

Abstract
The airway epithelium is comprised of specialized cell types that play key roles in protecting the lungs from environmental insults. The cellular composition of the murine respiratory epithelium is established during development and different cell types populate specific regions along the airway. Here we show that E2f4-deficiency leads to an absence of ciliated cells from the entire airway epithelium and the epithelium of the submucosal glands in the paranasal sinuses. This defect is particularly striking in the nasal epithelium of E2f4−/− mice where ciliated cells are replaced by columnar secretory cells that produce mucin-like substances. In addition, in the proximal lung, E2f4-loss causes a reduction in Clara cell marker expression indicating that Clara cell development is also affected. These defects arise during embryogenesis and, in the nasal epithelium, appear to be independent of any changes in cell proliferation, the principal process regulated by members of the E2f family of transcription factors. We therefore conclude that E2f4 is required to determine the appropriate development of the airway epithelium. Importantly, the combination of no ciliated cells and excess mucous cells can account for the chronic rhinitis and increased susceptibility to opportunistic infections that causes the postnatal lethality of E2f4 mutant mice.
Keywords: E2f4, Epithelium, Nasal, Respiratory, Ciliated, Goblet, Clara cells, Mucin
Introduction
The E2f family of transcription factors has primarily been implicated in the regulation of genes required for proliferation and passage through the cell cycle (Attwooll et al., 2004; Dimova and Dyson, 2005). To date, nine members of the E2f family have been identified. These can be divided into a number of subgroups based on differences in both their regulation and transcriptional activity. Notably, individual E2f subgroups appear to be predominantly involved in either the repression or activation of E2f-responsive genes. E2f4 and 5 appear to function primarily as transcriptional repressors and their nuclear localization seems to be dependent upon their association with members of the pocket protein family that includes the retinoblastoma protein (pRb) tumor suppressor, p107 and p130 (Apostolova et al., 2002; Gaubatz et al., 2001; Rayman et al., 2002; Verona et al., 1997). E2f5 specifically associates with p130 (Hijmans et al., 1995; Sardet et al., 1995), while E2f4 binds to all three of the pocket proteins in vivo (Moberg et al., 1996). The resultant E2f/pocket protein complexes can recruit histone deacetylases (HDACS) to the promoters of E2f-responsive genes to enforce their active repression (Giangrande et al., 2004; Rayman et al., 2002; Takahashi et al., 2000; Wells et al., 2000). In response to mitogenic signaling, the pocket proteins are phosphorylated by the cyclin dependent kinases (Dynlacht et al., 1994; Knudsen and Wang, 1997; Rubin et al., 2005; Xiao et al., 1996). This causes the repressive E2f/pocket protein complexes to dissociate and E2f4 and 5 are exported to the cytoplasm.
E2f4 is the most abundant E2f family member in vivo, so its loss was expected to cause de-repression of E2f-responsive genes. However, E2f4-deficient MEFs have no detectable defects in either target gene or cell cycle regulation (Humbert et al., 2000; Landsberg et al., 2003; Rempel et al., 2000). Since the combined loss of E2f4 and 5 can impair these processes, this likely reflects compensation by E2f5 (Gaubatz et al., 2000). Notably, E2f4 mutant mice do exhibit several defects including embryonic anemia, defects in red blood cell maturation, altered craniofacial morphology, reduced fertility, chronic rhinitis and poor postnatal survival (Humbert et al., 2000; Kinross et al., 2006; Rempel et al., 2000). The neonatal lethality of the E2f4 mutants is caused by the chronic rhinitis and associated opportunistic bacterial infections (Humbert et al., 2000). Adoptive transfer experiments indicated that this was not due to a defect in the immune system of E2f4 mutant mice (Humbert et al., 2000). The chronic rhinitis was thought to result from reduced or blocked drainage of mucus from the nasal cavity but the underlying cause of this was unclear.
To understand the cause of the chronic rhinitis in the E2f4 mutant mice, we have investigated the development of the nasal passages and, in particular, the epithelium that normally plays key roles in preventing bacterial infections of the upper respiratory tract. Most studies of respiratory epithelia growth and development have looked at the trachea and more distal areas of the lung (Perl and Whitsett, 1999; Rawlins and Hogan, 2006). Fewer studies have examined development of the nasal respiratory epithelium and these studies largely address the response of the epithelium to environmental insults (Harkema et al., 2006). The nasal respiratory epithelium is predominantly comprised of ciliated columnar cells, goblet cells and basal cells. Coordinated beating of cilia moves mucus towards the esophagus removing trapped or absorbed particulate matter, allergens, toxic chemicals and pathogens from the air prior to its passage into the lungs. Goblet cells produce mucins, a component of mucus, both constitutively and in an inducible manner following various environmental stimuli (Perez-Vilar et al., 2003; Rogers, 1994). This induction can occur at both the level of secretion and the cellular level, i.e. an increase in the population of mucin secreting cells. It is unclear what the precursor cells for ciliated or goblet cells are: basal cells were proposed to fulfill this role, however, recent studies challenge this model (Evans et al., 2001; Rawlins and Hogan, 2006). Here we show that mutation of E2f4 results in airway epithelia that lack cilia and this can occur independently from any detectable defect in cell proliferation indicating that E2f4 plays a key role in determining the cellular composition of the airway epithelium.
Materials and Methods
Animal maintenance and histological analyses
The generation of the E2f4 mutant mouse strain and genotyping protocols have been described previously (Humbert et al., 2000). In brief, the first five exons (apart from the first four codons) encoding the DNA binding and dimerization domains were deleted and replaced with stop codons in all three reading frames and a neo resistance cassette by homologous recombination in embryonic stem cells. This resulted in a recessive null mutation in E2f4. No phenotypes have been detected in mice heterozygous for this mutant allele in comparison with wild-type mice. For this study mice were maintained on a mixed C57BL/6 × 129Sv background in a conventional facility. All animal procedures followed protocols approved by the Institute’s Committee on Animal Care. Gestation was dated by detection of a vaginal plug. For analyses of cell proliferation pregnant mice were injected with 10μl/gm body weight of 6mg/ml 5-Bromo-2′-deoxyuridine (BrdU) in phosphate buffered saline (PBS) two hours prior to tissue collection. Collected embryo tissues were fixed overnight at room temperature in either Bouin’s Fixative, 3.7% formaldehyde in PBS or at 4°C overnight in PBS containing 4% paraformaldehyde as appropriate and then dehydrated via an ethanol series prior to embedding in paraffin for sectioning. For each embryo at least three different levels along the proximal-distal or dorsal-ventral axis were examined. From each level one section was stained with haematoxylin and eosin (HNE) using standard procedures and adjacent sections (all 5μm) stained as described below.
Histochemistry and immunohistochemistry
For all procedures slides were re-hydrated through an ethanol series following de-waxing in xylenes and rinsed in water or PBS as required. For the periodic acid Schiff (PAS) reaction slides were incubated in 0.5% periodic acid for 5 minutes, rinsed in water, incubated in Schiff reagent (Poly Scientific) for 15 minutes, washed in tap water for 10 minutes, counterstained with Harris hematoxylin and mounted using standard protocols. For the Alcian blue pH2.5 staining, slides were incubated in 3% acetic acid for 3 minutes then in Alcian blue solution (Alcian Blue 8GK 1% w/v, 3% acetic acid v/v, pH2.5) for 45 minutes at room temperature, washed in tap water for 5 minutes, counterstained with Harris hematoxylin and mounted using standard protocols.
