

Summary
HU and IHF are prokaryotic proteins that induce very large bends in DNA. They are present in high concentrations in the bacterial nucleoid and aid in chromosomal compaction. They also function as regulatory cofactors in many processes such as site-specific recombination and the initiation of replication and transcription. HU and IHF have become paradigms for understanding DNA bending and indirect readout of sequence. While IHF shows significant sequence specificity, HU binds preferentially to certain damaged or distorted DNAs. However, none of the structurally diverse HU substrates previously studied in vitro are identical to the distorted substrates in the recently published Anabaena HU(AHU) -DNA cocrystal structures. Here we report binding affinities for AHU and the DNA in the cocrystal structures. The binding free energies for formation of these AHU-DNA complexes range from ~−10 to −14.5 kcal/mol, representing Kds in the nM to low pM range, and a maximum stabilization of at least ~6.3 kcal/mol relative to complexes with undistorted, nonspecific DNA. We also investigated IHF binding and found that appropriate structural distortions can greatly enhance its affinity as well. Based on the coupling of structural and relevant binding data, we estimate the amount of conformational strain in an IHF-mediated DNA kink that is relieved by a nick (at least 0.76 kcal/mol) and pinpoint the location of the strain. In addition, we show that AHU has a sequence preference for an AT-rich region in the center of its DNA binding site, correlating with an unusually narrow minor groove. This is similar to sequence preferences shown by the eukaryotic nucleosome.
Keywords: Anabaena HU, IHF, DNA bending, DNA-dependent distortability, indirect readout
Introduction
HU and IHF are closely related eubacterial, DNA-bending proteins characterized by two overlapping and functionally relevant modes of DNA binding: a non-specific mode and a specific mode. In their capacity as histone-like architectural factors, HU and IHF bind to DNA nonspecifically and are found in high concentrations in the bacterial nucleoid1; 2, though their exact role in nucleoid compaction is still debated3. Sequence-specific binding, largely by IHF4, plays a major role in facilitating processes such as replication initiation, site-specific recombination and transcription, when sharp bends or DNA loops are required for assembly of multi-component nucleoprotein complexes 5. HU is able to regulate such processes by binding DNA in a topology-dependent or structure-specific manner6; 7.
These proteins form dimers with compact, positively charged body and two long beta ribbon arms that track along the DNA’s minor groove. They induce flexible bends in the DNA of ~105° to >180° (Figure 1a)8–11. Most of the bending is concentrated in two large kinks, spaced nine base pairs apart, where the base stacking is disrupted by a large roll angle and a highly conserved proline residue at the tip each arm is intercalated. IHF recognizes specific DNA sequences through indirect readout of the sequence-dependent conformational parameters of its binding site. Although HU binding is generally described as non-sequence specific, DNA modifications that stabilize the particular unusual conformation seen in the crystal would be expected to enhance binding.
Figure 1. IHF-and AHU-DNA cocrystal structures.
