

Abstract
The strength of hippocampal synapses can be persistently increased by signals that activate Ca2+/calmodulin-dependent protein kinase II (CaMKII). This CaMKII-dependent long-term potentiation is important for hippocampal learning and memory. In this work we show that CaMKII exhibits an intriguing switch-like activation that likely is important for changes in synaptic strength. We found that autophosphorylation of CaMKII by itself showed a steep dependence on Ca2+ concentration [Hill coefficient (nH) ≈ 5]. However, an even steeper Ca2+ dependence (nH ≈ 8) was observed when autophosphorylation is balanced by the dephosphorylation activity of protein phosphatase 1 (PP1). This autophosphorylation-dephosphorylation switch was found to be reversible because PP1 dephosphorylates CaMKII when Ca2+ is lowered to a basal level. The switch-like response of a CaMKII-PP1 system suggests that CaMKII and PP1 may function together as a simple molecular device that specifically translates only strong Ca2+ signals into all-or-none potentiation of individual hippocampal synapses.
In the CA1 region of the hippocampus, postsynaptic Ca2+ influx can elicit different effects on synaptic plasticity including long-term potentiation (LTP), long-term depression (LTD), or no change in synaptic strength (1). Different biochemical signaling pathways are responsible for these diverse changes, with the magnitude of Ca2+ stimulus largely determining the pathway that becomes activated (2). However, the dynamic range of [Ca2+] within dendritic spines is modest compared with other systems, varying from ≈100 nM under basal conditions to ≈10 μM on strong stimulation (3). How different pathways are activated with specificity over this relatively small range of Ca2+ is not well understood.
The primary effector of Ca2+ signals that elicit LTP is Ca2+/calmodulin (CaM)-dependent protein kinase II (CaMKII) (4-6). CaMKII is a multifunctional serine/threonine kinase that is highly abundant in neurons, particularly dendritic spines (7, 8). Binding of Ca2+/CaM both activates CaMKII toward downstream effectors such as the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptor (9) and recruits it to the synapse (10). In addition, CaM binding facilitates autophosphorylation of CaMKII at Thr-286 (in αCaMKII). This autophosphorylation has the important consequence of allowing CaMKII to retain enzymatic activity in the absence of Ca2+ (autonomy), thus sustaining CaMKII activity after diminution of the initial Ca2+ signal (11). The biological significance of CaMKII autophosphorylation at Thr-286 has been demonstrated by using a CaMKII “knock-in” mouse that cannot autophosphorylate (12). This mouse does not display hippocampal LTP and is deficient in spatial learning.
In contrast to LTP, Ca2+ influx elicited by LTD-inducing stimuli seems not to activate CaMKII but rather the Ca2+-sensitive phosphatase calcineurin (13). How might multiple pathways at the synapse share Ca2+ as their activator yet achieve nearly opposite responses? One mechanism for amplitude-based response specificity is for CaMKII to respond in a switch-like fashion to Ca2+ signals (14, 15). This type of response would narrow the range of Ca2+ where CaMKII becomes activated and hence would provide a distinct Ca2+ activation region for CaMKII compared with other Ca2+-activated enzymes at the synapse. In fact, it has been hypothesized that the signaling network controlling CaMKII autophosphorylation is a bistable type of switch that allows CaMKII to remain autophosphorylated long after Ca2+ returns to a basal level (16).
In this work we demonstrate experimentally that CaMKII responds in a switch-like fashion to Ca2+: CaMKII transitions rapidly from little to near-total autophosphorylation over a narrow range of Ca2+. Interestingly, this switch-like response was enhanced by the presence of protein phosphatase 1 (PP1). Together, CaMKII and PP1 allow CaMKII to remain dephosphorylated in response to subthreshold Ca2+ signals (such as those that cause LTD) but fully activate in response to strong signals (such as those that cause LTP). This provides a mechanism to help explain how modestly different Ca2+ levels can selectively activate different signaling pathways at the synapse. Furthermore, a CaMKII-PP1 molecular switch may provide a biochemical basis for the observed all-or-none potentiation of individual hippocampal synapses.
Materials and Methods
Protein Purification. αCaMKII and CaM were purified as described (17, 18). Purified, unstimulated CaMKII shows no evidence of basal Thr-286 autophosphorylation as evaluated by both a lack of autonomous activity and a failure to be recognized by a Thr-286 phospho-specific antibody.