Immunohistochemistry was performed using the following mouse monoclonal antibodies: acetylated α-tubulin (1:8000 T6793, Sigma), BrdU and Ki67 (1:50 347580 and 550609, BD biosciences), PCNA (1:2000 sc56, Santa Cruz), cytokeratin 8 (1:100 PRO61038, RDI), Foxa1 and Foxj1 (1:1000 WMAB2F83 and 319, Seven Hills Bioreagents), p63 (1:500 sc8431, Santa Cruz), E2f4 (1:1 LLF4.2 Moberg et al., 1996), MUC5AC (1:100, MS-145, LabVision). CC10 (1:100 goat polyclonal sc9772, Santa Cruz), SP-C (1:100 rabbit polyclonal sc13979 and goat polyclonal sc7706, Santa Cruz) and T1α (1:500 hamster monoclonal #8.1.1, Developmental Studies Hybridoma Bank, University of Iowa). Slides were washed in PBS 0.15% Triton X-100 followed by inactivation of endogenous peroxidases by incubation with 3% H202 in PBS or 0.5% H202 in methanol (Ki67). Antigen retrieval was performed either by heating for 20 minutes in 10mM sodium citrate, 0.05% Tween 20, pH6.0 in a boiling water bath or by heating in a 800W microwave for 6.5 minutes at full power followed by three rounds of 5 minutes at 60% power using a solution 8.2mM sodium citrate, 1.8mM citric acid, pH6.0 (Ki67, p63, E2f4 and Foxj1). Slides were blocked with PBS containing 5% of the appropriate serum and incubated overnight with the primary antibody diluted in PBS 0.15% Triton X-100 or this buffer alone or a non-specific antiserum as controls. Secondary antibodies (Vectastain ABC kits, Vector laboratories) were diluted 1:200 in PBS containing 0.4% of the appropriate blocking serum and detected using a DAB substrate following the manufacturers instructions (Vector Laboratories). A MOM kit (Vector Laboratories) was used according to the manufacturers instructions for PCNA and MUC5AC staining and an UltraVision LP Detection System (Lab Vision Corporation) for Foxj1 and E2f4 staining. BrdU immunohistochemistry was performed as described previously (Tsai et al., 2002). Following the detection reaction slides were counterstained with Harris hematoxylin and mounted using standard protocols. In some cases slides were stained with Alcian Blue pH2.5 prior to counter staining. For each marker analyzed, unless stated otherwise, a minimum of four pairs of control (wild-type or E2f4+/−) and E2f4−/− littermate embryo sections matched for their position along the proximal-distal axis were stained and, unless stated otherwise, all scored with the described phenotype.
To assess the levels of proliferation, control and E2f4−/− sections from littermates matched for their level along the proximal-distal axis were selected and the percentage of positive nuclei was determined by counting 100–600 (depending on the stage of development) nuclei within the respiratory epithelium per section. For each of the indicated gestational stages four to eight pairs of embryos were analyzed for BrdU, Ki67 and PCNA. Images were captured on a Nikon Eclipse E600 using a SPOT RTdigital camera.
Transmission electron microscopy
Tissue samples were fixed directly in a solution 2.5% gluteraldehyde, 2.5% formaldehyde, 0.1M cacodylic acid pH7.2 for 60–90 minutes at 4° C. Samples were then postfixed with 2% osmium tetroxide for 60 min, dehydrated via an alcohol series up to 70% and en bloc stained with 0.2% uranyl acetate in 70% alcohol for 60 min, at 4°C. The samples were then processed for epon embedding. Thin epon sections (400 nm) were cut with a diamond knife and post-stained with uranyl acetate and Reynold’s lead citrate and viewed with a FEI –Tecnai™ G2Spirit BioTWIN operated at 80 kV. Digital images were taken with an AMT 2k CCD camera.
Results
The airway epithelium of E2f4 mutant mice lacks cilia
Mutation of E2f4 results in greater than 85% percent of the E2f4−/− pups dying prior to three weeks of age as a consequence of chronic rhinitis and associated opportunistic bacterial infections. Adoptive transfer experiments showed that this phenotype was independent of the immune system and that it correlated with excess accumulation of mucus in the nasal cavities allowing for bacterial colonization (Humbert et al., 2000). However, the underlying basis for this defect was unclear. To understand this defect we conducted a retrospective analysis of the nasal structures of 12 E2f4−/− mice in comparison with 12 age matched littermate controls (either wild-type or E2f4+/− mice, since these are phenotypically indistinguishable with respect to all criteria) at P0, P3, P10, P14, P20, 2.5 months, 3 months, 7 months, 10 months and 11 months. All 12 wild-type or E2f4+/− littermates had a typical, highly ciliated nasal epithelium as judged by both morphology and by using immunohistochemistry to detect acetylated α-tubulin, a component of motile cilia (for example see Figure 1A). In contrast we observed a dramatic defect in the vast majority of E2f4−/− animals: in 7/12 (at P0, P3, P10 and P20) the epithelium completely lacked ciliated cells (data not shown) and 4/12 (P14, 2.5 months, 3 months and 11 months) had a dramatic reduction in number of ciliated cells (Figure 1B). Only one E2f4−/− animal (10 months) had a relatively normally ciliated epithelium. We believe that the partial penetrance of this phenotype results from variations in the mixed genetic background of this older E2f4 colony since, as we describe below, our current colony shows complete penetrance of this cilia defect.
Fig. 1.

Mutation of E2f4 leads to a dramatic reduction in the number of ciliated cells only in the airway epithelium. The presence of motile cilia on epithelia was assessed by immunohistochemistry using an antibody against acetylated α-tubulin that is a component of motile cilia (brown stain). Nasal epithelium from a control 3 months old E2f4+/− mouse (A) is highly ciliated in contrast to that of an E2f4−/− age matched littermate (B). Cilia were not detected in the bronchioles of 2.5 months old E2f4−/− mice (D) in contrast to an age matched wild-type littermate (C). The ciliated epithelium in the fallopian tube (E,F), at 7 months, the efferent ducts of the epididymis (G,H) and ependymal lining of the brain ventricles (I,J) at 2.5 months, of E2f4−/− mice appeared similar to that of the age matched littermate controls. Sperm flagella (indicated by the *) appear to be normal in E2f4−/− mice at 2.5 months (L) in comparison with control mice (K). Original magnification ×40. Arrowheads point to ciliated cells in B and C.
To determine if E2f4 was involved in the development of other ciliated epithelia we examined additional tissues in E2f4 mutants and their matched controls. Analyses of the bronchioles and trachea of E2f4−/−mice also showed a dramatic reduction in the number of ciliated cells relative to the wild-type or E2f4+/− littermates (n=7; Figure 1C. D). In contrast we did not observe any loss of cilia in the fallopian tube (n=3; Figure 1E,F), the efferent ducts of the epididymis (n=3, Figure 1G, H) or brain ependymal epithelium (n=5, Figure 1I,J). In addition sperm flagella, which are similar in composition to cilia, appeared completely normal in E2f4−/− male mice (n=3; Figure 1K.L). Thus the occurrence of epithelium lacking cilia in the E2f4−/− animals appeared to be restricted to the airway epithelium.
