

Abstract
DNA replication is an asymmetric process involving concurrent DNA synthesis on leading and lagging strands. Leading strand synthesis proceeds concomitantly with fork opening, whereas synthesis of the lagging strand essentially takes place on a single-stranded template. The effect of this duality on DNA damage processing by the cellular replication machinery was tested using eukaryotic cell extracts and model DNA substrates containing site-specific DNA adducts formed by the anticancer drug cisplatin or by the carcinogen N-2-acetylaminofluorene. Bypass of both lesions was observed only with fork-like substrates, whereas complete inhibition of DNA synthesis occurred on damaged single-stranded DNA substrates. These results suggest a role for additional accessory factors that permit DNA polymerases to bypass lesions when present in fork-like DNA.
Keywords: double- and single-stranded DNA replication, cell extracts, translesion synthesis, cisplatin and N-2-acetylaminofluorene adducts
The opposing orientation of the two DNA strands in the double helix imposes an asymmetry upon the replication process (1). Since polymerases only synthesize DNA in one direction (5′ → 3′), one strand (the leading strand) is synthesized continuously in the direction in which the helix unzips, whereas the other (the lagging strand) is synthesized discontinuously in the opposite direction in numerous short pieces known as Okazaki fragments. Therefore, leading strand synthesis proceeds concomitantly with fork opening from a double-stranded template, whereas synthesis of the lagging strand essentially takes place on a single-stranded template. Single-stranded and fork-like DNA templates have been previously suggested as models for lagging and leading strand replication, respectively (2, 3), although current models of DNA replication propose a mechanism in which leading and lagging strand replication are coupled. Genotoxic agents that induce bulky lesions in DNA often hamper progression of the replication fork and result in either non- or miscoding nucleotides. In a process known as translesion synthesis (TLS), the replication machinery will eventually proceed through some lesions with an increased error rate, resulting in the induction of mutations (4, 5, 6). We wished to investigate the effect of the asymmetry of DNA replication on the efficiency of translesion synthesis. For this purpose, we have compared TLS in vitro by eukaryotic cell extracts on forked or single-stranded (ss) DNA templates containing single site-specific adducts formed by a chemotherapeutic agent (cisplatin) or a strong chemical carcinogen (N-2-acetylaminofluorene or AAF). For both adducts, we observed TLS only when the adduct was located in the forked template.
MATERIALS AND METHODS
Materials.
The DNA damage-inducible CDK inhibitor p21 protein, purified as described (7), was a generous gift from Prof. Bernard Ducommun (Institut de Pharmacologie et de Biologie Structurale, Toulouse, France). Calf thymus DNA polymerases α, δ, and ɛ were purified as reported (8). All oligonucleotides were synthesized on a Cyclone Plus DNA synthesizer from MilliGen/Biosearch and purified on denaturing 20% polyacrylamide gels. [γ-32P]ATP and [α-32P]dATP were from DuPont/NEN and T4 polynucleotide kinase was from United States Biochemical. AciI restriction enzyme was purchased from New England Biolabs. N-Ethylmaleimide was from Sigma.
Construction and Purification of the ssDNA and Forked-DNA Templates.
The cisplatin-modified 90-mer was prepared by ligating a 14-mer, an 8-mer containing the Pt-d(GpG) adduct purified as reported (9), and a 68-mer, using a 65-mer as scaffold. The resulting 90-mer-Pt oligonucleotide was purified on a 20% polyacrylamide/7 M urea/30% formamide denaturing gel and hybridized to a 5′ 32P-labeled 17-mer primer for ssDNA synthesis assays. For double-stranded DNA replication assays, the ssDNA template was hybridized to a 59-mer partially complementary to the Pt-90-mer template (forked DNA) to mimic a DNA replication fork. Control unplatinated templates were prepared and purified starting with an intact 8-mer oligonucleotide following the same procedure. The AAF-modified 44-mer oligonucleotide was prepared as reported (10) and annealed to a 5′ 32P-labeled 15-mer primer for ssDNA assay. The forked DNA was obtained by hybridizing to the AAF modified ssDNA a partially complementary 29-mer oligonucleotide. Control undamaged templates were prepared following the same procedure.
Preparation of Cell Extracts.
