

Abstract
Enfuvirtide (ENF), the first approved fusion inhibitor (FI) for HIV, is a 36-aa peptide that acts by binding to the heptad repeat 1 (HR1) region of gp41 and preventing the interaction of the HR1 and HR2 domains, which is required for virus–cell fusion. Treatment-acquired resistance to ENF highlights the need to create FI therapeutics with activity against ENF-resistant viruses and improved durability. Using rational design, we have made a series of oligomeric HR2 peptides with increased helical structure and with exceptionally high HR1/HR2 bundle stability. The engineered peptides are found to be as much as 3,600-fold more active than ENF against viruses that are resistant to the HR2 peptides ENF, T-1249, or T-651. Passaging experiments using one of these peptides could not generate virus with decreased sensitivity, even after >70 days in culture, suggesting superior durability as compared with ENF. In addition, the pharmacokinetic properties of the engineered peptides were improved up to 100-fold. The potent antiviral activity against resistant viruses, the difficulty in generating resistant virus, and the extended half-life in vivo make this class of fusion inhibitor peptide attractive for further development.
Keywords: coiled coil, gp41 HIV-1, viral entry, drug design
HIV type 1 (HIV-1) fusion is mediated by a complex set of interactions involving cellular receptors and viral glycoproteins (1, 2). The precise nature of these interactions remains unclear, but in the current model, viral attachment occurs via an interaction between gp120 and CD4, along with chemokine receptors (such as CCR5 or CXCR4) that act as viral coreceptors for HIV-1. A conformational change in the gp41/gp120 oligomer allows the fusion peptide sequence, located on the N terminus of gp41, to insert into the membrane of the target cell (3). The gp41 ectodomain is then thought to undergo a series of conformational changes to form the fusion-active state, which is believed to bring the viral and cellular membranes into closer proximity to facilitate membrane fusion (2, 3). Crystallographic studies provide support for a model where gp41 adopts a six-helix bundle structure late in the fusion process (4–6), and this structure is likely involved in the final steps before fusion.
In peptide models of gp41, the heptad repeat 1 (HR1) region forms a trimeric inner coiled-coil core, which creates the binding interface for peptides from the HR2 region. A highly conserved portion of this groove, termed the “deep pocket,” has been shown to be an attractive target for blocking the viral fusion process (7–9). Peptides derived from the HR2 region of gp41 have been shown to have potent antiviral activity (7, 10) and enfuvirtide (ENF, also known as T-20 or DP-178), the first approved fusion inhibitor (FI) for HIV, is a 36-aa peptide derived from this region (11). Although ENF has demonstrated excellent efficacy in clinical trials (11), novel fusion inhibitors that are active against ENF-resistant viruses and exhibit a higher genetic barrier to resistance could be important drug candidates.
Biophysical and structural analysis has shown that HR2 peptides are typically unstructured in solution but adopt an α-helical conformation upon binding (5, 12, 13). Furthermore, several groups have demonstrated increased antiviral activity of peptides containing motifs that stabilize helical structure (9, 10, 14, 15). Various covalent strategies have been used to stabilize helical structure in peptides derived from gp41 (10, 16); however, these synthetic approaches may not be amenable for large-scale manufacturing of peptide therapeutics. Several strategies exist for increasing helical structure through protein engineering. The rules that govern the stability of the α-helix have been extensively studied, and a variety of methods have been used to estimate the helical propensity of amino acids (17–20). Introducing ion pair interactions, or “salt bridges,” has also been suggested to increase helical structure in peptides and stabilize coiled coils (21–24). Some of these noncovalent methods have been successful in enhancing antiviral activity of monomeric HR2 peptides (15, 25).
Using rational design, we have designed a series of HR2 peptides with greatly enhanced helical structure that self-associate into stable oligomeric structures. The peptides are shown to have high affinity to HR1 targets, potent antiviral activity against HR2-resistant viruses, and improved pharmacokinetics in cynomolgus monkeys. Interestingly, viruses appear to require more mutations to decrease sensitivity to the engineered peptides, and the impact of these mutations on activity is significantly less than that observed for ENF. Taken together, these peptides are attractive for further clinical development.
Results
Helix Stabilization of HR2 Peptides.
Peptide design began with T-651 and T-2410, peptides that are derived from a gp41 HR2 region slightly N-terminal to that from which ENF is derived (Fig. 1). T-651 and T-2410 each have potent antiviral activity against IIIB (8 ng/ml) and the primary isolate 098 (≈33 ng/ml), as well as viruses that are resistant to ENF and T-1249 (39–151 ng/ml) (Table 1). Although the antiviral results for T-2410 are virtually identical to those of T-651 (Table 1), the stability results for the HR1-HR2 bundles (74°C for T-2410 and 68°C for T-651) highlight a potential advantage for T-2410. Therefore, T-2410 was chosen as the starting point for the current design strategy. T-2410 includes two additional residues (EL) on the C-terminal end of T-651 so that the C-terminal residue is at an “a” position in the heptad repeat. The heptad repeat, present in most coiled-coil structures, is characterized by a preference for hydrophobic amino acids within the core “a” and “d” positions of the heptad, and hydrophilic residues at the outside “b,” “c,” “e,” “f,” and “g” positions. This pattern places hydrophobic side chains along one face of the α-helix, which interact with other helicies to form the coiled-coil structural motif.
Fig. 1.

Design of helix-stabilized, oligomeric, HR2 peptides. Starting with the T-651 analog, T-2410, charged residues, alanines, and hydrophobic residues were added to generate self-associating, helical HR2 peptides. Residues that are underlined indicate a change from the previous peptide. The percent helicity as determined by circular dichroism is indicated for each design.
Table 1.