The His-tagged PP1α catalytic subunit in the vector pDR540 (Pharmacia) was a gift of Angus Nairn (Yale University, New Haven, CT). Escherichia coli cells were grown at room temperature in 0.5 liters of LB with ampicillin (50 μg/ml) and 100 μM CoCl2 until OD = 0.4 nm, induced overnight with 0.5 mM isopropyl α-D-thiogalactoside, and harvested. Cells were resuspended in 10 mM Tris·HCl, pH 8.0/30 mM imidazole/10% glycerol/300 mM NaCl/1 mM CoCl2 (resuspension buffer), disintegrated by French press, and centrifuged at 30,000 × g for 30 min. The supernatant was loaded onto a 5-ml nickel-nitrilotriacetic acid agarose column (Qiagen, Valencia, CA) equilibrated in resuspension buffer. The column was washed in 20 mM Hepes, pH 7.4/200 mM KCl/2 mM Mg2+/10% glycerol (wash buffer) and eluted with wash buffer plus 400 mM imidazole. Fractions were pooled to 95% purity at 16.6 μM PP1 and immediately snap-frozen.
PP1 catalytic subunit purified from E. coli has been shown to have properties that differ from PP1 purified from tissue (19). However, the PP1 purified here in CoCl2 resulted in decreased p-nitrophenyl phosphate activity compared with PP1 purified in the absence of CoCl2, suggesting that CoCl2-purified PP1 is enzymatically more similar to tissue-purified PP1 than PP1 purified from E. coli in the absence of CoCl2.
CaMKII Autophosphorylation and the CaMKII-PP1 Signaling System. CaMKII phosphorylation experiments were performed in 40 μl of wash buffer plus 50 μM CaM, 2 mM ATP, and varying Ca2+ at 0°C. Eight microliters of a 5× solution of CaM, Mg2+, and ATP was first added to 4 μl of a 10× solution of Ca2+ stock buffer. Solutions of wash buffer and, if applicable, PP1 then were added to 20 μl. Twenty microliters of 2× CaMKII solution was added to initiate the reaction. Experiments were performed at 0°C to avoid the time-dependent decrease in enzyme activity during autophosphorylation that CaMKII exhibits at higher temperatures. Performing experiments at 0°C prevents autophosphorylation from occurring at the inhibitory sites on CaMKII (Thr-305 and Thr-306); this simplifies the CaMKII activation process but does not explore the role of Thr-305 and Thr-306 in the Ca2+-dependent activation of CaMKII.
For pairs of experiments where CaMKII was initially either dephosphorylated or phosphorylated, two stock Ca2+ buffers (”high” and “low”) were used. It was calculated that the combination of these two Ca2+ solutions would give the final desired [Ca2+]. For experiments where CaMKII was initially dephosphorylated, 2 μl of both Ca2+ stocks was added to 8 μl of the 5× solution of ATP/MgCl2/CaM; the additional solution components then were added to initiate the reaction. However, for experiments where CaMKII was initially phosphorylated, only the 2-μl high Ca2+ stock was initially added to 8 μl of ATP/MgCl2/CaM. Twenty microliters of CaMKII then was added to allow CaMKII autophosphorylation. Subsequently, the 2-μl low Ca2+ stock, the buffer, and PP1 were added to bring the solution to the final condition.
Autophosphorylation reactions were stopped by dilution of CaMKII to 100 nM into 20 mM Hepes, pH 7.4/200 mM KCl/50 mM EDTA/1 mM EGTA/0.1% BSA/500 nM microcystin-LR (quench buffer). The amount of autophosphorylation then was assessed by determining the level of autonomous CaMKII activity (17). The Ca2+/CaM-dependent activity of CaMKII was also determined as described in ref. 17. CaMKII assay conditions were 50 mM Pipes, pH 7.0, 20 mM Mg2+, 200 μM ATP, 0.1% BSA, 50 μM AC-2 substrate, 1 μCi of [32P]ATP (1 Ci = 37 GBq), and either 1 mM EGTA (autonomous) or 2 μM CaM and 500 μM Ca2+ (+Ca2+/CaM). The values of autonomy were fit by using the program SCIENTIST (Micromath, Salt Lake City) to the Hill equation
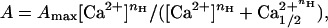 |
1 |
where A is the percent autonomy, Amax is the maximal autonomy, Ca1/22+ is the half-maximal Ca2+ value, and nH is the Hill coefficient.
Phosphatase Activity. To assess the activity of PP1 toward CaMKII, 25 μM CaMKII was first autophosphorylated with 500 μM Ca2+/50 μM CaM/2 mM MgCl2/500 μM ATP at 0°C. The reaction was quenched with 20 mM EDTA/5 mM EGTA. Autophosphorylated CaMKII was diluted to eight different concentrations, and PP1 then was added at both 5- and 10-fold lower concentrations than CaMKII. CaMKII autonomy was assessed after 10, 20, and 30 min. From these data, a plot of autophosphorylation versus time was generated that gave the initial velocity at each [CaMKII]. The velocities were fit to the Michaelis-Menton equation to determine the maximal velocity (Vmax) and Michaelis constant (Km).