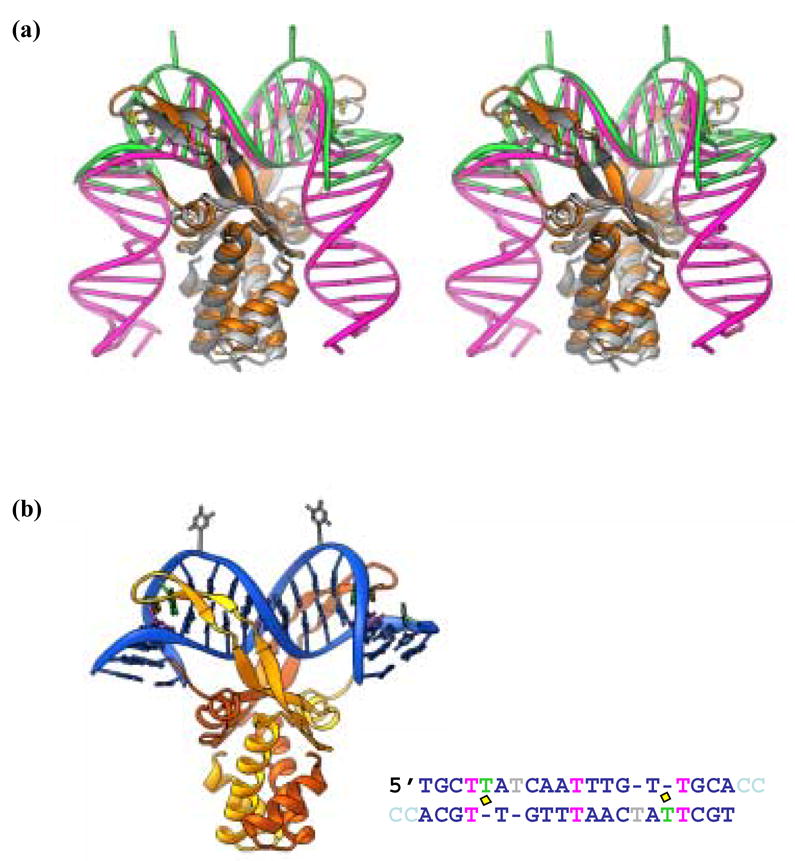
(a) Stereoview of a superposition of the IHF- and AHU-DNA complexes. IHF protein is shown in grey and white while the IHF DNA is pink (1IHF). AHU is gold and the bound DNA is green (1P71). Prolines at the tips of arm-like β -ribbon extension are in yellow. (b) AHU-DNA complex. The AHU-DNA complex (1P71) is color coded as in Table 1. The protein subunits are gold and orange and the intercalating prolines are in yellow. Canonical DNA is blue while unpaired bases are green (stacked) or grey (flipped) and mismatches are pink. Part b reprinted from Figure 3 of Swinger and Rice, 200310.
Structure-specific binding of HU to DNA in vitro is in fact well-documented12–18, and, given genetic data showing that HU-deficient cells are quite sensitive to UV damage19; 20, it suggests that HU may be involved in DNA repair. However, in cases where high-affinity binding by HU to non-canonical (e.g nicked or mispaired) duplexes has been described, accompanying structural data has been absent and models have been based on the IHF-DNA cocrystal structure6; 7; 14,8. Here we present affinity data for a set of distorted duplexes based on the one found in our recent cocrystal structures of Anabaena HU (AHU) –DNA complexes10. These data can thus be used to estimate the strain in known DNA conformations.
Results
The DNA duplexes in the IHF- and AHU-DNA cocrystal structures are shown in Figure 2a and 2b. The AHU DNA duplex was one of four oligonucleotides (TR3 for Top Right 3) that were annealed to create the doubly-nicked IHF site for crystallization. The fact that AHU crystallized with low amounts of TR3 in the presence of higher concentrations of a less distorted DNA duplex led us to predict that the TR3 duplex would bind very tightly to AHU in vitro. In fact, the affinity of AHU for a TR3-related duplex was so high, it became clear that coupling binding data with relevant structural data could enhance our understanding of the strain induced in DNA upon bending and the structural determinants for binding of HU and IHF to DNA.
Figure 2. Origin of DNA duplexes used in this binding study.