Sequences, biophysical properties, pharmacokinetics, and antiviral activity of designed peptides
| Peptide | Sequence | % hel | HR2 Tm, °C* | HR1/HR2 Tm 8M urea, °C | PK |
Antiviral activity, IC50, μg/ml |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cl, liter/h/kg | T1/2, h | IIIB | 098 | 098-T20 | 098-T1249 | 098-T651 | |||||
| ENF (T-20) | YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF | 12 | <5 | <5 | 0.04 | 8.8 | 0.006 | 0.050 | 0.526 | 54.958 | 47.822 |
| 1249 | WQEWEQKITALLEQAQIQQEKNEYELQKLDKWASL WEWF | 49 | <5 | 59 | 0.01 | 5.9 | 0.003 | 0.013 | 0.022 | 0.363 | 8.140 |
| 651 | MTWMEWDREINNYTSLIHSLIEESQNQQEKNEQELL | 7 | <5 | 68 | 0.432 | 0.3 | 0.008 | 0.033 | 0.060 | 0.151 | 7.599 |
| 2410 | MTWMEWDREINNYTSLIHSLIEESQNQQEKNEQELLEL | 7 | <5 | 74 | 0.372 | 1.2 | 0.008 | 0.032 | 0.039 | 0.137 | 4.975 |
| 2429 | TTWEEWDREINEYTSRIESLIRESQEQQEKNEQELREL | 19 | <5 | 80 | — | — | 0.012 | 0.021 | 0.056 | 0.037 | 0.167 |
| 2638 | MTWMAWDRAIANYAALIHALIEAAQNQQEKNEAALLEL | 51 | <5 | — | — | — | 0.061 | 0.079 | 0.079 | 0.120 | 0.250 |
| 290676 | TTWEAWDRAIAEYAARIEALIRASQEQQEKNEAELREL | 47 | <5 | 78 | 0.086 | 1.2 | 0.006 | 0.013 | 0.022 | 0.072 | 1.314 |
| 2635 | TTWEAWDRAIAEYAARIEALIRAAQEQQEKNEAALREL | 75 | 42 | 86 | 0.043 | 16.3† | 0.007 | 0.018 | 0.025 | 0.015 | 0.021 |
| 2544 | MTWEAWDRAIAEYAARIEALIRAAQEQQEKNEAALREL | 69 | 47 | — | 0.032 | 5.1 | 0.007 | 0.026 | 0.033 | 0.014 | 0.021 |
| 267209 | TTWEAWDRAIAEYAARIEALIRAAQEQQEKNEAILREL | 90 | 51 | 89 | 0.066 | 3.4 | 0.007 | 0.021 | 0.034 | 0.024 | 0.039 |
| 267221 | TTWEAWDRAIAEYAARIEALIRALQEQQEKNEAALREL | 97 | 65 | 82 | 0.010 | 15.2 | 0.011 | 0.035 | 0.028 | 0.035 | 0.050 |
| 267226 | TTWEAWDRAIAEYAARIEALIRAAQELQEKNEAALREL | 94 | 59 | 89 | 0.026 | 7.6 | 0.007 | 0.021 | 0.035 | 0.030 | 0.020 |
| 267225 | TTWEAWDRAIAEYAARIEALIRALQEQQEKNEAILREL | 103 | 72 | 85 | 0.012 | 12.4 | 0.019 | 0.043 | 0.079 | 0.038 | 0.196 |
| 267227 | TTWEAWDRAIAEYAARIEALIRAAQEQQEKLEAALREL | 95 | 75 | 86 | 0.009 | 17.9 | 0.012 | 0.045 | 0.028 | 0.025 | 0.044 |
| 291022 | TTWEAWDRAIAEYAARIEALIRAAQEQQEKLEAVLREL | 89 | 80 | 85 | 0.004 | 28.6 | 0.022 | 0.049 | 0.093 | 0.046 | 0.242 |
| 290822 | TTWEAWDRAIAEYAARIEALIRALQELQEKNEAALREL | 96 | 83 | 82 | 0.008 | 8.6 | 0.030 | 0.054 | 0.084 | 0.147 | 0.242 |
| 290821 | TTWEAWDRAIAEYAARIEALIRALQELQEKNEAILREL | 94 | 95 | 84 | 0.009 | 9.6 | 0.065 | 0.118 | 0.154 | 0.192 | >20 |
| 267228 | TTWEAWDRAIAEYAARIEALIRAAQELQEKLEAALREL | 98 | >100 | 92 | 0.002 | 30.8 | 0.172 | 0.600 | 0.416 | 0.555 | 0.987 |
| 267229 | TTWEAWDRAIAEYAARIEALIRALQELQEKLEAILREL | 93 | >100 | ND | 0.003 | 35.4 | 3.162 | >20 | >20 | >20 | >20 |
ND, due to the high stability of 267229, the bundle Tm could not be reliably determined.
*Helicity and Tm of HR2 peptides was done at a concentration of 10 μM. HR1/HR2 bundle stability was determined using T865 as the HR1 at an equal molar concentration of 1 μM in PBS pH 7.0 + 8 M urea.
†Due to one animal (of six) with markedly different PK for T2635, we also report the median Cl as 0.045 (range 0.008–0.070) and median half-life as 5.9 (range 4.1–68.6).
