

Abstract
Malignant gliomas are one of the leading causes of cancer deaths worldwide, but chemoprevention strategies for them are few and poorly investigated. Here, we show that cholera toxin, the traditional biotoxin and well known inducer of accumulation of cellular cAMP, is capable of inducing differentiation on malignant gliomas in vitro with rat C6 and primary cultured human glioma cells. Cholera toxin-induced differentiation was characterized by typical morphological changes, increased expression of glial fibrillary acid protein, decreased expression of Ki-67, inhibition of cellular proliferation, and accumulation of cells in the G1 phase of the cell cycle. Cholera toxin also triggered a significant reduction in the G1 cell-cycle regulatory proteins cyclin D1 and Cdk2 along with an overexpression of cell-cycle inhibitory proteins p21Cip1 and p27Kip1. Abrogation of cAMP-dependent protein kinase A activity by protein kinase A inhibitor or silencing of cAMP-responsive element binding proteins by RNA interference resulted in suppressed differentiation. These findings imply the attractiveness of cholera toxin as a drug candidate for further development of differentiation therapy. Furthermore, activation of the protein kinase A/cAMP-responsive element binding protein pathway may be a key and requisite factor in glioma differentiation.
Astroglial cells are important components of the mammalian central nervous system (CNS), outnumbering neurons several times in the adult brain (1). Glioma derived from astrocytes or astroglial precursors is the most common malignant cancer affecting the CNS, accounting for >60% of primary brain tumors (2). Current therapy with surgery, radiation, and chemotherapy rarely, if ever, cures the disease and infrequently prolongs life for >1 year (3, 4).
Differentiation therapy, using agents that modify cancer cell differentiation, has shown promise in the spectrum of agents used against tumors (5). Wang and Chen (6) demonstrated the clinical application for differentiation therapy by introducing all-trans-retinoic acid to clinical use for the treatment of acute promyelocytic leukemia (APL) (7). Notably, the inorganic toxicant arsenic trioxide (As2O3), a well known environmental carcinogen, has also proven to be an effective drug in the treatment of APL patients by triggering apoptosis and differentiation of APL cells in a dose-dependent manner (8, 9). Such excellent effects, however, were not reproduced in other hematological and, particularly, solid tumors. Differentiation agents for malignant gliomas remain a real challenge.
Cholera toxin is the major virulent factor of Vibrio cholerae and is the most recognizable enterotoxin causing diarrhea, the disease second only to cardiovascular disease as causes of death (10). Cholera toxin catalyzes ADP-ribosylation of Gs protein and results in accumulation of cellular cAMP (11, 12). Ample evidence indicates that cAMP-elevating stimuli such as N-substituted cAMP analogues and cAMP-increasing reagents can induce cell differentiation in gliomas (13, 14). Guerrant et al. (15) reported that active whole cholera toxin, but not inactive choleragenoid, produces elevation of cAMP and parallel morphological changes in CHO cells. Ganglioside GM1 reaction with the B subunit of cholera toxin was reported to induce neuron-like differentiation of PC12 and neuroblastoma cells (16, 17). All of the findings mentioned above revealed the potential of cholera toxin, a kind of biotoxin, in the differentiation induction of tumor cells. Surprisingly, a possible biological effect of cholera toxin in cancer therapy has, to our knowledge, never been investigated previously. An exploration for the differentiation-inducing and possible therapeutic potential of cholera toxin on malignant glioma is therefore essential.
In this study, we demonstrate that the traditional biotoxin cholera toxin is capable of inducing cellular differentiation in both rat C6 and primary cultured human malignant glioma cells. This effect could be suppressed by cAMP-dependent protein kinase A (PKA) blocker and siRNA knockdown of cAMP-responsive element binding (CREB) protein expression, indicating that differentiation triggered by cholera toxin is effected through the PKA/CREB pathway and suggesting the requisite role of PKA and transcription factor CREB for cholera toxin-induced differentiation.
Results
Cholera Toxin Induces Morphological Transformation of C6 Cells.
Differentiation of rat C6 glioma cells toward astrocyte type is characterized by morphological transformation from a flat polygonal appearance to spindle shape with processes. Microscopic observation of C6 glioma cells treated with 10 ng/ml cholera toxin revealed major alterations in their morphology. Unlike the mainly polygonal morphology of control, the shape of cholera toxin-treated C6 cells was similar to that of mature astrocytes, with smaller round cell bodies and much longer, fine, tapering processes (Fig. 1 A and B). This finding indicates that cholera toxin has the ability to induce glioma cells to differentiate into the maturation process of astrocytes.
Fig. 1.

Cholera toxin induces morphological transformation of glioma cells. C6 (A and B) and primary cultured human glioma cells (C and D) were incubated with (B and D) or without (A and C) 10 ng/ml cholera toxin for 48 h. (Original magnification: ×200.)
