

Abstract
FHIT is a novel tumor suppressor gene located at human chromosome 3p14.2. Restoration of wild-type FHIT in 3p14.2-deficient human lung cancer cells inhibits cell growth and induces apoptosis. In this study, we analyzed potential upstream/downstream molecular targets of the FHIT protein and found that FHIT specifically targeted and regulated death receptor (DR) genes in human non-small-cell lung cancer (NSCLC) cells. Exogenous expression of FHIT by a recombinant adenoviral vector (Ad)-mediated gene transfer upregulated expression of DR genes. Treatment with a recombinant TRAIL protein, a DR-specific ligand, in Ad-FHIT-transduced NSCLC cells considerably enhanced FHIT-induced apoptosis, further demonstrating the involvement of DRs in FHIT-induced apoptosis. Moreover, we also found that FHIT targeted downstream of the DR-mediated signaling pathway. FHIT overexpression disrupted mitochondrial membrane integrity and activated multiple pro-apoptotic proteins in NSCLC cell. These results suggest that FHIT induces apoptosis through a sequential activation of DR-mediated pro-apoptotic signaling pathways in human NSCLC cells.
Keywords: FHIT, tumor suppressor, apoptosis, death receptor, caspase, lung cancer
Introduction
Lung cancer is the leading cause of cancer-related deaths. The pathogenesis of lung cancer involves a multistep process of genetic and molecular changes. Studies have shown that a genomic aberration in human chromosome 3p is the most frequent and earliest genetic event in lung tumorigenesis and that it affects several tumor suppressor genes and oncogenes in this region [1–3]. Researchers have identified the fragile histidine triad (FHIT) gene at chromosome 3p14.2 and characterized it as a tumor suppressor gene [1–3]. This gene is very vulnerable to environmental carcinogens and is frequently involved in allele loss, genomic rearrangement, and cytogenetic abnormalities in human cancer [4]. The FHIT protein consists of 147 amino acids and is a member of the histidine triad nucleotide-binding protein superfamily. Inactivation of the FHIT protein plays an important role in the development of several human cancers, including lung cancer [5–7]. Several lines of in vitro and in vivo experimental evidence have demonstrated the tumor suppression function of the FHIT gene [8–12]. Exogenous expression of the FHIT gene in 3p14.2-deficient cancer cells induces apoptosis and alters cell cycle kinetics in various types of cancer cells [13–15]. Also, a previous study implicated the FHIT-induced tumor suppression in FHIT-mediated inactivation of MDM2 and subsequent stabilization of p53 protein in human lung cancer cells [9]. Although the proapoptotic function of FHIT is well documented, the precise molecular mechanism by which FHIT exerts potent tumor suppression activity remains largely unclear. We hypothesized that FHIT specifically targets and activates certain cellular proteins involved in the apoptosis signaling pathway. In this study, we investigated cellular responses to exogenous expression of wild-type (wt) FHIT and explored the molecular mechanism involved in the FHIT-induced apoptotic pathway in human non-small cell lung cancer (NSCLC) cells transduced with a recombinant adenoviral vector carrying the FHIT gene (Ad-FHIT). Using a sensitive and quantitative multiprobe ribonuclease protection assay (RPA) with gene-specific probes for the most important factors involved in cell proliferation, apoptosis, and cell cycle regulation, we identified potential upstream and downstream molecular targets of the FHIT protein and found that FHIT specifically targeted and upregulated death receptors (DRs). Overexpression of FHIT in Ad-FHIT-transduced NSCLC cells considerably upregulated the expression of DR3, DR4, and DR5. We also showed that FHIT overexpression activated downstream of the DR-mediated apoptotic pathway, disrupting mitochondrial membrane integrity and activating caspase signaling pathway. Therefore, our study indicated that the mechanism by which FHIT induces apoptosis is mediated by activation of the DR and caspase cascade signaling pathways in human lung cancer cells. Our findings also provide insight into the molecular mechanism of FHIT in tumor suppression and suggest novel gene therapy strategies for lung cancer.
Materials and methods
Cell lines and recombinant adenoviral vectors
The human NSCLC cell lines H1299 and A549 were purchased from the American Type Culture Collection (Manassas, VA). H1299 was maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS). A549 was maintained in Ham’s F-12 medium with 10% FBS. The construction and production of Ad-FHIT, an adenoviral vector carrying the p53 gene (Ad-p53), and an empty adenoviral vector (Ad-EV) were described previously (9).
