

Abstract
The loss of cell volume or cell shrinkage has been a morphological hallmark of the programmed cell death process known as apoptosis. This isotonic loss of cell volume has recently been term apoptotic volume decrease or AVD to distinguish it from inherent volume regulatory responses that occurs in cells under anisotonic conditions. Recent studies examining the intracellular signaling pathways that result in this unique cellular characteristic have determined that a fundamental movement of ions, particularly monovalent ions, underlie the AVD process and plays an important role on controlling the cell death process. An efflux of intracellular potassium was shown to be a critical aspect of the AVD process, as preventing this ion loss could protect cells from apoptosis. However, potassium plays a complex role as a loss of intracellular potassium has also been shown to be beneficial to the health of the cell. Additionally, the mechanisms that a cell employs to achieve this loss of intracellular potassium vary depending on the cell type and stimulus used to induce apoptosis, suggesting multiple ways exist to accomplish the same goal of AVD. Additionally, sodium and chloride have been shown to play a vital role during cell death in both the signaling and control of AVD in various apoptotic model systems. This review examines the relationship between this morphological change and intracellular monovalent ions during apoptosis.
Keywords: Apoptosis, Cell Shrinkage, Apoptotic Volume Decrease (AVD), Ion Channels, Volume Regulation, Potassium, Sodium
Introduction
Cells are known to be very efficient in every physiological process they undertake. For example cell division, migration, and differentiation occurs with remarkable accuracy, such that errors in these processes are relatively uncommon. Thus it is not surprising that the cells have also developed an efficient programmed cell death process known as apoptosis to eliminate unwanted or dying cells from the body. Apoptosis is a physiological mode of cell death that removes cells at a given time or in response to a given stimulus in the absence of an inflammatory response. It is defined by unique morphological and biochemical characteristics including cell shrinkage, nuclear condensation, internucleosomal DNA fragmentation, and the eventual formation of apoptotic bodies. While other characteristics have been ascribed to the apoptotic process such as externalization of membrane phosphatidylserine, depolarization of the mitochondrial membrane potential, and activation of specific proteases known as caspases, the loss of volume or cell shrinkage has been the single characteristic observed in all physiologic models of programmed cell death (Fig. 1).
Fig. 1.
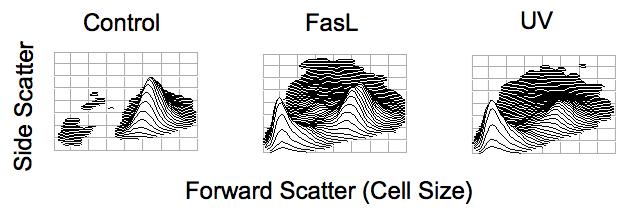
Apoptotic volume decrease (AVD) as observed by changes in the light scattering properties of a cell during apoptosis. Jurkat T-cells were treated with 25 ng/ml Fas Ligand or 30 mJ/cm2 UV for 4 hours. Cells were examined for changes in their light scattering properties by flow cytometry. Forward scattered light is a direct measure of cell size, with cells having a high forward scatter value being of a larger cell size, while cells having a lower forward scatter value are of a smaller cell size. Side scatter is measure of cellular granularity or complexity, with cells having a higher side scatter value being more granular. Upon apoptotic stimulation, a decrease in the cells ability to scatter light in the forward direction, along with a transient increase in the cells ability to scatter light at a side angle is observed indicating a loss of cell volume characteristic of apoptosis.
Early studies noted the unique morphological occurrence of cell shrinkage in dying cells that has become a hallmark of apoptosis [1.2]. Thomas and Bell [3] reported a significant decrease in cell size by electronic sizing in freshly isolated rat thymocytes upon glucocorticoid (dexamethasone) treatment. Dexamethasone treatment of the T-cell line CEM-C7A resulted in a 42% decrease in cell size after 48 hours [4]. Radiation-induced cell death of rat thymocytes resulted in rapid 25% decrease in cell size, followed by a slower reduction in cell volume to 57% their initial cell size compared to untreated controls [5]. Additionally, human eosinophils upon cytokine withdrawal had a 63.2% reduction in cell volume after 48 hours [6]. These studies highlight the predominance of a significant decrease in cell size as a key characteristic of apoptosis under various apoptotic conditions. However, it is not just this change in cell size that is important, but also the critical movement of ions that underlies this visual morphological observation.
Potassium as a Key Player in the Regulation of Apoptosis
When one considers the ions that may play a critical role in apoptosis, specifically that result in cell shrinkage, potassium has received considerable attention due to being the most abundant, osmotically important cation inside the cell [7]. In most mammalian cells, there is a high concentration of intracellular potassium and a low concentration of intracellular sodium. In contrast, outside of the cell there is a high concentration of extracellular sodium and a low concentration of extracellular potassium. Thus a concentration gradient exists for the loss of intracellular potassium and the gain of intracellular sodium in the cell. However, this normal concentration gradient of ions is maintained through the constitutive activation of various channels and ionic transporters, most notably the activity of the Na+/K+-ATPase. Additionally, the importance of maintaining this concentration gradient is highlighted in the cells ability to adapt to anisotonic conditions by inherent volume regulatory mechanisms that counter either a hypertonic environment via regulatory volume increase (RVI) response or a hypotonic environment via a regulatory volume decrease (RVD) response [8,9].
The loss of cell volume during apoptosis is unique in that it occurs in the absence of any osmotic imbalance. Thus cell shrinkage as observed during apoptosis occurs in an isotonic environment. The term “apoptotic volume decrease” or AVD has since been coined to refer to this unique characteristic of cell death to separate apoptotic cell shrinkage from normal volume regulatory responses resulting from anisotonic conditions [10]. Flow cytometry has been a simple and straightforward method to analyze AVD during apoptosis. Based on the light scattering properties of a cell, a decrease in forward-scattered light indicates cells of a shrunken or smaller cell size (Fig. 1). Since most cells can respond to changes in cell size via inherent volume regulatory responses, these mechanisms must be inhibited or overridden during the apoptotic process allowing for AVD. We showed in T-cells lacking a normal RVI response that hypertonic conditions resulted in the occurrence of apoptosis [11]. In contrast, we observed that COS-7 cells, L-cells, and PC12 cells, all known to have a functional RVI response, were resistant to hypertonic-induced apoptosis. However, it is known that these cells can undergo apoptosis given an appropriate stimulus. Therefore, the inherent RVI that would tend to counter AVD during the cell death process must be inactivated or overridden to permit apoptosis. Recently, Shimiu et al., [12] reported that inhibition of hypertonicity-induced cation channels sensitized HeLa cells to shrinkage-induced apoptosis.
Several studies have suggested that physical cell shrinkage as described above not only mimics AVD as observed during apoptosis, but also activates various signaling cascades involved in programmed cell death. Mannitol-induced apoptosis in bovine aortic endothelial cells was accompanied by activation of tyrosine and stress kinases, phosphorylation of focal adhesion contact-associated proteins paxillin and FAK, and elevation of intracellular free calcium [13], suggesting the activation of multiple signaling pathways under hyperosmotic conditions. Hyperosmolality was shown to activate the Akt pathway that prevented mild hyperosmolality-induced apoptosis in MDCK cells [14]. Hyperosmotic conditions were also shown to traffic the death receptor CD95 to the plasma membrane in rat hepatocytes, sensitizing cells to CD95L-induced apoptosis [15]. This sensitivity to CD95L-induced apoptosis upon hyperosmotic treatment was abolished with cyclic adenosine monophosphate, PKC, or JNK inhibition. These authors extended this study to show that the signal for CD95 trafficing to the plasma membrane involved oxidative stress and EGFR activation in a JNK-dependent receptor association [16]. Finally, acute elevation of osmolarity using either sucrose or NaCl resulted in an increase in the phosphorylation of the stress activated p38 protein kinase in NIH 3T3 fibroblasts that was further enhanced in the presence of cells expressing the constitutively active G protein Rac [17]. These studies highlight the activation of multiple signaling pathways that can occur under hypertonic conditions leading to or preventing apoptosis.
Initial studies on cell shrinkage and apoptosis revealed a critical loss of intracellular potassium as an important feature for the programmed cell death process that was independent of cell type or stimulus used to induce death [4,6, 18-21]. These early studies showed an efflux of intracellular ions, especially potassium, which was a defining event for the reduction of cell size. However, the efflux of intracellular potassium may not be the only ionic mechanism that contributes to AVD, as an inability of cells to take up extracellular potassium may also play a critical role in certain model systems. We have shown in anti-Fas treated Jurkat cells that the enhanced efflux of intracellular potassium is coupled to an early inability to take up potassium [22]. These data suggest that inhibition of the Na+-K+-ATPase may be an important ionic transport mechanism during apoptosis. Additionally, ouabain, a known inhibitor of the Na+-K+-ATPase was shown to dramatically enhance anti-Fas induced apoptosis in Jurkat cells, suggesting that alterations in the balance of ions can regulate the cell death process [22,23].
We have shown that many of the characteristics of apoptosis correlate with the shrunken population of cells [19,20,24,25]. For example, only the shrunken population of cells was shown to have degraded DNA [19]. This degraded DNA could be prevented in the presence of high extracellular potassium, which specifically impedes the efflux of this ion by diminishing its concentration gradient across the cell membrane. Interestingly, this study also showed that if cells were exposed to hypotonic conditions, where an initial loss of intracellular potassium occurs during the RVD response as cells regulate back to a near normal cell size, a resulting apoptotic stimulus enhanced the amount of cell death. Together, these studies support the hypothesis that alterations in the balance of intracellular ions, specifically potassium, can regulate apoptosis.
Recently, in a study of early morphological changes during staurosporine-induced apoptosis using atomic force microscopy in human epidermoid carcinoma (KB) cells, AVD was shown to precede key biochemical hallmarks of apoptosis including loss of the mitochondrial membrane potential, externalization of membrane phosphatidylserine, nuclear fragmentation, and measurable caspase-3 activity [26]. Additionally, it has been shown that AVD once initiated occurs within 12 to 20 minutes depending on the cell type and stimulus studied [27; Bortner and Cidlowski, unpublished data]. Apoptosis is known to be a very stochastic process, therefore only a small window of opportunity exists to capture individual apoptotic events as they relate to the loss of cell volume during apoptosis.
The study of AVD has now been evaluated in several types of cells. In rat epithelial cells, potassium channels blockers such as TEA, 4-AP, stromatoxin, chromanol 293B, and 48F10 were shown to prevent AVD and apoptosis [28]. Early studies on neuronal cell death also demonstrated the importance of potassium as a key regulator of apoptosis [29-31]. Murine cultured cortical neurons induced by either serum deprivation, staurosporine, or ß-amyloid fragments showed an enhanced potassium current that was sensitive to the potassium channel blocker TEA or high extracellular potassium. This protective effect of TEA or high extracellular potassium was initially thought to result from the subsequent activation of voltage-gated Ca2+ channels, however inhibition of Ca2+ influx with the broad-spectrum Ca2+ channel antagonist (gadolinium) or an L-type Ca2+ channel antagonist (nifedipine) did not alleviate the protective effect of TEA or high extracellular potassium [30]. Additionally, Bossy-Wetzel et al. [27] recently showed that application of exogenous nitric oxide to cerebrocortical neurons resulted in a loss in intracellular potassium in the distal neuritis, and this preceded a loss of potassium from the cell body. However in these cells, only the loss of potassium from the cell body was associated with cell shrinkage and membrane blebbing characteristic of apoptosis.