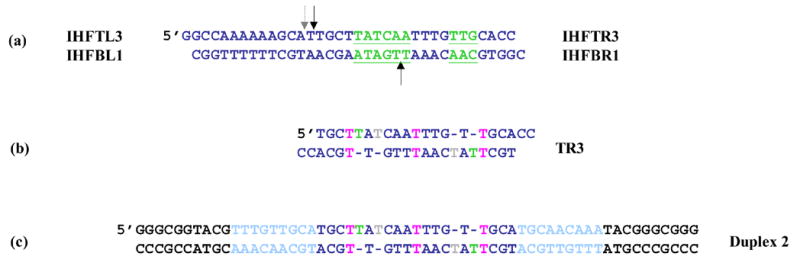
(a) IHF’s cognate site, H’. The IHF consensus site is underlined in green. The black arrows show the location of backbone nicks placed 9 base pairs apart in an effort to phase AHU binding for homogeneous crystal formation. The dashed arrow shows the location of the nick in the IHF-DNA crystal structure (1IHF). (b) DNA observed in an AHU-DNA cocrystal (1P71). Two copies of the top right oligonucleotide bound to each other forming a duplex with 4 unpaired Ts and 3 T • T mismatches. Color coding is as follows: green, analogous base in crystal structure is stacked; grey, analogous base in crystal structure is flipped; pink, T • T base pair (c) DNA duplex for gel shift experiments. The middle of the site is identical to duplex TR3 from the crystal. The flanking DNA in light blue is identical to DNA from neighboring protein-DNA complexes that contact AHU in the crystal. The black flanking sequence destabilizes a hairpin that forms in its absence and complicates binding experiments.
Measuring high-affinity binding of AHU to DNA containing multiple distortions
The crystallized sequence (Figure 2b) was modified to facilitate measurement of relevant binding constants by electrophoretic mobility shift assays (EMSA). Within the crystal each AHU dimer contacts DNA from neighboring complexes in addition to its own10. These sequences were appended to the substrate and are shown in (light blue in Figure 2c), however, hairpin formation in the resulting substrate complicated binding measurements. Additional flanking sequences were added to destabilize the hairpin. The resulting DNA duplex 2 (Figure 2c), binds to AHU with an apparent Kd of 13 ± 0.4 pM (Table 1). Duplex 2 contains all of the distortions found in the cocrystal structure: 3 T • T mismatches and 2 extra Ts on each strand. Though less physiologically relevant than substrates previously described, its affinity for AHU is up to 50-fold higher than that of any tested to date. Further studies used duplexes with slightly shorter flanking sequences that also prevented hairpin formation (duplexes 1 and 3–14 in Table 1; Figure 3).
Table 1.
Affinities of AHU for various DNA substrates

|
Figure 3. Representative EMSA experiment.
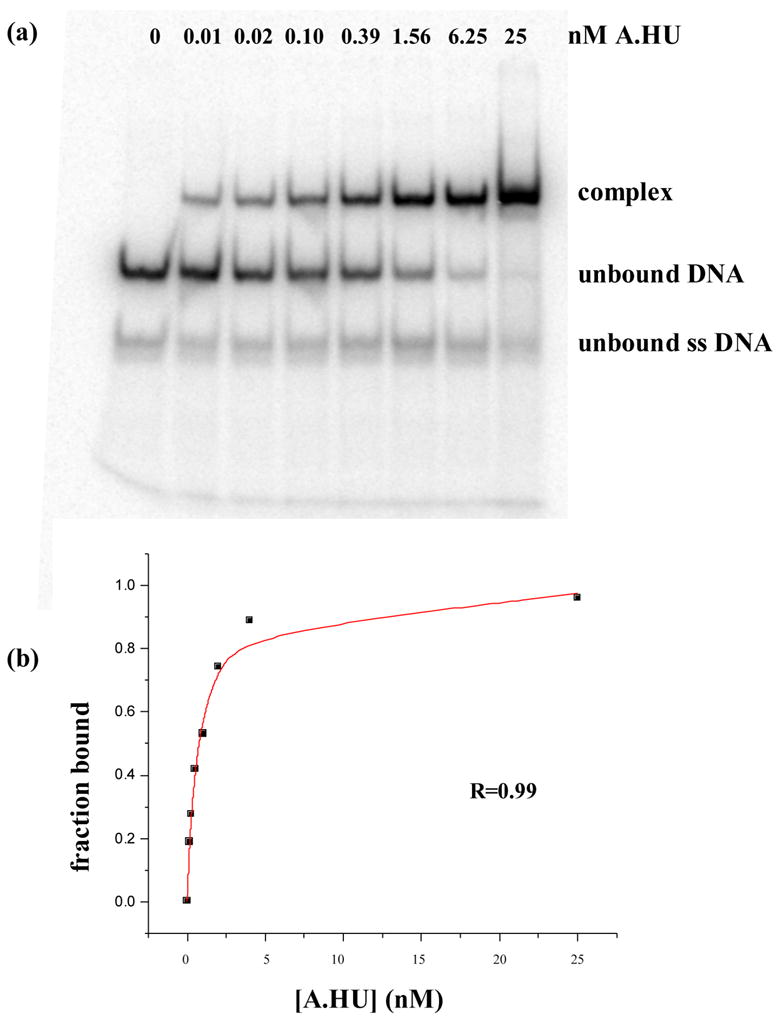
Panel (a) shows a polyacrylamide native gel shift experiment between duplex 6 and AHU. Assays were performed in binding buffer, 20mM Tris–HCl (pH 8.0), 70mM NaCl, 1 μ g/ml of salmon sperm DNA, and 5% glycerol, at 4° C by incubation of the 32P-labeled DNA various concentrations of protein. Further details are described in Materials and methods. Panel (b) shows the curve fit for data from panel (a) that was used to extract an apparent binding constant. The R value represents the square root of the difference between the total variation (t) and the unexplained variation (u) divided by the total variation or R = √((t-u)/t).