Several amino acid substitutions were used to increase the helical structure of T-2410 (Fig. 1). Residues for replacement were selected based on results from alanine scanning of T-651, which highlighted the contribution of each amino acid to six-helix bundle stability and antiviral activity (J.J.D., K.L.W., D.K.D., M.L.G., and M.K.D., unpublished data). Amino acids that were important for activity or bundle stability were not considered for replacement. Charged glutamic acid and arginine residues were introduced into non-core positions so that the spacing (i, i + 4) favored the formation of an ion pair in the helical conformation. Orientation of the ion pairs placed the basic side chain on the C-terminal side of the ion pair (Fig. 1). This spacing and orientation has been shown to be more stabilizing in peptide models, presumably due to favorable interactions with the helix dipole (21). These substitutions created at least five additional potential ion pairs. Roughly equal numbers of acidic and basic amino acids were added so that the isoelectric point and net charge of the peptide at pH 7 was similar to T-2410. The N-terminal methionine was changed to threonine to eliminate potential complications caused by oxidation. Measurement of the helicity of the resulting peptide, T-2429, indicated that the introduction of ion pairs increased the helicity from 7% to 19%. The antiviral results for T-2429 indicate that potency against the wild-type strains IIIB and 098, and the virus resistant to ENF (098-T20) has been maintained relative to the starting peptide T-2410. However, there is a significant increase in potency against the viruses resistant to T-1249 and T-651 (37 and 167 ng/ml for T-2429 vs. 137 and 4,975 ng/ml for T-2410).
Alanine has been shown to promote helix formation in peptides and has one of the highest helix propensities, as determined from host–guest experiments (24, 26–30). Based on the alanine scan data, eight noncritical residues that were not part of an ion pair were substituted with alanine (Fig. 1). The resulting peptide, T-290676, was found to be significantly more helical than the parent peptide T-2410 (47% vs. 7%). T-290676 maintained activity against IIIB, 098, 098-T20, and 098-T1249 relative to T-2410, and showed increased potency against 098-T651 (1,314 ng/ml) relative to T-2410 (4,975 ng/ml). T-2638, a peptide with the alanine substitutions but no ion pairs, was 51% helical (Table 1), suggesting that the helix-promoting alanine residues are sufficient for increased structure. However, the antiviral activity of T-290676 against IIIB (6 ng/ml) is superior to that of T-2638 (61 ng/ml), making the former a better candidate for further design.
Design of HR2 Oligomeric Peptides.
Alanine substitution at S649 and E659, which are residues located at the a and d positions in T-290676 and make contact with the HR1 groove, were found via alanine scanning to increase the stability of the six-helix bundle, even though antiviral activity against IIIB was unchanged (J.J.D., K.L.W., D.K.D., M.L.G., and M.K.D., unpublished data). Increasing hydrophobic contact at protein–protein interfaces is often associated with enhanced binding affinity (31). T2635 had potent antiviral activity against all viruses tested and was 10- to 200-fold more potent than T2410 against the resistant isolates 098-T1249 and 098-T651. Remarkably, T-2635 is >3,600-fold more potent than ENF against 098-T1249 and nearly 2,300-fold more active against 098-T651.
To more fully characterize the activity of the peptides against primary cells, several peptides were selected for testing against a primary isolate in peripheral blood mononuclear cells (PBMCs). T-2635 was found to have an IC50 of 0.214 μg/ml as compared with 22.96 μg/ml for ENF. T-267221 and T-267227 had IC50 values of 0.387 and 0.572 μg/ml, respectively (data not shown).
T-2635 Self-Associates Into a Trimer.
The peptide T-2635 was found to be 75% helical by circular dichroism. The thermal unfolding transition (Tm) for T-2635 was determined to be 42°C at a peptide concentration of 10 μM (Fig. 2). The Tm was found to depend on peptide concentration (data not shown), which is indicative of a self-associating species. The cooperative transition observed in the thermal unfolding experiments suggested that T-2635 self-associates. Analytical ultracentrifugation was used to examine the association behavior at several concentrations. Fig. 3a shows an example of a radial scan for a solution of T-2635 at a concentration of 200 μM. Fitting this data to the single ideal species model and averaging data from several experiments, a molecular mass (MW) of 14,190 (±382) Da was obtained, which is consistent with a trimer (expected MW of 13950). As indicated in Fig. 3b, a trimer is observed over the concentration range 50–1,000 μM. Below 50 μM, the weight-averaged MW decreases, suggesting that T-2635 exists in both monomeric and trimeric states in this concentration range. In accordance with the CD data, T-651 and T-2410 were found to be monomeric, as was T-290676 in this concentration range (data not shown).
Fig. 2.
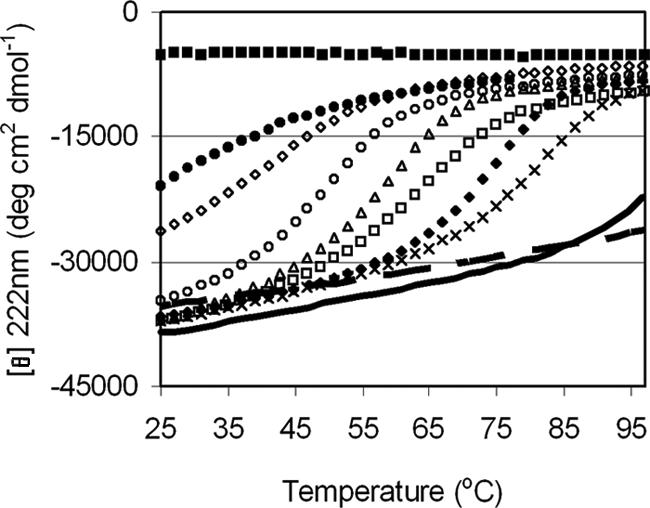
Thermal stability of HR2 oligomers. Thermal unfolding transitions of engineered peptides as determined by CD at a peptide concentration of 10 μM in PBS (pH 7.0). (From top to bottom) T-2410 (filled squares), T-290676 (filled circles), T-2635 (open diamonds), T-267209 (open circles), T-267226 (open triangles), T-267221 (open squares), T-267227 (filled circles), T-290822 [multiplication symbols (×)], T-267228 (thick line), T-267229 (dashed line).