Interestingly, Bossy-Wetzel et al. [27] also showed in exogenous nitric oxide treated cerebrocortical neurons that the presence of the pan-caspase inhibitor z-VAD did not prevent potassium efflux and the associated AVD, suggesting that the loss of cell volume during apoptosis is not entirely dependent on caspase activity. An earlier study by our group also showed that apoptotic cell shrinkage could occur in the presence of caspase inhibitors in a stimulus-dependent manner [24]. Berg et al. [32] suggested that upon apoptotic stimulation, AVD in mature erythrocytes devoid of caspase-9, Apaf-1, and cytochrome c resulted from the activation of calpain, not caspases. Furthermore, several reports indicate that the mechanisms of volume loss or AVD during apoptosis can occur independent of the cell nucleus and other organelles [33,34]. For example, in erythrocytes, the Ca2+ ionophore ionomycin was shown to induce a cell death process analogous to apoptosis with cell shrinkage, cell membrane blebbing, and externalization of phosphatidylserine [34]. These studies suggest that AVD can be caspase-independent and independent of other intracellular organelles.
Potassium channels in Apoptosis
The importance of potassium efflux in regulating apoptosis has resulted in many studies examining the role of various potassium channels in regards to the loss of cell volume during apoptosis [35]. Potassium channels are a very diverse group with many different types contributing to the complex ionic flux observed during normal cellular homeostasis, as well as during apoptosis. The identification of a single potassium channel that is universally responsible for the efflux of intracellular potassium in all apoptotic model systems has yet to be reported. This suggests that distinct potassium channels or potassium transport mechanisms may be involved depending on the cell type or stimulus used to induce apoptosis (Table 1). Manikkam et al. [36] demonstrated this diversity in potassium channels where different effects on cellular proliferation, differentiation, and apoptosis occurred depending on the specific potassium antagonists applied to porcine granulosa cells. Dupart et al. [37] showed that diverse types of K+ channels are differently modified in response to reactive oxygen species (ROS), a known stimulus of apoptosis in many cell types. Additionally, differences in the expression patterns of various potassium channels can occur depending on the state of a given cell as illustrated for voltage-gated versus Ca2+-activated potassium channels in activated or differentiated lymphocytes [38]. These studies highlight some of the complexities in studying the role of potassium channels during apoptosis.
Table 1.
K+ channels shown to delay or prevent apoptosis for various cell types, along with the specific apoptotic inducer and K+ channel inhibitor.
| Cell Type | Type of K+ Channel | Apoptotic Inducer | Inhibitor | Ref |
|---|---|---|---|---|
| Eosinophils | Not Defined | Cytokine withdrawl | 4-AP | 6 |
| Murine in vivo model | Not Defined | D-galactosamine/Lipopolysaccharide | Quinine | 117 |
| Mouse Neocortical Neurons | Delayed retifier K+ channel | Serum deprivation, Staurosporine | TEA high extracellular K+ | 29 |
| HL-60 cells | Ca2+-activated K+ channels | UV-irradiation | TPA | 21 |
| Cholinergic Neurons (SN56) | Delayed retifier K+ channel | Amyloid ß-peptide | TEA high extracellular K+ | 118 |
| Mouse Cortical Neurons | Delayed retifier K+ channel | Amyloid ß-peptide | TEA high extracellular K+ | 119 |
| Mouse Cortical Neurons | Delayed retifier K+ channel | C2-ceramide Sphingomyelinase | TEA, clofilium high extracellular K+ | 120 |
| Thymocytes | Not Defined | Glucocorticoids Etoposide γ-irradiation Ceramide |
TPA | 121 |
| Myeloblastic leukemia cells (ML-1) | Voltage-gated K+ channels | UV irradiation | 4-AP | 122 |
| Rat Liver Cells (HTC) | Inward rectifier K+ channel | TNF-α | Barium Quinine | 109 |
| HeLa cells PC12 cells Lymphoid cells (U937) Neuronal cells (NG108-15) |
Inward rectifier K+ channel | Staurosporine TNFα/cycloheximide | Barium Quinine | 10 |
| Mouse embryos | Not Defined | Staurosporine H2O2 Oxidizing agent (Diamide) |
TEA | 123 |
| Mouse Cortical Neurons | Delayed retifier K+ channel | Staurosporine | Tetrapentylammonium (TPeA) Tetrahexylammonium (THA) |
124 |
| Mouse Cortical Neurons | Not Defined | Oxidizing agent 2,2-dithiodipyridine (DTDP) | TEA high extracellular K+ | 125 |
| Pulmonary artery smooth muscle cells | Voltage-gated K+ channels | Staurosporine | 4-AP high extracellular K+ | 126 |
| Pulmonary artery smooth muscle cells | Voltage-gated K+ channels | Staurosporine | 4-AP, overexpression Bcl-2 | 74 |
| CA1 pyramidal neurons | Not Defined | Forebrain ischemia | TEA | 76 |
| Pulmonary artery smooth muscle cells | Maxi-K+ channels | Carbonyl cyanide (FCCP) | TEA, Iberiotoxin high extracellular K+ | 127 |
| Mouse embryos | Two-Pore K+ channels (TREK) | H2O2 | Quinine | 57 |
| Rat sympathetic neurons | Voltage-gated K+ channels Kv7 (M-type) | Nerve Growth Factor (NGF) withdrawl | Linopirdine XE991 | 128 |
| Pulmonary artery smooth muscle cells | Large conductance Ca2+-activated K+ channels and Voltage-gated K+ channels | S-nitroso-acetylpenicillamine (SNAP), NO donor | TEA, Iberiotoxin 4-AP high extracellular K+ | 129 |
| Mouse cytoxic T lymphocyte line (CTLL-2 cells) | Voltage-gated K+ channels (Kv1.3 expression in CTLL-2 cells) | Actinomycin D | Kv1.3-deficient CTLL-2 cells | 43 |
| Human mammary gland adenocarcinoma cells (SK-BR-3), Neuroblastoma cells (SH-SY5Y), Rat atrial tumor cells (HL-1) | Delayed rectifier K+ channels (HERG) | H2O2 TNF-α | Dof | 51 |
| Neuron - Glia cultures | Delayed rectifier K+ channels | Ouabain | TEA high extracellular K+ | 130 |
| Erythrocytes | Ca2+-sensitive Gardos K+ channels | Ionomycin | Charybdotoxin Clotrimazole high extracellular K+ | 50 |
| CD4+ lymphocytes | Ca2+-activated K+ channel | Calcimycin (A23187) calcium ionophore | Quinine Clotrimazole Charybdotoxin | 49 |
| Rat granule neurons | TASK K+ channels | Low K+ medium | Ruthenium Red Extracellular acidosis | 55 |
| Cortical Neurons | Voltage-gated K+ channels Kv2.1 | Staurosporine Oxidant | Dominant-Negative Kv2.1 constructs | 45 |
| Human erythroleukemia cells (K-562) | Small-conductance Ca2+-activated K+ channels | Ionomycin | Antisense oligonucleotide against hSK4 (KCNN4) | 50 |
| Mouse Cortical Neurons | Delayed retifier K+ channel | Hypoxia/Ischemia | TEA Clofilium | 31 |
| Rabbit corneal epithelial cells (RCE) Primary cultured rabbit corneal epithelial cells (PRCE) |
Voltage-gated K+ channels | UV-irradiation | 4-AP | 71 |
| Cardiomyocytes (H9c2 cells) | Voltage-gated K+ channels | Staurosporine | 4-AP and ARC (apoptotic repressor with caspase recruitment domain) | 131 |
| Cultured Cortical Neurons | Not Defined | Staurosporine C2-ceramide serum deprivation | TEA Clofilium | 68 |
| Neuron - Glia cultures | Voltage-gated K+ channels Kv2.1 | Oxidizing agent 2,2-dithiodipyridine (DTDP) | 48F10 | 46 |
| COS-7 cells / Pulmonary artery smooth muscle cells | Voltage-gated K+ channels (KCNA5 overexpression) | Staurosporine | 4-AP | 48 |
| HEK293 cells expressing HERG channels | Delayed rectifier K+ channels (HERG) | H2O2 | Dofetilide E-4031 | 53 |
| Cerebrocortical Neurons | Delayed rectifier K+ channel | S-nitrosocysteine (SNOC), NO donor | TEA | 27 |
| Rat epithelial cells (IEC-6) | Not Defined | Proteasome inhibitor MG-132 | TEA, 4-AP stromatoxin chromanol 293B 48F10 | 28 |
| Non-dopaminergic rat cortical neurons expressing dopamine transporter | Voltage-gated K+ channels Kv2.1 | 6-Hydroxydopamine (6-OHDA) | TEA Scyllatoxin (ScTX) | 132 |
| HL-60 cells | Not Defined | Tachyplesin (cationic peptide) | Ba2+ | 133 |
| Rat Hippocampal Neurons | Voltage-gated K+ channels | Gluamate | TEA | 134 |
| Cerebellar Granule Neurons | Voltage-gated K+ channels | Serum-free medium, Low depolarizing K+ | 4-AP | 135 |
| Type I Spiral Ligament Fibrocytes | Voltage-gated K+ channels, Big-conductance Ca2+-activated K+ channels | Cisplatin | TEA Iberiotoxin | 136 |
| Cortical Neurons | Delayed rectifier K+ channels | Amyloid ß-peptide | TEA | 137 |
The n-type potassium channel Kv1.3 has long been a channel of interest for the activation and execution of apoptosis, especially in T lymphocytes [39-41]. A stimulation of Kv1.3 was shown to occur upon ligation of the Fas receptor in Jurkat T-cells undergoing apoptosis [41]. This study suggested that a key step in Kv1.3 activation is the inhibition of the Na+-K+-ATPase, signifying that changes in the plasma membrane potential provides the signal for activation of this channel that results in apoptosis. Subsequent inactivation of Kv1.3 during CD95/Fas-induced apoptosis was shown to result via tyrosine phosphorylation in Jurkat T lymphocytes [39]. Additionally, ceramide generated upon Fas receptor ligation was shown to inhibit n-type K+ channel activity [42]. This ceramide-induced Kv1.3 channel inactivation in Jurkat T lymphocytes was also shown to occur by tyrosine phosphorylation through the activation of p56lck. Thus, the eventual inhibition of Kv1.3 may lead to apoptosis through its inability to regulate changes in cell volume and a sustained level of plasma membrane potential.