Deconvoluting the structural determinants of high-affinity binding to HU AHU binds tightly to single-base insertions that coincide with the DNA kinks
The DNA distortions in the crystal structure are highlighted in Figure 1b. We investigated the contribution of each one (separately and in combinations) to the overall affinity (Table 1). The greatest individual contribution to binding was provided by the existence of 2 single-T insertions spaced 9 base pairs apart: Kd for duplex 4 was 0.35nM, at least 3 orders of magnitude tighter than our estimate of 400nM – 2uM for nonspecific binding21; 22. When the spacing is reducted to 7 base pairs (duplex 9), a weak discretely shifted band is obscured by a nonspecific smear, suggesting that specific binding is weak, at best. Combining all 4 insertions (duplex 7) actually results in slightly weaker binding (Kd = 2.1nM) than that observed for the 2 properly phased insertions (duplex 4). These data are consistent with what is observed structurally. The single T insertions that coincide with the large kinks the DNA (9 base pairs aparat) are stacked in the duplex at the site of proline intercalation. The other two insertions, 7 base pairs apart, lie in a region whose structure is otherwise much closer to canonical B-form in both the HU- and IHF-DNA structures. They are flipped out of the double helix, and the flanking bases stack on one another.
Single-base insertions and nicks similarly relieve strain in kinked DNA
Comparison of the AHU structure to 2 different IHF cocrystal structures (Figure 4) reveals that both a nick and a single-base insertion relieve strain in the bound DNA and help to pinpoint the location of the strain. One DNA kink from the published IHF cocrystal structure (1IHF) is shown in pink in Figure 4a, 8. The DNA at this kinked site is fully intact, though there is a nick in the backbone 1 base pair away from the other kink (not shown) in this same structure. That nick facilitates crystal packing and does not enhance binding significantly23. However, the first IHF-DNA cocrystals grown contained a second nick directly at the kink site shown in Figure 4. Refinement of that structure was set aside in favor of data from a singly nicked complex. However, it was recently completed as an interesting comparison to our HU-DNA structures. A nick at this position (directly at a kink) enhances binding by ~4x (Table 3). This implies that a nick at the kink relieves backbone strain. This is supported by the structures: when the DNA is nicked, the ends spring apart (compare the pink DNAs in Figures 4a vs. 4b). The bases flanking the nick relax to positions remarkably similar to those of the bases flanking the stacked single T insertion in the HU-DNA complex. Taken together, the binding and structural data provide a clearer picture of backbone strain. The binding data suggests that a nick directly at the kink in IHF-bound DNA relieves at least ~0.76 kcal/mol of strain (Δ Δ G for IHF binding duplexes +/− the nick), and the structural data illustrates how.