Multiprobe ribonuclease protection assay (RPA)
RPA was performed according to the manufacturer’s instructions (Pharmingen, San Diego, CA). Briefly, total RNA was isolated from A549 and H1299 cells transduced with Ad-FHIT, Ad-p53, or Ad-EV or treated with phosphate-buffered saline (PBS) with TRIzol reagent (Life Technologies, Grand Island, NY). A multiprobe template set containing human DR genes and their effectors, including caspase 8 (Casp 8), FASL, FAS, DCR1, DR3, DR4, DR5, tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL), TNF receptor (TNFR), TNFR-associated death domain (TRADD), RIP, and L32, was used for T7 polymerase-directed synthesis of a high-specific-activity, 32P-labeled antisense RNA probe set. The radiolabeled probe set was hybridized in excess to the target RNA in solution (10–20 μg) at 56 °C for 16 h. After hybridization, free probes and unprotected single-stranded RNAs were digested with RNases. The remaining “RNase-protected” RNA fragments were purified and resolved on a 5% denaturing polyacrylamide gel. The RPA images and intensity of the bands for the selected genes were analyzed using phosphorimaging with a Storm 860 analysis system (Molecular Dynamics, Sunnyvale, CA). The expression of the selected genes was quantified as the percentage relative to that of the housekeeping gene GAPDH.
Apoptosis analysis
Induction of apoptosis in NSCLC cells was analyzed using a fluorescence-activated cell sorter (FACS) with a terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) reaction (Roche Molecular Biochemicals, Mannheim, Germany). Briefly, cells were plated in six-well plates (1 × 106 cells/well); 24 h later, they were transduced with Ad-FHIT, Ad-p53, or Ad-EV and treated with a recombinant TRAIL protein (R&D Systems, Minneapolis, MN) at a final concentration of 10 ng/ml. PBS was used as a mock control. At designated times after transduction, cells were harvested and apoptosis were analyzed according to the manufacturer’s instructions (Roche Molecular Biochemicals).
Analysis of mitochondrial membrane potential
Changes in mitochondrial membrane potential in adenoviral vector-transduced cells were measured using flow cytometry with 5, 5', 6, 6'-tetrachloro-1, 1', 3, 3'-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) staining (Molecular Probes, Eugene, OR). JC-1 exists as a monomer at low concentrations or at low membrane potential and emits green florescence at 527 nm. However, at higher concentrations or higher membrane potential, JC-1 forms J-aggregates and emits red fluorescence near its emission maximum of about 590 nm. Measurement of the ratio of the red to green JC-1 fluorescence in cells using flow cytometry is a sensitive and specific method for monitoring changes in mitochondrial potential in living cells during induction of apoptosis by various agents. Cells were cultured in six-well plates and, after reaching approximately 70% confluence, transduced with various adenoviral vectors at various multiplicities of infection. Cells were collected using centrifugation for 5 min at 1500 × g at 4 °C and resuspended in complete medium containing 10 μg/ml JC-1 at a density of 5 × 105 cells/ml. Cells were incubated for 10 min at room temperature in the dark. Cells were then washed twice with cold PBS, resuspended in 400 μl of PBS, and analyzed immediately using flow cytometry.
Western blot analysis
Western blots were probed with a specific antibody against FHIT, p53, BID poly(ADP-ribose) polymerase (PARP), caspase 3, caspase 8, caspase 9, or β-actin. The protein bands were detected using enhanced chemiluminescence. Antibodies against p53, BID, caspase 8, caspase 9, and β-actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA); FHIT polyclonal antiserum was purchased from Zymed (South San Francisco, CA); and antibodies against caspase 3 and PARP were purchased from BD Pharmingen (Los Angeles, CA).
Statistical analyses
All experiments were performed at least three times with duplicate samples. The mean values and standard errors were calculated. The StatView 5.0 (Abacus Concepts, Berkeley, CA) and SAS (SAS Institute, Cary, NC) software programs were used for all statistical analyses. Each bar denotes mean ± SEM of three experiments.