Molecular Evidence of Differentiation Induced by Cholera Toxin in C6 Cells.
We further examined the expressions of glial fibrillary acid protein (GFAP), a well established marker of mature astrocytes and Ki-67, the reliable marker of proliferating cells. As indicated in Fig. 2A, Western blotting analysis confirmed a significant up-regulation of GFAP protein expression in cholera toxin-treated cells compared with the controls in a concentration-dependent manner with a maximal induction effect at 10 ng/ml. Moreover, we found that the effect of cholera toxin on GFAP expression is time-dependent. It begins to increase at day 1, reaches the peak at day 4, and then maintains at a significant high level (Fig. 2B), whereas the expression of both isoforms (345 and 395 kDa) of Ki-67 protein were notably restrained by cholera toxin in a similar concentration- and time-dependent manner (Fig. 2 E and F), indicating that a subset of cells actually exit from the cell cycle and then differentiate.
Fig. 2.

Concentration- and time-dependent effect of cholera toxin on GFAP and Ki-67 expression in glioma cells. C6 (A, B, E, and F) and primary cultured human glioma cells (C, D, G, and H) were incubated with cholera toxin. (A and C) Concentration-dependent effect of cholera toxin on GFAP expression. Cells were incubated with 0, 5, 7.5, and 10 ng/ml cholera toxin for 48 h. (B and D) Time-dependent effect of cholera toxin on GFAP expression. Cells were incubated with 10 ng/ml cholera toxin for 1, 2, 4, and 6 days. (E and G) Concentration-dependent effect of cholera toxin on Ki-67 expression. Cells were incubated with 0, 5, 7.5, and 10 ng/ml cholera toxin for 48 h. (F and H) Time-dependent effect of cholera toxin on GFAP expression. Cells were incubated with 10 ng/ml cholera toxin for 1, 2, 4, and 6 days. Total protein was extracted and subjected to Western blot analysis of GFAP, Ki-67, and β-actin. Results are means ± SD (n = 3). Statistical differences compared with the controls are given as *, P < 0.01.
Cholera Toxin Causes Proliferation Inhibition and Cell-Cycle Arrest in Glioma Cells.
Cholera toxin also causes a significant subdued proliferation rate in a time-dependent manner compared with the control. After 48 h of incubation, the proliferation rate was inhibited 81.2% by cholera toxin in C6 cells (P < 0.01) (Fig. 3A).
Fig. 3.

Cholera toxin induces proliferation inhibition but does not induce cell death of glioma cells. Cells were incubated with 10 ng/ml cholera toxin for 48 h. (A) 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) absorbance was measured and proliferation rate was calculated. (B and C) Fluorescent staining of nuclei by Hoechst 33258 (original magnification: ×400) in C6 cells treated with (C) or without (B) cholera toxin. (D) LDH absorbance in C6 cells. Results are means ± SD (n = 3). Statistical differences compared with the controls are given as *, P < 0.05 and **, P < 0.01.
The reduction in proliferation rate could be explained either by cell death or reduced proliferation. Hoechst staining and lactate dehydrogenase (LDH) assay were used to investigate whether treatment of cholera toxin causes apoptosis or necrosis. Apoptotic bodies were not observed in significant numbers in either cholera toxin-treated or control cells with Hoechst staining (Fig. 3 B and C). There was no statistically significant difference in the mean LDH absorbance between cells treated with cholera toxin or the control (Fig. 3D). Hence, cytotoxicity was not contributing to reduction in proliferation rate, and this reduction appears to be at least partly caused by decreased proliferation.
Table 1 shows that a 48-h treatment with 10 ng/ml cholera toxin leads to a C6 accumulation in the G0/G1 phase to reach ≈88.2%, whereas that in control was 63.6% (P < 0.05). Concomitantly, there was a remarkable decrease in S-phase cell fraction (after 48 h of treatment, the percentage of S-phase cells was 7.9% in cholera toxin group compared with 27.7% in the control) (P < 0.01).
Table 1.
Cholera toxin causes a G0/G1-phase cell cycle arrest in C6 and human primary glioma cells.
| Groups | C6 glioma cells |
Human primary glioma cells |
||
|---|---|---|---|---|
| G0/G1 | S | G0/G1 | S | |
| Control | 63.6 ± 7.4 | 27.7 ± 5.9 | 51.5 ± 11.8 | 36.0 ± 8.2 |
| Cholera toxin | 88.2 ± 6.8* | 7.9 ± 3.9** | 81.3 ± 3.1** | 7.7 ± 2.9** |
Results are expressed as means ± SD (n=3) for control and cholera toxin groups.
*, P < 0.05;
**, P < 0.01 compared with control, respectively.
Effect of Cholera Toxin on Cell-Cycle Regulatory Molecules.