Figure 4. Comparisons of IHF- and AHU- kinked DNA.
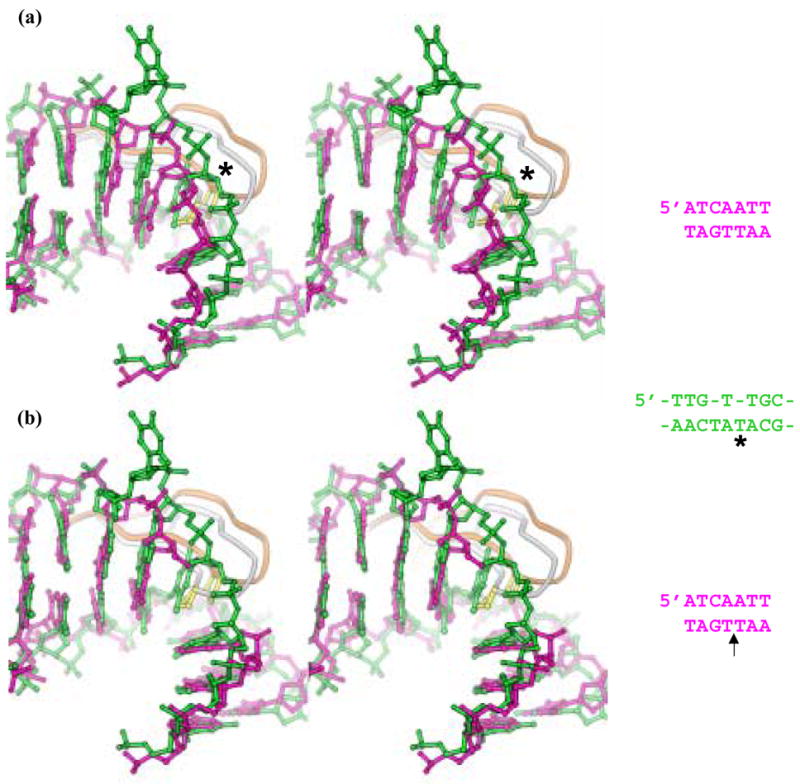
Panel (a) shows a stereoview of a superposition of kinked DNA from the previously published IHF-DNA structure in pink (1IHF), the sharper of the kinks in the AHU-DNA structure in green (1P78). The tips of the β -ribbon protein arms are shown in white (IHF) and gold (AHU) with the intercalating prolines in yellow. Bases at the kink in these two structures superimpose remarkably well considering the differences in sequences and the presence of an extra T in the AHU structure (marked by an asterisk). The sequences for the portions of the structures shown are in green for AHU and pink for IHF. The asterisk marks the intercalated T.
Panel (b) shows a stereoview of a superposition of kinked DNA from a nicked IHF-DNA structure recently deposited to the Protein Data Bank in pink (2HT0) and the AHU-DNA structure in green. The nick in the IHF DNA backbone in this structure is directly at the site of kinking. Proteins are color coded as in panel (a). The superposition of paired bases in these two structures is even closer than that observed in panel (a). The arrow by the IHF sequence in pink marks the location of the nick. This view illustrates that a nick and an extra T similarly relieve strain in the kinked DNA backbone.
Table 3.
Affinities of IHF for various DNA substrates

|
T • T mismatches enhance the effect of single-T insertions
The duplex containing 2 T insertions with 9-base pair spacing flanked only their 5’ sides by T • T mismatches (duplex 1) promotes the highest affinity AHU binding of all the duplexes tested: Kd = 4pM. The enhancement from 0.35nM for 2 single-base insertions only (duplex 4) reflects a Δ Δ G° of 2.5kcal/mol. This effect is quite specific: T • T mismatches on the 3’ side of the single T insertions (duplex 3) does not significantly enhance binding over that observed for insertions alone (duplex 4), and a substrate containing the outer T • T mismatches in the absence of the single T insertions (duplex 13) does not produce a discrete shifted band.