Fig. 3.

Sedimentation equilibrium of T-2635. (Left) Radial scan of 200 μM T-2635 in PBS pH 7 spun at 22,000 rpm at 4°C. The data are fit to the single ideal species model and yielded a weight-averaged molecular weight (MW) of 14088. (Right) The observed MW as a function of T-2635 concentration. Each point is an average of at least three determinations and the error in the MW is <10%.
Additional substitutions were made at other a and d positions of T-2635, which resulted in a family of HR2 oligomers with Tm values ranging from <25°C to >100°C (Fig. 2, Table 1). Several of these HR2 oligomers were analyzed by sedimentation equilibrium, and the MWs of these peptides were also generally consistent with a trimeric species (data not shown). Peptides with thermal stabilities >60°C were generally found to be fully oligomeric to the low micromolar range (the lower limit for analytical centrifugation of these peptides), consistent with a shift in equilibrium toward the oligomer in these stable designs (data not shown). The antiviral activity was found to be similar to T-2635 until the stability exceeded 70°C (Fig. 4). Peptides with Tm values >100°C were found to have significantly reduced antiviral activity.
Fig. 4.

Impact of HR2 thermal stability on antiviral activity against the virus IIIB. The midpoint of the thermal unfolding transition (Tm) of the engineered peptides was determined from the data in Fig. 2 and also includes data for T-651. The Tm values for 267228 and 267229 are graphed with a Tm value of 100°C, although Fig. 2 indicates that this is an underestimate. Antiviral activity is from Table 1.
Relationship Between Activity and Six-Helix Bundle Stability.
Antiviral activity of peptides is presumed to be related to the stability of the HR1/HR2 bundle (8, 16) although more recent results have demonstrated that this relationship may be complex (32, 33). Using circular dichroism, the stability of the engineered HR2 peptides was determined in complex with the 51-aa HR1 target, T-865 (13). The midpoint of the thermal unfolding transition (Tm) was found to greatly exceed 100°C in PBS (data not shown), so CD thermal melting experiments were performed in the presence of 8 M urea. Under these conditions, the HR1 oligomer (in the absence of HR2) is completely unfolded (Fig. 5), so that the thermal transition more accurately reflects the stability of the HR1/HR2 complex (13). Even in 8 M urea, the engineered peptide T-2635 forms a remarkably stable bundle (Tm of 86°C, Fig. 5 and Table 1). Fig. 5 also shows the thermal unfolding of T865/T2410 and T865/T290676. For T865/T2635, the Tm is 12°C higher than T865/T2410 and 18°C higher than T865/T651 (Table 1). For comparison, ENF does not form a stable complex with any HR1 target under these conditions (Table 1), demonstrating the greatly enhanced stability of the helix stabilized series. Interestingly, the Tm of the T865/HR2 bundle was similar for peptides engineered to have higher HR2 stability (Table 1), including T-290821 and T-267228, which were found to have reduced antiviral activity.
Fig. 5.

Stability of HR1/HR2 bundles. Thermal unfolding transitions by circular dichroism for HR2 peptides in complex with the HR1 peptide, T-865. Transitions were measured at a peptide concentration of 1 mM each in PBS (pH 7) in the presence of 8M urea. Data are shown for T-865 alone (filled squares), T-865 + T-2410 (open squares), T-865 + T290676 (open circles), and T-865 + T-2635 (filled circles), and the Tm values for these and other HR1/HR2 bundles are listed in Table 1.
We have attempted to determine the kinetic on- and off-rates of binding for these analogs by using surface plasmon resonance. Due to the extremely high affinity of the peptides, rate constants are difficult to measure accurately because of mass transport limitations and rebinding artifacts; however, the increased binding affinity seems to come primarily from a slower off-rate (not shown).
Improved Genetic Barrier to Resistance.
Given the impressive antiviral properties of T-2635 and its analogs, a series of passaging experiments were initiated to generate virus that was resistant to these peptides. As shown in Table 2, the durability of T-2544 (a precursor to T-2635 where the N-terminal residue is Met) is captured by several quantitative measures. At these conditions, culturing in the presence of increasing concentrations of peptides over a period of several weeks is sufficient to generate virus resistant to ENF. In contrast, T-651 and T-2544 require longer culturing to generate resistant virus. In addition, the maximum T-2544 peptide concentration for which virus will replicate is >50-fold lower than ENF and over 20-fold lower than T-651. Despite repeated efforts, virus could not be cultured in the presence of greater than 2 μg/ml T-2544.
Table 2.
In vitro passaging results for ENF, T-651, and T-2544.
| Peptide | N* | Max. ending concentration, μg/ml† | Avg. no. of passages‡ | Avg. days in culture | Avg. no. of mutations | Fold-decrease in susceptibility (Geo-mean) |
|---|---|---|---|---|---|---|
| ENF | 12 | 50 | 6 (17) | 38 | 1.6 | 81 |
| T-651 | 7 | 20 | 13 (74) | 106 | 4.9 | 164 |
| T-2544 | 3 | 3.0 | 16 (129) | 227 | 3.0 | 8.3 |
*N is the number of independent in vitro selections.
†Highest concentration tested under which virus would replicate.
‡A passage is defined as when virus is harvested and put on fresh cells with a different peptide concentration. The number in parentheses is the average number of times the cell culture was split to maintain cell concentration within the linear growth phase.