It is well established that ciliated epithelia can be damaged as a consequence of respiratory bacterial infections (Cole, 1997). However, since the lack of ciliated cells was observed in pups at P0, it seemed unlikely that infections could fully account for this defect. Instead, we wondered whether the loss of E2f4 might disrupt the development of the respiratory epithelium in utero. To address this question, we examined wildtype and E2f4 mutant embryos at 18.5 days post coitum (dpc). In stark contrast to their wild-type and E2f4+/− sibs, every E2f4−/− embryo (n>20) lacked ciliated cells throughout the airway epithelium including the entire nasal cavity, the nasopharynx and the serous/submucosal glands in the paranasal sinuses (Figure 2A–H). The lack of cilia was verified by using immunohistochemistry to detect acetylated α-tubulin (Figure 2I–P). The general morphology of the E2f4−/− respiratory epithelium also seemed quite different from that of the littermate controls. In contrast, no gross changes in the morphology of the adjacent olfactory epithelium were observed (data not shown). The epithelia of the trachea (n=5; Figure 2Q, R) and the bronchioles (n=7; Figure 2S,T) of 18.5 dpc E2f4−/− embryos were also completely devoid of ciliated cells in contrast to control embryos. To determine whether cilia were assembled on the cell surface in E2f4−/− embryos but then lost at later gestational stages, we examined acetylated α-tubulin staining at 16.5 dpc, the time at which we first detected cilia in wild-type or E2f4+/− embryo nasal epithelium. Staining for acetylated α-tubulin was not detected in the nasal epithelium of E2f4−/− embryos at 16.5 dpc indicating that cilia assembly on the cell surface was not initiated in E2f4−/− embryos (Figure 2U,V). These results show that mutation of E2f4 disrupts the development of the ciliated cells throughout the entire airway epithelium during embryogenesis.
Fig. 2.
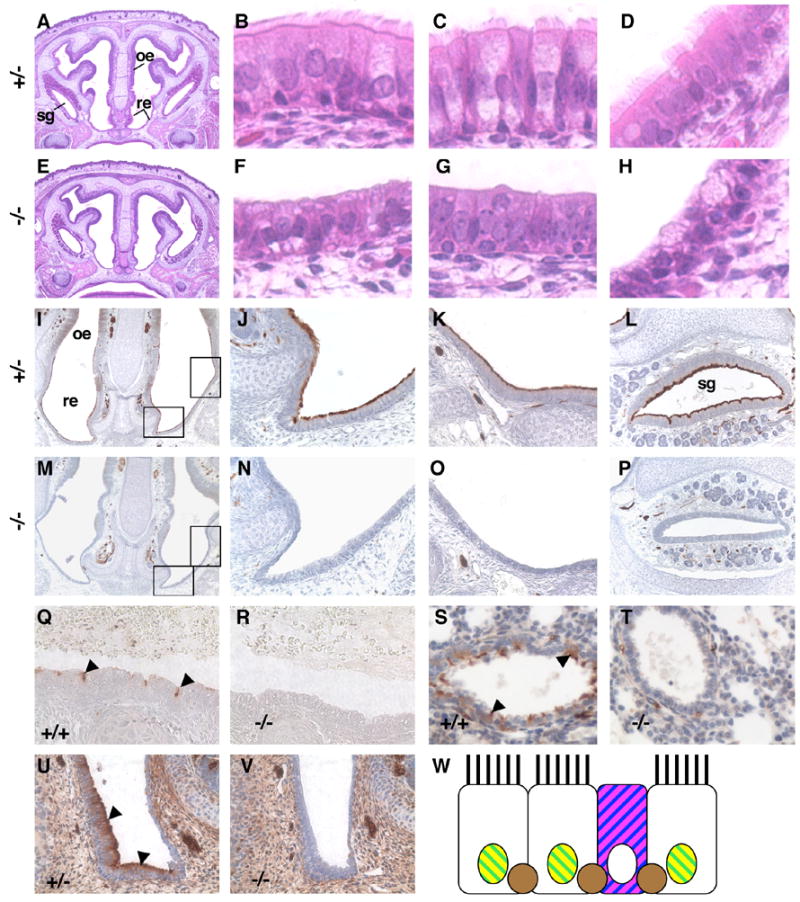
E2f4 mutant embryos lack ciliated cells in the airway epithelium. (A–H) Hematoxylin and eosin stained sections from E2f4+/− (A–D) and E2f4 mutant (E–H) 18.5 dpc littermate embryos. Panels A and E, coronal sections (original magnification ×4) showing the respiratory epithelium (re), olfactory epithelium (oe) and paranasal sinus serous gland (sg). High power images (original magnification ×40) of the respiratory epithelium (B, F), serous gland epithelium (C, G) and nasopharynx epithelium (D, H). (I–V) Acetylated α-tubulin, a component of cilia, was visualized by immunohistochemistry (brown stain). Panels I and M, coronal sections (original magnification ×10) with rectangles showing positions of the two adjacent panels. No staining was observed in the epithelium of E2f4 mutant embryos (M–O) compared with control embryos (I–K) (original magnification ×40) at 18.5 dpc. This phenotype was also observed in the serous gland (L, P) (original magnification ×20), in the trachea (Q, R) and bronchioles (S, T) at 18.5 dpc and in the nasal respiratory epithelium at 16.5 dpc (U, V) (original magnification ×40). Control genotypes are indicated (Q, S and U), E2f4−/− (R, T and V). Note that acetylated α-tubulin is also detected in neurons. Arrowheads point to ciliated cells in Q, S and U. W, Schematic of wild-type nasal respiratory epithelium with cell specific markers. Ciliated cells express Foxj1 (yellow) and Foxa1 (green). Goblet cell mucins stain with PAS (magenta) and Alcian Blue (blue). Basal cells express p63 (brown).
The disruption of airway epithelium development is not associated with dramatic alterations in cell proliferation
Since members of the E2f family are known to influence both cell proliferation and apoptosis, we wished to determine whether changes in either process contributed to the defects in the respiratory epithelium. First, by analyzing HNE stained sections for pyknotic nuclei, an indicator of apoptosis, we determined that there was a similar, extremely low level of apoptosis in the nasal respiratory epithelium of wildtype, E2f4+/− and E2f4−/− sibs at 14.5 dpc, 15.5 dpc, 16.5 dpc and 18.5 dpc (data not shown). Thus, the loss of ciliated cells within the E2f4 mutants is not due to apoptosis. Second, we analyzed the incorporation of the nucleotide analog, BrdU, by immunohistochemical analysis of matched sections taken from a minimum of four pairs of control (wild-type or E2f4+/−) and E2f4−/− littermate embryos at 14.5 dpc, 15.5 dpc, 16.5 dpc and 18.5 dpc. Regardless of the developmental stage, quantification showed that there was no statistically significant difference in the percentage of BrdU-positive nuclei in the respiratory epithelium of control versus E2f4−/− littermates apart from at 18.5 dpc after the phenotype is well established (Figure 3A–D and M). This was true for cells located adjacent to the basement membrane (including basal cells) as well as in the upper epithelial layer. To further expand this analysis, we also screened 15.5 dpc, 16.5 dpc and 18.5 dpc littermates for the presence of Ki67 and PCNA. These proteins are both well-documented markers of cellular proliferation and also the products of E2f-responsive genes. At 15.5 dpc and 18.5 dpc, we detected comparable levels of Ki67 staining in control versus E2f4 mutant epithelium (Figure 3E–H and M). At 16.5 dpc, the E2f4−/− embryos had a slightly lower level of Ki67 staining than the controls but this was not statistically significant (Figure 3E–H and M). Again this was true for cells located adjacent to the basement membrane (including basal cells) as well as in the upper epithelial layer. In contrast, the intensity of PCNA staining was somewhat higher in the E2f4−/− embryos versus littermate controls at all timepoints (Figure 3I–L; data not shown). Notably, this increase was not restricted to the respiratory epithelium but occurred through the embryo (Figure 3I–L; data not shown). Taken together, these data show that E2f4 loss causes a subtle derepression of at least one E2f-responsive gene (PCNA) but others (e.g. Ki67) are regulated normally and there is no significant alteration in the proliferative properties of cells within the E2f4−/− airway epithelium. We therefore conclude that the loss of ciliated cells in the E2f4−/− mutants does not result from any detectable change in either apoptosis or proliferation.
Fig. 3.