AHU was crystallized with AT base pairs (PDB IDs 1P78 and 1P51) as well as T • T base pairs (PDB ID 1P71) flanking the single T insertions on their 5’ sides. Despite the large difference in affinity, little structural difference was observed other than the phosphate backbones being a bit closer together in the structures with the TT base pairs,. These results suggest that an unpaired but stacked T with a T • T mismatch on its 5’ side may be particularly susceptible to intercalation. Interactions that stabilize the duplex may be weakened in just the right places to greatly facilitate kinking. It is also possible that the role of the adjacent T • T mismatches in duplex 1 is to stabilize the stacked form (as opposed to the flipped form) of the unpaired Ts in the unbound duplex.
A pair of appropriately phased insertions is required for tight binding
While there has been some debate over whether multiple HU dimers bind cooperatively to DNA, here we observe strong intramolecularly cooperative binding of 2 arm domains within an AHU homodimer to multiple distortions on single DNA sites. Whereas AHU binds to the 2 phased insertions of duplex 4 with an apparent Kd of 0.35nM, binding to a single insertion in the same overall sequence (duplex 10) is too weak to be measured accurately in this assay. A sharp band representing a specific AHU-DNA complex is observed; however, as the protein concentration is increased, a smeared band (representing nonspecific binding) is observed before the free duplex is fully shifted to complex. Similarly, binding to a single-T insertion with an adjacent T • T mismatch (duplex 11) is too weak to measure while binding to phased copies of this tandem single T-mismatch distortion (duplex 1) is 4.0pM. This intramolecular cooperativity of binding to distorted sites is not at all surprising. Grove and coworkers also observed tight binding of AHU and IHF to phased distortions (4-nucleotide loops); however, binding to a single 4-nucleotide loop was not reported17. Tight binding to phased distortions makes sense because once a single distortion is bound by one arm of an HU homodimer, the propensity for the second arm to bind a distortion at an appropriate distance from the first is likely to be greatly enhanced.
AHU shows sequence preference reflecting a narrow groove width at the center of the binding site
Structural data (summarized in Table 2) led us to predict a sequence preference for AHU. The 2 base pair steps surrounding the central T • T mismatch in the AHU cocrystal structure are overtwisted (39° vs. 36° for B-form) and the minor groove in this region is narrower than a canonical B-form minor groove (8.7Å vs. 10.7Å). This trend is also observed in the same position of the duplex bound to IHF in the cocrystal (44° and 9.2Å). Duplexes with a central T • T or GC base pair (duplexes 5 & 6) bind to AHU with less than twofold lower affinity than the duplex with a central AT pair (duplex 4); however, substitution of three adjacent GC pairs (duplex 8) in this region weakens binding significantly: about 17-fold. AT-rich regions of DNA are generally characterized by a narrow minor groove, whereas GC substitutions tend to widen the minor groove24. These results imply that a narrow and/or highly twistable minor groove in the center of the DNA site is an energetically favorable feature for binding. This agrees with the known sequence preferences of IHF, where AT base pairs are also preferred in this region25.
Table 2.
Twist and minor groove width for AHU-, IHF- and B-form DNA
IHF binding can also be driven by structurally appropriate DNA distortions
The structural data comparing IHF- and AHU- DNA kink geometries (Figure 4) led us to predict that distortions driving high-affinity binding by HU might also be favored by IHF. As shown in these figures, the rise distance between base pairs at the kink is large. The rise distance between base pairs at the kink in the AHU cocrystal structures ranges from 7.8 to 8.3Å calculated using 3DNA26, compared to 3.4Å for B-form DNA27. This is not surprising because an extra T is inserted in the kink. However, in the IHF cocrystal structure where there are no unpaired bases and the backbone is fully ligated, the rise is still relatively large, 6.8Å. IHF does show a preference over B-form DNA for the phased single-T insertions in the duplex 4 (Figure 5). The affinity for this distorted site (Kd=42 nM) is only slightly weaker than that for cognate IHF DNA-binding sequences, (Kd~2 nM-20nM)28. This result highlights a key feature of DNA recognition by proteins that rely on indirect readout. Since distortability of the DNA sequence is recognized (as opposed to specific functional groups), DNA sequences or structures that are quite different from each other can all potentially make suitable binding partners.