Results
FHIT overexpression targets and upregulates DR genes
We previously showed that exogenous expression of the wt-FHIT gene induced apoptosis in human NSCLC cells [4, 9]. To further characterize the response of NSCLC cells to exogenous expression of FHIT protein and analyze the molecular mechanism of FHIT-mediated apoptosis, we induced overexpression of wt-FHIT by transducing the human NSCLC cell lines H1299 and A549 with Ad-FHIT. We next used a multiprobe RPA to identify the potential upstream and downstream molecular targets of the FHIT protein in Ad-FHIT-transduced H1299 and A549 cells. RPA is a highly sensitive and specific method for the detection and quantitation of mRNA species. We used various sets of probes that could detect the primary factors involved in cell proliferation, apoptosis, and cell cycle regulation to quantify the transcription of genes in the apoptotic pathway that may be affected and regulated by overexpression of FHIT in Ad-FHIT-transduced NSCLC cells. The results showed that exogenous expression of wt-FHIT specifically targeted DR genes, resulting in marked upregulation of the expression of DR3, DR4, and DR5 in Ad-FHIT-transduced NSCLC cells (Fig. 1). We also found that the activation of DR3, DR4, and DR5 was independent of p53 gene status in these NSCLC cells, although the effect was enhanced in the presence of p53 expression in Ad-FHIT-transduced A549 cells with wt-p53. The FAS gene was also activated by FHIT protein overexpression in A549 cells with wt-p53 but not in H1299 cells negative for p53, indicating that regulation of FAS expression by FHIT depends on the p53 status of NSCLC cells (Fig. 1). In contrast, overexpression of FHIT did not alter the expression of other cell death-related genes, such as DCR1, TNFR, TRADD, and L32, in Ad-FHIT-transduced A549 (seen supplementary Figure-S1A) and H1299 cells (supplementary Figure-S1B). Details of the multi-probe RPA analysis with all probes for the cell death pathway and their quantifications were presented in Figure-S1A and S1B. Taken together, these results showed that FHIT specifically targeted DR3, DR4, and DR5, thereby activating a DR-mediated apoptotic pathway in NSCLC cells.
Fig. 1.
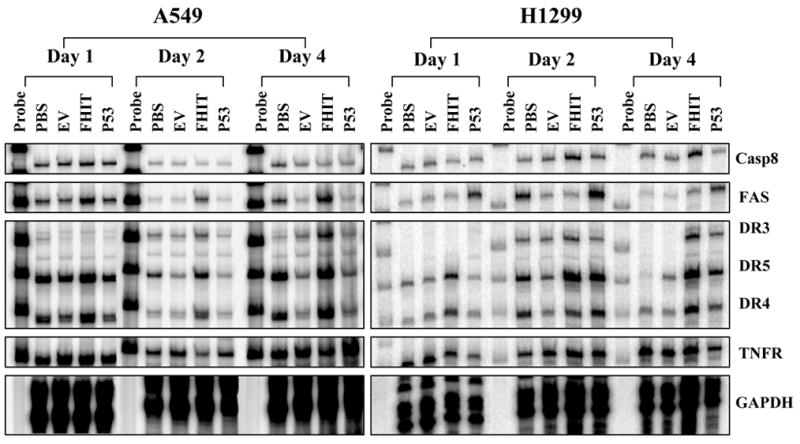
FHIT specifically targets and upregulates apoptosis-related DRs. The regulation of gene expression by FHIT was analyzed using a multiprobe RPA with specific probes for human DR genes and their effectors, caspase 8 (Casp-8), FASL, FAS, DCR1, DR3, DR4, DR5, TRAIL, TNFR, TRADD, RIP, and L32. The NSCLC cell lines A549 and H1299 were transduced with Ad-FHIT. PBS and an empty vector (EV) were used as negative controls, and Ad-p53 was used as a positive control. On days 1, 2, and 4 after transduction, cells were harvested, and the total RNAs were isolated and RPA was performed. Representative RPA images indicated the expression of the selected genes. Three separate experiments were performed.