Analyses of proliferation in the E2f4 mutant epithelium. Representative adjacent coronal sections from 16.5 dpc wild-type (A,B, E,F and I,J) or E2f4 mutant littermate embryos (C, D, G,H, and K,L) analyzed by immunohistochemistry (brown stain) to assess the incorporation of BrdU (A–D), expression of Ki67 (E–H) and PCNA (I–L). Dorsal is to the left, original magnification ×40. The line marks the junction between the olfactory epithelium (oe) and respiratory epithelium (re). The outlined rectangle marks the area displayed in the adjacent panel. Slightly higher intensity PCNA staining was detected throughout E2f4 −/− embryos. M, Quantification of BrdU and Ki67 staining is shown at the embryonic stages indicated. A minimum of four pairs of wild-type and E2f4+/− (Ct) or E2f4 −/− (−/−) littermate embryos were analyzed at each stage. The means with standard deviation error bars are shown. Performance of the paired Student’s t-test indicated that none of the pairs was significantly different (all p values >0.05) apart from the 18.5 dpc BrdU time point (p = 0.025).
Cells comprising the E2f4−/− nasal respiratory epithelium contain mucins
Prior analyses indicated that the nasal cavities of surviving E2f4−/− mice contained eosinophilic purulent exudate that often harbored bacterial infections and the associated invading neutrophils and macrophages (Humbert et al., 2000). This indicated that cells producing mucins are present within the epithelium of these animals. To assess this directly, sections matched for their position along the proximal-distal axis from control (wild-type or E2f4+/−) and E2f4−/− 18.5 dpc littermate embryos were subjected to the periodic acid Schiff (PAS) reaction that can detect neutral mucins (Figure 4A–F) or stained with Alcian Blue pH2.5 stain that can detect acidic mucins (Figure 4G–L). Remarkably, almost all of the cells within the E2f4−/− nasal epithelium and nasopharynx contain mucins. This is quite distinct from the normal epithelium where the staining is restricted to the goblet cells. Quantification showed that the percentage of PAS or Alcian Blue staining cells in the nasal respiratory epithelium was 4 to 5 fold higher in the E2f4−/− embryos than the wild-type or E2f4+/− control embryos (Table 1). To further characterize this defect, we also conducted immunohistochemistry with an antibody raised against mucin 5ac, MUC5AC (Figure 4M–R). Notably, in the case of MUC5AC expression some cells in the E2f4−/− epithelium stain intensely similarly to normal goblet cells whilst others exhibit a more diffuse staining. This suggests that they represent two distinct cell types, possibly bona fide goblet cells and a second cell type that also contains mucins. Specific mucin staining was not detected in the nasal respiratory epithelium of either wildtype or E2f4 mutants at 16.5 dpc, or earlier stages of development (data not shown).
Fig. 4. Cells comprising the E2f4 mutant nasal respiratory epithelium contain mucins.

Adjacent matched sections from wild-type or E2f4 +/− (A–C, G–I and M–O) or E2f4−/−embryos (D–F, J–L and P–R) at 18.5 dpc were subjected to the PAS reaction (A–F) that stains neutral mucins (pink stain), Alcian Blue pH2.5 stain (G–L) that stains acidic mucins and immunohistochemistry to detect MUC5AC, brown stain (M–R). Panels A,D,G,J,M and P lower power (original magnification ×20) images of coronal sections containing respiratory epithelium. Panels B,E,H,K,N and Q higher power images (original magnification ×40) of the area encompassed by the rectangle in the adjacent panel. Panels C,F,I,L,O and R sections of the nasopharynx from the same embryo as in the adjacent panels. In the control samples (wild-type or E2f4 +/−) only goblet cells stain for mucins whilst most of the cells within the E2f4 mutant epithelium stain for a mucin marker. PAS also stains bone and Alcian Blue also stains cartilage.
Table 1.
Mutation of E2f4 results in an increase in the percentage of nasal respiratory epithelium cells that stain with PAS and Alcian Blue.
| PAS | Alcian Blue | |
|---|---|---|
| Wild-type and E2f4 +/− | 16.3 +/− 6.6 | 14.4 +/− 4.3 |
| E2f4 −/− | 81 +/− 5.1 | 76 +/− 6.8 |
| n (pairs) | 5 | 4 |
| p value | <0.0001 | 0.001 |
For each stain pairs of matched sections from an 18.5 dpc E2f4−/− embryo and a wild-type or E2f4+/− littermate were analyzed. A minimum of 120 epithelial nuclei were counted per section and each cell scored for staining with either PAS or Alcian Blue. The number of pairs of independent embryos analyzed is indicated (n). The mean % of positive cells +/− standard deviation is presented and the data shown to be highly significant in both cases using a paired Student’s t-test.
We also examined mucin staining in the other epithelia that are affected by E2f4-loss at 18.5 dpc (data not shown). As judged by PAS and Alcian Blue-positive staining, the E2f4−/− paranasal serous gland epithelium had the same low number of goblet cells as the wildtype controls and no ectopic staining of mucin markers. Similarly, there was no detectable difference in either the distribution or level of PAS, (n=7 pairs), Alican Blue (n=8), or MUC5AC (n=5) staining in the trachea and proximal bronchus of control versus E2f4−/− embryos. Finally, regardless of genotype, the distal lungs showed no detectable signal with any of these three mucin stains/markers. We therefore conclude that E2f4 mutation alters the composition of the nasal respiratory epithelium, from ciliated cells to cells that produce mucins or mucin-like substances. In contrast, the proximal respiratory epithelia and the serous glands in the paranasal sinuses of E2f4−/− embryos also lack ciliated cells but there is no obvious increase in mucin staining.
Characterization of cells within the E2f4−/− epithelium
To further understand the nature of the cells comprising the nasal respiratory epithelium of E2f4−/− embryos, we also examined the expression of developmental markers (see Figure 2W). First, we showed that cells within the E2f4-deficient epithelium retained expression of both a general epithelial cell marker, cytokeratin 8 (Figure 5A, B), and one specific for the nasal respiratory epithelium, Foxa1 (Besnard et al., 2004;)(Figure 5C, D). However, the Foxa1 staining revealed a clear anomaly: in the control epithelium, the Alcian Blue pH2.5-positive goblet cells had very low, or no, detectable Foxa1 staining (Figure 5C) whilst the E2f4-deficient epithelium contained numerous cells that co-stained for both Foxa1 and Alcian Blue (Figure 5D). This suggests that a fraction of the E2f4 deficient epithelial cells adopt an alternate fate in which they retain respiratory epithelial cell markers and, without displaying true goblet cell characteristics, produce molecules that stain with Alcian Blue. Notably, we did detect some cells in the E2f4 deficient epithelium that stained strongly with Alcian Blue but not Foxa1 and we believe that these represent the bona fide complement of goblet cells.
Fig. 5.

