOI] [PubMed] [Google Scholar]
- Tourrel C, Bailbe D, Meile MJ, Kergoat M, Portha B. Glucagon-like peptide-1 and exendin-4 stimulate beta-cell neogenesis in streptozotocin-treated newborn rats resulting in persistently improved glucose homeostasis at adult age. Diabetes. 2001;50:1562–1570. doi: 10.2337/diabetes.50.7.1562. [DOI] [PubMed] [Google Scholar]
- Trucco M. Regeneration of the pancreatic beta cell. J Clin Invest. 2005;115:5–12. doi: 10.1172/JCI23935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudeau JD, Dutz JP, Arany E, Hill DJ, Fieldus WE, Finegood DT. Neonatal beta-cell apoptosis: a trigger for autoimmune diabetes? Diabetes. 2000;49:1–7. doi: 10.2337/diabetes.49.1.1. [DOI] [PubMed] [Google Scholar]
- Trumper A, Trumper K, Trusheim H, Arnold R, Goke B, Horsch D. Glucose-dependent insulinotropic polypeptide is a growth factor for beta (INS-1) cells by pleiotropic signaling. Mol Endocrinol. 2001;15:1559–1570. doi: 10.1210/mend.15.9.0688. [DOI] [PubMed] [Google Scholar]
- Trumper J, Ross D, Jahr H, Brendel MD, Goke R, Horsch D. The Rap-B-Raf signalling pathway is activated by glucose and glucagon-like peptide-1 in human islet cells. Diabetologia. 2005;48:1534–1540. doi: 10.1007/s00125-005-1820-5. [DOI] [PubMed] [Google Scholar]
- Trumper K, Trumper A, Trusheim H, Arnold R, Goke B, Horsch D. Integrative mitogenic role of protein kinase B/Akt in beta-cells. Ann N Y Acad Sci. 2000;921:242–250. doi: 10.1111/j.1749-6632.2000.tb06972.x. [DOI] [PubMed] [Google Scholar]
- Tsuboi T, da Silva Xavier G, Holz GG, Jouaville LS, Thomas AP, Rutter GA. Glucagon-like peptide-1 mobilizes intracellular Ca2+ and stimulates mitochondrial ATP synthesis in pancreatic MIN6 beta-cells. Biochem J. 2003;369:287–299. doi: 10.1042/BJ20021288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttle RL, Gill NS, Pugh W, Lee JP, Koeberlein B, Furth EE, Polonsky KS, Naji A, Birnbaum MJ. Regulation of pancreatic beta-cell growth and survival by the serine/threonine protein kinase Akt1/PKBalpha. Nat Med. 2001;7:1133–1137. doi: 10.1038/nm1001-1133. [DOI] [PubMed] [Google Scholar]
- Ungermann C, Langosch D. Functions of SNAREs in intracellular membrane fusion and lipid bilayer mixing. J Cell Sci. 2005;118:3819–3828. doi: 10.1242/jcs.02561. [DOI] [PubMed] [Google Scholar]
- Vazquez P, Roncero I, Blazquez E, Alvarez E. The cytoplasmic domain close to the transmembrane region of the glucagon-like peptide-1 receptor contains sequence elements that regulate agonist-dependent internalisation. J Endocrinol. 2005a;186:221–231. doi: 10.1677/joe.1.06179. [DOI] [PubMed] [Google Scholar]
- Vazquez P, Roncero I, Blazquez E, Alvarez E. Substitution of the cysteine 438 residue in the cytoplasmic tail of the glucagon-like peptide-1 receptor alters signal transduction activity. J Endocrinol. 2005b;185:35–44. doi: 10.1677/joe.1.06031. [DOI] [PubMed] [Google Scholar]
- Walker MD, Edlund T, Boulet AM, Rutter WJ. Cell-specific expression controlled by the 5′-flanking region of insulin and chymotrypsin genes. Nature. 1983;306:557–561. doi: 10.1038/306557a0. [DOI] [PubMed] [Google Scholar]