Treatment with DR-ligand TRAIL protein enhance FHIT-induced apoptosis
The tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) protein is the specific ligand for DRs [16, 17]. We reasoned that because DRs were molecular targets of FHIT tumor suppressor, the presence of their ligands should enhance FHIT-mediated apoptosis induction. To further provide evidence that FHIT-induced apoptosis is mediated by activation of the DR-associated apoptotic pathway, we exogenously supplied recombinant TRAIL proteins into Ad-FHIT-transduced NSCLC cell lines A549 and H1299 and examined the apoptotic cell population using TUNEL-based FACS analysis. PBS-treated cells were used as a negative control. The results showed that induction of FHIT-mediated apoptosis in Ad-FHIT-transduced A549 and H1299 cells was markedly enhanced by the addition of the biologically active recombinant TRAIL protein both after 2 days and after 4 days (Fig. 2), confirming the role of DRs in FHIT-induced apoptosis.
Fig. 2.
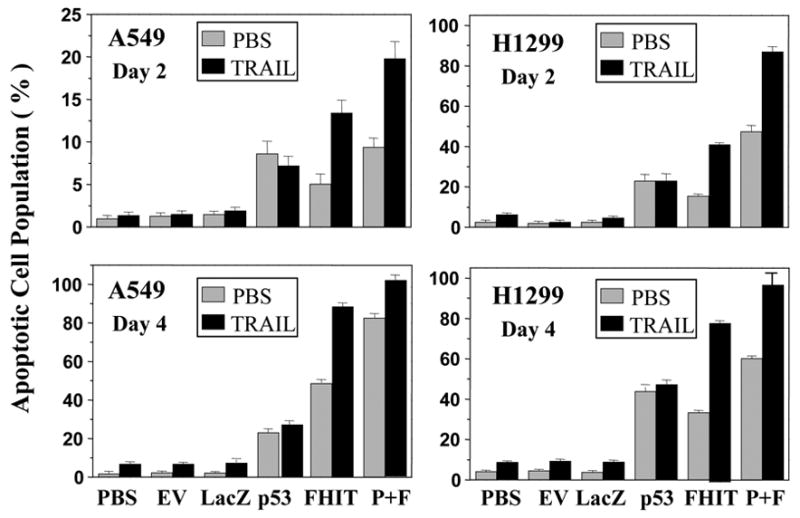
Treatment of TRAIL proteins enhances FHIT-mediated apoptosis. The NSCLC cell lines A549 and H1299 were transduced with Ad-FHIT and treated with a recombinant TRAIL protein (10 ng/ml). On days 2 and 4 after transduction, cells were harvested, and apoptosis was examined using TUNEL-based FACS analysis. PBS, an empty vector (EV), and LacZ were used as negative controls, and Ad-p53 was used as a positive control. Apoptosis is shown as the relative percentage of apoptotic cells. Three separate experiments were performed in duplicate and each bar denotes mean ± SEM of three experiments. P + F means p53 + FHIT.
FHIT overexpression disrupts the integrity of the mitochondrial membrane
The integrity of the mitochondrial membrane plays an important role in living cells. Loss of mitochondrial membrane potential is an important event in the DR-mediated apoptotic pathway. To determine whether FHIT-induced apoptosis also involves disruption of integrity of mitochondrial membrane, we analyzed the effect of overexpression of wt-FHIT on mitochondrial membrane potential using FACS analysis with JC-1 fluorescent staining. Comparable with the activation of DR gene expression by FHIT, exogenous expression of wt-FHIT resulted in disruption of the integrity of mitochondrial membrane, as demonstrated by the loss of mitochondrial membrane potential in Ad-FHIT-transduced A549 and H1299 cells (Figs. 3A and B). By contrast, treatment cells with negative control, PBS or empty vector, did not affect the integrity of mitochondrial membrane. Overexpression of p53, which we used as the positive control, also disrupted the integrity of mitochondrial membrane. Thus, these results showed that FHIT targeted DRs and consequently affected the event involved in DR-mediated apoptotic pathway by disrupting integrity of mitochondrial membrane in human NSCLC cells.
Fig. 3.