Immunohistochemical analyses of epithelium specific markers. Matched sections from 18.5 dpc wild-type or E2f4+/− as indicated (A,C) or E2f4−/− littermate embryos (B,D) were stained for cytokeratin 8 (A,B), a general marker for epithelial cells, and Foxa1 (C,D) a specific marker for proximal respiratory epithelial cells (brown stain). Note that Foxa1 is not expressed in goblet cells (stained with Alcian Blue) in the control samples. Expression of Foxj1 at 18.5 dpc (E,F) and 15.5 dpc (G,H) a marker of ciliated epithelial cells was absent from the nuclei of the E2f4−/− embryo epithelium (F,H) in contrast to the control samples (E,G), brown stain. Basal cells were detected by analyzing the expression of p63 at 18.5 dpc (I,J) and 16.5 dpc (K,L). In controls (I,K), in addition to the basal cells, nuclei in the columnar epithelial cells stained for p63 protein (brown stain) whereas this staining was not detected in the epithelium of E2f4−/− embryos (J,L). E2f4 was detected in many of the nuclei of control (wild-type or E2f4+/−) embryo epithelial cells at 18.5 dpc (M) and 15.5 dpc (O), brown stain. Staining was absent from matched sections of E2f4−/− embryos (N,P). E2f4 was also detected in most nuclei in the lung at 18.5 dpc including those in the bronchiole epithelium (Q) and staining was absent from the E2f4−/− embryo lung (R). At 18.5 dpc CC10, a marker of Clara cells, was observed in the bronchioles of E2f4−/− embryos (T) at a reduced level in comparison with E2f4+/− embryos (S). T1α, a marker for type I alveolar cells was expressed on the apical membrane at similar levels in wild-type (U) and E2f4−/− embryos (V) at 18.5 dpc. Cells staining for SP-C (black stain), a marker for type II alveolar cells are detected at similar frequency in E2f4+/− (W) and E2f4−/− embryos (X) at 18.5 dpc. Sections in panels C–F,I,J were additionally stained using the Alcian Blue pH2.5 method to identify cells containing acidic mucins. Original magnification ×40.
We also stained for Foxj1, a marker of ciliated cells (Blatt et al., 1999; Tichelaar et al., 1999). Nuclear Foxj1 can first be detected in the nasal epithelium at 15.5 dpc, prior to the formation of cilia (Blatt et al., 1999). Consistent with the absence of ciliated cells, the E2f4−/− nasal respiratory epithelium completely lacked nuclear Foxj1 staining at both 18.5 dpc (Figure 5E, F) and 15.5 dpc (Figure 5G, H) indicating that E2f4 loss prevents the formation of ciliated cells at an early stage of the differentiation process. Normal respiratory epithelium also contains basal cells. To look at this cell type, we stained for a well-known basal cell marker, p63. Cells within the basal layer of both the control (wild-type and E2f4+/−) and E2f4−/− epithelium showed strong p63 staining at 18.5 dpc (Figure 5I, J) and 16.5 dpc (Figure 5K,L) suggesting that basal cells are present at similar levels. In the controls, we also detected nuclei above the basal layer that had a lower intensity of p63 than the basal cells, but these cells were not present in the E2f4−/− epithelium at either developmental stage. It seems likely that this change is somehow related to the early loss of ciliated cells or their precursors. No expression of Clara cell 10kDa protein (CC10, also known as Scgb1a1, CCA or CCSP) or surfactant protein C (SP-C, also known as Sftpc), known markers of Clara cells or cuboidal type II cells respectively, was observed in the control or E2f4−/− nasal respiratory epithelium (n=5) indicating that the epithelium was not expressing markers of either proximal or distal lung fate (data not shown). Importantly, E2f4 was detected in the nuclei of many cells in the nasal epithelium at both 18.5 dpc and 15.5 dpc (Figure 5M, O). In the lung at 18.5 dpc E2f4 was detected in nearly all nuclei (Figure 5Q,R).
To assess if mutation of E2f4 affected cells in other areas of the respiratory tract we also examined Clara cell, type I and type II alveolar epithelial cell specific markers. At 18.5 dpc, we observed a reproducible decrease in the staining intensity of the Clara cell marker CC10 in the trachea and bronchi of E2f4−/− embryos (n=8; Figure 5S, T). Quantification of strongly CC10 positive cells in the bronchi showed a statistically significant reduction in CC10 staining in the E2f4−/− embryos (Table 2). In contrast, there was no difference in the distribution or expression levels of either T1α, a marker of type I alveolar epithelial cells (Millien et al., 2006; Williams et al., 1996), or SP-C a marker of cuboidal type II alveolar cells in the lungs of control versus the E2f4−/− embryos at 18.5 dpc (Figure 5U-X, Table 2). These data indicate that loss of E2f4 interferes with CC10 expression in Clara cells but it has no effect on the alveolar type I or type II cell markers suggesting that these cell types are unaffected.
Table 2.
Quantification of CC10 and SP-C marker expression.
| CC10 | SP-C | |
|---|---|---|
| Wild-type and E2f4 +/− | 64.4 +/− 10.1 | 7.7+/− 2.3 |
| E2f4 −/− | 34.1 +/− 17.1 | 8 +/− 2.8 |
| n (pairs) | 8 | 4 |
| p value | 0.00013 | 0.31 |
For each stain pairs of matched sections from an 18.5 dpc E2f4−/− embryo and a wild-type or E2f4+/− littermate were analyzed. A minimum of 200 bronchiolar (CC10) or 350 lung (SP-C) nuclei were counted per section and each cell scored for strong staining with CC10 or staining with SP-C antibodies. The number of pairs of independent embryos analyzed is indicated (n). The mean % of positive cells +/− standard deviation is presented and the data analyzed using a paired Student’s t-test and the p values indicated.
Taken together, our analysis shows that E2f4-loss causes a change in the composition of the nasal respiratory epithelium that involves the absence of ciliated cells and, in their place, mucin-producing cell types whose characteristics distinguish them from normal goblet cells. To further understand the nature of these cells, we performed transmission electron microscopy on nasal epithelia dissected from 18.5 dpc embryos. Wild-type or E2f4+/− epithelium (n=3 embryos), contained ciliated columnar cells and intermittent goblet cells both of which had microvilli projecting into the lumen (Figure 6A). The goblet cells contained mucin granules exactly as expected. In contrast, consistent with our light microscopy studies, no cilia were detected on the cells of E2f4−/−epithelium and only microvilli were observed projecting into the lumen (n=4 embryos; Figure 6B–D). In addition, these cells completely lacked cilia basal bodies in the cytoplasm. More than 95% (27/28) of cells contained structures resembling mucin granules that were localized to the apical surface or distributed throughout the cytoplasm (Figure 6B and C) and the cytoplasm contained high amounts of rough endoplasmic reticulum. These latter cells resembled columnar secretory epithelial cells. In 14% (4/28) of cells the rough endoplasmic reticulum was dilated with, presumably, proteinaceous material that also appeared to be released into the lumen (Figure 6C and D). This analysis indicates that at least two morphologically distinct cell types exist in the E2f4−/−epithelium. One resembles bona fide goblet cells and the other a secretory cell with abnormal characteristics. In combination with the staining studies, these data show that E2f4-loss causes a major change in the composition of the nasal epithelium: it blocks the formation of ciliated cells at an early point in the differentiation process and instead allows the formation of an aberrant, columnar secretory cell type that contains mucin like substances. In addition, loss of E2f4 impinges upon another respiratory epithelial cell type, the Clara cells in that they express low levels of CC10, a normally strongly expressed Clara cell specific protein.
Fig. 6. Transmission electron microscopy analyses of E2f4−/− nasal epithelium.

Control (wild-type or E2f4+/−) embryo epithelium contained ciliated columnar cells interspersed with goblet cells (arrowhead), * denotes cilia and the arrow points to microvilli (A). Epithelium of E2f4−/− embryos lacks cilia and is comprised of cells resembling columnar secretory cells that contained high amounts of rough endoplasmic reticulum and secretory vesicles (arrowheads) distributed apically (B) and throughout the cell, arrow indicates microvilli (C). D, higher power image of the area delineated by the box in C showing dilated rough endoplasmic reticulum. Scale bar in A, B and C 2μm, in D 500nm.