To compare the activity of PP1 toward CaMKII in the presence and absence of Ca2+, 5 μM CaMKII was first autophosphorylated as described above except that a limiting amount of ATP (5 μM) was used. PP1 at 500 nM then was either added alone or along with 5 mM EGTA. The autonomy of CaMKII was assessed at 2, 5, 15, and 30 min.
Ca2+/EGTA Buffer. In all experiments [Ca2+] was controlled precisely by using a 10-fold concentrated system of Ca2+/EGTA buffer (Molecular Probes). The volume of K+/EGTA and Ca2+/EGTA stock solutions to be combined was calculated based on the determined dissociation constant of Ca2+ for EGTA of 60 nM under the used experimental conditions (20). To confirm the [Ca2+], a fluorescence titration with Fluo-4 (Molecular Probes) was performed (Fig. 6, which is published as supporting information on the PNAS web site, www.pnas.org).
Computational Models. We used a six-subunit biochemistry-based mathematical model of the CaMKII holoenzyme (21). Our experimental autophosphorylation conditions (0°C) allowed us to reduce the dimensionality of the previously published model by assuming (i) no phosphorylation at Thr-305 and Thr-306 and (ii) no CaM dissociation from autophosphorylated subunits. Except for the Km value of PP1 (which was experimentally determined here), parameter values were the same as those used previously (21).
Results
Ultrasensitivity in CaMKII Autophosphorylation. Our goal in this study was to better understand the dynamic response of CaMKII autophosphorylation at the synapse to changes in [Ca2+]. In the first experiments, CaMKII was incubated with the necessary buffer components (Mg2+, ATP, CaM, and Ca2+) to achieve phosphorylation at Thr-286. Here [Ca2+] was precisely varied in the micromolar range by using a Ca2+/EGTA buffer system (20). The state of CaMKII autophosphorylation after a 5-min incubation was evaluated by determining the level of autonomous activity of CaMKII, the activity of CaMKII in the absence of Ca2+. This value has been demonstrated previously to show a 1:1 correspondence with the level of Thr-286 autophosphorylation (17, 22). CaMKII autophosphorylation was observed to steeply transition from minimal to maximal phosphorylation over a small Ca2+ range (Fig. 1). For instance, CaMKII was only 10% autophosphorylated at 1.0 μM Ca2+ but 88% phosphorylated at 1.8 μM Ca2+. To estimate the level of cooperativity displayed by the autophosphorylation level, the data were fit to the Hill equation (Eq. 1). The nH of the curve was 4.4 ± 0.6 with a Ca1/2 2+value of 1.6 ± 0.1 μM. Hence, CaMKII demonstrates ultrasensitivity (cooperativity) to [Ca2+].
Fig. 1.

CaMKII autophosphorylation is cooperative to Ca2+. Plotted is the CaMKII autophosphorylation level (indicated by the maximum CaMKII autonomy) after 5 min versus [Ca2+] [error bars are ± SD; the number of autonomous activity values determined (n) was 2]. The solid line is the best fit to the Hill equation (Eq. 1). Here, [CaMKII] = 0.2 μM. All shown Ca2+ titration data are representative data obtained from at least two, and often three or more, data sets collected at each condition.
To better understand the dynamics of CaMKII autophosphorylation, we monitored the time course of autophosphorylation at each Ca2+ level (Fig. 7, which is published as supporting information on the PNAS web site). CaMKII generally showed a rapid initial autophosphorylation rate that then slowed as autophosphorylation continued.
The Mechanism of CaMKII Ultrasensitivity. What is the molecular basis by which CaMKII responds cooperatively to Ca2+? We hypothesized that several factors could potentially play a role: (i) cooperative binding of Ca2+ to CaM, (ii) cooperative binding of Ca2+-CaM to CaMKII, (iii) and the requirement of Ca2+-CaM to bind two different CaMKII subunits for Thr-286 autophosphorylation to occur. We first examined whether cooperative binding of CaM to CaMKII contributes to the ultrasensitive response. An evaluation of how the Ca2+/CaM-dependent enzymatic activity of CaMKII varied with [CaM] revealed a noncooperative curve with an nH value of 1.1 ± 0.2 and a CaM1/2 value of 48 ± 6 nM (Fig. 2A). This finding of noncooperative interaction between CaM and CaMKII corroborates previous reports (23) and indicates that the interaction between CaM and CaMKII does not contribute to the observed Ca2+ ultrasensitivity.
Fig. 2.