The cholera toxin-induced G1 arrest was further confirmed by examining the cellular levels of the G1 cell-cycle control proteins cyclin D1 and Cdk2 in C6 cells. Western immunoblot analysis confirmed that treatment with 10 ng/ml cholera toxin down-regulated the levels of cyclin D1 and Cdk2 proteins. In addition, the expression of p21Cip1 and p27Kip1 was substantially up-regulated stimulated by cholera toxin after 48 h (Fig. 4). Based on these findings, cholera toxin is likely to block the cell-cycle progression through G1 to S phase.
Fig. 4.

Effect of cholera toxin on cell-cycle regulatory molecules. C6 cells were incubated with 0, 5, 7.5, or 10 ng/ml cholera toxin for 48 h. Total protein was extracted and blotted with antibodies against cyclin D1, Cdk2, p21cip1, and p27kip1. Blots are representative of three independent experiments.
PKA/CREB Pathway Is Requisite for Cholera Toxin-Induced Differentiation of C6 Glioma Cells.
To elucidate the molecular basis for cholera toxin-induced cell differentiation, we measured the expression of major proteins of the PKA/CREB signaling pathway. As shown in Fig. 5A, the PKA activity accelerated between 1 and 3 h after cholera toxin exposure and then activated moderately. Significant phosphorylation of CREB at Ser-133 (p-CREBser133) was detected within 3 h and reached maximum after 6 h of treatment (Fig. 5B). In addition, we have found that activation of the PKA pathway by doses of forskolin (Sigma, St. Louis, MO) or db-cAMP (Sigma) mimic the differentiation effects of cholera toxin on cell morphology, GFAP expression, cell proliferation, and cell-cycle distributions in C6 glioma cells [supporting information (SI) Fig. 7]. However, the morphological changes induced by cholera toxin were attenuated by a PKA inhibitor (PKI) (Sigma) (Fig. 5C). Exposure alone to PKI did not alter the morphology of C6 cells (Fig. 5C). Moreover, the up-regulation of GFAP protein induced by cholera toxin was weakened by PKI (Fig. 5D). Cyclin D1 plays a critical role in the regulation of differentiation by regulating the cell-cycle control machinery (18). The cholera toxin-triggered degradation of cyclin D1 was also prevented by PKI (Fig. 5D). The findings indicate that the differentiation effect of cholera toxin was mediated by PKA-dependent mechanisms.
Fig. 5.

PKA activity mediates for cholera toxin-induced differentiation in C6 cells. (A and B) C6 cells were incubated with 10 ng/ml cholera toxin for the time indicated. (A) PKA activity was measured. (B) Total protein was extracted and blotted with antibodies against p-CREB and CREB. (C and D) C6 cells were pretreated with 10 μM PKI for 2 h and then treated with 10 ng/ml cholera toxin for an additional 48 h for morphology of C6 cells (C) (original magnification: ×200) and for Western blotting to evaluate GFAP, cyclin D1, and CREB phosphorylation (D).
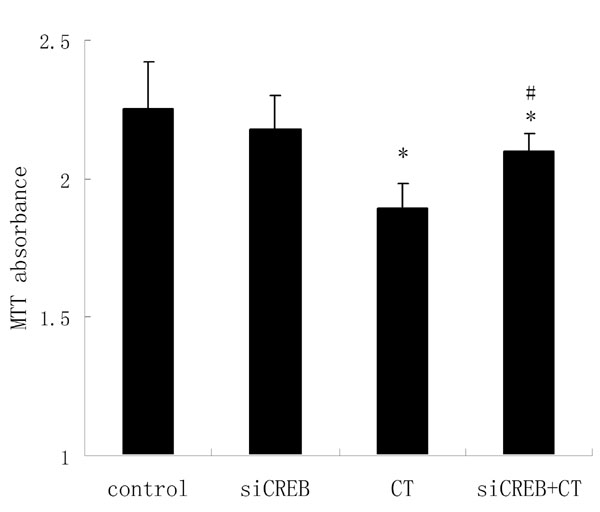
To further study the requirement for CREB, we used siRNA to selectively knock down this gene. After 48 h of transfection, the CREB level was greatly reduced compared with the control (Fig. 6A). Then cholera toxin was added, and cells were further incubated for 48 h. Similar effect to PKI was obtained (Fig. 5 B and C), and the proliferation inhibition induced by cholera toxin was also relieved (SI Fig. 8), indicating that CREB and the PKA/CREB pathway are directly involved in the process of differentiation in glioma cells.
Fig. 6.