- Wan QF, Dong Y, Yang H, Lou X, Ding J, Xu T. Protein kinase activation increases insulin secretion by sensitizing the secretory machinery to Ca2+ J Gen Physiol. 2004;124:653–662. doi: 10.1085/jgp.200409082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Brubaker PL. Glucagon-like peptide-1 treatment delays the onset of diabetes in 8 week-old db/db mice. Diabetologia. 2002;45:1263–1273. doi: 10.1007/s00125-002-0828-3. [DOI] [PubMed] [Google Scholar]
- Wang Q, Li L, Xu E, Wong V, Rhodes C, Brubaker PL. Glucagon-like peptide-1 regulates proliferation and apoptosis via activation of protein kinase B in pancreatic INS-1 beta cells. Diabetologia. 2004;47:478–487. doi: 10.1007/s00125-004-1327-5. [DOI] [PubMed] [Google Scholar]
- Wang X, Cahill CM, Pineyro MA, Zhou J, Doyle ME, Egan JM. Glucagon-like peptide-1 regulates the beta cell transcription factor, PDX-1, in insulinoma cells. Endocrinology. 1999;140:4904–4907. doi: 10.1210/endo.140.10.7158. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhou J, Doyle ME, Egan JM. Glucagon-like peptide-1 causes pancreatic duodenal homeobox-1 protein translocation from the cytoplasm to the nucleus of pancreatic beta-cells by a cyclic adenosine monophosphate/protein kinase A-dependent mechanism. Endocrinology. 2001;142:1820–1827. doi: 10.1210/endo.142.5.8128. [DOI] [PubMed] [Google Scholar]
- Wang Y, Egan JM, Raygada M, Nadiv O, Roth J, Montrose-Rafizadeh C. Glucagon-like peptide-1 affects gene transcription and messenger ribonucleic acid stability of components of the insulin secretory system in RIN 1046-38 cells. Endocrinology. 1995;136:4910–4917. doi: 10.1210/endo.136.11.7588224. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Yaegashi H, Koizumi M, Toyota T, Takahashi T. Changing distribution of islets in the developing human pancreas: a computer-assisted three-dimensional reconstruction study. Pancreas. 1999;18:349–354. doi: 10.1097/00006676-199905000-00004. [DOI] [PubMed] [Google Scholar]
- Wheeler MB, Lu M, Dillon JS, Leng XH, Chen C, Boyd AE., 3rd Functional expression of the rat glucagon-like peptide-I receptor, evidence for coupling to both adenylyl cyclase and phospholipase-C. Endocrinology. 1993;133:57–62. doi: 10.1210/endo.133.1.8391428. [DOI] [PubMed] [Google Scholar]
- White MF. Insulin signaling in health and disease. Science. 2003;302:1710–1711. doi: 10.1126/science.1092952. [DOI] [PubMed] [Google Scholar]
- Wicksteed B, Herbert TP, Alarcon C, Lingohr MK, Moss LG, Rhodes CJ. Cooperativity between the preproinsulin mRNA untranslated regions is necessary for glucose-stimulated translation. J Biol Chem. 2001;276:22553–22558. doi: 10.1074/jbc.M011214200. [DOI] [PubMed] [Google Scholar]
- Widmann C, Dolci W, Thorens B. Agonist-induced internalization and recycling of the glucagon-like peptide-1 receptor in transfected fibroblasts and in insulinomas. Biochem J. 1995;310(Pt 1):203–214. doi: 10.1042/bj3100203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmann C, Dolci W, Thorens B. Desensitization and phosphorylation of the glucagon-like peptide-1 (GLP-1) receptor by GLP-1 and 4-phorbol 12-myristate 13-acetate. Mol Endocrinol. 1996a;10:62–75. doi: 10.1210/mend.10.1.8838146. [DOI] [PubMed] [Google Scholar]