Figure 5. EMSA experiment shows that IHF binds to phased single-base insertions.
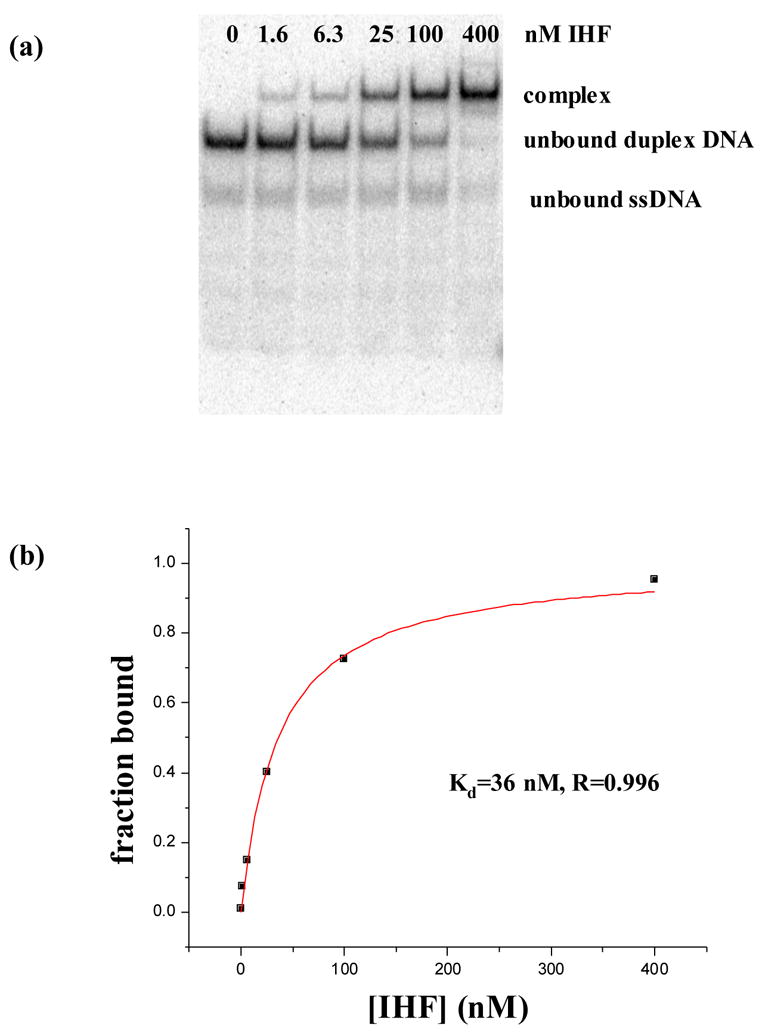
Panel (a) shows a polyacrylamide native gel shift experiment between duplex 4 containing 2 single-T insertions and IHF. Assays were performed in binding buffer, 20mM Tris–HCl (pH 8.0), 70mM NaCl, 1 μ g/ml of salmon sperm DNA, and 5% glycerol, at 4°C by incubation of the 32P-labeled DNA various concentrations of protein. Further details are described in Materials and methods. Panel (b) is the curve fit used to extract an apparent binding constant of 36nM for the gel in panel (a). After three repetitions of the experiment, an apparent Kd of 42±6nM was determined as reflected in Table 1.