Bock et al. [43] observed in CTLL-2 T-leukemic cells genetically deficient for Kv1.3, a resistance to actinomycin D-induced apoptosis. Re-transfection of Kv1.3 into these cells restored sensitivity to actinomycin D-induced cell death. In a study looking at the expression of voltage-gated potassium channels in normal and human prostate cancer specimens, Kv1.3 was highly expressed in normal prostate epithelium cells and in approximately half of the prostate cancer cells, with the other half of prostate cancer cells having moderate to low expression of Kv1.3 [44], suggesting a correlation of K+ channel expression and the probability of metastatic disease. In this study, K+ channel openers (minoxidil, 1-Ethyl-2-benzimidazolinone, and diazoxide) were shown to stimulate cell growth in four tested human prostate cell lines, while K+ channel blockers (dequalinium, amiodrone, and glibenclamide) had a significant growth suppressive effect resulting in apoptosis within 4 hours of treatment. Therefore in contrast to the conventional role of K+ channels during apoptosis, these results suggest that depending on the cell type, K+ channel activity can be beneficial to cellular health, while K+ channel inhibition results in cell death, perhaps simply by serving as a death signal.
Other voltage-gated potassium channels have also been shown to be key players in certain apoptotic model systems. Kv2.1 was shown to be necessary in cortical neurons during oxidant or staurosporine-induced apoptosis [45]. Using a yeast-based high-throughput screening assay, Zaks-Makhina et al. [46] reported a novel Kv2.1 channel blocker that prevented in vitro neuronal apoptosis at nontoxic concentrations. Kv1.2 expression was shown to be elevated in adult rat brain model of cell death following middle cerebral artery occlusion and in SH-SY5Y neuroblastoma cells following hypoxia and glucose deprivation [47]. The overexpression of Kv1.5 potassium channels in COS-7 or pulmonary artery smooth muscle cells (PASMC) resulted in an enhancement of staurosporine-induced apoptosis by accelerating the apoptotic volume decrease and increasing caspase-3 activity [48]. Thus numerous voltage-gated potassium channels have been suggested to play a role in apoptosis.
Voltage-gated channels have not been the only type of potassium channel implicated in apoptosis. For example, AVD mediated by an IKCa1 potassium channel upon calcium-induced apoptosis was reported in lymphocytes and thymocytes [49]. This study also showed that inhibition of IKCa1 with quinine, clotrimazole, or charybdotoxin prevented the externalization of phosphatidylserine, a classic characteristic of apoptosis, suggesting that ionic fluxes may play a critical role in the occurrence of other apoptotic characteristics beyond solely the loss of cell volume. In erythrocytes induced with ionomycin, AVD, membrane blebbing, and externalization of phosphatidylserine were shown to be dependent in part on the activation of the Ca2+-sensitive Gardos K+ channels [50]. Charybdotoxin or clotrimazole, both Gardos K+ channel blockers, or the presence of high extracellular potassium was shown to blunt these classical features of apoptosis during ionomycin-induced erythrocyte cell death. The human ether-a-go-go related gene (HERG) that encodes a delayed rectifier K+ channel was shown to promote H2O2-induced apoptosis in a variety of tumor cells expressing this channel, in direct contrast to cells that lacked HERG expression [51]. Additionally, cell lines with high HERG potassium channel expression have been shown to be more sensitive to chemotherapeutic drugs than cells with a lower HERG expression [52]. Over-expression of HERG in low expressing cells resulted in an increase in sensitivity to drug treatment, again suggesting that potassium channels can be major players for the control and/or activation of apoptosis. Furthermore, in HEK293 cells stably expressing HERG, an increase in apoptosis was observed upon H2O2 treatment that was absent in wild-type cells [53].
Potassium leak channels, such as 2P-domain channels TASK, TALK, and TWIK, have also been reported to play an important role in the loss of potassium and apoptosis [54], specifically in cultured cerebellar granule neurons [55] and in exocrine pancreatic cells [56]. In mouse embryos it was shown that recruitment of a two-pore domain K+ (K2P) channel underlies AVD that was prevented with the K+ channel blocker quinine [57]. However, these tandem pore domain K+ channels may also play a protective role as shown by Liu et al. [58], where overexpression of these channels enhanced cell viability by inhibiting the activation of intracellular apoptotic pathways. Thus the loss of intracellular potassium as observed during AVD can be accomplished through a variety of mechanisms depending on the cell type or stimulus used to induce apoptosis, and may be beneficial or detrimental to the overall health of the cell.
Thus one can imagine that not a single channel, but multiple potassium channels may be involved in cell death, providing a complementary array of ionic transport mechanisms to ensure the death of the cell. Therefore, this complex movement of ion fluxes makes if difficult to determine the exact molecular mechanisms involved in apoptosis. For instance, if one potassium channel is the primary route of K+ efflux during apoptosis, inhibition of this channel may be compensated via secondary channels, obscuring the identification of the key ionic efflux pathway. Compensation of this nature has been shown in the generation of Kv1.3 deficient mice [59]. Thymocytes from Kv1.3 deficient mice showed no detectable voltage-dependent potassium current, however the level of chloride current in these cells was increased approximately 50-fold over that in wild-type mice. There were no abnormalities in lymphocyte types or numbers, no obvious defects in thymocyte apoptosis or T cell proliferation, and no apparent immune system defects in the Kv1.3 deficient mice, suggesting a compensatory effect of increased chloride conductance in the absence of detectable potassium current. As chloride channels also play an important role in the regulation of cell volume, several ions may participate in the AVD response to an apoptotic stimulus.
Potassium as a Signal for Apoptosis
What have evolved from studies on AVD and cell death are not only a link between apoptotic cell shrinkage and loss of intracellular potassium, but also a critical role that intracellular ions play in regulating this programmed cell death process. The high intracellular potassium concentration found in most mammalian cells has focused studies of AVD and cell shrinkage during apoptosis on this particular ion. Consequently these studies have shown that altering the intracellular concentration of potassium has a dramatic effect on the apoptotic process. Enhancing the efflux of potassium has been shown to augment apoptosis [19, 60], suggesting that the normal intracellular concentration of potassium has a repressive effect on the death process. In a separate study, apoptotic nuclease activity was shown in vitro to be prevented by increasing concentrations of potassium [20]. In this study, it was also reported that the activation of caspase-3-like enzymes was also under the influence of the intracellular ionic composition of the cell, where both apoptotic nuclease activity and caspase-3-like activation were restricted to the shrunken population of cells. Interestingly, only the activation of caspases was sensitive to the potassium concentration, as in vitro experiments showed that once activated, caspase activity was not inhibited by the presence of high potassium. Thus by simply controlling the level of intracellular potassium, one could regulate apoptotic nuclease activity and caspase activation during apoptosis.
Elevated extracellular potassium was shown to prevent apoptosis in both Jurkat T-cells [19] and in the human monocytic tumour cell line THP.1 [61] under a variety of apoptotic conditions prior to externalization of phosphatidylserine, mitochondrial depolarization, cytochrome c release, caspase activation, and DNA degradation. Additionally, Cain et al. [62] showed that oligomerization of Apaf-1 and assembly of the active apoptosome complex is suppressed by normal intracellular concentrations of potassium, thus preventing caspase-9 activation. Interestingly, once assembled the apoptosome was shown to be relatively insensitive to the ionic effects surrounding this complex, similar to caspase-3. Therefore, the normal intracellular concentration of potassium acts to repress the apoptotic machinery by preventing the inappropriate formation of the apoptosome, caspase activation, and apoptotic nuclease activity.
Since it is well established that the loss of intracellular potassium results in AVD during apoptosis, many recent studies have focused on how the loss of this ion occurs and how it may signal other critical processes that regulate cell death. Gating characteristics of potassium channels are known to come under the influence of a variety of cytoplasmic factors such as kinases, phosphatases, small GTPases, as well as other intracellular second messengers. The activation of p38 was shown to be upstream and required for early potassium current in thiol oxidant-induced neuronal apoptosis [63]. This TEA sensitive current was attenuated by p38 inhibition, but not by caspase inhibition, suggesting that early kinase activity can have a direct effect on potassium channels involved in apoptosis. Additionally, src family tyrosine kinases are known to constitutively activate known apoptotic potassium channels such as Kv1.5 and Kv2.1 in mouse Schwann cells [64]. These currents are significantly downregulated with tyrosine kinase inhibitors such as herbimycin A and genistein, and upregulated upon introduction of recombinant Fyn kinase. In contrast, in SH-SY5Y neuroblastoma cells following hypoxia and glucose deprivation, Qiu et al. [47] showed that tyrosine phosphorylation of Kv1.2 via VEGF attenuated cell death, suggesting that the phosphorylation status of various potassium channels can directly control the apoptotic process. Structural-based functional analysis of Kv1.2 potassium channels revealed the importance of the N-terminal domain as an interacting partner with these second messengers in controlling the gating properties of these channels [65].
Similar to the diverse nature of K+ channels themselves, the exact role K+ channel phosphorylation may play in regards to apoptosis is equally complex. Han et al. [53] suggested that enhanced H2O2 apoptotic sensitivity in HEK293 cells stably expressing HERG K+ channels was due in part to the activation of p38, SAPKs, and ERKs MAP kinases. However, these kinase activities could be blocked with the HERG channel inhibitors dofetilide or E-4031. This suggests that in certain model systems, K+ loss promotes a later increase in kinase activity to enhance the activation of the apoptotic machinery, as opposed to the kinases directly regulating the loss of intracellular potassium via interacting with the K+ channels themselves.
A relatively unexplored area in regard to cell shrinkage during apoptosis are proteins that can modulate ion channel expression to boost ionic currents that might also be involved in the AVD process. These modulatory proteins do not directly affect the gating properties of the channel, but interact with various transcription factors to achieve their effect. The potassium channel-associated protein/protein inhibitor of activated STAT (KChAP/PIAS3B) is one such example. This protein channel modulator was shown to increase potassium efflux and reduce the cell size when expressed in the prostate cancer cell line LNCaP [66]. Additionally, these cells had an increased sensitivity to staurosporine-induced cell death, suggesting that apoptosis can be enhanced through ion channel modulation.
The level of intracellular potassium may affect not only K+ channels, but also the regulation and activity of other ion channels or events involved in AVD and apoptosis. As a general example of this type of regulation, it has recently been shown that declining intracellular potassium deactivates via dephosphorylation the cystic fibrosis transmembrane conductance regulator (CFTR) Cl- channel limiting the apical influx of Cl- and Na+ until the intracellular concentration of potassium is stabilized [67]. In regards to AVD and apoptosis, cultured cortical neurons treated with staurosporine or ceramide resulted in an inhibition of chloride currents preventing apoptotic cell shrinkage. However, these blockers did not significantly prevent caspase activation or DNA fragmentation [68]. In contrast, inhibition of potassium currents prevented cell shrinkage, caspase activation, and DNA fragmentation, suggesting distinct roles for individual ions in regulating different apoptotic events.