Up-regulation of CREB is required for cholera toxin-induced differentiation in C6 cells. (A) Western blot was used to estimate the levels of CREB after transfection with 20 nM siCREB for 48 h. (B and C) C6 cells were pretreated with siCREB for 48 h and then treated with 10 ng/ml cholera toxin for an additional 48 h for morphology (B) (original magnification: ×200) and Western blotting to evaluate GFAP and cyclin D1 (C).
However, there is a small fraction of cells with processes that mimic cholera toxin-treated cells in the small interfering CREB (siCREB) group. This finding may be attributed to the low-serum culture for a long time (19). Nonetheless, the majority of cells in this group are more similar to those in the control group. Moreover, the level of CREB was not knock downed absolutely to 0% by siRNA (seen in Fig. 5A); thus, it was not surprising that several cells in the siCREB plus cholera toxin group do not look like control but still look differentiated.
Cholera Toxin Induces Differentiation of Primary Cultured Human Glioma Cells.
To test whether our findings extended to human glioma cells, we prepared primary cultures of human glioma cells from human glioma tissues. After exposure to cholera toxin as low as 10 ng/ml for 24 h, the primary human glioma cells also displayed all of the differentiated characteristics with a stellar shape with pronounced elongation of filamentous processes, whereas the controls were flattened and spindle-shaped (seen in Fig. 1 C and D). Furthermore, the primary cultured human glioma cells displayed the same alteration panel as rat C6 cell line did in the GFAP and Ki-67 levels (Fig. 2 C and G). Consistent with the C6 cells, primary human glioma cells also showed a concentration- and time-dependent manner in response to cholera toxin inducing up- and down-regulation of expression of GFAP and Ki-67 protein (Fig. 2 C, D, G, and H). By flow cytometry analysis, we observed that cholera toxin leads to an accumulation of cells in the G0/G1 phase (81.3% in treated cells compared with 51.1% in controls) (P < 0.01). In contrast, cells in S phase were dramatically decreased from 36.0% to 7.7% after exposure to cholera toxin for 48 h (P < 0.01) (Table 1). All of the above findings seem to demonstrate that cholera toxin is able to induce differentiation in both glioma cell line C6 and the human glioma cells of primary cultures.
Discussion
Rat C6 glioma cells are one of the well established glioma cell lines with an undifferentiated phenotype and oligodendrocytic, astrocytic, and neuronal properties, constituting a useful model in the studies of glial cell differentiation (20). In our study, we used C6 in conjunction with primary cultured human glioma cells, which are much more clinically relevant, to characterize the effect of cholera toxin on the key malignant phenotypes of malignant glioma cells to see whether it induces differentiation in them. Cholera toxin triggered cell transformation indicative of the cells' differentiation into a more mature astrocytic state. This differentiation potential was further confirmed by an increased expression of GFAP, a 50-kDa type III intermediate filament protein considered to be a reliable differentiation marker of normal astrocytes (20), and a lowered amount of Ki-67 protein. The Ki-67 antigen presents during all active phases of the cell cycle but is absent from resting cells exclusively in the nuclei of cycling cells; the defined period of nuclear expression makes it a reliable marker of malignant proliferating cells (21, 22). However, the B subunit of cholera toxin (choleragenoid) (Sigma) at doses of 10 ng/ml and as high as 10 μg/ml does not cause any alterations on cell morphology, GFAP expression, and proliferation in C6 glioma cells (SI Fig. 9). A mechanism that cholera toxin specifically activates ganglioside GM1 and induces differentiation in C6 glioma cells is therefore eradicated. Our results indicate that some percentage of malignant glioma cells dose exit from the proliferating cell cycle and then might be induced to differentiate by active whole cholera toxin but not inactive choleragenoid.
The regulation of cell proliferation and terminal differentiation is a critical aspect of normal development and homeostasis, but is frequently disturbed during tumorigenesis. Cell proliferation and differentiation are specifically controlled in the G1 phase and the G1/S phase transition in the cell cycle (23). We found that induction of differentiation triggered by cholera toxin was accompanied by cellular proliferation inhibition but not significant cell death and accumulation of cells in the G0/G1 phase of the cell cycle at multiple points within the machinery governing the G1/S transition. The expression of G1 control proteins cyclin D1 and Cdk2 was down-regulated and associated with profound increased p21Cip1 and p27Kip1 protein levels.
Cyclin D1 is a critical regulator involved in cell-cycle progression through the G1 phase into the S phase, thereby contributing to cell proliferation. Cumulative evidence indicates that, among G1 cyclins, cyclin D1 is most strongly implicated in tumorigenesis (24, 25). Inappropriate overexpression of the cyclin D1 protein and gene has been found in human gliomas (26–28). Cyclin D1 expression is significantly correlated with the degree of malignancy, invasion, and prognosis of patients in a variety of human carcinomas, including glioma (27, 29–31). In combination with cyclin E, Cdk2 is necessary for the G1-to-S-phase transition. Cdk2 activity is also necessary for entry into mitosis because it activates mitotic cyclin-cdc2 kinase activity (32). We demonstrate that cholera toxin inhibits the expression of cyclin D1 and Cdk2 and exerts other inhibitory effects in glioma cells, further encouraging the use of this drug in the chemoprevention and treatment of malignant glioma.