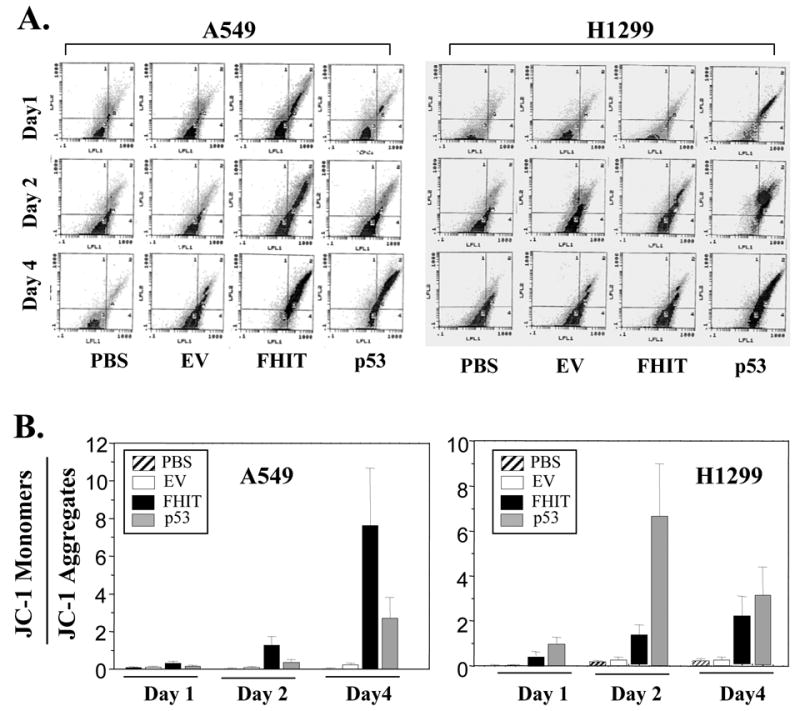
FHIT overexpression disrupts the integrity of the mitochondrial membrane. The NSCLC cell lines A549 and H1299 were transduced with Ad-FHIT. PBS and an empty vector (EV) were used as negative controls, and Ad-p53 was used as a positive control. On days 1, 2, and 4 after transduction, cells were harvested, and the mitochondrial membrane potential was assessed using JC-1 staining by flow cytometry. Panel (A) shows representative images of JC-1 staining. The mitochondrial membrane potential (B) in cells is shown as the relative value of JC-1 monomers and JC-1 aggregates. Each bar denotes mean ± SEM of three experiments.
FHIT overexpression activates caspases
Caspases are the downstream molecules in the DR-mediated apoptotic signaling pathway. Activation of caspases plays a key role in DR-mediated apoptosis. We next analyzed the influence of exogenous expression of FHIT on activation of caspases in NSCLC cells. Western blot analysis of cell lysates from Ad-FHIT-transduced NSCLC cells showed a correlation between the upregulation of DR expression by FHIT and the activation of caspase 3, caspase 8, caspase 9, and the executor poly(ADP-ribose) polymerase (PARP) proteins. These pro-apoptotic proteins were strongly cleaved from their inactive precursor form into active fragments in the Ad-FHIT-transduced A549 (Fig. 4A) and H1299 cells (see supplementary Fig. S2A). FHIT overexpression also activated the apoptosis initiator BID in A549 cells, as indicated by the truncated BID (tBID) bands (Fig. 4A). However, the FHIT-mediated activation of BID appeared to be p53-depedent, as the tBID was only detected in Ad-FHIT-transduced A549 cells that contain wt-p53 (Fig. 4A), but not in H1299 cells that have homozygous deletion of p53 (see supplementary Fig. S2A).
Fig. 4.
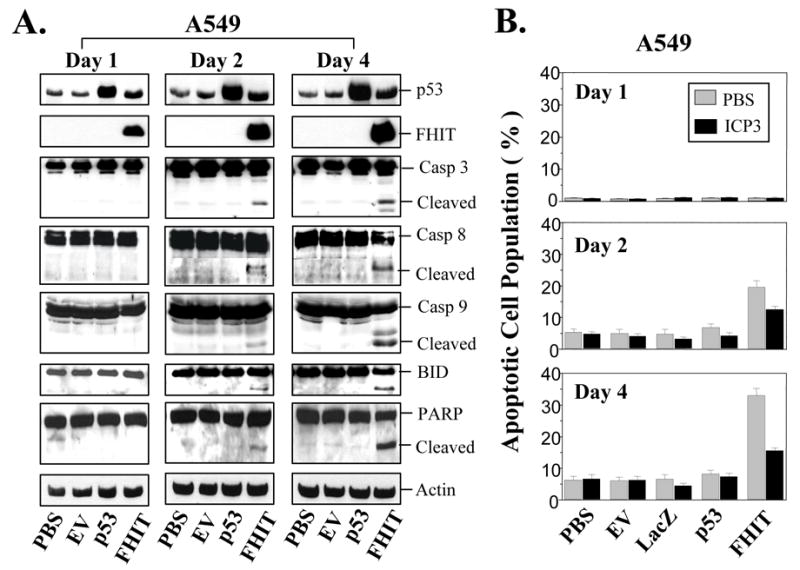
FHIT overexpression activates caspase-dependent signaling pathway. (A), FHIT-mediated activation of caspases. The NSCLC cell line A549 was transduced with Ad-FHIT. On days 1, 2, and 4 after transduction, the activation of caspases was assessed using Western blot analysis with specific antibodies. The activation of caspase 3, caspase 8, caspase 9 and PARP is indicated by detected cleavage of these proteins. (B), Inhibition of ICP3 on FHIT-mediated apoptosis. A549 cells were transduced with Ad-FHIT and treated with or without 10 μM ICP3. After 1, 2, or 4 days, apoptosis was examined using TUNEL-based FACS analysis. Apoptosis is shown as the relative percentage of apoptotic cells. Three separate experiments were performed in duplicate. Each bar denotes mean ± SEM of three experiments.