Chinese hamster ovary (CHO) cells were grown to subconfluence as monolayer cultures on 145-mm plates at 37°C in a 5% CO2 humidified incubator in MEM (GIBCO/BRL) supplemented with glutamine (2 mM), 8% fetal calf serum (GIBCO/BRL), penicillin (40 units/ml), and streptomycin (40 μg/ml). The cells were washed three times with ice-cold PBS. All subsequent steps were carried out at 4°C. Excess buffer was removed, and the cells were scraped off the plates with ice-cold PBS buffer and harvested by centrifugation (1000 × g for 5 min). The cell pellet was suspended in two volumes of hypotonic buffer (10 mM Tris·HCl, pH 7.5/10 mM KCl/10 mM MgCl2/1 mM DTT) containing protease inhibitors (1 mM polymethylsulfonyl fluoride and 5 μg/ml each of leupeptin, pepstatin, chymostatin, and aprotinin). The cells were allowed to swell on ice for 10 min and disrupted with 20 strokes of the tight-fitting pestle in a Dounce homogenizer. The cell disruption and the integrity of the nuclei was examined by light microscopy. Nuclei were harvested by centrifugation for 10 min at 3300 × g at 0°C and cytosolic supernatants were kept on ice. The nuclear pellet was suspended in hypotonic buffer, and NaCl was added with gentle mixing to a final concentration of 350 mM. After 30 min of extraction at 0°C, the nuclear extracts were centrifuged at 15,000 × g for 20 min. Cytosolic and nuclear extracts were mixed and precipitated by the addition of ammonium sulfate (0.33 g/ml) and gentle stirring for 30 min at 0°C. The precipitates were collected by centrifugation, resuspended in dialysis buffer (50 mM Tris·HCl, pH 7.5/1 mM EDTA/100 mM potassium glutamate/1 mM DTT/1 mM polymethylsulfonyl fluoride/10% glycerol), and dialyzed for 2 h at 0°C. The extracts were frozen in liquid nitrogen and stored at −70°C. Protein concentrations were determined with the Micro BCA system using BSA as a standard. One microliter of extract typically contained 3–12 μg of protein. HeLa cell extracts were prepared with the same procedure as for the CHO cells.
In Vitro DNA Replication Assays.
Standard 15-μl reaction mixtures contained 5 ng of labeled intact or damaged substrate and 3 μg of cell extract protein in reaction buffer containing 45 mM Hepes·KOH (pH 7.8), 7 mM MgCl2, 1 mM DTT, 0.4 mM EDTA, 2 mM ATP, 50–500 μM each dATP, dCTP, dGTP, and dTTP, 3.4% glycerol, 65 mM potassium glutamate, and 18 μg of BSA. At the end of the reaction, 5 μl of stopping buffer (90% formamide/0.1% xylene cyanol/0.1% bromophenol blue/0.1 mM EDTA) was added. Samples were denatured for 10 min at 70°C and loaded onto a 15% polyacrylamide/7 M urea/30% formamide gel.
Quantification of TLS Frequency.
Determination of TLS frequency was achieved by excising from polyacrylamide gels the regions that contained TLS products and determining their radioactivity in a Beckman scintillation counter. These values were compared with the ones obtained for the products arrested at the site of the lesion.
RESULTS AND DISCUSSION
For the present work we constructed templates that contain either the most abundant platinum adduct, an intrastrand crosslink between two adjacent guanines [Pt-d(GpG)], or the major AAF adduct at the C(8) position of guanine (G-AAF). Both unmodified and adduct containing templates were constructed. Oligonucleotides containing the single adduct (90-mer-Pt adduct and a 44-mer-AAF) were used as templates for DNA synthesis. In ssDNA replication assays, annealing of a short primer to the 3′ end of the template places the adduct in the 5′ single-stranded region of the template (Figs. 1A and 2A). Fork-like template replication was modeled by annealing an additional oligonucleotide to the substrate used for single-strand replication (Figs. 1B and 2B). The second oligonucleotide is partially complementary to the 5′ end of the template strand, which upon annealing will form a fork-like structure containing both a double-stranded region, within which the adduct is located, and a 5′ single-stranded tail. DNA synthesis was monitored using 5′ 32P-end-labeled primer elongation reactions in the presence of extracts prepared from CHO and human HeLa cells.