The Cdk inhibitors p21Cip1 and p27Kip1 play an important role in mediating growth arrest and are thought to function as brakes of the cell cycle (33). Considerable studies have been published on the roles of p21Cip1 and p27Kip1 in carcinogenesis of tumors, including glioma (34–36). The increase in p21Cip1 could, by inhibiting cyclin D1/Cdk4 or Cdk6 kinase activity, explain, at least in part, the increase of cells in the G1 phase by cholera toxin treatment in our study. The subsequent decrease in cyclin D1 may cooperate with the early induction of p21Cip1 and p27Kip1 to arrest cells in the G1 phase and thereby further contribute to cholera toxin-induced growth inhibition. In addition, blockade of cyclin D1-Cdk4 association prevented the sequestration of p21Cip1 and p27Kip1, inhibited cyclin A, and led to the abolition of Cdk2 activity, which is also necessary for the G1-to-S-phase transition in combination with cyclin E. The overexpression of p21Cip1 and p27Kip1 might therefore be a pivotal candidate for the perturbed cell-cycle progression and differentiation induced by cholera toxin.
PKA signaling has been reported to play important roles in multiple physiological processes, including growth, differentiation, gene regulation, and apoptosis (37). Disruption of the PKA catalytic subunit causes destabilization of the diploid cell cycle, and the cells start meiosis under conditions repressive for wild-type meiosis (38). One of the potential downstream signaling targets of PKA is the CREB protein, an inducible transcription factor. Phosphorylation of Ser-133 is a critical event in CREB activation, leading to an increase of transcriptional activation by the recruitment of additional coactivators. The phosphorylation of CREB by PKA-mediated cAMP-dependent signaling cascade responds to a variety of external signals and appears to play crucial roles in differentiation and neurite outgrowth in multiple cell lines (39–41). Cells from transgenic mice expressing a dominant-negative form of CREB showed a profound proliferative defect and G1 cell-cycle arrest in response to a number of different activation signals (42). It remains to be explored, however, whether PKA/CREB signaling participates in the differentiation of malignant glioma cells. Here, we observed that a cholera toxin-induced cAMP increase was able to trigger the activation of downstream effectors, as evidenced by PKA activation and CREB transcription factor phosphorylation. In addition, the essential role of PKA activation in the cholera toxin-induced differentiation process is supported by experiments showing that differentiation is impaired if PKA activity is inhibited by PKI added before the differentiating agent cholera toxin or depletion of CREB by siRNA.
Cyclin D1 expression may be regulated at the transcriptional level, and posttranscriptional mechanisms are also involved. However, cyclin D1 mRNA was not modified by cholera toxin in our study (data not shown), and there may be intermediary repressive factors beyond transcriptional regulation by CREB involved in the cyclin D1 down-regulation in our experiment. Recent evidence supports a role for ubiquitin-proteasome degradation of phosphorylated cyclin D1 in the control of cyclin D1 (43). There may be kinases that phosphorylate cyclin D1 to accelerate its destruction via an ubiqutin pathway in cholera toxin-induced differentiation and cyclin D1 down-regulation in glioma cells.
In conclusion, we confirmed that cholera toxin might be an astrocytic differentiation-inducing agent in rat C6 and primary cultured human glioma cells of intensified therapeutic interest in the treatment of glioma. This effect may be caused by G1 arrest in the cell cycle ascribed to the restrain of cyclin D1 and Cdk2 activities by augmented expression of p21Cip1 and p27Kip1 proteins. Of the various possibilities for cholera toxin-induced differentiation, elevated PKA/CREB signaling pathway represents a plausible candidate. Significantly, cholera toxin, the traditional bacterial toxin, may be a novel agent with substantial differentiation potential in the therapy of malignant glioma. Future studies, however, should extend these findings to in vivo tumor models.
Materials and Methods
Cell Culture and Drug Treatment.
C6 rat glioma cells were obtained from the American Type Culture Collection (Manassas, VA) and maintained in DMEM (Invitrogen, Grand Island, NY) supplemented with 10% FBS in a humidified atmosphere of 5% CO2 at 37°C. Human glioma tissues were obtained immediately after surgical removal with informed consent from each patient. Tissues were washed with PBS solution and minced into pieces of ≈2 mm in diameter. The tissue fragments were suspended in 0.25% trypsin (Invitrogen) and digested for 10 min at 37°C with a magnetic stirring bar. Supernatant cells were harvested, washed, filtered with a 200-μm mesh, and then resuspended in RPMI medium 1640 (Invitrogen) containing 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin. The remaining tissues were redigested for 10-min periods until cells were well dispersed. The cultures were kept at 37°C in a humidified atmosphere of 5% CO2, and the medium was replaced every third day.