Inositol 1,2-cyclic 4,5-triphosphate (ICP3) is a specific caspase inhibitor. To verify the existence of a caspase-associated apoptotic pathway in Ad-FHIT-transduced NSCLC cells, we analyzed the effect of ICP3 on FHIT-induced apoptosis. Treatment with ICP3 considerably attenuated the induction of apoptosis by wt-FHIT in Ad-FHIT-transduced A549 (Fig. 4B) and H1299 cells (see supplementary Fig-S2B), confirming that caspase activation, the downstream of the DRs-mediated signaling pathway, is involved in FHIT-induced apoptosis in NSCLC cells.
DISCUSSION
In this study, we showed that the FHIT tumor suppressor specifically targets and upregulates death receptors in Ad-FHIT-transduced NSCLC cells. Exogenous expression of FHIT selectively mediates upregulation of the expression of DR3, DR4, and DR5 and stimulates the DR-associated apoptotic pathway. Although several lines of evidence showed that FHIT exhibits potent tumor suppression function via diverse mechanisms, previous reports focused primarily on the classical pathways, such as cell cycle arrest and apoptosis induction [5, 6, 8, 11, 13–15]. The precise molecular mechanism involved in the antitumor function of FHIT remains largely unclear. To our knowledge, this is the first report of the involvement of death receptors in the FHIT-mediated apoptosis induction and tumor suppression in human lung cancer cells. Recent evidence suggests that DR3, DR4, and DR5 occupy a unique position in the DR-mediated apoptotic pathway and are key molecular targets for the development of novel therapeutics for human cancers [16, 17]. Thus, the finding that FHIT selectively targets and upregulates the expression of DR3, DR4, and DR5 is of considerably therapeutic importance.
DRs, also known as TRAIL receptors, are activated by their physiologic ligand, TRAIL [17]. Recent studies suggested that DRs also activate the nuclear factor κB and c-Jun N-terminal kinase pathways [18, 19]. Because TRAIL is a specific ligand for DRs, we also postulated that co-treatment with biologically active TRAIL protein ligands in Ad-FHIT- transduced NSCLC cells would enhance FHIT-mediated apoptosis. The present study confirmed this effect and further demonstrated the role of DRs in FHIT-mediated apoptosis induction. TRAIL has shown significant promise as a novel anticancer agent that can augment the effects of other anticancer agents that also upregulate the expression of DRs. Thus, targeting DRs with TRAIL in combination with other pro-apoptotic anticancer agents such as FHIT could prove superior to using TRAIL or FHIT alone for some cancers, including lung cancer. A better understanding of the signaling events derived from DRs could therefore prove beneficial in improving the value of FHIT in cancer therapy.
The results of our study also showed that activation of FAS is involved in FHIT-mediated apoptosis in a p53-dependent manner. Overexpression of FHIT induced upregulation of FAS expression in wt-p53-bearing A549 cells but not in p53-deficient H1299 cells. A previous study identified FAS as one of the downstream targets of p53 [20], which mediates its apoptotic effects partly by activating FAS, which in turn recruits intracellular adaptor molecules and proximal caspase 8. Because p53 function is frequently altered in cancer cells, p53-dependent activation of FAS could be an important pathway of FHIT-mediated apoptosis.