Dissecting the molecular basis of Ca2+ ultrasensitivity. (A) Lack of significant cooperativity in CaM activation of CaMKII. Plotted is the Ca2+-dependent CaMKII activity versus [CaM] (±SD, n = 2). (B) Modest Ca2+ ultrasensitivity (nH ≈ 3) caused by cooperative binding of Ca2+ to CaM. Plotted is the CaM-dependent CaMKII activity versus [Ca2+] (±SD, n = 2). (C) Greater Ca2+ ultrasensitivity (nH ≈ 5) induced by the dual requirement of CaM for autophosphorylation. Plotted is the maximum autonomy versus [Ca2+](±SD, n = 2). Experiments were performed at 0.2 μM (triangle, solid black line), 1.0μM (diamond, dotted line), and 5.0 μM (square, solid gray line) CaMKII.
Second, to evaluate the role of cooperative interactions between Ca2+ and CaM, we examined the Ca2+ dependence of how CaM activates the peptide phosphorylation activity of CaMKII. However, monitoring the time course of peptide phosphorylation at low Ca2+ revealed a nonlinear time course of product formation, complicating the determination of CaMKII activity at low Ca2+. We determined that this nonlinearity was due to progressive autophosphorylation of CaMKII during the assay (see Fig. 8, which is published as supporting information on the PNAS web site). Because nonlinear product formation precludes a simple, rigorous determination of CaMKII activity, we simply estimated the initial CaMKII activity at low Ca2+. Plotting the estimated CaM-dependent activity versus [Ca2+] revealed a modestly cooperative curve with an nH value of 3.0 ± 0.2 and a Ca1/22+ value of 5.6 ± 0.2 μM (Fig. 2B). The nH value of 3.0 ± 0.2 is identical to the known Hill coefficient for binding of Ca2+ to CaM, and hence cooperative binding interactions between Ca2+ and CaM partially contribute to the overall ultrasensitivity of CaMKII autophosphorylation.
Finally, we examined how the ultrasensitivity of CaMKII autophosphorylation depended on [CaMKII]. CaMKII at 5, 1, and 0.2 μM was allowed to autophosphorylate at different Ca2+ values for 5 min, and the autonomy generated was monitored (Fig. 2C). Analysis of the data (Tables 1 and 2) revealed that the cooperativity of CaMKII autophosphorylation (nH ≈ 5) is greater than that of Ca2+/CaM activity (nH ≈ 3). Furthermore, increasing [CaMKII] had little effect on either nH or Ca1/22+. We attribute the greater cooperativity in the autophosphorylation of CaMKII compared with its Ca2+/CaM-dependent activity as reflecting the requirement of two bound CaMs for each autophosphorylation event (24). In total, it seems that the Ca2+ ultrasensitivity of CaMKII alone arises from two main factors: (i) the cooperative binding of Ca2+ to CaM and (ii) the dual requirement of CaM for CaMKII autophosphorylation.
Table 1. Hill coefficients for the Ca2+ dependence of CaMKII autophosphorylation at varying concentrations of CaMKII and PP1.
|
[CaMKII], μM
|
|||
|---|---|---|---|
| [PP1], μM | 0.2 | 1 | 5 |
| 0 | 4.4 ± 0.6 | 4.9 ± 0.3 | 5.4 ± 0.4 |
| 0.5 | 7.7 ± 0.8 | 6.6 ± 1.7 | 8.9 ± 1.8 |
| 1.25 | 6.5 ± 0.5 | 8.6 ± 2.5 | 6.9 ± 1.6 |
| 2.5 | — | 5.9 ± 0.3 | 8.9 ± 2.2 |
Each nH value represents the mean obtained from at least two Ca2+ titrations. nH values were often obtained at several time points, and these values were averaged to obtain the final value. Uncertainties are the SD.
Table 2. Ca2+½ (μM) values of CaMKII autophosphorylation at varying concentrations of CaMKII and PP1.
|
[CaMKII], μM
|
|||
|---|---|---|---|
| [PP1], μM | 0.2 | 1 | 5 |
| 0 | 1.6 ± 0.1 | 1.4 ± 0.2 | 1.4 ± 0.1 |
| 0.5 | 2.6 ± 0.2 | 2.3 ± 0.5 | 1.9 ± 0.1 |
| 1.25 | 3.6 ± 0.2 | 3.1 ± 0.6 | 3.8 ± 0.9 |
| 2.5 | — | 5.8 ± 0.1 | 5.2 ± 0.1 |
Each Ca2+½ value is the mean from at least two experiments. Uncertainties are the SD.
Switch-Like Behavior of a CaMKII-PP1 System. At the synapse, CaMKII autophosphorylation is regulated by the action of phosphatases, particularly PP1 (5). Theoretical models of the interplay between kinases and phosphatases have suggested that these dynamic phosphorylation-dephosphorylation systems under certain circumstances could create switch-like responses to stimuli such as Ca2+ (14, 15). Hence, here we investigated how a CaMKII-PP1 system responds to Ca2+.