C6 cell differentiation was induced with the indicated concentration of cholera toxin (Sigma) in DMEM containing 1.5% FBS. Control was treated with an equivalent volume of DMEM containing 1.5% FBS. For primary human glioma cells, indicated concentration of cholera toxin was added to the 10% FBS medium.
Morphological Evaluation.
The morphologies of untreated and cholera toxin-treated cells were studied during a time course of 48 h with an IX71 inverted microscope (Olympus, Melville, NY) and a DP70 CCD camera (Olympus).
Proliferation and Survival Assays.
Cell proliferation was evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay with a cell counting kit (CCK-8; Dojindo Molecular Technologies, Gaithersburg, MD). The cell proliferation rate = ODexperiment/ODcontrol × 100%.
Apoptosis was observed by Hoechst 33258 staining as described (44). Apoptotic cells were characterized by morphological alteration as condensed nuclei and cell shrinkage.
LDH release was quantified with a CytoTox 96 nonradioactive cytotoxicity assay kit (Promega, Madison, WI) according to the manufacturer's instructions. Plates were incubated at room temperature for 30 min in the dark, and absorbance was then measured at 490 nm with a EXL800 microimmunoanalyzer (BioTek, Burlington, VT).
Cell-Cycle Analysis.
A flow cytometry analysis of DNA content of cells was performed to assess the cell-cycle phase distributions as described (45) with minor modifications. In brief, the cells were collected by trypsinization, washed in PBS, and fixed in 70% ethanol for 30 min at 4°C. After washing with PBS, cells were incubated with the DNA-binding dye propidium iodide (50 μg/ml) and RNase (1.0 mg/ml) for 30 min at 37°C in the dark. Finally, cells were washed and red fluorescence was analyzed by a FACSCalibur flow cytometer (BD, Heidelberg, Germany) using a peak fluorescence gate to discriminate aggregates.
Measurement of PKA Activity.
C6 cells were plated in 100-mm dishes and allowed to attach and grow overnight. Medium was changed to DMEM containing 1.5% FBS, and 10 ng/ml cholera toxin was added to cultures for varying treatment periods. Medium was aspirated, and cell layers were washed twice in PBS and disrupted with cold lysis buffer containing 25 mM Tris·HCl, 0.5 mM EDTA, 0.5 mM EGTA, 10 mM 2-mercaptoethanol, 1 μg/ml leupeptin, and 1 μg/ml aprotinin followed by centrifugation at 14,000 × g at 4°C for 5 min. This supernatant was assayed for PKA activity by using a fluorescence-based kemptide phosphorylation assay kit (PepTag; Promega) following the manufacturer's instructions. Phosphorylated fluorescent substrate bands were visualized under UV illumination, excised, and quantified at 570 nm by spectrofluorimetry.
siRNA-Mediated Knockout of CREB Expression.
The CREB ShortCut siRNA Mix kit (New England Biolab, Ipswich, MA) was used to depletion CREB. Cells were transfected with siCREB (20 nM, 48 h) by using Lipofectamine 2000 (Invitrogen). Down-regulation of total CREB was evaluated by Western blotting. Total cell lysates were collected and analyzed for CREB, GFAP, and cyclin D1 protein expression by Western blotting.
Western Blot Analysis.
Western blot was performed as described (46). The following antibodies were used: antibodies against GFAP, cyclin D1, p-CREB, CREB, p-Akt, Akt, p-GSK-3β (1:1,000; Cell Signaling Technology, Beverly, MA), Cdk2, p21, p-Rb (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA), and β-actin (1:2,000; New England Biolabs). After incubation with horseradish peroxidase-labeled secondary antibody (1:1,000; Cell Signaling Technology), visualization was performed by enhanced chemiluminescence (Amersham Pharmacia Biotech, Piscataway, NJ) and exposure to autoradiographic film (Kodak, Rochester, NY).
Statistical Analysis.
Data are presented as mean ± SD of three separate experiments. Statistical significance was determined by Student's t test. A result with a P value of < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Pengxin Qiu, Ying Guo, Lijun Chen, Xingwen Su, and Yanqiu Ou for excellent technical assistance and continued support of this work. This research was supported by National Natural Science Foundation of China Grant 30472010 and Project of Natural Science Foundation of Investigation Team, Guangdong Province of China Grant 039191.
Abbreviations
- CREB
cAMP-responsive element binding
- GFAP
glial fibrillary acid protein
- LDH
lactate dehydrogenase
- PKI
PKA inhibitor
- siCREB
small interfering CREB.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0701990104/DC1.