ENF-resistant viruses generated by in vitro selection contained an average of 1.6 mutations and showed an 82-fold increase in the geometric mean IC50. In contrast, virus selected by using T-651 and T-2544 and carried out substantially longer are found to have an average of 4.9 and 3.7 mutations, respectively. Despite extensive passaging in vitro and the appearance of an average of 3.7 mutations, the geometric mean IC50 of T-2544 is increased by only 3.3-fold.
Effects of HR2 Design on Pharmacokinetics in Cynomolgus Monkeys.
Although T-651 itself has excellent activity against ENF- and T-1249-resistant virus, the observed half-life of this peptide in cynomolgus monkeys is ≈20 min (Table 1). To determine whether the helix-stabilization strategy resulted in peptides with altered in vivo clearance, we measured the pharmacokinetics after IV administration to cynomolgus monkeys. Unexpectedly, the engineered peptides were generally found to have significantly extended half-lives and reduced clearance values relative to T-2410. For example, T-2635 has an ≈10-fold lower clearance than T-651 and the family of HR2 oligomers maintain or exceed the improvement seen in T-2635. The most improvement in pharmacokinetic paramenters was seen in T-267229, with a clearance value of 0.003 liter/h/kg and a half-life of 35 h, which represents a >100-fold improvement in clearance and a 100-fold increase in half-life over T-651. Another characteristic of the improved pharmacokinetics is the observation that enhanced oligomer stability is associated with longer half-lives (Fig. 6) and lower clearance, suggesting that the substitutions used to stabilize the oligomeric structure were also affecting pharmacokinetics.
Fig. 6.

Correlation between HR2 thermal stability and half-life in cynomolgus monkeys. The Tm values were obtained from Fig. 2. The Tm values for 267228 and 267229 are indicated a value of 100°C, although Fig. 2 indicates that this is an underestimate. The R2 value of the fit was 0.71 (267228 and 267229 are excluded from the fit).
Discussion
ENF, the first fusion inhibitor approved for the treatment of HIV in treatment-experienced patients, has shown excellent efficacy in clinical use. When resistance to ENF does occur, it typically renders the virus less fit (34). Therefore, peptides with a higher genetic barrier to resistance may result in virus with limited ability for replication and pathogenesis, and may provide sustained viral suppression in vivo. We have designed a series of HR2 peptides with helix stabilization motifs that bind to HR1 targets with markedly higher affinity in vitro and that show potent activity against virus resistant to HR2 peptides such as ENF and T-1249. In vitro passaging experiments suggest that it takes a longer period to generate virus with significantly reduced sensitivity to the engineered peptides. In addition, based on data with a T-2635 precursor, more than three mutations are required to alter the activity of these peptides, with a resulting 3.3-fold increase in the geometric mean IC50. Compared with the 150-fold loss of sensitivity for T-651, this finding suggests that the potency of the engineered peptides may be significantly less impacted by viral mutations.
The helix stabilization motifs have significantly increased the thermal stability of the six-helix bundle relative to ENF and T-651, demonstrating that these peptides bind with higher affinity to the HR1 target. A similar design strategy was very recently described in a series of HR2 peptides from SARS coronavirus (25). Some of these analogs were found to stabilize bundle formation; however, the impact of the changes on the inhibitory action against SARS virus was not reported. Although the antiviral activity against IIIB is not improved in our design, the peptides show up to a 250-fold improvement in activity as compared with T-2410 and as much as a 3,600-fold increase as compared with ENF. We speculate that the enhanced activity against resistant isolates and the higher genetic barrier are due to the greatly enhanced affinity of these peptides. One mechanism by which resistance mutations reduce sensitivity to ENF is by decreasing the affinity of HR2 peptide fusion inhibitors to the viral HR1 target (35). Engineering the HR2 peptide to recover this lost affinity should act to restore the antiviral activity. Similarly, generating a virus with resistance to a high affinity peptide may be difficult, because more than one or two mutations may be required to reduce the affinity of the peptide and/or alter fusion kinetics to escape the action of the peptide. We propose that the enhanced genetic barrier is related to the inability of the virus to find solutions that permit multiple mutations while maintaining a high degree of replicative fitness.
Although the engineered HR2 peptides significantly enhance bundle stability, the antiviral activity of these peptides against both the lab-adapted strain IIIB and the primary isolate 098 is generally similar to ENF and T-2410. A correlation between bundle stability and IC50 has been demonstrated for HXB2 in both a viral entry and cell–cell fusion assay (8). At a bundle Tm of ≈65°C, the reported antiviral activity was ≈1 nM, suggesting that the peptides described here (with Tm values well over 100°C under similar conditions) might show greatly enhanced activity. However, our experiments highlight potency improvements against resistant strains but not in fully sensitive strains, suggesting that these peptides may have reached an affinity threshold for maximum activity in sensitive strains. During our analysis of the alanine scan data for T-651 and related peptides, we also noted that antiviral activity against IIIB appears to plateau over a broad range of bundle stability. This phenomena has also been observed in other studies, where a nonlinear relationship between binding affinity and activity has been described (33) or a complete lack of correlation (32). The reason for the complex relationship between binding and activity is not clear, although it may be related to the relative kinetics of binding of the peptide to the viral target versus the kinetics of fusion. For example, if a high-affinity peptide has a sufficiently slow off-rate from the viral HR1 so that it remained bound throughout the kinetic window of gp41 fusion, enhancing the affinity of the peptide may offer no further activity advantage. It is likely that the modest change in activity observed for the engineered peptides against the fully sensitive strains IIIB and 098 reflects this affinity “threshold.”