In these experiments, 1 μM CaMKII (initially dephosphorylated) and 1.25 μM PP1 were incubated at varying Ca2+ along with the necessary buffer components (Mg2+, ATP, CaM, and Ca2+) to allow competitive phosphorylation and dephosphorylation of CaMKII. It was determined that, after an initial equilibration time, the phosphorylation state of CaMKII was stable with time. Fitting the equilibrium values under these conditions to the Hill equation revealed an nH value of 8.6 ± 2.5 with a Ca1/22+ value of 3.1 ± 0.6 μM (Fig. 3A). Hence, the CaMKII-PP1 system demonstrates even more dramatic switch-like behavior to [Ca2+] than CaMKII alone.
Fig. 3.

A CaMKII-PP1 system is a reversible switch. (A) Plotted is the CaMKII autophosphorylation level (or percent of maximum CaMKII autonomy) at equilibrium versus [Ca2+] (±SD, n = 2). Open squares and black diamonds represent experiments initiated with dephosphorylated and phosphorylated CaMKII, respectively. The solid line is the best fit of the initially dephosphorylated CaMKII data to the Hill equation (Eq. 1). Here, [CaMKII] = 1 μM and [PP1] = 1.25 μM. (B-D) Role of varying [CaMKII] and [PP1] on the CaMKII-PP1 switch. Plotted is the CaMKII autonomy at equilibrium versus [Ca2+]. Squares, diamonds, and triangles represent experiments performed with 0.5, 1.25, and 2.5 μM PP1, respectively. The solid gray, dashed, and solid black lines represent the best fit of the 0.5, 1.25, and 2.5 μM PP1 data, respectively, to the Hill equation (Eq. 1). B-D show data at 0.2, 1, and 5 μM CaMKII, respectively.
Because the magnitude of CaMKII-PP1 ultrasensitivity could be infuenced by experimental conditions (particularly [CaMKII] and [PP1]), the equilibrium CaMKII autophosphorylation values were determined as a function of Ca2+ for a panel of eight different conditions between 0.2 and 5 μM CaMKII and 0.5 and 2.5 μM PP1 (Fig. 3 B-D). Each curve demonstrates a relatively similar steep transition from little autophosphorylation to full autophosphorylation over a small range of Ca2+. We observed that Ca1/22+ increases with increasing [PP1], which is expected because greater [PP1] should more effectively suppress autophosphorylation (Table 2). However, Ca1/22+ changes little with [CaMKII]. The average nH values for each condition vary between 5.9 and 8.9, with the average from all experiments being 7.5 (Table 1). Surprisingly, no trend in nH with [CaMKII] or [PP1] was evident from the data. However, comparing the nH values for all experiments performed either with or without PP1 reveals a statistically significant increase in switch-like behavior in the presence of PP1 (P < 0.02 by Mann-Whitney test).
CaMKII and PP1 Form an Ultrasensitive Type of Switch. The switch-like Ca2+ response of the CaMKII-PP1 system could potentially be attributed to two different mechanisms: ultrasensitivity or bistability (see ref. 25). These mechanisms differ regarding whether CaMKII autophosphorylation is maintained once Ca2+ returns to a basal level. An ultrasensitive switch would be “turned off” when [Ca2+] is reduced, whereas a bistable switch could allow CaMKII autophosphorylation to be indefinitely maintained at low Ca2+ levels, even considering both the action of phosphatases and protein turnover at the synapse (16). Several computational investigations have suggested that a CaMKII-PP1 system, or a signaling network focused on CaMKII and PP1, could form a bistable type of switch (26, 27).
To identify the type of switching exhibited by CaMKII and PP1, we performed pairs of experiments initiated under identical conditions except that CaMKII was initially either in the dephosphorylated or phosphorylated state. It was examined whether the two conditions equilibrated to a single (ultrasensitive switch) or different (bistable switch) steady-state values. Initial trials at 1 μM CaMKII and 1.25 μM PP1 indicated that the CaMKII-PP1 system equilibrated to the same autophosphorylation level irrespective of [Ca2+] or the initial state of autophosphorylation (Fig. 3A). Comparing the time courses of equilibration indicated that the CaMKII-PP1 systems at the same [Ca2+] reached the identical level of autophosphorylation after 2.5 h, and this value remained constant through 7.5 h (Fig. 9, which is published as supporting information on the PNAS web site); the time course of equilibration appeared similar for all Ca2+ conditions. Subsequent experiments under eight different conditions between 0.2 and 5 μM CaMKII and 0.5 and 2.5 μM PP1 all showed equilibration to the same autophosphorylation level, demonstrating that the CaMKII-PP1 system is an ultrasensitive-type switch under all these conditions.