Additionally, cellular potassium content was also shown to selectively affect the DNA binding activity of transcription factors in vitro where a low potassium concentration promoted DNA binding of pro-apoptotic factors such as p53 and Forkhead, while inhibiting the anti-apoptotic transcription factor cAMP-responsive element-binding protein in regulating gene expression [69]. In cortical neurons, low intracellular potassium enhanced the binding of NF-κB to target gene DNA thus increasing its transcriptional activity without effecting its activation or nuclear translocation [70].
Conversely, the initial loss of intracellular potassium during apoptosis may not involve an ion channel modification, but an enhanced intracellular efflux of potassium through potassium leak channels. A unique feature of these two-pore domain K+ channels, suggested to play a role in various apoptotic model systems [48], is their independence from modulation by various intracellular messengers such as calcium, ATP, or arachidonic acid. Additionally, these channels function independent of changes in the transmembrane voltage and the cytoskeleton. Thus it has been suggested that the absence of cellular modulation of these channels allows them to be uniquely suited to participate in the early phases of apoptosis. It is important to remember that the particular potassium channels involved in AVD may be cell type or stimulus specific. In corneal epithelial cells, UV irradiation was shown to activate an early and robust K+ channel conductance resulting in cell death [71]. These authors showed that 4-AP suppressed this channel activity and prevented apoptosis. Interestingly, 4-AP had no effect on etoposide-induced cell death in epithelial cells, suggesting a specificity of a given type of potassium channel in response to a specific stimulus during apoptosis.
The expression of various pro- and anti-apoptotic proteins has been shown to have a direct effect on changes in intracellular potassium and thus on AVD during cell death. Cytoplasmic dialysis of cytochrome c in pulmonary vascular smooth muscle cells resulted in a rapid K+ current that occurred prior to nuclear condensation and independent of caspase-9 activity [72], suggesting that other components of the apoptotic process may regulate ion fluxes and AVD during the cell death process. An in vitro model of apoptosis was established in cholangiocytes using beauvericin, a K+ ionophore that bypassed the protective effect of the anti apoptotic oncoprotein Bcl-2 [73]. Interestingly, in this model system, abolishing the K+ gradient did not prevent apoptosis, though omission of extracellular Ca2+ or the addition of interleukin-1ß-converting enzyme (ICE) inhibitor did protect cholangiocytes from beauvericin-induced apoptosis, suggesting a Ca2+-dependent protease sensitive apoptotic pathway in the presence of Bcl-2 that results in AVD. In a separate study, Bcl-2 was shown to significantly diminish the activity of endogenous voltage-gated potassium channels in pulmonary artery smooth muscle cells [74]. Specifically, staurosporine was shown to enhance Kv currents and these currents were completely blocked by over-expressing Bcl-2 or with the potassium channel blocker 4-AP, both protecting the cells from apoptosis. Bcl-2 over-expression decreased the mRNA expression of several pore-forming Kv channel α-subunits such as Kv1.1, Kv1.5, and Kv2.1, suggesting that the protection afforded by Bcl-2 may also depend in part on the regulation of the expression of these ion channels.
An inhibition of potassium efflux may not be universally beneficial for cell survival, as the opening of K+ channels has been shown to promote cell survival in some apoptotic model systems. Protection of global ischemia-induced neuronal cell death in pyramidal cells of the CA1 field of the hippocampus was prevented in the presence of K+ channel openers such as cromakalim, nicorandil, and pinacidil [75]. A specific blocker of ATP-sensitive K+ channels abolished the beneficial effects of the K+ channel openers, demonstrating the advantageous effect of potassium efflux in protecting cells from apoptosis. Furthermore, the timing of K+ opening may play an important role for promoting either cell death or cell survival. Following transient forebrain ischemia, Huang et al. [76] showed that TEA, but not 4-AP, prevented CA1 hippocampal neuronal cell death if administered post-ischemic treatment, and neither K+ channel blocker prevented cell injury given pre-ischemic treatment. This data suggests not only a specific type of K+ channel activity is involved during apoptosis, but the timing of various K+ channel activity may also play an important role in the programmed cell death process.
The idea of ionic transport proteins such as exchangers or co-transporters as signal transducers during apoptosis has recently been demonstrated for the Na+, K+-ATPase by Zhang et al. [77]. Here the authors showed the direct interaction of the N-terminal tail of the catalytic α-subunit of the Na+, K+-ATPase with the N-terminus of the inositol 1,4,5-triphosphate receptor. This interaction induced by low concentrations of ouabain resulted in low frequency calcium oscillations that activated NF-κB and protected cells from apoptosis. Recently lipid rafts have been found to play an important role for the control of ionic transport proteins, receptor molecules, and ion channels [78-80]. These plasma membrane microdomains enriched in (glyco)sphingolipids and cholesterol can be fused by ceramide to form large platforms that play a role in activation or inhibition of various cell signals. In regards to apoptosis, Bock et al. [81] suggested that the potassium channel Kv1.3 is regulated by the composition of the cell membrane such that clustering of Kv1.3 into large lipid rafts or disruption of the smaller lipid rafts prevents channel activity. Thus K+ channel activity during apoptosis may be regulated by the size of these lipid microdomains.
What may be also important to consider during apoptosis is not the individual intracellular concentration of a single ion such as potassium, but the overall ionic strength of the intracellular environment. Recently, the importance of intracellular ionic strength has been reported to control many diverse cellular functions. For example, ionic strength was shown to control a conformation-dependent protease resistance of the mis-folded protein termed PrPSc, an essential component of infectious prions [82]. Here high ionic strength buffers conferred resistance to protease digestion, where as protease digestion in low ionic strength buffers was shown to be approximately 20 times more sensitive. In regards to apoptosis, Kahlenberg and Dubyak [83] showed that treatment of intact macrophage with extracellular ATP accelerated the in vitro processing of caspase-1 through the activation of the P2X7 receptor, a non-desensitizing cation channel that collapsed the intracellular ionic gradient resulting in an efflux of intracellular potassium. Cystathionine-ß-synthase (CBS) domains found in many proteins have recently been described as sensors for intracellular ionic strength [84]. These domains have been found in numerous channels and transporters, cation efflux systems, enzymes, and transcription factors. CBS domains in tandom were shown to control the transport activity in the osmoregulatory ABC transporter OpuA through an electrostatic switching mechanism based in the surface charge of the membrane and internal ionic strength. Thus these domains serve as a gating mechanism for membrane ionic transport for various transporters, and may play a role in the complex series of events leading to AVD and apoptosis.
Sodium as a Signal during Apoptosis
Sodium is another monovalent ion that has been shown to play a role during apoptosis. However, due to its low intracellular concentration in most mammalian cells, sodium has not received as much scientific attention as potassium. What has separated sodium from potassium during apoptosis is the occurrence of an early increase in sodium during the cell death process. Several initial studies on ions and apoptosis have reported this early increase in intracellular sodium [85-88]. Additionally, we have shown that apoptotic cells have a significant increase in intracellular sodium that occurs prior to the dramatic loss of cell volume with a variety of apoptotic agents in Jurkat T-cells [89]. Furthermore, in the prostate cancer cell line PC3, X-ray microanalysis of etoposide-induced apoptosis showed a progressive increase in intracellular sodium and magnesium along with the decrease in intracellular potassium and chloride in parallel with changes in cell volume [90].
The increase in intracellular sodium has been suggested to occur through the activation of various Na+ channels, although other mechanisms may exist. Sodium entry through voltage-sensitive sodium channels was shown to contribute to neuronal apoptosis [91]. A sodium influx was shown to largely mediate nitric oxide-induced cell death of cultured hepatocytes [92]. Hypoxia-induced apoptosis in cultured rat neocortical neurons was attenuated with the sodium channel blocker tetrodotoxin as accessed by a reduction in caspase-3 activation. Additionally, the sodium channel opener veratridine induced neuronal cell death, suggesting in certain model systems, sodium can be a key player in the activation of apoptosis. Veratridine was also a used to induce apoptosis in hippocampal neurons by preventing the inactivation of voltage-dependent sodium channels thus resulting in a sustained state of depolarization [93]. This insult resulted in the production of reactive oxygen species and the activation of p53, therefore linking initial changes in intracellular ions to downstream signaling events for apoptosis. A novel sodium channel blocking and free radical scavenging drug, AM-36 was shown to prevent neuronal apoptosis upon a veratridine insult by inhibiting both the increase in intracellular sodium and the production of reactive oxygen species [94].
Similar to potassium channels, the involvement or activation of sodium channels during apoptosis may come under specific regulation depending again on the stimulus and cell type employed. The increase in intracellular sodium during apoptosis may result from a shrinkage-activated sodium conductance as described by Böhmer et al. [95] in regards to the regulatory volume increase response in rat hepatocytes. The amiloride sensitivity of these channels in hepatocytes suggested they might be related to the epithelial Na+ channels (ENaCs). An interesting aspect of this study was that the coexpression of serine/threonine protein kinase SGK with ENaC in Xenopus oocytes altered the sensitivity of the channel to various drugs. Specifically, expression of SGK decreased the channel affinity to amiloride, while increasing its affinity to ethylisopropylamiloride (EIPA) a specific inhibitor of the Na+/H+ exchanger. Thus similar to the potential regulation of potassium channels, the activation or upregulation of various kinases may have a profound effect on the movement of sodium during apoptosis.
Ion channels may not be the only routes for sodium entry into the cell, as various sodium transporters are also thought to play a role during apoptosis. As mentioned earlier, the Na+/K+-ATPase has been shown to be critical component of programmed cell death in certain apoptotic model systems. An early inhibition of the Na+/K+-ATPase was shown in anti-Fas induced apoptosis in Jurkat T-cells [23, 89], as well as a loss in primary rat thymocytes treated with dexamethasone [96]. Interestingly saxitoxin, a sodium channel blocker, was also shown to prevent anti-Fas induced apoptosis in Jurkat T-cells [97]. As neither veratridine nor ouabain alone resulted in cell death, inhibition of the Na+/K+-ATPase coupled with an increase in sodium channel activity probably both contributed to the early increase in intracellular sodium. The result of this increase in intracellular sodium was a sustained state of cellular depolarization without repolarization, which again may be a critical signal for downstream apoptotic events.
Several early studies on apoptosis and ions have suggested cellular depolarization as an early event coinciding with AVD [98-100]. Nolte et al. [101] showed that a sodium influx contributed to cellular depolarization in the early stages of arsenic trioxide-induced apoptosis in human histiocytic lymphoma (U-937) cells. Interestingly, while sodium was shown to be the major ion involved in early cellular depolarization, this study also showed that chloride was the main ion responsible for the change in membrane potential during the later stages of apoptosis. However, neither of these ions was shown to play a role in cellular depolarization upon anti-Fas treatment of U-937 cells, highlighting that different mechanisms for apoptotic plasma membrane depolarization may exist even within a single cell type.