- Widmann C, Dolci W, Thorens B. Heterologous desensitization of the glucagon-like peptide-1 receptor by phorbol esters requires phosphorylation of the cytoplasmic tail at four different sites. J Biol Chem. 1996b;271:19957–19963. doi: 10.1074/jbc.271.33.19957. [DOI] [PubMed] [Google Scholar]
- Widmann C, Dolci W, Thorens B. Internalization and homologous desensitization of the GLP-1 receptor depend on phosphorylation of the receptor carboxyl tail at the same three sites. Mol Endocrinol. 1997;11:1094–1102. doi: 10.1210/mend.11.8.9959. [DOI] [PubMed] [Google Scholar]
- Wiederkehr A, Wollheim CB. Minireview: implication of mitochondria in insulin secretion and action. Endocrinology. 2006;147:2643–2649. doi: 10.1210/en.2006-0057. [DOI] [PubMed] [Google Scholar]
- Wilmen A, Goke B, Goke R. The isolated N-terminal extracellular domain of the glucagon-like peptide-1 (GLP)-1 receptor has intrinsic binding activity. FEBS Lett. 1996;398:43–47. doi: 10.1016/s0014-5793(96)01214-8. [DOI] [PubMed] [Google Scholar]
- Wilmen A, Van Eyll B, Goke B, Goke R. Five out of six tryptophan residues in the N-terminal extracellular domain of the rat GLP-1 receptor are essential for its ability to bind GLP-1. Peptides. 1997;18:301–305. doi: 10.1016/s0196-9781(96)00321-x. [DOI] [PubMed] [Google Scholar]
- Wilson ME, Kalamaras JA, German MS. Expression pattern of IAPP and prohormone convertase 1/3 reveals a distinctive set of endocrine cells in the embryonic pancreas. Mech Dev. 2002;115:171–176. doi: 10.1016/s0925-4773(02)00118-1. [DOI] [PubMed] [Google Scholar]
- Wilson ME, Scheel D, German MS. Gene expression cascades in pancreatic development. Mech Dev. 2003;120:65–80. doi: 10.1016/s0925-4773(02)00333-7. [DOI] [PubMed] [Google Scholar]
- Winzell MS, Ahrén B. Glucagon-like Peptide-1 and Islet Lipolysis. Horm Metab Res. 2004;36:795–803. doi: 10.1055/s-2004-826166. [DOI] [PubMed] [Google Scholar]
- Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren JM, Previs S, Zhang Y, Bernal D, Pons S, Shulman GI, Bonner-Weir S, White MF. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. 1998;391:900–904. doi: 10.1038/36116. [DOI] [PubMed] [Google Scholar]
- Wrede CE, Dickson LM, Lingohr MK, Briaud I, Rhodes CJ. Protein kinase B/Akt prevents fatty acid-induced apoptosis in pancreatic beta-cells (INS-1) J Biol Chem. 2002;277:49676–49684. doi: 10.1074/jbc.M208756200. [DOI] [PubMed] [Google Scholar]
- Wu KL, Gannon M, Peshavaria M, Offield MF, Henderson E, Ray M, Marks A, Gamer LW, Wright CV, Stein R. Hepatocyte nuclear factor 3beta is involved in pancreatic beta-cell-specific transcription of the pdx-1 gene. Mol Cell Biol. 1997;17:6002–6013. doi: 10.1128/mcb.17.10.6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Q, Jeng W, Wheeler MB. Characterization of glucagon-like peptide-1 receptor-binding determinants. J Mol Endocrinol. 2000;25:321–335. doi: 10.1677/jme.0.0250321. [DOI] [PubMed] [Google Scholar]