Computational Modeling of CaMKII-PP1 Switching. We next characterized several aspects of CaMKII dephosphorylation by PP1. Michaelis-Menton analysis revealed that the Vmax and Km for dephosphorylation were 31 ± 8 nmol·min−1 per mg and 11 ± 5 μM, respectively (Fig. 4A). This Km value is similar to, albeit slightly larger than, that of PP1 for other substrates (19, 28) and close to the estimated synaptic [CaMKII]. Furthermore, the time course of dephosphorylation was identical in either Ca2+ or EGTA (Fig. 4B), indicating that the PP1 catalytic subunit dephosphorylates the autophosphorylated, CaM-bound form of CaMKII at the same rate as the autophosphorylated, CaM-free form.
Fig. 4.

Characterization of CaMKII dephosphorylation by PP1. (A) Michaelis-Menton plot of CaMKII dephosphorylation by PP1. Plotted is the velocity of dephosphorylation versus [CaMKII] (±SD, n = 2). The line represents the best fit to the Michaelis-Menton equation. (B) Time course of CaMKII dephosphorylation in the presence and absence of Ca2+. Plotted is the CaMKII autonomy versus time (±SD, n = 2).
We used this PP1 characterization together with a biochemically based computational model of the CaMKII-PP1 interaction (see ref. 21 and Materials and Methods) to better understand the molecular basis of Ca2+ ultrasensitivity. We simulated the equilibrium CaMKII autophosphorylation values as a function of [Ca2+] and found that the simulated data reproduce the experimentally observed Ca2+ ultrasensitivity of the CaMKII-PP1 system both in the large magnitude of ultrasensitivity and in the manner that Ca1/22+ depends on [PP1] (Fig. 10, which is published as supporting information on the PNAS web site). Hence, a computational model that takes into account each of the molecular processes required for CaMKII autophosphorylation is able to produce a similar Ca2+ ultrasensitivity as we observed experimentally.
Discussion
CaMKII is the central enzyme at hippocampal synapses for recognizing LTP-inducing Ca2+ signals and transducing this information to downstream targets (5). Despite the key function of CaMKII autophosphorylation for changes in synaptic strength such as LTP, the dynamics of how CaMKII autophosphorylation changes with [Ca2+] and the level of phosphatase activity have not been characterized previously. Here we demonstrate that CaMKII autophosphorylation occurs in a switch-like fashion over a narrow and physiologically relevant range of [Ca2+]. The causes of this behavior are the cooperative binding of Ca2+ to CaM, the dual requirement of Ca2+-CaM for CaMKII autophosphorylation, and competing the CaMKII autophosphorylation reaction with dephosphorylation.
The Ca2+ ultrasensitivity of CaMKII helps explain how distinct synaptic signaling pathways can be activated by Ca2+ while still maintaining pathway specificity. The prevailing model of synaptic Ca2+ signaling is that low to moderate levels of Ca2+ cause LTD by activating calcineurin, whereas high Ca2+ induces LTP primarily through CaMKII activation (29). However, Ca2+ imaging studies have shown the dynamic range of [Ca2+] in dendritic spines is modest compared with other systems, varying from ≈100 nM under basal conditions to ≈10 μM after strong stimulation (3). If CaMKII did not respond with ultrasensitivity to Ca2+, an 81-fold change in [Ca2+] would be required to transition CaMKII from 10% to 90% activation (15); this type of response would encompass the entire physiological range of Ca2+ and hence would certainly overlap the calcineurin activation region (Fig. 5). Ultrasensitive Ca2+ switching by CaMKII permits CaMKII to transition from active to inactive over a 300 nM range of [Ca2+], providing for multiple pathways to be specifically activated within the physiological range of Ca2+ (Fig. 5).
Fig. 5.

How a CaMKII-PP1 switch facilitates specificity in synaptic signaling. Shown are the molecular events causing the CaMKII-PP1 switch (A) and the effect of these events on the Ca2+ dependence of CaMKII autophosphorylation (B). Plotted in the graph is the percent CaMKII autophosphorylation versus the range of [Ca2+] in dendritic spines. If CaMKII activation were noncooperative, the activation curve would encompass the entire range of physiological Ca2+ values [dashed blue line and (0)]. Ultrasensitivity in CaMKII activation is imposed by both the cooperative binding of Ca2+ to CaM [dashed green line and (1)] and the dual requirement of CaM for autophosphorylation [dashed orange line and (2)]. Competitive dephosphorylation by PP1 further enhances the switch-like behavior and increases the Ca1/22+ value to the physiological [Ca2+] corresponding to LTP [solid red line and (3)]. The Ca2+ activation regions corresponding to LTP and LTD were determined based on the Ca2+ level in dendritic spines under depolarizing and resting potentials, respectively (3).