Sodium has also been shown to play a role in other events associated with apoptosis. Externalization of phosphatidylserine (PS) during apoptosis was shown to be responsive to changes in intracellular sodium [102]. In various thymocyte populations treated with extracellular ATP, rapid PS exposure occurred under conditions of physiological external sodium in the absence of external calcium. When the external concentrations of sodium and calcium were reversed, a submaximal response of PS exposure was observed, suggesting that sodium is more potent than calcium in inducing PS exposure. Additionally, intracellular sodium was shown to activate muscarinic K+ channels [103], and promote the dissociation between GαGDP and Gβγ to activate G protein-gated K+ channels [104]. These studies suggest that the influx of sodium during apoptosis may lead directly to the eventual loss of intracellular potassium and AVD.
One may consider that the increase in intracellular sodium during apoptosis is solely to balance the loss of intracellular potassium during the cell death process. However, this is not necessarily the case as a significant ionic imbalance has been shown to occur under a variety of apoptotic conditions. In rat thymocytes treated with dexamethasone or etoposide [105] or U-937 cells treated with staurosporine [106], apoptotic cell shrinkage was paralleled by a decrease in intracellular potassium that exceeded the increase in intracellular sodium, thus resulting in a significant decrease in total sodium and potassium content. In contrast, when U-937 cells, that showed an ionic imbalance with staurosporine, were treated with etoposide to induce apoptosis, the intracellular potassium to sodium ratio decreased, but the total sodium and potassium content remained virtually the same. Thus the loss of potassium is balanced by the gain in sodium, suggesting that other ions or osmolytes must be involved in the AVD process.
The increase in intracellular sodium as suggested in many of these studies might not be restricted to an early phase of the apoptotic process. Depending on the model system employed to study cell death, a late increase in intracellular sodium has also been reported during apoptosis. It was recently shown by x-ray microanalysis that staurosporine-induced [107] and UV-induced [108] apoptosis in U-937 cells resulted in an increase in intracellular sodium that occurs late in the cell death process. In these studies, the investigators could divide the changes in cellular elemental composition into two stages: an early stage characterized by a decrease in intracellular potassium concomitant with cell shrinkage and prior to caspase-3 activity or nuclear changes; and a later stage of decreased potassium content coupled with an increase in sodium content resulting from the activation of caspase-3. This late increase in intracellular sodium was suggested to result from a late inactivation of the Na+/K+-ATPase, thus different ionic transport mechanisms may be employed at various stages of the apoptotic process.
While it is generally expected that potassium can control the activation of the apoptotic machinery, our lab has revealed a unique role for sodium in controlling the change in cell size or AVD during apoptosis [97]. In this study, Jurkat T-cells under sodium-substituted conditions were treated with anti-Fas to induce apoptosis. Apoptotic stimulation in the absence of physiological levels of extracellular sodium resulted in cell swelling upon anti-Fas treatment. However, these swollen cells had many of the classical characteristics of apoptosis including chromatin condensation, externalization of phosphatidylserine, caspase activity, PARP cleavage, and internucleosomal DNA fragmentation. Interestingly, upon reintroduction of extracellular sodium to these swollen, apoptotic cells, the cells immediately shrank and displayed the characteristic apoptotic morphology. This shows that cell shrinkage or AVD can be distinguished from other characteristics of apoptosis and the movement of ions plays a defining role in both the morphological and biochemical aspects of this cell death process.
Chloride as a Counter-Ion
While the focus of this review has been on the role of monovalent cations during AVD and apoptosis, a growing amount of evidence also suggests that anions, particularly chloride plays an equally important role during the cell death process. Cells maintain given cell size by maintaining a consistent ratio of positive and negative ions inside the cell relative to their extracellular environment. Thus the loss or gain of a cation is usually countered by the loss or gain of an anion to maintain this homeostatic balance of ions and to remain electrochemically neutral. The loss of intracellular potassium during AVD as described above has also been shown in several studies to be paralleled by the activation of a chloride current [10,68,109] in a variety of cell types and apoptotic stimuli [reviewed in 110]. Similar to the protective effect of various K+ channels inhibitors; pharmacological blockade of Cl- channels has also been shown to prevent AVD and apoptosis [110].
Early studies on ions and apoptosis showed the activation of an outwardly rectifying chloride current (ORCC) upon CD95 receptor stimulation in Jurkat T-cells [111]. This current was shown to be mediated by Src-like tyrosine kinases, and was abolished by the tyrosine kinase inhibitor herbimycin or absent in cells genetically deficient in p56lck. Additionally, a swelling-induced chloride current was present in Xenopus oocytes upon treatment with pro-apoptotic sphingolipids that could be inhibited with NPPB or lanthanum, but not niflumic acid [112], suggesting a potential role for the movement of chloride during apoptosis. Additionally, Myssina et al. [113] showed that the Cl- channel blockers NPPB and niflumic acid blunted AVD and externalization of phosphatidylserine in ionomycin treated erythrocytes.
While parallel activation of K+ and Cl- channels has been reported during apoptosis, the roles of each of these currents during apoptosis may be different. In cultured cortical neurons, CLC-2 and CLC-3 chloride channel activity was detected upon staurosporine, C2-ceramide, or serum deprivation resulting in AVD and apoptosis [68]. Addition of the Cl- channel blockers DIDS, SITS, or NPPB prevented AVD, however caspase activation and/or DNA fragmentation were still present. In contrast, addition of the K+ channel blockers TEA or clofilium prevent AVD along with all other characteristics of apoptosis. These results not only suggest the occurrence of apoptosis in the absence of cell shrinkage, but distinct roles for K+ and Cl- during apoptosis. Interestingly, it has also been proposed that Cl- channels may play a dual role depending on the specific type of cell death, such that Cl- channels have a protective effect if activated during necrosis, while having a cell-killing effect if activated during apoptosis [114]. Recently, Heimlich and Cidlowski [115] showed that the Cl- channel blocker SITS or medium with reduced chloride concentration diminished UV-C induced apoptosis in Jurkat T-cells, but was ineffective in preventing apoptosis induced through the death receptor pathway.
The most extensively studied Cl- channel for its role in AVD has been the volume-sensitive outwardly rectifying (VSOR) Cl- channel, a swelling-activated channel characterized by the activation and inhibition by cellular swelling and shrinkage, respectively [110,116]. Maeno et al. [10] showed in HeLa, lymphoid, neuronal, and PC12 cells stimulated with a variety of apoptotic agents that the inhibition of this Cl- channel with either NPPB, DIDS, SITS or glibenclamide prevented AVD. Additionally they showed that phloretin, a volume-sensitive Cl- channel blocker know inhibit VSOR Cl- channels at low concentrations but not other types of Cl- channels, was also effective in preventing AVD. Thus while the major focus of ionic studies during AVD and apoptosis has centered on the roles of potassium and sodium, the movement of anions particularly chloride cannot be discounted.
Conclusion
A hallmark of apoptosis is the morphological occurrence of cell shrinkage, known as the apoptotic volume decrease or AVD response. For many years this distinguishing characteristic of AVD during programmed cell death was thought to be inconsequential in relation to the other events associated with this mode of cell death. However, it is now general accepted that the movement of ions that underlies this loss of cell volume, executes an important role in controlling the apoptotic process, not only in regards to AVD, but also in the control of the apoptotic machinery. This universal loss of cell volume during apoptosis would appear to occur by a straightforward and uncomplicated mechanism, common in all cell types and stimuli used to study this mode of cell death. However from numerous studies probing the mechanism(s) behind this apparent simple apoptotic event, a complex series of events has emerged. Surprisingly, the activation and regulation of specific ionic transport mechanisms that results in AVD appears to be different for each cell type and stimulus used to induce apoptosis. Therefore, multiple ways exist to accomplish the same goal of AVD or cell shrinkage during apoptosis.
In summary, figure 2 shows that upon apoptotic stimulation a cell goes through a series of complex cellular events, most notably a deregulation of its intracellular ionic balance that results in AVD. A flux of ions, particularly the efflux of intracellular potassium has been shown in general to underlie this morphological event. Coupled with this loss of intracellular ions, the cell may also lose the ability to take up ions, as exemplified by an early inhibition of the Na+/K+-ATPase in certain model systems. This dramatic decrease in intracellular ions results in a cellular ionic environment permitting the activation of various cell death enzymes including caspases and apoptotic nucleases. The presence of high extracellular potassium prevents AVD by inhibiting the efflux of this ion, indicating that the normal intracellular ionic environment has a repressive effect on the apoptotic process (Fig. 2). While AVD and the accompanying loss of intracellular ions is the customary result of apoptotic stimulation, the presence of extracellular sodium has been shown to control the morphological appearance of shrunken cells (Fig. 2). Therefore, multiple ions act in concert to achieve AVD. Consequently, it is not the physical observation of cell shrinkage that is vital in our understanding of apoptosis, but the underlying movement of ions that challenges our knowledge of this common apoptotic characteristic.
Fig. 2.

Ionic regulation of apoptosis. Cells use numerous ionic transport mechanisms including channels, co-transporters, and exchangers to maintain a constant cell volume. Under anisotonic conditions, these ionic transport mechanisms are activated to combat an increase or decrease in cell size through inherent RVD and RVI responses respectively. However upon apoptotic stimulation, a change in intracellular ions results from the activation or inhibition of stimulus or cell type specific ionic transport mechanisms including ion channels and ionic transport proteins. Under normal conditions of high extracellular sodium the apoptotic-induced change in intracellular ions, specifically the loss of intracellular potassium, results in a loss of cell volume or AVD permitting the activation of apoptotic enzymes. Interestingly, in the absence of extracellular sodium, this change in intracellular ions does not induce cell shrinkage or AVD, but results in cell swelling. However the change in intracellular ions still permits the activation of the apoptotic machinery. Thus, the presence or absence of extracellular sodium (red arrows) has been shown to control whether an apoptotic cell physically shrinks or swells, respectively during cell death. In contrast and independent of the presence or absence of extracellular sodium, high extracellular potassium (green arrows) can inhibit AVD along with preventing the activation of the apoptotic machinery to protect cells from apoptosis.