Discussion
The data presented here shows that E2f4 is required for ciliated cell development throughout the airway epithelium and the epithelium of the submucosal glands in the paranasal sinuses. This phenotype arises during embryogenesis indicating that it is a defect in development. Unexpectedly, we did not detect any gross changes in the rate of cell proliferation or apoptosis in the nasal epithelium, suggesting that E2f4 has functions apart from those involved in regulating proliferation and apoptosis. Clearly, we cannot rule out the possibility that, at early stages of development, the proliferation of ciliated cell precursors was severely reduced and other cell types proliferated in their place. However, we can be sure that E2f4-loss leads to a profound change in the composition of the airway epithelium. Specifically, the nasal epithelium contains basal and goblet cells, but the ciliated cells that normally account for the majority of the surface epithelium are replaced by columnar secretory cell types. These cells have goblet cell-like characteristics in that they express mucin like substances, as judged by histochemical stains and immunohistochemistry. However, they are not bona fide goblet cells since they stain with Alcian Blue, a mucin marker, and Foxa1, a protein that is normally expressed in ciliated, but not goblet cells. These observations strongly suggest that E2f4-loss somehow disrupts the specification of cells within the nasal epithelium.
Our data indicate that some, but not all, aspects of this epithelial defect are observed in the serous/submucosal glands of the paranasal sinuses, the trachea and the bronchioles. We see the complete loss of cilia from these epithelia. However, the serous/submucosal gland epithelium shows no evidence of ectopic mucin production. The tracheas of control and E2f4−/− embryos show positive staining with PAS, which is known to detect multiple molecules other than mucins, but no significant staining with other mucin markers, including Alcian Blue and MUC5AC. In the remaining respiratory tract, i.e. bronchioles and alveolar epithelium, no staining for PAS, Alcian Blue or MUC5AC was detected. Thus, in these tissues, E2f4-loss causes the loss of ciliated cells but it does not trigger the appearance of a cell type expressing mucins. This yields two important conclusions. First, the most prevalent defect appears to be the loss of ciliated cells as opposed to the preferential survival/expansion of the mucous secreting cell type. Second, these defects clearly distinguish the E2f4 mutant mouse from other mouse models that have respiratory epithelium defects. Specifically, mutation of E2f4 does not result in the gross goblet cell hyperplasia that has been reported, for example, in Foxa2 mutant mice (Wan et al., 2004). In addition, the E2f4 mutant phenotype is different from those cases where cilia fail to assemble on the cell surface, as seen in the Foxj1 mutant (Brody et al., 2000; Gomperts et al., 2004) since, contrary to this mutant, cilia basal bodies are not detected in the cytoplasm of cells comprising the E2f4 mutant airway epithelium. Thus, the defects we have observed in E2f4 mutant mice represent a novel class of airway epithelium defects.
The loss of ciliated cells and continued production of mucins, and possibly other proteinaceous molecules contributes to the build up of fluids in the nasal cavity and the chronic rhinitis observed in the E2f4 mutant mice. Chronic rhinitis is also observed in humans with the genetic disorders Primary Ciliary Dyskinesia and Kartagener’s Syndrome in which cilia have been found to be dysfunctional (Geremek and Witt, 2004; Ibanez-Tallon et al., 2003). Mapping of the genetic loci involved in these syndromes has identified candidate genes, several of which encode components of the cilia dynein arms. We are not aware of any loci mapping to E2F4. These syndromes can also be associated with other symptoms including situs inversus (Kartagener’s syndrome), male and female infertility, hydrocephalus and polycystic kidney disease. Surviving E2f4 mutant mice exhibit reduced fertility but we do not observe loss of cilia from the epithelium of the fallopian tube or the efferent ducts of the epididymis suggesting that this does not account for the defect. Moreover, we have never observed situs inversus or hydrocephalus in E2f4 mutant mice suggesting that E2f4 is not required for all ciliated epithelium development. Interestingly, E2f5-deficient mice do develop hydrocephalus (Lindeman et al., 1998). This defect was proposed to result from increased secretion of cerebrospinal fluid from the choroid plexus independent of changes in proliferation (Lindeman et al., 1998). However, it should be noted that hydrocephalus may also arise from defects in immotile cilia on the choroid plexus epithelium (Banizs et al., 2005). Since E2f4 and 5 are known to play overlapping roles in other processes, including the repression of E2f-responsive genes, this raises the possibility that both these proteins contribute to the development of ciliated epithelia and may play redundant roles in some tissues. Indeed, E2f4 is detected in the nuclei of cells within the ciliated ependymal epithelium of the brain as well as the ciliated epithelium of the fallopian tube and the efferent ducts of the epididymis (Supplementary Figure 1).
The mechanism whereby E2f4 mutation disrupts respiratory ciliated epithelium development may involve a change in the expression level of one or more E2f target genes. This could occur through a variety of mechanisms. First, E2f4 could directly regulate the transcription of one, or more, genes that are required for ciliated cell development in either a pocket protein (pRb, p107 or p130) dependent, or independent manner. Second, the loss of E2f4 could have an indirect affect on gene transcription by releasing the normally E2f4 bound pocket proteins (pRb, p107 or p130) and allowing them to bind to, and modulate the activity of, other E2fs or other transcription factors. As a first step towards distinguishing between these models, we retrospectively examined double mutant embryos lacking E2f4 and p107, E2f4 and p130, E2f4 and pRb, as well as triple mutant embryos lacking E2f4, p107 and p130 to determine whether the loss of the pocket proteins could either suppress or enhance the epithelial defects of the E2f4 mutants (P.S.D., E.Y. Lee, H. Richter and R. L. Landsberg, unpublished observations). Removal of p107 and/or p130 had no detectable effect on this phenotype suggesting that these proteins are not playing a role. In contrast, we found that E2f4+/−;Rb−/− and E2f4+/−;E2f5−/− mice showed the same nasal epithelial defects, including the loss of ciliated cells and presence of aberrant mucous cells, as the E2f4 mutant mice (P.S.D., E.Y. Lee and J. Sero, unpublished observations). Since this defect is never observed in the E2f4+/− mice, we conclude that pRb and E2f5 both act synergistically with E2f4 to control epithelial development. The best-documented, shared role of pRb, E2f4 and E2f5 is the transcriptional repression of cell cycle genes. However, while E2f4-loss caused the upregulation of one known E2f-responsive target, PCNA, others appear to be regulated normally and there is no evidence of a proliferation defect in the E2f4-deficient developing nasal epithelium. Notably, there is mounting evidence that E2f4 and/or pRb can influence the transcription and/or activity of transcription factors that regulate terminal differentiation processes, for example adipogenesis (Chen et al, 1996; Fajas et al., 2002; Landsberg et al., 2003). In this instance, pRb-loss promotes a switch in cell fate from white to brown adipocytes (Hansen et al., 2004; Puigserver et al., 1998). Given these observations, it is possible that E2f4 is required for the appropriate regulation of one, or more, factors that control the differentiation of ciliated epithelial cells. Importantly, E2f4 is present at high levels in the nucleus of ciliated cells, consistent with the notion that it acts directly in this cell-type. However, it is also possible that E2f4 is required in the progenitor cells or in the underlying mesenchyme. We believe that this phenotype is less likely to involve a defect in the basal cells since the analysis of the p63 mutant mouse shows that basal cells can be lost from the tracheal epithelium without disrupting ciliated cell development (Daniely et al., 2004).