What additional functions might be served by having a CaMKII-PP1 switch? Molecular switches have been found in many biological circumstances where varying levels of input must be translated into an all-or-none decision by the cell (15). In the case of synaptic plasticity, it has been found that LTP induction at individual CA3-CA1 hippocampal synapses is also a highly cooperative, possibly all-or-none, process (30). Hence, a CaMKII-PP1 switch provides a biochemical mechanism to explain how individual synapses could respond in an all-or-none fashion to varying levels of Ca2+ stimulation.
In this work we experimentally found the CaMKII-PP1 switch to be a reversible rather than bistable type of switch. We attribute this result largely to the fact that the Km of PP1 for CaMKII (11 ± 5 μM) is larger than the experimentally used [CaMKII]. These conditions likely do not provide sufficient phosphatase saturation to create a bistable type of switch (14).
Within neurons, CaMKII is located both in the cytosol and also at a cytoskeletal specialization known as the postsynaptic density (PSD). The experimental conditions used in this study more closely resemble cytosol than PSD, and thus our finding of ultrasensitive Ca2+ sensing relates more directly to the cytosolic pool of CaMKII. It is likely that CaMKII within the PSD also responds in a switch-like fashion to Ca2+. However, as described below, the specialized environment of the PSD may modulate the Ca2+ ultrasensitivity of CaMKII in several ways from the behavior we observed in vitro.
One factor that may affect the Ca2+ ultrasensitivity of CaMKII is binding to the NR2B subunit of the N-methyl-D-aspartate (NMDA) receptor. This interaction has been demonstrated to bestow CaMKII with autonomous activity independent of Thr-286 autophosphorylation (31). Subunits of the CaMKII holoenzyme that directly bind NR2B will partially lose their dependence on Ca2+ for activity. However, computational modeling indicates that any subunits of the CaMKII holoenzyme that translocate to, but do not directly bind, the NMDA receptor should demonstrate an increase in the switch-like character of their Ca2+ response. Hence, binding to the NMDA receptor could alter the Ca2+ ultrasensitivity of CaMKII in multiple ways.
The context of the PSD will likely affect not only the properties of CaMKII but also those of PP1. At the PSD, PP1 binds to targeting subunits such as spinophilin, and these interactions modulate the catalytic activity of the enzyme (19). Furthermore, postsynaptic PP1 is regulated by Ca2+ through a “gating” mechanism whereby protein kinase A and calcineurin control the activity of PP1 through phosphorylation and dephosphorylation of the PP1 inhibitor protein I-1 (32). Gating of PP1 activity could enhance the switch-like properties of the CaMKII-PP1 system significantly if PP1 activity decreases with Ca2+ in the same range where CaMKII activity is increasing with Ca2+.
How might the Ca2+ ultrasensitivity of CaMKII be exhibited within the PSD? Because synaptic changes in [Ca2+] often occur as spikes of increased Ca2+ that recur with a specific frequency, the Ca2+ ultrasensitivity of CaMKII may often be manifested as a highly tuned sensitivity to the frequency of Ca2+ oscillations. Indeed, 2.5-Hz Ca2+ pulses have been demonstrated to cause rapid CaMKII autophosphorylation, whereas pulses at the slightly lower frequency of 1 Hz fail to initiate significant autophosphorylation (23).
The demonstration here that a CaMKII-PP1 system functions as a Ca2+-sensitive switch adds to the growing understanding that signaling molecules are organized into switches and networks so as to convey information in ways that are often nonlinear. The way that most of these networks are constructed and how they function is still not well understood. Further quantitative studies of the interplay between signaling molecules will be required to provide a better understanding of how different signaling pathways function.