- Xu G, Kaneto H, Lopez-Avalos MD, Weir GC, Bonner-Weir S. GLP-1/exendin-4 facilitates beta-cell neogenesis in rat and human pancreatic ducts. Diabetes Res Clin Pract. 2006;73:107–110. doi: 10.1016/j.diabres.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Xu G, Stoffers DA, Habener JF, Bonner-Weir S. Exendin-4 stimulates both beta-cell replication and neogenesis, resulting in increased beta-cell mass and improved glucose tolerance in diabetic rats. Diabetes. 1999;48:2270–2276. doi: 10.2337/diabetes.48.12.2270. [DOI] [PubMed] [Google Scholar]
- Yaekura K, Julyan R, Wicksteed BL, Hays LB, Alarcon C, Sommers S, Poitout V, Baskin DG, Wang Y, Philipson LH, Rhodes CJ. Insulin secretory deficiency and glucose intolerance in Rab3A null mice. J Biol Chem. 2003;278:9715–9721. doi: 10.1074/jbc.M211352200. [DOI] [PubMed] [Google Scholar]
- Yagi T, Nishi S, Hinata S, Murakami M, Yoshimi T. A population association study of four candidate genes (hexokinase II, glucagon-like peptide-1 receptor, fatty acid binding protein-2, and apolipoprotein C-II) with type 2 diabetes and impaired glucose tolerance in Japanese subjects. Diabet Med. 1996;13:902–907. doi: 10.1002/(SICI)1096-9136(199610)13:10<902::AID-DIA242>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Yan L, Figueroa DJ, Austin CP, Liu Y, Bugianesi RM, Slaughter RS, Kaczorowski GJ, Kohler MG. Expression of voltage-gated potassium channels in human and rhesus pancreatic islets. Diabetes. 2004;53:597–607. doi: 10.2337/diabetes.53.3.597. [DOI] [PubMed] [Google Scholar]
- Yan SZ, Huang ZH, Andrews RK, Tang WJ. Conversion of forskolin-insensitive to forskolin-sensitive (mouse-type IX) adenylyl cyclase. Mol Pharmacol. 1998;53:182–187. doi: 10.1124/mol.53.2.182. [DOI] [PubMed] [Google Scholar]
- Yaney GC, Civelek VN, Richard AM, Dillon JS, Deeney JT, Hamilton JA, Korchak HM, Tornheim K, Corkey BE, Boyd AE., 3rd Glucagon-like peptide 1 stimulates lipolysis in clonal pancreatic beta-cells (HIT) Diabetes. 2001;50:56–62. doi: 10.2337/diabetes.50.1.56. [DOI] [PubMed] [Google Scholar]
- Yaney GC, Korchak HM, Corkey BE. Long-chain acyl CoA regulation of protein kinase C and fatty acid potentiation of glucose-stimulated insulin secretion in clonal beta-cells. Endocrinology. 2000;141:1989–1998. doi: 10.1210/endo.141.6.7493. [DOI] [PubMed] [Google Scholar]
- Yang Y, Gillis KD. A highly Ca2+-sensitive pool of granules is regulated by glucose and protein kinases in insulin-secreting INS-1 cells. J Gen Physiol. 2004;124:641–651. doi: 10.1085/jgp.200409081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yew KH, Hembree M, Prasadan K, Preuett B, McFall C, Benjes C, Crowley A, Sharp S, Tulachan S, Mehta S, Tei E, Gittes G. Cross-talk between bone morphogenetic protein and transforming growth factor-beta signaling is essential for exendin-4-induced insulin-positive differentiation of AR42J cells. J Biol Chem. 2005;280:32209–32217. doi: 10.1074/jbc.M505465200. [DOI] [PubMed] [Google Scholar]
- Yew KH, Prasadan KL, Preuett BL, Hembree MJ, McFall CR, Benjes CL, Crowley AR, Sharp SL, Li Z, Tulachan SS, Mehta SS, Gittes GK. Interplay of glucagon-like peptide-1 and transforming growth factor-beta signaling in insulin-positive differentiation of AR42J cells. Diabetes. 2004;53:2824–2835. doi: 10.2337/diabetes.53.11.2824. [DOI] [PubMed] [Google Scholar]