List of Abbreviations
- 4-AP
4-aminopyridine
- ABC
ATP-binding cassette
- AVD
apoptotic volume decrease
- CBS
cystathionine-β-synthase
- CFTR
cystic fibrosis transmembrane conductance regualator
- DIDS
4,4’-diisothiocyanatostilbene-2,2’-disulfonic acid
- EIPA
ethylisopropylamiloride
- ENaC
epithelial Na+ channel
- ERK
extracellular signal-related protein kinase
- ICE
interleukin-1β-converting enzyme
- MAP
mitogen-activated protein kinase
- NF-κB
nuclear factor kappa B
- NPPB
5-nitro-2-(3-phenylpropylamino)-benzoic acid
- PARP
poly ADP-ribose polymerase
- PS
phosphatidylserine
- ROS
reactive oxygen species
- RVD
regulatory volume decrease
- RVI
regulatory volume increase
- SAPK
stress-activated protein kinase
- SITS
4-acetamido-4’-isothiocyanatostilbene-2,2’disulfonic acid
- SGK
serum and glucocorticoid-inducible kinase
- STAT
signal transducers and activators of transcription
- TEA
tetraethylammonium
- VEGF
vascular endothelial growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Kerr JF, Wyllie AH, Currie AR. Br. J. Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wyllie AH. Nature. 1980;284:555–556. doi: 10.1038/284555a0. [DOI] [PubMed] [Google Scholar]
- [3].Thomas N, Bell PA. Mol. Cell. Endocrinol. 1981;22:71–84. doi: 10.1016/0303-7207(81)90103-9. [DOI] [PubMed] [Google Scholar]
- [4].Benson RSP, Heer S, Dive C, Watson JM. Am. J. Physiol. Cell Physiol. 1996;270:C1190–C1203. doi: 10.1152/ajpcell.1996.270.4.C1190. [DOI] [PubMed] [Google Scholar]
- [5].Klassen NV, Walker PR, Ross CK, Cygler J, Lach B. Int. J. Radiat. Biol. 1993;64:571–581. doi: 10.1080/09553009314551791. [DOI] [PubMed] [Google Scholar]
- [6].Beauvais F, Michel L, Dubertret L. J. Leukoc. Biol. 1995;57:851–855. doi: 10.1002/jlb.57.6.851. [DOI] [PubMed] [Google Scholar]
- [7].Franco R, Bortner CD, Cidlowski JA. J. Membr. Biol. 2006;209:43–58. doi: 10.1007/s00232-005-0837-5. [DOI] [PubMed] [Google Scholar]
- [8].Wehner F, Olsen H, Tinel H, Kinne-Saffran E, Kinne RK. Rev. Physiol. Biochem. Pharmacol. 2003;148:1–80. doi: 10.1007/s10254-003-0009-x. [DOI] [PubMed] [Google Scholar]
- [9].Pedersen SF, Hoffmann EK, Mills JW. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2001;130:385–399. doi: 10.1016/s1095-6433(01)00429-9. [DOI] [PubMed] [Google Scholar]
- [10].Maeno E, Ishizaki Y, Kanaseki T, Hazama A, Okada Y. Proc. Natl. Acad. Sci. U.S.A. 2000;97:9487–9492. doi: 10.1073/pnas.140216197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bortner CD, Cidlowski JA. Am. J. Physiol. Cell Physiol. 1996;271:C950–C961. doi: 10.1152/ajpcell.1996.271.3.C950. [DOI] [PubMed] [Google Scholar]
- [12].Shimizu T, Wehner F, Okada Y. Cell. Physiol. Biochem. 2006;18:295–302. doi: 10.1159/000097607. [DOI] [PubMed] [Google Scholar]
- [13].Melek AM, Goss GG, Jiang L, Izumo S, Alper SL. Stoke. 1998;29:2631–2640. doi: 10.1161/01.str.29.12.2631. [DOI] [PubMed] [Google Scholar]
- [14].Terada Y, Inoshita S, Hanada S, Shimamura H, Kuwahara M, Ogawa W, Kasuga M, Sasaki S, Marumo F. Kidney Int. 2001;60:553–567. doi: 10.1046/j.1523-1755.2001.060002553.x. [DOI] [PubMed] [Google Scholar]
- [15].Reinehr R, Graf D, Fischer R, Schliess F, Haussinger D. Hepatology. 2002;36:602–614. doi: 10.1053/jhep.2002.35447. [DOI] [PubMed] [Google Scholar]
- [16].Reinehr R, Schliess F, Haussinger D. FASEB J. 2003;17:731–733. doi: 10.1096/fj.02-0915fje. [DOI] [PubMed] [Google Scholar]
- [17].Friis MB, Friborg CR, Schneider L, Nielsen M-B, Lambert IH, Christensen ST, Hoffmann EK. J. Physiol. 2005;567:427–443. doi: 10.1113/jphysiol.2005.087130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Barbiero G, Duranti F, Bonelli G, Amenta JS, Baccino FM. Exp. Cell Res. 1995;217:410–418. doi: 10.1006/excr.1995.1104. [DOI] [PubMed] [Google Scholar]
- [19].Bortner CD, Hughes FM, Jr., Cidlowski JA. J. Biol. Chem. 1997;272:32436–32442. doi: 10.1074/jbc.272.51.32436. [DOI] [PubMed] [Google Scholar]
- [20].Hughes FM, Jr., Bortner CD, Purdy GD, Cidlowski JA. J. Biol. Chem. 1997;272:30567–30576. doi: 10.1074/jbc.272.48.30567. [DOI] [PubMed] [Google Scholar]
- [21].McCarthy JV, Cotter TG. Cell Death Diff. 1997;4:756–770. doi: 10.1038/sj.cdd.4400296. [DOI] [PubMed] [Google Scholar]
- [22].Bortner CD, Cidlowski JA. J. Biol. Chem. 2001;276:4304–4313. doi: 10.1074/jbc.M005171200. [DOI] [PubMed] [Google Scholar]
- [23].Nobel CSI, Aronson JK, van den Dobbelsteen DJ, Slater AFG. Apoptosis. 2000;5:153–163. doi: 10.1023/a:1009684713784. [DOI] [PubMed] [Google Scholar]
- [24].Bortner CD, Cidlowski JA. J. Biol. Chem. 1999;274:21953–21962. doi: 10.1074/jbc.274.31.21953. [DOI] [PubMed] [Google Scholar]
- [25].Gomez-Angelats M, Bortner CD, Cidlowski JA. J. Biol. Chem. 2000;275:19609–19619. doi: 10.1074/jbc.M909563199. [DOI] [PubMed] [Google Scholar]
- [26].Hessler JA, Budor A, Putchakayala K, Mecke A, Rieger D, Hall MMB, Orr BG, Bielinska A, Beals J, Baker J., Jr. Langmuir. 2005;21:9280–9286. doi: 10.1021/la051837g. [DOI] [PubMed] [Google Scholar]
- [27].Bossy-Wetzel E, Talantova MV, Lee WD, Schölzke MN, Harrop A, Matthews E, Götz T, Han J, Ellisman MH, Perkins GA, Lipton SA. Neuron. 2004;41:351–365. doi: 10.1016/s0896-6273(04)00015-7. [DOI] [PubMed] [Google Scholar]
- [28].Grishin A, Ford H, Wang J, Li H, Salvador-Recatala V, Levitan ES, Zaks-Makhina E. Am. J. Physiol. Gastrointest. Liver Physiol. 2005;289:G815–G821. doi: 10.1152/ajpgi.00001.2005. [DOI] [PubMed] [Google Scholar]
- [29].Yu SP, Yeh C-H, Sensi SL, Gwag BJ, Canzoniero LMT, Farhangrazi ZS, Ying HS, Tian M, Dugan LL, Choi DW. Science. 1997;278:114–117. doi: 10.1126/science.278.5335.114. [DOI] [PubMed] [Google Scholar]
- [30].Yu SP, Yeh C-H, Strasser U, Tian M, Choi DW. Science. 1999;284:336–339. doi: 10.1126/science.284.5412.336. [DOI] [PubMed] [Google Scholar]
- [31].Wei L, Yu SP, Gottron F, Snider BJ, Zipfel GJ, Choi DW. Stroke. 2003;34:1281–1286. doi: 10.1161/01.STR.0000065828.18661.FE. [DOI] [PubMed] [Google Scholar]
- [32].Berg CP, Engels IH, Rothbart A, Lauber K, Renz A, Schlosser SF, Schulze-Osthoff K, Wesselborg S. Cell Death Diff. 2001;8:1197–1206. doi: 10.1038/sj.cdd.4400905. [DOI] [PubMed] [Google Scholar]
- [33].Bratosin D, Estaquier J, Petit F, Arnoult D, Quatannens B, Tissler J-P, Slomianny C, Sartiaux C, Alonso C, Huart J-J, Montreuil J, Ameisen JC. Cell Death Diff. 2001;8:1143–1156. doi: 10.1038/sj.cdd.4400946. [DOI] [PubMed] [Google Scholar]
- [34].Lang F, Lang KS, Wieder T, Myssina S, Birka C, Lang PA, Kaiser S, Kempe D, Duranton C, Huber SM. Pflugers Arch. 2003;447:121–125. doi: 10.1007/s00424-003-1150-8. [DOI] [PubMed] [Google Scholar]
- [35].Burg ED, Remillard CV, Yuan JX-J. J. Membr. Biol. 2006;209:3–20. doi: 10.1007/s00232-005-0838-4. [DOI] [PubMed] [Google Scholar]
- [36].Manikkam M, Li Y, Mitchell BM, Mason DE, Freeman LC. Biol. Reprod. 2002;67:88–98. doi: 10.1095/biolreprod67.1.88. [DOI] [PubMed] [Google Scholar]
- [37].Duprat F, Guillemare E, Romey G, Fink M, Lesage F, Lazdunski M. Proc. Natl. Acad. Sci. U.S.A. 1995;92:11796–11800. doi: 10.1073/pnas.92.25.11796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Chandy KG, Wulff H, Beeton C, Pennington M, Gutman GA, Cahalan MD. Trends Pharmacol. Sci. 2004;25:280–289. doi: 10.1016/j.tips.2004.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Szabó I, Gulbins E, Apfel H, Zhang X, Barth P, Busch AE, Schlottmann K, Pongs O, Lang F. J. Biol. Chem. 1996;271:20465–20469. doi: 10.1074/jbc.271.34.20465. [DOI] [PubMed] [Google Scholar]
- [40].Lang F, Föller M, Lang KS, Lang PA, Ritter M, Gulbins E, Vereninov A, Huber SM. J. Membr. Biol. 2005;205:147–157. doi: 10.1007/s00232-005-0780-5. [DOI] [PubMed] [Google Scholar]
- [41].Storey NM, Gómez-Angelates M, Bortner CD, Armstrong DL, Cidlowski JA. J. Biol. Chem. 2003;278:33319–33326. doi: 10.1074/jbc.M300443200. [DOI] [PubMed] [Google Scholar]