References
- 1.Yiu G, He Z. Nat Rev Neurosci. 2006;7:617–627. doi: 10.1038/nrn1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeAngelis LM. N Engl J Med. 2001;344:114–123. doi: 10.1056/NEJM200101113440207. [DOI] [PubMed] [Google Scholar]
- 3.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 4.Curran WJ, Jr, Scott CB, Horton J, Nelson JS, Weinstein AS, Fischbach AJ, Chang CH, Rotman M, Asbell SO, Krisch RE, et al. J Natl Cancer Inst. 1993;85:704–710. doi: 10.1093/jnci/85.9.704. [DOI] [PubMed] [Google Scholar]
- 5.Leszczyniecka M, Roberts T, Dent P, Grant S, Fisher PB. Pharmacol Ther. 2001;90:105–156. doi: 10.1016/s0163-7258(01)00132-2. [DOI] [PubMed] [Google Scholar]
- 6.Wang ZY, Chen Z. Lancet Oncol. 2000;1:101–106. doi: 10.1016/s1470-2045(00)00017-6. [DOI] [PubMed] [Google Scholar]
- 7.Huang ME, Ye YC, Chen SR, Chai JR, Lu JX, Zhoa L, Gu LJ, Wang ZY. Blood. 1988;72:567–572. [PubMed] [Google Scholar]
- 8.Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, Zhu J, Tang W, Sun GL, Yang KQ, et al. Blood. 1997;89:3354–3360. [PubMed] [Google Scholar]
- 9.Kinjo K, Kizaki M, Muto A, Fukuchi Y, Umezawa A, Yamato K, Nishihara T, Hata J, Ito M, Ueyama Y, et al. Leukemia. 2000;14:431–438. doi: 10.1038/sj.leu.2401646. [DOI] [PubMed] [Google Scholar]
- 10.Goodman L, Segreti J. Dis Mon. 1999;45:268–299. doi: 10.1016/S0011-5029(99)90000-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moss J, Vaughan M. Annu Rev Biochem. 1979;48:581–600. doi: 10.1146/annurev.bi.48.070179.003053. [DOI] [PubMed] [Google Scholar]
- 12.Guerrant RL, Fang GD, Thielman NM, Fonteles MC. Proc Natl Acad Sci USA. 1994;91:9655–9658. doi: 10.1073/pnas.91.20.9655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Kolen K, Slegers H. J Neurochem. 2004;89:442–453. doi: 10.1111/j.1471-4159.2004.02339.x. [DOI] [PubMed] [Google Scholar]
- 14.Takanaga H, Yoshitake T, Hara S, Yamasaki C, Kunimoto M. J Biol Chem. 2004;279:15441–15447. doi: 10.1074/jbc.M311844200. [DOI] [PubMed] [Google Scholar]
- 15.Guerrant RL, Brunton LL, Schnaitman TC, Rebhun LI, Gilman AG. Infect Immun. 1974;10:320–327. doi: 10.1128/iai.10.2.320-327.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kimura M, Hidari KI, Suzuki T, Miyamoto D, Suzuki Y. Glycobiology. 2001;11:335–343. doi: 10.1093/glycob/11.4.335. [DOI] [PubMed] [Google Scholar]
- 17.Masco D, Van de Walle M, Spiegel S. J Neurosci. 1991;11:2443–2452. doi: 10.1523/JNEUROSCI.11-08-02443.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benda P, Lightbody J, Sato G, Levine L, Sweet W. Science. 1968;161:370–371. doi: 10.1126/science.161.3839.370. [DOI] [PubMed] [Google Scholar]
- 19.Helmbrecht K, Rensing L. Neurochem Res. 1999;24:1293–1299. doi: 10.1023/a:1020933308947. [DOI] [PubMed] [Google Scholar]
- 20.Roymans D, Vissenberg K, De Jonghe C, Grobben B, Claes P, Verbelen JP, Van Broeckhoven C, Slegers H. J Neurochem. 2001;76:610–618. doi: 10.1046/j.1471-4159.2001.00077.x. [DOI] [PubMed] [Google Scholar]
- 21.Schluter C, Duchrow M, Wohlenberg C, Becker MH, Key G, Flad HD, Gerdes J. J Cell Biol. 1993;123:513–522. doi: 10.1083/jcb.123.3.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, Bachmann MH, Borowsky AD, Ruebner B, Cardiff RD, et al. Nature. 2004;431:1112–1117. doi: 10.1038/nature03043. [DOI] [PubMed] [Google Scholar]
- 23.Nurse P. Cell. 2000;100:71–78. doi: 10.1016/s0092-8674(00)81684-0. [DOI] [PubMed] [Google Scholar]