The design of oligomeric HR2 peptides is a unique approach to stabilizing helical structure in HIV fusion inhibitor peptides. Two substitutions in the a and d core of the T289676 (S649A and E659A) were sufficient to direct oligomer formation of T2635. Interestingly, one of the two substitutions used in T-2635, S649A, has been postulated to be a compensatory mutation in response to ENF resistance in patients (36). The S649A and E659A substitutions in the T-2410 background do not form oligomers (data not shown), but polar residues in core positions of the HR2 region of gp41 could be required by the virus to modulate bundle stability during the fusion process.
An interesting finding with the engineered peptides presented here was the relationship between oligomerization of the HR2 peptides, driven by the changes to the a, d core positions, and the antiviral activity. Enhanced bundle stability, ranging from a Tm of 78–92°C, is observed for the helix-stabilized class. However, the stability of the HR2 oligomer ranges from <5°C to >100°C. Although substitutions within the a, d core are likely responsible for the enhanced bundle stability and HR2 oligomer formation, it is apparent that macromolecular recognition of the HR1 trimer requires a distinct set of interactions as compared with HR2 self-association. For extremely stable oligomers, the amount of monomeric peptide present in the antiviral assay is likely to be very low. As the Tm increases, the loss in antiviral activity is likely related to the diminishing concentration of monomeric peptide present at peptide concentrations near the IC50. The antiviral experiments are done in the presence of FCS and so it is also possible that changes in the extent of binding to serum proteins may contribute to the loss of activity. A full thermodynamic analysis of these peptides is underway to directly test these hypotheses.
A surprising and highly desirable finding was the enhanced pharmacokinetics observed in the designed HR2 peptides. The increase in half-life and decreased clearance in cynomolgus monkeys was striking, although the mechanism for the enhanced PK is not clear from these data. We have not measured the hydrophobicity of these peptides directly, but it is likely that the changes that were made to the a, d core, which resulted in enhanced HR2 oligomer stability, also increased the hydrophobicity of these peptides. The improved PK could be due to decreased renal clearance or decreased metabolism by proteases as a result of altered interactions between the peptide and serum proteins or by the enhanced structural stability. We also cannot exclude the possibility that serum proteins alter the equilibrium between monomer and oligomer. Understanding the complex equilibria that likely exists in the presence of serum will be necessary before a complete understanding of the pharmacokinetic properties of these self-associating peptides can be fully described.
The designed FI peptides presented here have potent antiviral activity against the lab-adapted IIIB strain as well as the primary isolate 098. Viruses that are resistant to other FI peptides, such as ENF, T-1249, and T-651 are fully sensitive to several peptides in the helix-stabilized series. Moreover, the durability of these peptides, as measured by passaging experiments, suggest that viruses may have more difficultly developing resistance to this class. Although alternative resistance mechanisms could eventually be observed, our results suggest that these peptides have a high genetic barrier to resistance. The superior antiviral properties coupled with the enhanced pharmacokinetics of these peptides make them attractive for further development.
Materials and Methods
Peptide Synthesis.
Peptides were synthesized on a Rainin Symphony multiplex peptide synthesizer using 9-fluorenylmethoxycarbonyl chemistry protocols. Peptides were prepared with blocked ends by using rink amide myxobacterial hemagglutinin resin for C-terminal amides, and acetylation on the N terminus using acetic anhydride and N-methyl maleimide in dimethylformamide. They were purified to >90% purity and peptide identity was confirmed by mass using mass spectrometry.
Circular Dichroism.
Lyophilized peptides were resuspended in PBS (pH 7) at ≈1 mg/ml. Concentrations were determined by using the method of Edelhoch (37). Thermal unfolding of HR2 oligomers was determined at 10 μM in PBS at 222 nm with 2°C increments from 25°C to 97°C with a 16-s averaging time on an AVIV 202-01 circular dichroism spectrophotometer. Tm values for HR1/HR2 complexes were obtained by combining each HR2 peptide with the HR1 peptide T-865 at equal 1 μM concentrations into a solution of PBS plus 8 M urea. The Tm was determined to be the value that corresponded to the maximum value of the first derivative of the thermal transition (13).
Analytical Ultracentrifugation.
Sedimentation equilibrium experiments were performed on a Beckman Optima XL-A analytical ultracentrifuge at 4°C as described (13). For reported MWs, there was no significant decrease in absorbance due to the sedimentation of very high MW aggregates. T-2635 was diluted by using 50 mM KH2PO4/100 mM KCl (pH 7.0) to final concentrations ranging from 1 to 1,000 μM. Data were collected at 18,000 and 22,000 rpm at wavelengths of 225–308 nm (depending on peptide concentration) and at 4°C. Weight-averaged molecular weights were obtained by fitting each data file individually using a single ideal species model in the Beckman Origin software (version 3.78 for Windows). The solvent density of the phosphate buffer (1.00894) and the partial specific volume of T-2635 (0.7291) was calculated by using the program SEDNTERP (38).
Antiviral Activity.
The antiviral activity for each HR2 peptide was determined by using a MAGI/cMAGI infectivity assay (39, 40). Peptides were resuspended at ≈1 mg/ml in PBS and serially diluted to final concentrations. The assay is based on a single cycle of infection. To ensure that secondary rounds of infection did not complicate the analysis, an active HR2 peptide was added at high concentration to all wells 24 h after infection. Seventy-two hours after infection, cells were fixed with formaldehyde and glutaraldehyde and stained with X-Gal. Infected nuclei were counted by using a charge-coupled device detector. The IC50 was determined by using the concentration required to inhibit viral replication by 50%.