- [42].Gulbins E, Szabo I, Baltzer K, Lang F. Proc. Natl. Acad. Sci. U.S.A. 1997;94:7661–7666. doi: 10.1073/pnas.94.14.7661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bock J, Szabó I, Jekle A, Gulbins E. Biochem. Biophys. Res. Comm. 2002;295:526–531. doi: 10.1016/s0006-291x(02)00695-2. [DOI] [PubMed] [Google Scholar]
- [44].Abdul M, Hoosein N. Can. Lett. 2002;186:99–105. doi: 10.1016/s0304-3835(02)00348-8. [DOI] [PubMed] [Google Scholar]
- [45].Pal S, Hartnett KA, Nerbonne JM, Levitan ES, Aizenman E. J. Neurosci. 2003;23:4798–4802. doi: 10.1523/JNEUROSCI.23-12-04798.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zaks-Makhina E, Kim Y, Aizenman E, Levitan ES. Mol. Pharmacol. 2004;65:214–219. doi: 10.1124/mol.65.1.214. [DOI] [PubMed] [Google Scholar]
- [47].Qiu M-H, Zhang R, Sun F-Y. J. Neurochem. 2003;87:1509–1517. doi: 10.1046/j.1471-4159.2003.02110.x. [DOI] [PubMed] [Google Scholar]
- [48].Brevnova EE, Platoshyn O, Zhang S, Yuan JX-J. Am. J. Physiol. Cell Physiol. 2004;287:C715–C722. doi: 10.1152/ajpcell.00050.2004. [DOI] [PubMed] [Google Scholar]
- [49].Elliott JI, Higgins CF. EMBO Rep. 2003;4:189–194. doi: 10.1038/sj.embor.embor722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Lang PA, Kaiser S, Myssina S, Wieder T, Lang F, Huber SM. Am. J. Physiol. Cell Physiol. 2003;285:C1553–C1560. doi: 10.1152/ajpcell.00186.2003. [DOI] [PubMed] [Google Scholar]
- [51].Wang H, Zhang Y, Cao L, Han H, Wang J, Yang B, Nattel S, Wang Z. Can. Res. 2002;62:4843–4848. [PubMed] [Google Scholar]
- [52].Chen S-Z, Jiang M, Zhen Y-S. Can. Chemother. Pharmacol. 2005;56:212–220. doi: 10.1007/s00280-004-0960-5. [DOI] [PubMed] [Google Scholar]
- [53].Han H, Wang J, Zhang Y, Long H, Wang H, Xu D, Wang Z. Cell. Physiol. Biochem. 2004;14:121–134. doi: 10.1159/000078104. [DOI] [PubMed] [Google Scholar]
- [54].Patel AJ, Lazdunski M. Pflugers Arch. 2004;448:261–73. doi: 10.1007/s00424-004-1255-8. [DOI] [PubMed] [Google Scholar]
- [55].Lauritzen I, Zanzouri M, Honoré E, Duprat F, Ehrengruber MU, Lazdunski M, Patel AJ. J. Biol. Chem. 2003;278:32068–32076. doi: 10.1074/jbc.M302631200. [DOI] [PubMed] [Google Scholar]
- [56].Duprat F, Girard C, Jarretou G, Lazdunski M. J. Physiol. 2005;562:235–244. doi: 10.1113/jphysiol.2004.071266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Trimarchi JR, Liu L, Smith PJS, Keefe DL. Am. J. Physiol. Cell Physiol. 2002;282:C588–C594. doi: 10.1152/ajpcell.00365.2001. [DOI] [PubMed] [Google Scholar]
- [58].Liu C, Cotton JF, Schuyler JA, Fahlman CS, Au JD, Bickler PE, Yost CS. Brain Res. 2005;1031:164–173. doi: 10.1016/j.brainres.2004.10.029. [DOI] [PubMed] [Google Scholar]
- [59].Koni PA, Khanna R, Chang MC, Tang MD, Kaczmarek LK, Schlichter LC, Flavell RA. J. Biol. Chem. 2003;278:39443–39451. doi: 10.1074/jbc.M304879200. [DOI] [PubMed] [Google Scholar]
- [60].Marklund L, Anderson B, Behnam-Motlagh P, Sandström P-E, Henriksson R, Grankvist K. Basic Clin. Pharmacol. Toxicol. 2004;94:245–251. doi: 10.1111/j.1742-7843.2004.pto940508.x. [DOI] [PubMed] [Google Scholar]
- [61].Thompson GJ, Langlais C, Cain K, Conley EC, Cohen GM. Biochem. J. 2001;357:137–145. doi: 10.1042/0264-6021:3570137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Cain K, Langlais C, Sun X-M, Brown DG, Cohen GM. J. Biol. Chem. 2001;276:41986–41990. doi: 10.1074/jbc.M107419200. [DOI] [PubMed] [Google Scholar]
- [63].McLaughlin B, Pal SP, Tran MP, Parsons AA, Barone FC, Erhardt JA, Aizenman E. J. Neurosci. 2001;21:3303–3311. doi: 10.1523/JNEUROSCI.21-10-03303.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Sobko A, Peretz A, Attali B. EMBO J. 1998;17:4723–4734. doi: 10.1093/emboj/17.16.4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Yi BA, Minor DL, Jr., Lin Y-F, Jan YN, Jan LY. Proc. Natl. Acad. Sci. U.S.A. 2001;98:11016–11023. doi: 10.1073/pnas.191351798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wible BA, Wang L, Kuryshev YA, Basu A, Haldar S, Brown AM. J. Biol. Chem. 2002;277:17852–17862. doi: 10.1074/jbc.M201689200. [DOI] [PubMed] [Google Scholar]
- [67].Reddy MM, Quinton PM. Am. J. Physiol. Cell Physiol. 2006;291:C122–C129. doi: 10.1152/ajpcell.00134.2005. [DOI] [PubMed] [Google Scholar]
- [68].Wei L, Xiao AY, Jin C, Yang A, Lu ZY, Yu SP. Pflugers Arch. 2004;448:325–334. doi: 10.1007/s00424-004-1277-2. [DOI] [PubMed] [Google Scholar]
- [69].Yang Q, Yan D, Wang Y. Neuroreport. 2006;17:1199–1204. doi: 10.1097/01.wnr.0000224. [DOI] [PubMed] [Google Scholar]
- [70].Tao Y, Yan D, Yang Q, Zeng R, Wang Y. Mol. Cell. Biol. 2006;26:1038–1050. doi: 10.1128/MCB.26.3.1038-1050.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Wang L, Li T, Lu L. Invest. Ophthalmol. Vis. Sci. 2003;44:50095–5101. doi: 10.1167/iovs.03-0590. [DOI] [PubMed] [Google Scholar]
- [72].Platoshyn O, Zhang S, McDaniel SS, Yuan JX-J. Am. J. Physiol. Cell Physiol. 2002;283:C1298–C1305. doi: 10.1152/ajpcell.00592.2001. [DOI] [PubMed] [Google Scholar]
- [73].Que FG, Gores GJ, LaRusso NF. Am. J. Physiol. Gastrointest. Liver Physiol. 1997;272:G106–G115. doi: 10.1152/ajpgi.1997.272.1.G106. [DOI] [PubMed] [Google Scholar]
- [74].Ekhterae D, Platoshyn O, Krick S, Yu Y, McDaniel SS, Yuan JX-J. Am. J. Physiol. Cell Physiol. 2001;281:C157–C165. doi: 10.1152/ajpcell.2001.281.1.C157. [DOI] [PubMed] [Google Scholar]
- [75].Heurteauz C, Bertanina V, Widmann C, Lazdunski M. Proc. Natl. Acad. Sci. U.S.A. 1993;90:9431–9435. doi: 10.1073/pnas.90.20.9431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Huang H, Gao TM, Gong L-W, Zhuang Z-Y, Li X. Neurosci. Lett. 2001;305:83–86. doi: 10.1016/s0304-3940(01)01821-3. [DOI] [PubMed] [Google Scholar]
- [77].Zhang S, Malmersjö S, Li J, Ando H, Aizman O, Uhlén P, Mikoshiba K, Aperia A. J. Biol. Chem. 2006;281:21954–21962. doi: 10.1074/jbc.M601578200. [DOI] [PubMed] [Google Scholar]
- [78].Simons K, Ikonen E. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- [79].Janes PW, Ley SC, Magee AL. J. Cell Biol. 1999;147:447–461. doi: 10.1083/jcb.147.2.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Szabó I, Adams C, Gulbins E. Pflugers Arch. 2004;448:304–312. doi: 10.1007/s00424-004-1259-4. [DOI] [PubMed] [Google Scholar]
- [81].Bock J, Szabó I, Gamper N, Adams C, Gulbins E. Biochem. Biophys. Res. Comm. 2003;305:890–897. doi: 10.1016/s0006-291x(03)00763-0. [DOI] [PubMed] [Google Scholar]
- [82].Nishina K, Jenks S, Supattapone S. J. Biol. Chem. 2004;279:40788–40794. doi: 10.1074/jbc.M406548200. [DOI] [PubMed] [Google Scholar]
- [83].Kahlenberg JM, Dubyak GR. Am. J. Physiol. 2004;286:C1100–C1108. doi: 10.1152/ajpcell.00494.2003. [DOI] [PubMed] [Google Scholar]
- [84].Biemans-Oldehinkel E, Mahmood NABN, Poolman B. Proc. Natl. Acad. Sci. U.S.A. 2006;103:10624–10629. doi: 10.1073/pnas.0603871103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Jonas D, Walev I, Berger T, Liebetrau M, Palmer M, Bhakdi S. Infect. Immun. 1994;62:1304–1312. doi: 10.1128/iai.62.4.1304-1312.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Offen D, Ziv I, Gorodin S, Barzilai A, Malik Z, Melamed E. Biochim. Biophys. Acta. 1995;1268:171–177. doi: 10.1016/0167-4889(95)00075-4. [DOI] [PubMed] [Google Scholar]
- [87].Skepper JN, Karydis I, Garnett MR, Hegyi L, Hardwick SJ, Warley A, Michinson MJ, Cary NRB. J. Pathol. 1999;188:100–106. doi: 10.1002/(SICI)1096-9896(199905)188:1<100::AID-PATH306>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- [88].Fernández-Segura E, Cañzaris FJ, Cubero MA, Warley A, Campos A. Exp. Cell Res. 1999;253:454–462. doi: 10.1006/excr.1999.4657. [DOI] [PubMed] [Google Scholar]
- [89].Bortner CD, Gomez-Angelates M, Cidlowski JA. J. Biol. Chem. 2001;276:4304–4314. doi: 10.1074/jbc.M005171200. [DOI] [PubMed] [Google Scholar]
- [90].Salido M, Vilches J, Lopez A, Roomans GM. Cell Biol. Int. 2001;25:499–508. doi: 10.1006/cbir.2000.0763. [DOI] [PubMed] [Google Scholar]