Supplementary Material
Acknowledgments
We thank K. U. Bayer, A. Hudmon, and J. Tsui for critical reading of the manuscript. This work was supported by National Institutes of Health Grants GM40600 and GM30179. J.M.B. is a fellow of the Jane Coffin Childs Memorial Fund for Medical Research.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: LTP, long-term potentiation; LTD, long-term depression; CaM, calmodulin; CaMKII, Ca2+/CaM-dependent protein kinase II; PP1, protein phosphatase 1; PSD, postsynaptic density. www.pnas.org/cgi/
References
- 1.Sheng, M. & Kim, M. J. (2002) Science 298, 776-780. [DOI] [PubMed] [Google Scholar]
- 2.Franks, K. M. & Sejnowski, T. J. (2002) BioEssays 24, 1130-1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sabatini, B. L., Oertner, T. G. & Svoboda, K. (2002) Neuron 33, 439-452. [DOI] [PubMed] [Google Scholar]
- 4.Rongo, C. (2002) BioEssays 24, 223-233. [DOI] [PubMed] [Google Scholar]
- 5.Lisman, J., Schulman, H. & Cline, H. (2002) Nat. Rev. Neurosci. 3, 175-190. [DOI] [PubMed] [Google Scholar]
- 6.Fink, C. C. & Meyer, T. (2002) Curr. Opin. Neurobiol. 12, 293-299. [DOI] [PubMed] [Google Scholar]
- 7.Hudmon, A. & Schulman, H. (2002) Annu. Rev. Biochem. 71, 473-510. [DOI] [PubMed] [Google Scholar]
- 8.Soderling, T. R., Chang, B. & Brickey, D. (2001) J. Biol. Chem. 276, 3719-3722. [DOI] [PubMed] [Google Scholar]
- 9.Barria, A., Muller, D., Derkach, V., Griffith, L. C. & Soderling, T. R. (1997) Science 276, 2042-2045. [DOI] [PubMed] [Google Scholar]
- 10.Shen, K. & Meyer, T. (1999) Science 284, 162-166. [DOI] [PubMed] [Google Scholar]
- 11.Saitoh, T. & Schwartz, J. H. (1985) J. Cell Biol. 100, 835-842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giese, K. P., Fedorov, N. B., Filipkowski, R. K. & Silva, A. J. (1998) Science 279, 870-873. [DOI] [PubMed] [Google Scholar]
- 13.Mulkey, R. M., Endo, S., Shenolikar, S. & Malenka, R. C. (1994) Nature 369, 486-488. [DOI] [PubMed] [Google Scholar]
- 14.Goldbeter, A. & Koshland, D. E., Jr. (1982) Q. Rev. Biophys. 15, 555-591. [DOI] [PubMed] [Google Scholar]
- 15.Ferrell, J. E., Jr. (1996) Trends Biochem. Sci. 21, 460-466. [DOI] [PubMed] [Google Scholar]
- 16.Lisman, J. E. & Zhabotinsky, A. M. (2001) Neuron 31, 191-201. [DOI] [PubMed] [Google Scholar]
- 17.Bradshaw, J. M., Hudmon, A. & Schulman, H. (2002) J. Biol. Chem. 277, 20991-20998. [DOI] [PubMed] [Google Scholar]
- 18.Singla, S. I., Hudmon, A., Goldberg, J. M., Smith, J. L. & Schulman, H. (2001) J. Biol. Chem. 276, 29353-29360. [DOI] [PubMed] [Google Scholar]
- 19.Watanabe, T., Huang, H. B., Horiuchi, A., da Cruze Silva, E. F., Hsieh-Wilson, L., Allen, P. B., Shenolikar, S., Greengard, P. & Nairn, A. C. (2001) Proc. Natl. Acad. Sci. USA 98, 3080-3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bers, D. M., Patton, C. W. & Nuccitelli, R. (1994) Methods Cell Biol. 40, 3-29. [DOI] [PubMed] [Google Scholar]
- 21.Kubota, Y. & Bower, J. M. (2001) J. Comput. Neurosci. 11, 263-279. [DOI] [PubMed] [Google Scholar]
- 22.Ikeda, A., Okuno, S. & Fujisawa, H. (1991) J. Biol. Chem. 266, 11582-11588. [PubMed] [Google Scholar]
- 23.DeKoninck, P. & Schulman, H. (1998) Science 279, 227-230. [DOI] [PubMed] [Google Scholar]
- 24.Hanson, P. I., Meyer, T., Stryer, L. & Schulman, H. (1994) Neuron 12, 943-956. [DOI] [PubMed] [Google Scholar]
- 25.Ferrell, J. E., Jr. (1998) Trends Biochem. Sci. 23, 461-465. [DOI] [PubMed] [Google Scholar]
- 26.Zhabotinsky, A. M. (2000) Biophys. J. 79, 2211-2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okamoto, H. & Ichikawa, K. (2000) Biol. Cybern. 82, 35-47. [DOI] [PubMed] [Google Scholar]
- 28.Huang, H. B., Horiuchi, A., Goldberg, J., Greengard, P. & Nairn, A. C. (1997) Proc. Natl. Acad. Sci. USA 94, 3530-3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lisman, J. (1989) Proc. Natl. Acad. Sci. USA 86, 9574-9578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petersen, C. C., Malenka, R. C., Nicoll, R. A. & Hopfield, J. J. (1998) Proc. Natl. Acad. Sci. USA 95, 4732-4737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bayer, K. U., De Koninck, P., Leonard, A. S., Hell, J. W. & Schulman, H. (2001) Nature 411, 801-805. [DOI] [PubMed] [Google Scholar]
- 32.Blitzer, R. D., Connor, J. H., Brown, G. P., Wong, T., Shenolikar, S., Iyengar, R. & Landau, E. M. (1998) Science 280, 1940-1942. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.