Figure 1.
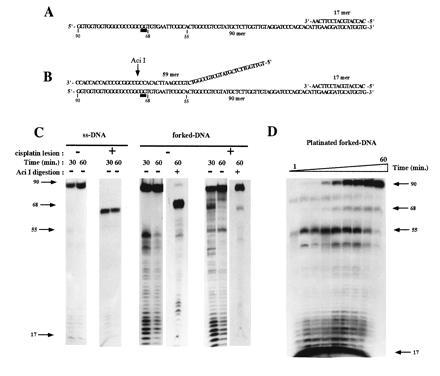
A single Pt-d(GpG) adduct inhibits replication of ssDNA but not forked DNA by CHO cell extracts. Sequences of the ssDNA (A) and the forked template (B) damaged with a unique cisplatin adduct used in the replication assays. ▪ indicates the position of the Pt-d(GpG) adduct. AciI shows the position of the cleavage by the restriction enzyme. (C) In vitro replication by CHO cell extract of the untreated and platinated ssDNA and forked DNA. Reaction conditions were as indicated in Materials and Methods. In forked-DNA synthesis assays, the replication products were digested with AciI when indicated. Positions of the 17-mer (primer), 55-mer (ss/ds DNA junction), 68-mer (product of synthesis to the dT base preceding the Pt lesion), and 90-mer (full-size product) are indicated by arrows. (D) Kinetics of DNA replication across the Pt-d(GpG) lesion on the forked-DNA substrates; Reactions were performed as described in C for 1, 2, 5, 7, 10, 15, 20, 30, and 60 min.
Full-length elongation products were seen with undamaged single-stranded and fork-like templates (Figs. 1C and 2C). The DNA synthesis activity monitored in our assay is most likely mediated by the replicative DNA polymerases δ and/or ɛ as we observed inhibition by 2 mM N-ethylmaleimide (1) and the p21 protein, which inhibits DNA replication elongation in vitro by interfering with proliferating cell nuclear antigen, a component of the replication apparatus (11) (data not shown). However, since inhibition by p21 was not complete, an additional role for DNA polymerase α cannot be excluded.
Elongation of the ssDNA templates was totally blocked at the site of the lesion for both platinum and AAF adducts (Figs. 1C and 2C). This observation extends our previous work showing that purified DNA polymerases α, δ, and ɛ from calf thymus were incapable of DNA synthesis beyond the Pt-d(GpG) lesion, even in presence of proliferating cell nuclear antigen or replication protein A (8). Mutagenic DNA synthesis past the platinum adduct was observed on single-stranded template only by using large amount of the purified repair enzyme DNA polymerase β (8, 12) or with a cell extract from a CHO cell line overproducing the rat DNA polymerase β (data not shown). Likewise, we found that a single AAF adduct blocked replication by Escherichia coli DNA polymerase I and III (13), T7 DNA polymerase (10) and calf thymus polymerase-α (data not shown) at the position preceding the damaged nucleotide.
Interestingly, TLS by cell extracts was observed for both adducts when using fork-like templates (Fig. 1 C and D; Fig. 2D). Comparable results were obtained with cell extracts prepared from human HeLa cells (data not shown). With both substrates, early time points in the reaction exhibited a pause site at the fork junction but increasing amounts of full-length replication products were formed as a function of time. However, the TLS reaction was found to be more efficient for the platinum adducted template as compared with the AAF template: after 60 min of incubation, the extent of TLS were equal to 95% and 15%, respectively (see Materials and Methods for the quantification procedure). This quantitative difference was not due to structural difference between the forked templates since we constructed a platinum template with the same geometry as the AAF template (with a 12-nucleotide-long tail and a distance between the lesion and the junction of five nucleotides) and observed very efficient TLS (data not shown). The difference between the two adducts in the kinetics of TLS can be rationalized in terms of major differences in the local distortion of the DNA structure. Covalent binding of platinum to the GpG sequence induces only a small local bend in the helix axis (14) while AAF binding to the C(8) position of guanine triggers a severe conformational change in the vicinity of the adduct according to the insertion-denaturation model (15, 16). It is therefore not surprising that platinum adducts are bypassed more readily than AAF adducts. The mutation specificity of the two adducts is also in agreement with this drastic difference in the conformational change induced in the DNA structure. Indeed, platinum adducts induce base pair substitutions (17, 18, 19) while AAF adducts essentially induce frameshift mutations (20, 21, 22).