- [91].Banasiak KJ, Burenkova O, Haddad GG. Neurosci. 2004;126:31–44. doi: 10.1016/S0306-4522(03)00425-1. [DOI] [PubMed] [Google Scholar]
- [92].Petrat F, Li T, Dehne N, deGroot H, Rauen U. Life Sci. 2006;79:1606–1615. doi: 10.1016/j.lfs.2006.05.025. [DOI] [PubMed] [Google Scholar]
- [93].Jordán J, Galindo MF, González-García C, Ceña V. Neurosci. 2003;122:707–715. doi: 10.1016/j.neuroscience.2003.08.030. [DOI] [PubMed] [Google Scholar]
- [94].Callaway JK, Beart PM, Jarrott B, Giardina SF. Br. J. Pharmacol. 2001;132:1691–1698. doi: 10.1038/sj.bjp.0704018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Böhmer C, Wagner CA, Beck S, Moschen I, Melzig J, Werner A, Lin J-T, Lang F, Wehner F. Cell. Physiol. Biochem. 2000;10:187–194. doi: 10.1159/000016349. [DOI] [PubMed] [Google Scholar]
- [96].Mann CL, Bortner CD, Jewell CM, Cidlowski JA. Endocrinol. 2001;142:5059–5068. doi: 10.1210/endo.142.12.8516. [DOI] [PubMed] [Google Scholar]
- [97].Bortner CD, Cidlowski JA. J. Biol. Chem. 2003;278:39176–39184. doi: 10.1074/jbc.M303516200. [DOI] [PubMed] [Google Scholar]
- [98].Deckers CLP, Lyons AB, Samuel K, Sanderson A, Maddy AH. Exp. Cell Res. 1993;208:362–370. doi: 10.1006/excr.1993.1257. [DOI] [PubMed] [Google Scholar]
- [99].Dallaporta B, Hirsch T, Susin SA, Zamzami N, Larochette N, Brenner C, Marzo I, Kroemer G. J. Immunol. 1998;160:5605–5615. [PubMed] [Google Scholar]
- [100].Mann CL, Cidlowski JA. Endocrinol. 2001;142:421–429. doi: 10.1210/endo.142.1.7904. [DOI] [PubMed] [Google Scholar]
- [101].Nolte F, Friedrich O, Rojewski M, Fink RHA, Schrezenmeier H, Körper S. FEBS Lett. 2004;578:85–89. doi: 10.1016/j.febslet.2004.10.075. [DOI] [PubMed] [Google Scholar]
- [102].Courageot M-P, Lépine S, Hours M, Giraud F, Sulpice J-C. J. Biol. Chem. 2004;279:21815–21823. doi: 10.1074/jbc.M401426200. [DOI] [PubMed] [Google Scholar]
- [103].Sui JL, Petit-Jacques J, Logothetis DE. Proc. Natl. Acad. Sci. U.S.A. 1998;95:1307–1312. doi: 10.1073/pnas.95.3.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Rishal I, Keren-Raifman T, Yakubovich D, Ivanina T, Dessauer CW, Slepak VZ, Dascal N. J. Biol. Chem. 2003;278:3840–3845. doi: 10.1074/jbc.C200605200. [DOI] [PubMed] [Google Scholar]
- [105].Yurinskaya VE, Moshkov AV, Rozanov YM, Shirokova AV, Vassilieva IO, Shumilina EV, Lang F, Volgareva EV, Vereninov AA. Cell Physiol. Biochem. 2005;16:15–22. doi: 10.1159/000087727. [DOI] [PubMed] [Google Scholar]
- [106].Yurinskaya VE, Goryachaya TS, Guzhova IG, Moshkov AV, Rozanov YM, Sakuta GA, Shirokova AV, Shuilina EV, Vassilieva IO, Lang F, Vereninov AA. Cell Physiol. Biochem. 2005;16:155–162. doi: 10.1159/000089841. [DOI] [PubMed] [Google Scholar]
- [107].Arrebola F, Cañizares J, Cubero MA, Crespo PV, Warley A, Fernández-Segura E. Apoptosis. 2005;10:1317–1331. doi: 10.1007/s10495-005-2718-x. [DOI] [PubMed] [Google Scholar]
- [108].Arrebola F, Fernández-Segura E, Campos A, Crespo PV, Skepper JN, Warley A. Am. J. Physiol. Cell Physiol. 2006;290:C638–C649. doi: 10.1152/ajpcell.00364.2005. [DOI] [PubMed] [Google Scholar]
- [109].Nietsch HH, Roe MW, Fiekers JF, Moore AL, Lidofsky SD. J. Biol. Chem. 2000;275:20556–20561. doi: 10.1074/jbc.M002535200. [DOI] [PubMed] [Google Scholar]
- [110].Okada Y, Shimizu T, Maeno E, Tanabe S, Wang X, Takahashi N. J. Membr. Biol. 2006;209:21–29. doi: 10.1007/s00232-005-0836-6. [DOI] [PubMed] [Google Scholar]
- [111].Szabó I, Lepple-Wienhues A, Kaba KN, Zoratti M, Gulbins E, Lang F. Proc. Natl. Acad. Sci. U.S.A. 1998;95:6169–6174. doi: 10.1073/pnas.95.11.6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Souktani R, Berdeaux A, Ghaleh B, Guidicelli JF, Guize L, Le Heuzey JY, Henry P. Am. J. Physiol. Cell Physiol. 2000;279:C158–C165. doi: 10.1152/ajpcell.2000.279.1.C158. [DOI] [PubMed] [Google Scholar]
- [113].Myssina S, Lang PA, Kempe DS, Kaiser S, Huber SM, Wieder T, Lang F. Cell Physiol. Biochem. 2004;14:241–248. doi: 10.1159/000080333. [DOI] [PubMed] [Google Scholar]
- [114].Okada Y, Maeno E, Shimizu T, Manabe K, Mori S-I, Nabekura T. Pflugers Arch. 2004;448:287–295. doi: 10.1007/s00424-004-1276-3. [DOI] [PubMed] [Google Scholar]
- [115].Heimlich G, Cidlowski JA. J. Biol. Chem. 2006;281:2232–2241. doi: 10.1074/jbc.M507367200. [DOI] [PubMed] [Google Scholar]
- [116].Okada Y. Am. J. Physiol. Cell Physiol. 1997;273:C755–C789. doi: 10.1152/ajpcell.1997.273.3.C755. [DOI] [PubMed] [Google Scholar]
- [117].Gantner F, Uhlig S, Wendel A. Eur. J. Pharmacol. 1995;294:353–355. doi: 10.1016/0014-2999(95)00724-5. [DOI] [PubMed] [Google Scholar]
- [118].Colon LV, Diaz ME, Beers DR, Neely A, Xie W-J, Appel SH. J. Neurochem. 1998;70:1925–1934. doi: 10.1046/j.1471-4159.1998.70051925.x. [DOI] [PubMed] [Google Scholar]
- [119].Yu SP, Farhangrazi ZS, Ying HS, Yeh CH, Choi DW. Neurobiol. Dis. 1998;5:81–88. doi: 10.1006/nbdi.1998.0186. [DOI] [PubMed] [Google Scholar]
- [120].Yu SP, Yeh CH, Gottron F, Wang X, Grabb MC, Choi DW. J. Neurochem. 1999;73:933–941. doi: 10.1046/j.1471-4159.1999.0730933.x. [DOI] [PubMed] [Google Scholar]
- [121].Dallaporta B, Marchetti P, de Pablo MA, Maisse C, Duc HT, Metivier D, Zamzami N, Geuskens M, Kroemer G. J. Immunol. 1999;162:6534–6542. [PubMed] [Google Scholar]
- [122].Wang L, Xu D, Dai W, Lu L. J. Biol. Chem. 1999;274:3678–2685. doi: 10.1074/jbc.274.6.3678. [DOI] [PubMed] [Google Scholar]
- [123].Trimarchi JR, Liu L, Smith PJ, Keefe DL. Biol. Reprod. 2000;63:851–857. doi: 10.1095/biolreprod63.3.851. [DOI] [PubMed] [Google Scholar]
- [124].Wang X, Xiao AY, Ichinose T, Yu SP. J. Pharmacol. Exp. Ther. 2000;295:524–530. [PubMed] [Google Scholar]
- [125].Aizenman E, Stout AK, Hartnett KA, Dineley KE, McLaughlin B, Reynolds IJ. J. Neurochem. 2000;75:1878–1888. doi: 10.1046/j.1471-4159.2000.0751878.x. [DOI] [PubMed] [Google Scholar]
- [126].Krick S, Platoshyn O, McDaniel SS, Rubin LJ, Yuan JX. Am. J. Physiol. Lung Cell Mol. Physiol. 2001;281:L887–L894. doi: 10.1152/ajplung.2001.281.4.L887. [DOI] [PubMed] [Google Scholar]
- [127].Krick S, Platoshyn O, Sweeney M, Kim H, Yuan JX. Am. J. Physiol. Cell Physiol. 2001;280:C970–C979. doi: 10.1152/ajpcell.2001.280.4.C970. [DOI] [PubMed] [Google Scholar]
- [128].Xia S, Lampe PA, Deshmukh M, Yang A, Brown BS, Rothman SM, Johnson EM, Jr., Yu SP. J. Neurosci. 2002;22:114–122. doi: 10.1523/JNEUROSCI.22-01-00114.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Krick S, Platoshyn O, Sweeney M, McDaniel SS, Zhang S, Rubin LJ, Yuan JX. Am. J. Physiol. Heart Circ Physiol. 2002;282:H184–H193. doi: 10.1152/ajpheart.2002.282.1.H184. [DOI] [PubMed] [Google Scholar]
- [130].Xiao AY, Wei L, Xia S, Rothman S, Yu SP. J. Neurosci. 2002;22:1350–1362. doi: 10.1523/JNEUROSCI.22-04-01350.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Ekhterae D, Platoshyn O, Zhang S, Remillard CV, Yuan JX. Am. J. Physiol. Cell Physiol. 2003;284:C1405–C1410. doi: 10.1152/ajpcell.00279.2002. [DOI] [PubMed] [Google Scholar]
- [132].Redman PT, Jefferson BS, Ziegler CB, Mortensen OV, Torres GE, Levitan ES, Aizenman E. Neurosci. 2006;143:1–6. doi: 10.1016/j.neuroscience.2006.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Zhang H-T, Wu J, Zhang H-F, Zhu Q-F. Acta Pharmacol. Sin. 2006;10:1367–1374. doi: 10.1111/j.1745-7254.2006.00377.x. [DOI] [PubMed] [Google Scholar]
- [134].Zhao Y-M, Sun L-N, Zhou H-Y, Wang X-L. Neurosci. Lett. 2006;398:22–27. doi: 10.1016/j.neulet.2005.12.073. [DOI] [PubMed] [Google Scholar]
- [135].Hu C-L, Liu Z, Zeng X-M, Liu Z-Q, Chen X-H, Zhang Z-H, Mei Y-A. Neuropharmacol. 2006;51:737–746. doi: 10.1016/j.neuropharm.2006.05.013. [DOI] [PubMed] [Google Scholar]
- [136].Liang F, Schulte BA, Qu C, Hu W, Shen Z. Neurosci. 2006;135:263–271. doi: 10.1016/j.neuroscience.2005.05.055. [DOI] [PubMed] [Google Scholar]
- [137].Yu H-B, Li Z-B, Zhang H-X, Wang X-L. J. Neurosci. Res. 2006;84:1475–1484. doi: 10.1002/jnr.21054. [DOI] [PubMed] [Google Scholar]