Our data clearly show that the loss of ciliated cells in the nasal respiratory epithelium is accompanied by the appearance of mucin-producing secretory cells that are not present in the wildtype controls. We have considered three possible explanations for this observation. The first possibility is that the population of a cell type that is indigenous to the nasal epithelium, such as nonciliated cuboidal/columnar epithelial cells (located in the transitional epithelium) or serous cells, has expanded at the expense of the ciliated cell population. We disfavor this hypothesis because the characteristics of the E2f4-deficient mucin-producing secretory cells (high levels of rough ER, secretory granules and mucin-expression) diverge considerably from those of either the transitional epithelial cells (no secretory granules and little mucosubstances) or the serous cells (smooth ER) in rodents. The second possibility is that mutation of E2f4 leads to activation of pathways normally induced in response to lung damage, infections or exposure to allergens. These are proposed to cause ciliated cells to lose their cilia and undergo transformation into mucus secreting cells (Harkema et al., 2006; Rawlins and Hogan, 2006). In some situations, epithelial cells have been shown to contain dilated rough ER (Harkema et al., 2006) that resembles our transmission EM micrographs. It seems highly unlikely that the epithelial changes result from an infection, since they occur in utero and are specific to the E2f4−/− embryos and not littermate controls. However, it is possible that E2f4-loss somehow activates a response pathway in the absence of any infection. Even in this case, it is unclear why this response would be specifically restricted to the nasal epithelium of the E2f4−/− mice since in other models the increase in mucus secreting cells occurs in the trachea and lungs, a phenotype not observed in E2f4−/− mutants. For example, Evans and coworkers report that antigen challenge causes Clara cells within the trachea and bronchus/bronchioles to produce multiple mucins without a significant decrease in the number of ciliated cells and other studies show a decrease in SP-C protein levels in response to lung damage or infection (Evans et al., 2004, Mulugeta and Beers, 2006). Neither of these responses occurs in E2f4−/− mutants. In addition we did not detect Interleukin-13, a known activator of mucous cell populations or the presence of phosphorylated STAT6, that is an indicator of activated interleukin signaling associated with inflammation (data not shown). In addition we did not detect the presence of Ym1/2(chitinase) proteins that are often found in cells with dilated endoplasmic reticulum in the respiratory tract following damage or inflammation (Harkema et al., 2006), in the E2f4 mutants (data not shown). The third possibility is that E2f4-loss disrupts the specification of the cell lineage in a manner that leads to the formation of an abnormal cell type. This hypothesis is supported by the fact that these cells stain with both Foxa1 and Alcian Blue pH2.5, markers that are not normally co-expressed. Clearly, the specification defect becomes less pronounced in the proximal respiratory epithelium: the ciliated cells are lost but the aberrant mucin containing cells are not found, and other ciliated epithelia, such as in the fallopian tubes, are completely normal. This variability could be due to the degree with which other members of the E2f family are expressed and able to compensate for the loss of E2f4. In addition to the absence of ciliated cells from the airway epithelium E2f4 loss results in aberrant Clara cells in the proximal lung epithelium in that the epithelial cells express CC10 at a reduced level in comparison with wild-type epithelium. Whether other aspects of Clara cell development are abnormal remains to be established. The fact that two airway epithelial cell types are affected supports our third hypothesis that E2f4-loss is somehow disrupts cell specification. It will be interesting to identify the E2f4 target genes and pathways involved in determining the ciliated and perhaps other cell lineages especially considering that they belong to a class of E2f4 target genes that appear not to be involved in proliferation.
Supplementary Figure 1
E2f4 is expressed in the nuclei of epithelial cells with motile cilia.
E2f4 was detected by immunohistochemistry in the ependymal epithelium of the brain ventricles (A), the fallopian tube epithelium (B) and epithelium of the efferent ducts of the epididymis (C) of adult mice. Parallel staining of the efferent ducts of the epididymis from an E2f4−/− mouse indicates that the antiserum used is specific for E2f4 (D). Original magnification ×40.
Acknowledgments
We wish to thank Dave Loudy, Scott Randell, Jack Harkema and members of the Lees laboratory for suggestions; Eunice Lee, Henrijette Richter, Rebecca Landsberg and Julia Sero for providing sections of tissues collected whilst they were in the laboratory of J.A.L and Cyril Fisher for advice on cell morphology. The monoclonal #8.1.1 developed by A. Farr was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa. This work was funded by NIH grants awarded to J.A.L. (GM53204, CA121921). J.A.L. is a Ludwig Scholar.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Apostolova MD, et al. Active nuclear import and export pathways regulate E2F-5 subcellular localization. J Biol Chem. 2002;277:34471–9. doi: 10.1074/jbc.M205827200. [DOI] [PubMed] [Google Scholar]
- Attwooll C, et al. The E2F family: specific functions and overlapping interests. Embo J. 2004;23:4709–16. doi: 10.1038/sj.emboj.7600481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banizs B, et al. Dysfunctional cilia lead to altered ependyma and choroid plexus function, and result in the formation of hydrocephalus. Development. 2005;132:5329–39. doi: 10.1242/dev.02153. [DOI] [PubMed] [Google Scholar]
- Besnard V, et al. Immunohistochemical localization of Foxa1 and Foxa2 in mouse embryos and adult tissues. Gene Expr Patterns. 2004;5:193–208. doi: 10.1016/j.modgep.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Blatt EN, et al. Forkhead transcription factor HFH-4 expression is temporally related to ciliogenesis. Am J Respir Cell Mol Biol. 1999;21:168–76. doi: 10.1165/ajrcmb.21.2.3691. [DOI] [PubMed] [Google Scholar]
- Brody SL, et al. Ciliogenesis and left-right axis defects in forkhead factor HFH-4-null mice. Am J Respir Cell Mol Biol. 2000;23:45–51. doi: 10.1165/ajrcmb.23.1.4070. [DOI] [PubMed] [Google Scholar]
- Chen PL, et al. Retinoblastoma protein positively regulates terminal adipocyte differentiation through direct interaction with C/EBPs. Genes Dev. 1996;10:2794–804. doi: 10.1101/gad.10.21.2794. [DOI] [PubMed] [Google Scholar]
- Cole P. The damaging role of bacteria in chronic lung infection. J Antimicrob Chemother. 1997;40(Suppl A):5–10. doi: 10.1093/jac/40.suppl_1.5. [DOI] [PubMed] [Google Scholar]
- Daniely Y, et al. Critical role of p63 in the development of a normal esophageal and tracheobronchial epithelium. Am J Physiol Cell Physiol. 2004;287:C171–81. doi: 10.1152/ajpcell.00226.2003. [DOI] [PubMed] [Google Scholar]
- Dimova DK, Dyson NJ. The E2F transcriptional network: old acquaintances with new faces. Oncogene. 2005;24:2810–26. doi: 10.1038/sj.onc.1208612. [DOI] [PubMed] [Google Scholar]
- Dynlacht BD, et al. Differential regulation of E2F transactivation by cyclin/cdk2 complexes. Genes Dev. 1994;8:1772–86. doi: 10.1101/gad.8.15.1772. [DOI] [PubMed] [Google Scholar]
- Evans CM, et al. Mucin is produced by clara cells in the proximal airways of antigen-challenged mice. Am J Respir Cell Mol Biol. 2004;31:382–94. doi: 10.1165/rcmb.2004-0060OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MJ, et al. Cellular and molecular characteristics of basal cells in airway epithelium. Exp Lung Res. 2001;27:401–15. doi: 10.1080/019021401300317125. [DOI] [PubMed] [Google Scholar]