We also found that the FHIT activated the downstream targets of DR-mediated apoptotic pathway. Because DR-mediated apoptotic signaling pathway involves loss of mitochondrial membrane potential and activation of caspases, we suspected that FHIT-induced apoptosis also dependent on these events. As expected, overexpression of FHIT affected mitochondrial membrane integrity and activate multiple pro-apoptotic proteins. We demonstrated a correlation between the upregulation of DRs and the loss of potential of mitochondrial membrane and the appearance of active forms of caspases and PARP protein in Ad-FHIT-transduced NSCLC cells. Thus, our results further confirmed that induction of apoptosis by FHIT is through DR-mediated signaling pathway.
We conclude that FHIT tumor suppressor induces apoptosis in NSCLC cells by targeting death receptors such as DR3, DR4, and DR5, disrupting the integrity of the mitochondrial membrane, and sequentially activating downstream targets caspases and RAPA protein, and resulting in cell death (see supplementary Fig. S3). Our findings have important pathophysiologic and therapeutic implications. Specifically, they provide valuable information regarding the mechanism by which FHIT induces apoptosis directly targeting DR genes and activating DR-mediated apoptotic signaling pathways. Because most human lung cancers are associated with a loss of function of 3p tumor suppressors such as FHIT, our findings may be very useful in understanding the precise mechanism of action of other 3p tumor suppressor genes. Furthermore, elucidation of the mechanism of action of FHIT may provide a foundation for the development of targeted molecular therapy for human lung cancer or other cancers.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health, National Cancer Institute Specialized Program of Research Excellence (SPORE) grants CA70907 and MMHCC U01CA10535201; the Department of Defense TARGET Lung Cancer Programs grant DAMD17-02-1-070; the National Cancer Institute M. D. Anderson Cancer Center Support (CORE) Grant CA16672; and a grant from the Tobacco Settlement Funds as appropriated by the Texas State Legislature.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Peto R, Lopez AD, Boreham J, Thun M, Heath C, Jr, Doll R. Mortality from smoking worldwide. Br Med Bull. 1996;52:12–21. doi: 10.1093/oxfordjournals.bmb.a011519. [DOI] [PubMed] [Google Scholar]
- 2.Zabarovsky ER, Lerman MI, Minna JD. Tumor suppressor genes on chromosome 3p involved in the pathogenesis of lung and other cancers. Oncogene. 2002;21:6915–6935. doi: 10.1038/sj.onc.1205835. [DOI] [PubMed] [Google Scholar]
- 3.Sozzi G, Pastorino U, Moiraghi L, Tagliabue E, Pezzella F, Ghirelli C, Tornielli S, Sard L, Huebner K, Pierotti MA, Croce CM, Pilotti S. Loss of FHIT function in lung cancer and preinvasive bronchial lesions. Adv Cancer Res. 1998;74:141–166. [PubMed] [Google Scholar]
- 4.Ji L, Fang B, Yen N, Fong K, Minna JD, Roth JA. Induction of apoptosis and inhibition of tumorigenicity and tumor growth by adenovirus vector-mediated fragile histidine triad (FHIT) gene overexpression. Cancer Res. 1999;59:3333–3339. [PubMed] [Google Scholar]
- 5.Pekarsky Y, Palamarchuk A, Huebner K, Croce CM. FHIT as tumor suppressor: mechanisms and therapeutic opportunities. Cancer Biol Ther. 2002;1:232–236. doi: 10.4161/cbt.73. [DOI] [PubMed] [Google Scholar]
- 6.Huebner K, Croce CM. Cancer and the FRA3B/FHIT fragile locus: it's a HIT. Br J Cancer. 2003;88:1501–1506. doi: 10.1038/sj.bjc.6600937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barnes LD, Garrison PN, Siprashvili Z, Guranowski A, Robinson AK, Ingram SW, Croce CM, Ohta M, Huebner K. Fhit, a putative tumor suppressor in humans, is a dinucleoside 5', 5'''-P1,P3-triphosphate hydrolase. Biochemistry. 1996;35:11529–11535. doi: 10.1021/bi961415t. [DOI] [PubMed] [Google Scholar]