- 24.Lukas J, Pagano M, Staskova Z, Draetta G, Bartek J. Oncogene. 1994;9:707–708. [PubMed] [Google Scholar]
- 25.Fu M, Wang C, Li Z, Sakamaki T, Pestell RG. Endocrinology. 2004;45:5439–5447. doi: 10.1210/en.2004-0959. [DOI] [PubMed] [Google Scholar]
- 26.Cuevas P, Diaz-Gonzalez D, Dujovny M. Neurol Res. 2003;25:691–693. doi: 10.1179/016164103101202165. [DOI] [PubMed] [Google Scholar]
- 27.Buschges R, Weber RG, Actor B, Lichter P, Collins VP, Reifenberger G. Brain Pathol. 1999;9:435–443. doi: 10.1111/j.1750-3639.1999.tb00532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holland EC. J Neurooncol. 2001;51:265–276. doi: 10.1023/a:1010609114564. [DOI] [PubMed] [Google Scholar]
- 29.Arato-Ohshima T, Sawa H. Int J Cancer. 1999;83:387–392. doi: 10.1002/(sici)1097-0215(19991029)83:3<387::aid-ijc15>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 30.Lamb J, Ramaswamy S, Ford HL, Contreras B, Martinez RV, Kittrell FS, Zahnow CA, Patterson N, Golub TR, Ewen ME. Cell. 2003;114:323–334. doi: 10.1016/s0092-8674(03)00570-1. [DOI] [PubMed] [Google Scholar]
- 31.Landis MW, Pawlyk BS, Li T, Sicinski P, Hinds PW. Cancer Cell. 2006;9:13–22. doi: 10.1016/j.ccr.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 32.Guadagno TM, Newport JW. Cell. 1996;84:73–82. doi: 10.1016/s0092-8674(00)80994-0. [DOI] [PubMed] [Google Scholar]
- 33.Sherr C, Roberts JM. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 34.Weiss RH. Cancer Cell. 2003;4:425–429. doi: 10.1016/s1535-6108(03)00308-8. [DOI] [PubMed] [Google Scholar]
- 35.Jackson RJ, Adnane J, Coppola D, Cantor A, Sebti SM, Pledger WJ. Oncogene. 2002;21:8486–8497. doi: 10.1038/sj.onc.1205946. [DOI] [PubMed] [Google Scholar]
- 36.Komata T, Kanzawa T, Takeuchi H, Germano IM, Schreiber M, Kondo Y, Kondo S. Br J Cancer. 2003;88:1277–1280. doi: 10.1038/sj.bjc.6600862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walsh DA, Van Patten SM. FASEB J. 1994;8:1227–1236. doi: 10.1096/fasebj.8.15.8001734. [DOI] [PubMed] [Google Scholar]
- 38.Maeda T, Watanabe Y, Kunitomo H, Yamamoto M. J Biol Chem. 1994;269:9632–9637. [PubMed] [Google Scholar]
- 39.Sato K, Suematsu A, Nakashima T, Takemoto-Kimura S, Aoki K, Morishita Y, Asahara H, Ohya K, Yamaguchi A, Takai T, et al. Nat Med. 2006;12:1410–1416. doi: 10.1038/nm1515. [DOI] [PubMed] [Google Scholar]
- 40.Impey S, McCorkle SR, Cha-Molstad H, Dwyer JM, Yochum GS, Boss JM, McWeeney S, Dunn JJ, Mandel G, Goodman RH. Cell. 2004;119:1041–1054. doi: 10.1016/j.cell.2004.10.032. [DOI] [PubMed] [Google Scholar]
- 41.Masson N, Hurst C, Lee KA. Nucleic Acids Res. 1993;21:1163–1169. doi: 10.1093/nar/21.5.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barton K, Muthusamy N, Chanyangam M, Fischer C, Clendenin C, Leiden JM. Nature. 1996;379:81–85. doi: 10.1038/379081a0. [DOI] [PubMed] [Google Scholar]
- 43.Diehl JA, Zindy F, Sherr CJ. Genes Dev. 1997;11:957–972. doi: 10.1101/gad.11.8.957. [DOI] [PubMed] [Google Scholar]
- 44.Jenkins JK, Suwannaroj S, Elbourne KB, Ndebele K, McMurray RW. Int Immunopharmacol. 2001;1:1897–1911. doi: 10.1016/s1567-5769(01)00114-x. [DOI] [PubMed] [Google Scholar]
- 45.Roz L, Gramegna M, Ishii H, Croce CM, Sozzi G. Proc Natl Acad Sci USA. 2002;99:3615–3620. doi: 10.1073/pnas.062030799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Akagi T, Murata K, Shishido T, Hanafusa H. Mol Cell Biol. 2002;22:7015–7023. doi: 10.1128/MCB.22.20.7015-7023.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}