Primary clinical virus isolates were obtained through coculture of CD8+ T cell-depleted PBMC with patient PBMC. Virus isolate stocks were subsequently generated by a single virus passage in either PBMC or MT-2 cells, and stored with 30% FBS at −80°C until use. CD8-depleted activated PBMCs (target cells) are infected at high MOI by spinoculation (41). Serially diluted test compounds and uninfected target cells are incubated with infected cells at 2 × 105 cells per well for 9 days. Samples are taken and fresh compound is added every 2–3 days. Viral replication in these samples is determined by reverse transcriptase (RT) assay (42), and IC50 estimates are calculated as decreases in RT signals in treated wells from RT production in infected but untreated control wells.
In Vitro Passaging.
CEM4, MT2, or MT4 cells were infected with the clinical isolate 098 and cultured in the presence of increasing concentrations of HR2 peptide (beginning at twice the IC50) to select for resistance, as described (40). When in vitro selections reached 20 or 50 μg/ml, virus stocks were expanded in the presence of 20 or 50 μg/ml of peptide and peptide-free harvests were collected. Phenotypic susceptibility was determined by using a MAGI/cMAGI infectivity assay (40).
Pharmacokinetic Studies in Cynomolgus Monkeys.
Male cynomolgus monkeys (Macaca fascicularis), were administered a single i.v. dose of each test peptide in isotonic phosphate-mannitol vehicle. Peptides were delivered in cassettes (typically n ≤ 8) at a dose of ≈1–2 mg of peptide per kg of body weight and administered at 1 ml/kg body weight. Pharmacokinetic parameters were calculated from the plasma peptide concentration versus time data using either mono- or biexponential mathematical models. Samples of whole blood were collected and processed for plasma. Before analysis, plasma samples were treated with 3 volumes of 0.5% (vol/vol) formic acid in acetonitrile to precipitate proteins. High-performance liquid chromatography was performed on an Agilent 1100 system (Palo Alto, CA) by means of reversed-phase gradient elution from Phenomenex (Torrance, CA) Luna (C8 or C18 50 × 2 mm i.d., 3 μm) or Synergi RP Hydro (C18, 75 × 2 mm i.d., 4 μm) columns. The mobile phase used to generate gradients consisted of 10 mM ammonium acetate (pH 6.8) (A), and 100% acetonitrile (B). Peptide quantification was performed by mass spectrometry on an Applied BioSystems API4000 QTrap, operated in the selected ion monitoring (SIM) mode with electrospray ionization.
Acknowledgments
We thank Doug Ahrens and Xuefang Bai for thoughtful discussions; Carrington Jackson, Robin Parham, and Lloyd Frick for help concerning the pharmacokinetics; and Dani Bolognesi, Carol Ohmstede, and our colleagues at Roche for their support. Financial support of this work was provided by Trimeris and Roche.
Abbreviations
- HR1
heptad repeat 1
- ENF
enfuvirtide
- FI
fusion inhibitor
- PBMC
peripheral blood mononuclear cell.
Footnotes
Conflict of interest statement: J.J.D., K.L.W., D.K.D., S.A.F., J.E.S., S.A.W., N.A.T., T.J.M., M.L.G., and M.K.D. are employees of Trimeris.
This article is a PNAS Direct Submission.
References
- 1.Helseth E, Olshevsky U, Furman C, Sodroski J. J Virol. 1991;65:2119–2123. doi: 10.1128/jvi.65.4.2119-2123.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weissenhorn W, Dessen A, Calder LJ, Harrison SC, Skehel JJ, Wiley DC. Mol Membr Biol. 1999;16:3–9. doi: 10.1080/096876899294706. [DOI] [PubMed] [Google Scholar]
- 3.Chan DC, Kim PS. Cell. 1998;93:681–684. doi: 10.1016/s0092-8674(00)81430-0. [DOI] [PubMed] [Google Scholar]
- 4.Chan DC, Fass D, Berger JM, Kim PS. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 5.Lu M, Blacklow SC, Kim PS. Nat Struct Biol. 1995;2:1075–1082. doi: 10.1038/nsb1295-1075. [DOI] [PubMed] [Google Scholar]
- 6.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. Nature. 1997;387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 7.Wild CT, Shugars DC, Greenwell TK, McDanal CB, Matthews TJ. Proc Natl Acad Sci USA. 1994;91:9770–9774. doi: 10.1073/pnas.91.21.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan DC, Chutkowski CT, Kim PS. Proc Natl Acad Sci USA. 1998;95:15613–15617. doi: 10.1073/pnas.95.26.15613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eckert DM, Malashkevich VN, Hong LH, Carr PA, Kim PS. Cell. 1999;99:103–115. doi: 10.1016/s0092-8674(00)80066-5. [DOI] [PubMed] [Google Scholar]
- 10.Judice JK, Tom JY, Huang W, Wrin T, Vennari J, Petropoulos CJ, McDowell RS. Proc Natl Acad Sci USA. 1997;94:13426–13430. doi: 10.1073/pnas.94.25.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matthews T, Salgo M, Greenberg M, Chung J, DeMasi R, Bolognesi D. Nat Rev Drug Discov. 2004;3:215–225. doi: 10.1038/nrd1331. [DOI] [PubMed] [Google Scholar]