- Fajas L, et al. E2Fs regulate adipocyte differentiation. Dev Cell. 2002;3:39–49. doi: 10.1016/s1534-5807(02)00190-9. [DOI] [PubMed] [Google Scholar]
- Gaubatz S, et al. E2F4 is exported from the nucleus in a CRM1-dependent manner. Mol Cell Biol. 2001;21:1384–92. doi: 10.1128/MCB.21.4.1384-1392.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaubatz S, et al. E2F4 and E2F5 play an essential role in pocket protein-mediated G1 control. Mol Cell. 2000;6:729–35. doi: 10.1016/s1097-2765(00)00071-x. [DOI] [PubMed] [Google Scholar]
- Geremek M, Witt M. Primary ciliary dyskinesia: genes, candidate genes and chromosomal regions. J Appl Genet. 2004;45:347–61. [PubMed] [Google Scholar]
- Giangrande PH, et al. A role for E2F6 in distinguishing G1/S- and G2/M-specific transcription. Genes Dev. 2004;18:2941–51. doi: 10.1101/gad.1239304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomperts BN, et al. Foxj1 regulates basal body anchoring to the cytoskeleton of ciliated pulmonary epithelial cells. J Cell Sci. 2004;117:1329–37. doi: 10.1242/jcs.00978. [DOI] [PubMed] [Google Scholar]
- Hansen JB, et al. Retinoblastoma protein functions as a molecular switch determining white versus brown adipocyte differentiation. Proc Natl Acad Sci U S A. 2004;101:4112–7. doi: 10.1073/pnas.0301964101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkema JR, et al. The nose revisited: a brief review of the comparative structure, function, and toxicologic pathology of the nasal epithelium. Toxicol Pathol. 2006;34:252–69. doi: 10.1080/01926230600713475. [DOI] [PubMed] [Google Scholar]
- Hijmans EM, et al. E2F-5, a new E2F family member that interacts with p130 in vivo. Mol Cell Biol. 1995;15:3082–9. doi: 10.1128/mcb.15.6.3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert PO, et al. E2F4 is essential for normal erythrocyte maturation and neonatal viability. Mol Cell. 2000;6:281–91. doi: 10.1016/s1097-2765(00)00029-0. [DOI] [PubMed] [Google Scholar]
- Ibanez-Tallon I, et al. To beat or not to beat: roles of cilia in development and disease. Hum Mol Genet. 2003;12(Spec No 1):R27–35. doi: 10.1093/hmg/ddg061. [DOI] [PubMed] [Google Scholar]
- Kinross KM, et al. E2f4 regulates fetal erythropoiesis through the promotion of cellular proliferation. Blood. 2006 doi: 10.1182/blood-2005-09-008656. [DOI] [PubMed] [Google Scholar]
- Knudsen ES, Wang JY. Dual mechanisms for the inhibition of E2F binding to RB by cyclin-dependent kinase-mediated RB phosphorylation. Mol Cell Biol. 1997;17:5771–83. doi: 10.1128/mcb.17.10.5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landsberg RL, et al. The role of E2F4 in adipogenesis is independent of its cell cycle regulatory activity. Proc Natl Acad Sci U S A. 2003;100:2456–61. doi: 10.1073/pnas.0138064100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeman GJ, et al. A specific, nonproliferative role for E2F-5 in choroid plexus function revealed by gene targeting. Genes Dev. 1998;12:1092–8. doi: 10.1101/gad.12.8.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millien G, et al. Alterations in gene expression in T1 alpha null lung: a model of deficient alveolar sac development. BMC Dev Biol. 2006;6:35. doi: 10.1186/1471-213X-6-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moberg K, et al. E2F-4 switches from p130 to p107 and pRB in response to cell cycle reentry. Mol Cell Biol. 1996;16:1436–49. doi: 10.1128/mcb.16.4.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulugeta S, Beers MF. Surfactant protein C: its unique properties and emerging immunomodulatory role in the lung. Microbes Infect. 2006;8:2317–23. doi: 10.1016/j.micinf.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Perez-Vilar J, et al. Making More MUCS. Am J Respir Cell Mol Biol. 2003;28:267–70. doi: 10.1165/rcmb.F262. [DOI] [PubMed] [Google Scholar]
- Perl AK, Whitsett JA. Molecular mechanisms controlling lung morphogenesis. Clin Genet. 1999;56:14–27. doi: 10.1034/j.1399-0004.1999.560103.x. [DOI] [PubMed] [Google Scholar]
- Puigserver P, et al. Involvement of the retinoblastoma protein in brown and white adipocyte cell differentiation: functional and physical association with the adipogenic transcription factor C/EBPalpha. Eur J Cell Biol. 1998;77:117–23. doi: 10.1016/s0171-9335(98)80079-4. [DOI] [PubMed] [Google Scholar]
- Rawlins EL, Hogan BL. Epithelial stem cells of the lung: privileged few or opportunities for many? Development. 2006;133:2455–65. doi: 10.1242/dev.02407. [DOI] [PubMed] [Google Scholar]
- Rayman JB, et al. E2F mediates cell cycle-dependent transcriptional repression in vivo by recruitment of an HDAC1/mSin3B corepressor complex. Genes Dev. 2002;16:933–47. doi: 10.1101/gad.969202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempel RE, et al. Loss of E2F4 activity leads to abnormal development of multiple cellular lineages. Mol Cell. 2000;6:293–306. doi: 10.1016/s1097-2765(00)00030-7. [DOI] [PubMed] [Google Scholar]
- Rogers DF. Airway goblet cells: responsive and adaptable front-line defenders. Eur Respir J. 1994;7:1690–706. [PubMed] [Google Scholar]
- Rubin SM, et al. Structure of the Rb C-terminal domain bound to E2F1-DP1: a mechanism for phosphorylation-induced E2F release. Cell. 2005;123:1093–106. doi: 10.1016/j.cell.2005.09.044. [DOI] [PubMed] [Google Scholar]
- Sardet C, et al. E2F-4 and E2F-5, two members of the E2F family, are expressed in the early phases of the cell cycle. Proc Natl Acad Sci U S A. 1995;92:2403–7. doi: 10.1073/pnas.92.6.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, et al. Analysis of promoter binding by the E2F and pRB families in vivo: distinct E2F proteins mediate activation and repression. Genes Dev. 2000;14:804–16. [PMC free article] [PubMed] [Google Scholar]
- Tichelaar JW, et al. HNF-3/forkhead homologue-4 (HFH-4) is expressed in ciliated epithelial cells in the developing mouse lung. J Histochem Cytochem. 1999;47:823–32. doi: 10.1177/002215549904700612. [DOI] [PubMed] [Google Scholar]
- Tsai KY, et al. ARF is not required for apoptosis in Rb mutant mouse embryos. Curr Biol. 2002;12:159–63. doi: 10.1016/s0960-9822(01)00659-5. [DOI] [PubMed] [Google Scholar]
- Verona R, et al. E2F activity is regulated by cell cycle-dependent changes in subcellular localization. Mol Cell Biol. 1997;17:7268–82. doi: 10.1128/mcb.17.12.7268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan H, et al. Foxa2 regulates alveolarization and goblet cell hyperplasia. Development. 2004;131:953–64. doi: 10.1242/dev.00966. [DOI] [PubMed] [Google Scholar]
- Wells J, et al. Target gene specificity of E2F and pocket protein family members in living cells. Mol Cell Biol. 2000;20:5797–807. doi: 10.1128/mcb.20.16.5797-5807.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MC, et al. T1 alpha protein is developmentally regulated and expressed by alveolar type I cells, choroid plexus, and ciliary epithelia of adult rats. Am J Respir Cell Mol Biol. 1996;14:577–85. doi: 10.1165/ajrcmb.14.6.8652186. [DOI] [PubMed] [Google Scholar]
- Xiao ZX, et al. Regulation of the retinoblastoma protein-related protein p107 by G1 cyclin-associated kinases. Proc Natl Acad Sci U S A. 1996;93:4633–7. doi: 10.1073/pnas.93.10.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
E2f4 is expressed in the nuclei of epithelial cells with motile cilia.
E2f4 was detected by immunohistochemistry in the ependymal epithelium of the brain ventricles (A), the fallopian tube epithelium (B) and epithelium of the efferent ducts of the epididymis (C) of adult mice. Parallel staining of the efferent ducts of the epididymis from an E2f4−/− mouse indicates that the antiserum used is specific for E2f4 (D). Original magnification ×40.