Figure 2.
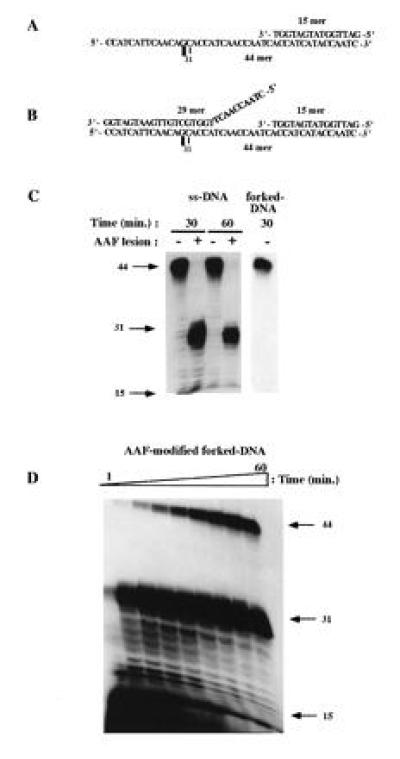
Differential inhibition by a unique AAF adduct of ssDNA and forked-DNA replication catalyzed by CHO cell extracts. Sequences of the ssDNA (A) and the forked-DNA template (B) damaged with a unique AAF adduct used in the replication assay. ▪ indicates the position of the AAF adduct. DNA synthesis conditions for ssDNA and forked DNA (C and D) were the same as those described in Fig. 1. Autoradiograph in D was intentionally overexposed to clearly detect TLS on damaged forked DNA. The position of the 15-mer (primer), 31-mer (product of synthesis to the dC base preceding the AAF adduct), and 44-mer (full-size product) are indicated by arrows.
To prove that the full-length products seen on the acrylamide gel resulted from TLS and not from an artifactual ligation reaction between a partial degradation product of the single-stranded tail oligonucleotide and a partial elongation product, the following control reaction was performed (see Fig. 3): platinum containing single-stranded template primed with a nonlabeled oligonucleotide was extended to the site of the lesion with cell extracts. Subsequently, the tailed fork was formed by adding the 59-mer oligonucleotide and elongation was continued in the presence of [α-32P]dATP. Further incubation yielded the formation of radioactively labeled full-length products (Fig. 3, lane 1), indicating that TLS took place in presence of the complementary strand.
Figure 3.
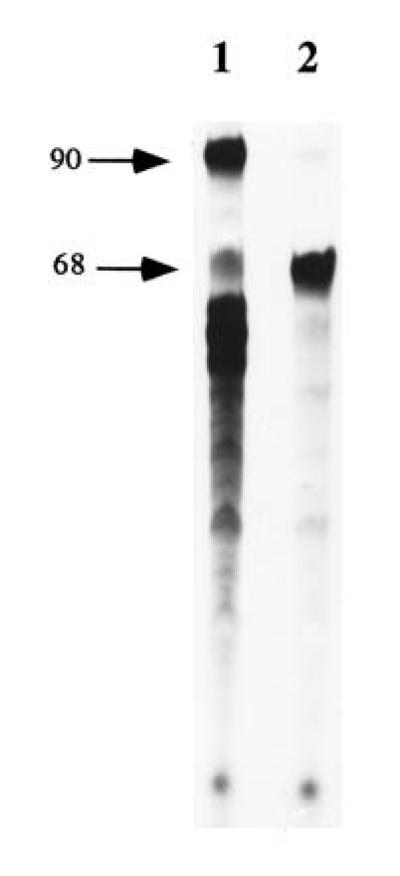
TLS of the cisplatin adduct detected on 90-mer by incorporation of [α-32P]dATP in presence of the 59-mer complementary strand. Nonradioactive primed single-stranded DNA modified site specifically with cisplatin (see Fig. 1A) was replicated for 30 min by cell extracts to allow complete arrest of replication at the site of the adduct, as monitored by a parallel reaction with labeled primer (not shown). Subsequently, the tailed fork was formed by annealing the 59-mer complementary strand and the reaction was continued in the presence of [α-32P]dATP for 30 min (lane 1). A control reaction without the 59-mer was conducted in parallel (lane 2). Positions of the 68-mer (product of synthesis to the dT base preceding the Pt lesion) and 90-mer (full-size product) are indicated by arrows.