- Yi Z, Yokota H, Torii S, Aoki T, Hosaka M, Zhao S, Takata K, Takeuchi T, Izumi T. The Rab27a/granuphilin complex regulates the exocytosis of insulin-containing dense-core granules. Mol Cell Biol. 2002;22:1858–1867. doi: 10.1128/MCB.22.6.1858-1867.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue F, Cui L, Johkura K, Ogiwara N, Sasaki K. Glucagon-Like Peptide-1 Differentiation of Primate Embryonic Stem Cells into Insulin-Producing Cells. Tissue Eng. 2006;12:2105–2116. doi: 10.1089/ten.2006.12.2105. [DOI] [PubMed] [Google Scholar]
- Zander M, Madsbad S, Madsen JL, Holst JJ. Effect of 6-week course of glucagon-like peptide 1 on glycaemic control, insulin sensitivity, and beta-cell function in type 2 diabetes: a parallel-group study. Lancet. 2002;359:824–830. doi: 10.1016/S0140-6736(02)07952-7. [DOI] [PubMed] [Google Scholar]
- Zhang J, Li L. BMP signaling and stem cell regulation. Dev Biol. 2005;284:1–11. doi: 10.1016/j.ydbio.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Cook JT, Hattersley AT, Firth R, Saker PJ, Warren-Perry M, Stoffel M, Turner RC. Non-linkage of the glucagon-like peptide 1 receptor gene with maturity onset diabetes of the young. Diabetologia. 1994;37:721–724. doi: 10.1007/BF00417698. [DOI] [PubMed] [Google Scholar]
- Zhao AZ, Zhao H, Teague J, Fujimoto W, Beavo JA. Attenuation of insulin secretion by insulin-like growth factor 1 is mediated through activation of phosphodiesterase 3B. Proc Natl Acad Sci U S A. 1997;94:3223–3228. doi: 10.1073/pnas.94.7.3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Egan JM. SNAP-25 is phosphorylated by glucose and GLP-1 in RIN 1046-38 cells. Biochem Biophys Res Commun. 1997;238:297–300. doi: 10.1006/bbrc.1997.7286. [DOI] [PubMed] [Google Scholar]
- Zhou J, Montrose-Rafizadeh C, Janczewski AM, Pineyro MA, Sollott SJ, Wang Y, Egan JM. Glucagon-like peptide-1 does not mediate amylase release from AR42J cells. J Cell Physiol. 1999a;181:470–478. doi: 10.1002/(SICI)1097-4652(199912)181:3<470::AID-JCP11>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Zhou J, Pineyro MA, Wang X, Doyle ME, Egan JM. Exendin-4 differentiation of a human pancreatic duct cell line into endocrine cells: involvement of PDX-1 and HNF3beta transcription factors. J Cell Physiol. 2002;192:304–314. doi: 10.1002/jcp.10143. [DOI] [PubMed] [Google Scholar]
- Zhou J, Wang X, Pineyro MA, Egan JM. Glucagon-like peptide 1 and exendin-4 convert pancreatic AR42J cells into glucagon- and insulin-producing cells. Diabetes. 1999b;48:2358–2366. doi: 10.2337/diabetes.48.12.2358. [DOI] [PubMed] [Google Scholar]
- Zhou YT, Shimabukuro M, Lee Y, Koyama K, Higa M, Ferguson T, Unger RH. Enhanced de novo lipogenesis in the leptin-unresponsive pancreatic islets of prediabetic Zucker diabetic fatty rats: role in the pathogenesis of lipotoxic diabetes. Diabetes. 1998;47:1904–1908. doi: 10.2337/diabetes.47.12.1904. [DOI] [PubMed] [Google Scholar]
- Zhu M, Breslin MB, Lan MS. Expression of a novel zinc-finger cDNA, IA-1, is associated with rat AR42J cells differentiation into insulin-positive cells. Pancreas. 2002;24:139–145. doi: 10.1097/00006676-200203000-00004. [DOI] [PubMed] [Google Scholar]