- 12.Lawless MK, Barney S, Guthrie KI, Bucy TB, Petteway SR, Jr, Merutka G. Biochemistry. 1996;35:13697–13708. doi: 10.1021/bi9606962. [DOI] [PubMed] [Google Scholar]
- 13.Dwyer JJ, Hasan A, Wilson KL, White JM, Matthews TJ, Delmedico MK. Biochemistry. 2003;42:4945–4953. doi: 10.1021/bi027283n. [DOI] [PubMed] [Google Scholar]
- 14.Jin BS, Ryu JR, Ahn K, Yu YG. AIDS Res Hum Retroviruses. 2000;16:1797–1804. doi: 10.1089/08892220050195757. [DOI] [PubMed] [Google Scholar]
- 15.Otaka A, Nakamura M, Nameki D, Kodama E, Uchiyama S, Nakamura S, Nakano H, Tamamura H, Kobayashi Y, Matsuoka M, et al. Angew Chem Int Ed Engl. 2002;41:2937–2940. doi: 10.1002/1521-3773(20020816)41:16<2937::AID-ANIE2937>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 16.Sia SK, Carr PA, Cochran AG, Malashkevich VN, Kim PS. Proc Natl Acad Sci USA. 2002;99:14664–14669. doi: 10.1073/pnas.232566599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chou PY, Fasman GD. Annu Rev Biochem. 1978;47:251–276. doi: 10.1146/annurev.bi.47.070178.001343. [DOI] [PubMed] [Google Scholar]
- 18.O'Neil KT, DeGrado WF. Science. 1990;250:646–651. doi: 10.1126/science.2237415. [DOI] [PubMed] [Google Scholar]
- 19.Lyu PC, Liff MI, Marky LA, Kallenbach NR. Science. 1990;250:669–673. doi: 10.1126/science.2237416. [DOI] [PubMed] [Google Scholar]
- 20.Padmanabhan S, Marqusee S, Ridgeway T, Laue TM, Baldwin RL. Nature. 1990;344:268–270. doi: 10.1038/344268a0. [DOI] [PubMed] [Google Scholar]
- 21.Marqusee S, Baldwin RL. Proc Natl Acad Sci USA. 1987;84:8898–8902. doi: 10.1073/pnas.84.24.8898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mayne L, Englander SW, Qiu R, Yang J, Gong Y, Spek E, Kallenbach NR. J Am Chem Soc. 1998;120:10643–10645. [Google Scholar]
- 23.Kammerer RA, Jaravine VA, Frank S, Schulthess T, Landwehr R, Lustig A, Garcia-Echeverria C, Alexandrescu AT, Engel J, Steinmetz MO. J Biol Chem. 2001;276:13685–13688. doi: 10.1074/jbc.M010492200. [DOI] [PubMed] [Google Scholar]
- 24.Spek EJ, Bui AH, Lu M, Kallenbach NR. Protein Sci. 1998;7:2431–2437. doi: 10.1002/pro.5560071121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yan Z, Tripet B, Hodges RS. J Struct Biol. 2006;155:162–175. doi: 10.1016/j.jsb.2006.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rohl CA, Chakrabartty A, Baldwin RL. Protein Sci. 1996;5:2623–2637. doi: 10.1002/pro.5560051225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rohl CA, Fiori W, Baldwin RL. Proc Natl Acad Sci USA. 1999;96:3682–3687. doi: 10.1073/pnas.96.7.3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu M, Shu W, Ji H, Spek E, Wang L, Kallenbach NR. J Mol Biol. 1999;288:743–752. doi: 10.1006/jmbi.1999.2707. [DOI] [PubMed] [Google Scholar]
- 29.Olson CA, Spek EJ, Shi Z, Vologodskii A, Kallenbach NR. Proteins. 2001;44:123–132. doi: 10.1002/prot.1079. [DOI] [PubMed] [Google Scholar]
- 30.Yang J, Spek EJ, Gong Y, Zhou H, Kallenbach NR. Protein Sci. 1997;6:1264–1272. doi: 10.1002/pro.5560060614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bogan AA, Thorn KS. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 32.Gallo SA, Sackett K, Rawat SS, Shai Y, Blumenthal R. J Mol Biol. 2004;340:9–14. doi: 10.1016/j.jmb.2004.04.027. [DOI] [PubMed] [Google Scholar]
- 33.Steger HK, Root MJ. J Biol Chem. 2006;281:25813–25821. doi: 10.1074/jbc.M601457200. [DOI] [PubMed] [Google Scholar]
- 34.Lu J, Sista P, Giguel F, Greenberg M, Kuritzkes DR. J Virol. 2004;78:4628–4637. doi: 10.1128/JVI.78.9.4628-4637.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mink M, Mosier SM, Janumpalli S, Davison D, Jin L, Melby T, Sista P, Erickson J, Lambert D, Stanfield-Oakley SA, et al. J Virol. 2005;79:12447–12454. doi: 10.1128/JVI.79.19.12447-12454.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu L, Pozniak A, Wildfire A, Stanfield-Oakley SA, Mosier SM, Ratcliffe D, Workman J, Joall A, Myers R, Smit E, et al. Antimicrob Agents Chemother. 2005;49:1113–1119. doi: 10.1128/AAC.49.3.1113-1119.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edelhoch H. Biochemistry. 1967;6:1948–1954. doi: 10.1021/bi00859a010. [DOI] [PubMed] [Google Scholar]
- 38.Laue TM, Shah BD, Ridgeway TM, Pelletier SL. Analytical Ultracentrifugation in Biochemistry and Polymer Science. Cambridge, U.K.: Royal Soc London; 1992. [Google Scholar]
- 39.Kimpton J, Emerman M. J Virol. 1992;66:2232–2239. doi: 10.1128/jvi.66.4.2232-2239.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu QX, Barry AP, Wang ZX, Connolly SM, Peiper SC, Greenberg ML. J Virol. 2000;74:11858–11872. doi: 10.1128/jvi.74.24.11858-11872.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O'Doherty U, Swiggard WJ, Malim MH. J Virol. 2000;74:10074–10080. doi: 10.1128/jvi.74.21.10074-10080.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen CH, Weinhold KJ, Bartlett JA, Bolognesi DP, Greenberg ML. AIDS Res Hum Retroviruses. 1993;9:1079–1086. doi: 10.1089/aid.1993.9.1079. [DOI] [PubMed] [Google Scholar]