We ruled out the possibility that full-length replication products was due to nucleotide excision repair of the adducts in the fork-like template followed by replication on an undamaged template since we found that extract from UV4 CHO cell line, a mutant deficient in nucleotide excision repair (23), was as efficient in bypass replication as the wild-type extract (data not shown). In addition, we took advantage of the fact that the platinum adduct is located within an AciI restriction site (Fig. 1B). Full-length replication product obtained on the platinum damaged template was resistant to this restriction enzyme (Fig. 1C) proving that the adduct was still present. On the contrary, the replication product generated from the undamaged template was Aci I sensitive.
It is not clear why the extracts used in the present work support TLS of bulky lesions on fork-like and not on single-stranded templates. One could argue that different polymerases with different accessory proteins are involved. For instance, replication of the fork-like template requires a DNA helicase to unwind the duplex region. Such activity is probably essential since we found that replication of fork-like templates by purified calf thymus polymerases α, δ, and ɛ was stopped at the double-stranded junction (data not shown). In this respect, it is of interest that the τ subunit of replicative polymerase III holoenzyme has been shown to be in direct contact with the dnaB helicase in E. coli. (24), resulting in a high rate of replication fork movement. In addition, a replicative helicase, the UL9 protein purified from herpes simplex virus, was shown to unwind past the Pt-d(GpG) lesion in conjunction with the viral single-stranded binding protein ICP8 (25). Affinity of human helicases to fork like DNA templates similar to the one used in this study has been reported (26, 27), suggesting that polymerases and helicases might interact to achieve replication across the lesions. Further work will be required to investigate this point.
Substantial replication of double-stranded DNA carrying UV lesions either in vivo (28) or in vitro by cell free extracts (29, 30) has been observed, but the molecular mechanisms underlying these processes remain to be elucidated. We think that the use of forked templates such as those employed here to generate strong lesion bypass signals may provide a critical new tool to investigate TLS mechanisms.
Acknowledgments
We thank Drs. Neil Johnson and Paul Boehmer for critical reading of the manuscript and Stéphane Vispe for advice regarding cell extract preparation. This work was supported by Grant 5036 from the Association pour la Recherche sur le Cancer (Villejuif, France) to G.V.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: TLS, translesion synthesis; AAF, N-2-acetylaminofluorene; ssDNA, single-stranded DNA; CHO, Chinese hamster ovary.
References
- 1.Kornberg A, Baker T. DNA Replication. New York: Freeman; 1992. [Google Scholar]
- 2.Spacciapoli P, Nossal N G. J Biol Chem. 1994;269:447–455. [PubMed] [Google Scholar]
- 3.Debyser Z. Methods Enzymol. 1995;262:457–466. doi: 10.1016/0076-6879(95)62037-0. [DOI] [PubMed] [Google Scholar]
- 4.Trinh T Q, Sinden R R. Nature (London) 1991;352:544–547. doi: 10.1038/352544a0. [DOI] [PubMed] [Google Scholar]
- 5.Thomas D C, Svoboda D L, Vos J M H, Kunkel T A. Mol Cell Biol. 1996;16:2537–2544. doi: 10.1128/mcb.16.5.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Veaute X, Fuchs R P P. Science. 1993;261:598–600. doi: 10.1126/science.8342022. [DOI] [PubMed] [Google Scholar]