- 8.Dumon KR, Ishii H, Fong LY, Zanesi N, Fidanza V, Mancini R, Vecchione A, Baffa R, Trapasso F, During MJ, Huebner K, Croce CM. FHIT gene therapy prevents tumor development in Fhit-deficient mice. Proc Natl Acad Sci USA. 2001;98:3346–3351. doi: 10.1073/pnas.061020098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nishizaki M, Sasaki J, Fang B, Atkinson EN, Minna JD, Roth JA, Ji L. Synergistic tumor suppression by coexpression of FHIT and p53 coincides with FHIT-mediated MDM2 inactivation and p53 stabilization in human NSCLC cells. Cancer Res. 2004;64:5745–5752. doi: 10.1158/0008-5472.CAN-04-0195. [DOI] [PubMed] [Google Scholar]
- 10.Ishii H, Dumon KR, Vecchione A, Fong LY, Baffa R, Huebner K, Croce CM. Potential cancer therapy with the fragile histidine triad gene: review of the preclinical studies. JAMA. 2001;286:2441–2449. doi: 10.1001/jama.286.19.2441. [DOI] [PubMed] [Google Scholar]
- 11.Ishii H, Dumon KR, Vecchione A, Trapasso F, Mimori K, Alder H, Mori M, Sozzi G, Baffa R, Huebner K, Croce CM. Effect of adenoviral transduction of the fragile histidine triad gene into esophageal cancer cells. Cancer Res. 2001;61:1578–1584. [PubMed] [Google Scholar]
- 12.Fong LY, Fidanza V, Zanesi N, Lock LF, Siracusa LD, Mancini R, Siprashvili Z, Ottey M, Martin SE, Druck T, McCue PA, Croce CM, Huebner K. Muir-Torre-like syndrome in Fhit-deficient mice. Proc Natl Acad Sci USA. 2000;97:4742–4747. doi: 10.1073/pnas.080063497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garinis GA, Gorgoulis VG, Mariatos G, Zacharatos P, Kotsinas A, Liloglou T, Foukas P, Kanavaros P, Kastrinakis NG, Vassilakopoulos T, Vogiatzi T, Field JK, Kittas C. Association of allelic loss at the FHIT locus and p53 alterations with tumour kinetics and chromosomal instability in non-small cell lung carcinomas. J Pathol. 2001;193:55–65. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH731>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 14.Roz L, Andriani F, Ferreira CG, Giaccone G, Sozzi G. The apoptotic pathway triggered by the Fhit protein in lung cancer cell lines is not affected by Bcl-2 or Bcl-x(L) overexpression. Oncogene. 2004;23:9102–9110. doi: 10.1038/sj.onc.1208142. [DOI] [PubMed] [Google Scholar]
- 15.Roz L, Gramegna M, Ishii H, Croce CM, Sozzi G. Restoration of fragile histidine triad (FHIT) expression induces apoptosis and suppresses tumorigenicity in lung and cervical cancer cell lines. Proc Natl Acad Sci USA. 2002;99:3615–3620. doi: 10.1073/pnas.062030799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheikh MS, Huang Y. Death receptors as targets of cancer therapeutics. Curr Cancer Drug Targets. 2004;4:97–104. doi: 10.2174/1568009043481597. [DOI] [PubMed] [Google Scholar]
- 17.Jeremias I, Debatin KM. TRAIL induces apoptosis and activation of NFkappaB. Eur Cytokine Netw. 1998;9:687–688. [PubMed] [Google Scholar]
- 18.Hu WH, Johnson H, Shu HB. Tumor necrosis factor-related apoptosis-inducing ligand receptors signal NF-kappaB and JNK activation and apoptosis through distinct pathways. J Biol Chem. 1999;274:30603–30610. doi: 10.1074/jbc.274.43.30603. [DOI] [PubMed] [Google Scholar]
- 19.Owen-Schaub LB, Zhang W, Cusack JC, Angelo LS, Santee SM, Fujiwara T, Roth JA, Deisseroth AB, Zhang WW, Kruzel E, Radinsky R. Wild-type human p53 and a temperature-sensitive mutant induce Fas/APO-1 expression. Mol Cell Biol. 1995;15:3032–3040. doi: 10.1128/mcb.15.6.3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bennett M, Macdonald K, Chan SW, Luzio JP, Simari R, Weissberg P. Cell surface trafficking of fas: a rapid mechanism of p53 mediated apoptosis. Science. 1998;282:290–293. doi: 10.1126/science.282.5387.290. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.