- 7.Goubin F, Ducommun B. Oncogene. 1995;10:2281–2287. [PubMed] [Google Scholar]
- 8.Hoffmann J S, Pillaire M J, Maga G, Podust V, Hübscher H, Villani G. Proc Natl Acad Sci USA. 1995;92:5356–5360. doi: 10.1073/pnas.92.12.5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pillaire M J, Villani G, Hoffmann J S, Mazard A M, Defais M. Nucleic Acids Res. 1992;20:6473–6479. doi: 10.1093/nar/20.24.6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lindsley J E, Fuchs R P P. Biochemistry. 1994;33:764–772. doi: 10.1021/bi00169a018. [DOI] [PubMed] [Google Scholar]
- 11.Waga S, Hannon G J, Beach D, Stillman B. Nature (London) 1994;369:574–578. doi: 10.1038/369574a0. [DOI] [PubMed] [Google Scholar]
- 12.Hoffmann J S, Pillaire M J, Garcia-Estefania D, Lapalu S, Villani G. J Biol Chem. 1996;271:15386–15392. doi: 10.1074/jbc.271.26.15386. [DOI] [PubMed] [Google Scholar]
- 13.Belguise-Valladier P, Maki H, Sekiguchi M, Fuchs R P P. J Mol Biol. 1994;236:151–164. doi: 10.1006/jmbi.1994.1125. [DOI] [PubMed] [Google Scholar]
- 14.Takahara P M, Rosenzweig A C, Frederick C A, Lippard S J. Nature (London) 1995;377:649–652. doi: 10.1038/377649a0. [DOI] [PubMed] [Google Scholar]
- 15.Fuchs R P P, Daune M P. Biochemistry. 1972;11:2659–2666. doi: 10.1021/bi00764a017. [DOI] [PubMed] [Google Scholar]
- 16.Fuchs R P P, Lefevre J F, Pouyet J, Daune M P. Biochemistry. 1976;15:3347–3351. doi: 10.1021/bi00660a027. [DOI] [PubMed] [Google Scholar]
- 17.Burnouf D, Gauthier C, Chottard J C, Fuchs R P P. Proc Natl Acad Sci USA. 1990;87:6087–6091. doi: 10.1073/pnas.87.16.6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yarema K J, Wilson J M, Lippard S J, Essigmann J M. J Mol Biol. 1994;236:1034–1048. doi: 10.1016/0022-2836(94)90010-8. [DOI] [PubMed] [Google Scholar]
- 19.Pillaire M J, Margot A, Villani G, Sarasin A, Defais M, Gentil A. Nucleic Acids Res. 1994;22:2519–2524. doi: 10.1093/nar/22.13.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fuchs R P P, Schwartz N, Daune M P. Nature (London) 1981;294:657–659. doi: 10.1038/294657a0. [DOI] [PubMed] [Google Scholar]
- 21.Thomas D C, Veaute X, Kunkel T A, Fuchs R P P. Proc Natl Acad Sci USA. 1994;91:7752–7756. doi: 10.1073/pnas.91.16.7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas D C, Veaute X, Fuchs R P P, Kunkel T A. J Biol Chem. 1995;270:21226–21233. doi: 10.1074/jbc.270.36.21226. [DOI] [PubMed] [Google Scholar]
- 23.Collins A R. Mutat Res DNA Repair. 1993;293:99–118. doi: 10.1016/0921-8777(93)90062-l. [DOI] [PubMed] [Google Scholar]
- 24.Kim S, Dallmann H G, McHenry C S, Marians K J. Cell. 1996;84:643–650. doi: 10.1016/s0092-8674(00)81039-9. [DOI] [PubMed] [Google Scholar]
- 25.Villani G, Pillaire M J, Boehmer P. J Biol Chem. 1994;269:21676–21681. [PubMed] [Google Scholar]
- 26.Seo Y S, Hurwitz J. J Biol Chem. 1993;268:10282–10295. [PubMed] [Google Scholar]
- 27.Tuteja N, Tuteja R, Ochem A, Taneja P, Huang N W, Simoncsits A, Susic S, Rahman K, Marusic L, Chen J, Zhang J, Wang S, Pongor S, Falaschi A. EMBO J. 1994;13:4991–5001. doi: 10.1002/j.1460-2075.1994.tb06826.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spivak G, Hanawalt P C. Biochemistry. 1992;31:6794–6800. doi: 10.1021/bi00144a021. [DOI] [PubMed] [Google Scholar]
- 29.Carty M P, Hauser J, Levine A S, Dixon K. Mol Cell Biol. 1993;13:533–542. doi: 10.1128/mcb.13.1.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Svoboda D L, Vos J M. Proc Natl Acad Sci USA. 1995;92:11975–11979. doi: 10.1073/pnas.92.26.11975. [DOI] [PMC free article] [PubMed] [Google Scholar]