Discussion
Among the substrates we tested, the free energies of binding Δ G° range from a rough estimate of ~−6.3 kcal/mol (or weaker) for nonspecific binding to −14.5 kcal/mol for the tightest specific complex (summarized in Table 1). The largest release of free energy upon binding occurs when the unbound duplex itself contains helix-destabilizing distortions and specifically, when those distortions are positioned so that both intercalating side chains on the protein can contact them. Duplexes 1 and 2, which both contain 2 single-T insertions each adjacent to a T • T mismatch, produce the lowest Kd values, reflecting binding that is ~100,000-fold tighter than nonspecific DNA (using a rough lower estimate of 400nM). As seen in Figure 4, when complexed with the protein, the resulting structure at the DNA kink is very similar to that of one with a simpler, more common type of damage, a nick. The fact that the unnicked IHF-DNA structure is slightly different immediately at the kink shows that the protein does not fully stabilize the bent DNA structure, and that both the single-T insertion and the nick appear to relieve similar strain in a similar manner.
In the eyes of HU, not all DNA distortions are equal. In our study, some distorted substrates bound with pM affinity whereas others were not preferred over undistorted DNA. Helix-destabilizing distortions that are not accommodated by protein, such as unpaired bases that are flipped out in the complex structure, are slightly destabilizing to the complex (e.g. duplex 4 vs. 7). Furthermore, even a seemingly subtle change to our tightest-binding substrate, the placement of a T • T mismatch on the 5’ vs. the 3’ side of a single-base insertion, results in either 4pM (duplex 1) or 250pM (duplex 3) binding respectively. This represents a Δ Δ G° of 2.3kcal/mol and clearly shows that, while generic “flexibility” at or near the kinks may enhance binding, structure-specific effects are also very important.
Our measurements are broadly consistent with previously published results. The most closely related substrates are those studied by Grove and coworkers17. They observed tight binding (3.5 nM) between AHU and tandem mismatches (4-nucleotide loops) spaced 9 base pairs apart. While we did not observe quantifiable specific binding to phased T • G or T • T mismatches (duplexes 12 and 13 respectively), the stacked insertions neighbored by mismatches found in duplexes 1, 2, and 3 are reminiscent of 4-nucleotide loops. The 4-nucleotide loops in the Grove et al. study were tested at a salt concentration of 10mM as opposed to 70mM used in our assays, so direct comparison is not possible. Binding by HU and IHF is somewhat weakened by increasing salt concentrations, although for IHF it has been noted to be less salt-sensitive than for many other protein-nucleic acid complexes (data not shown; 29; 30).
A wide variety of non-B-form DNA substrates have been tested, and an interesting and possibly more physiologically relevant example was observed by Kamashev et al.15. They observed the preference of E. coli HU for a duplex with a 3’ overhang as opposed to a 5’ overhang. These authors point out that the 3’ overhang is the more likely intermediate for double strand break repair. It is still unclear, however, whether the growing number of high-affinity distorted DNA substrates reveals something about a specific biological role for HU in DNA repair or whether differential preferences reflect only differences in how each substrate can be distorted into the particular conformation most preferred by HU. In addition, the idea that HUs from particular species specialize and prefer certain distortions has been put forth by Grove and coworkers18; 22. Relatively small differences in the proteins’ electrostatics and/or flexibility might alter the way in which DNA substrates can best adapt to them.
How do the energies measured here for stabilization of AHU- and IHF-DNA complexes by DNA distortions relate to the energetic cost of kinking DNA? Yan and Marko estimate the cost of a sharp kink as ~11kBT, or ~6.1kcal/mol at 4°C (our binding measurements were carried out at 4°C)31. Using a rough lower estimate of Kd=400nM for nonspecific HU binding, we calculate that the complex with duplex 1, containing properly spaced single-T insertions flanked by T:T mismatches, is ~6.4 kcal/mole more stable than one with canonical DNA, corresponding to ~3.2 kcal/mole of kinks. However, additional factors come into play here: First, even with this substrate, it is unlikely that the energetic cost of kinking is fully prepaid – some binding energy is probably still expended in distorting the DNA to the state seen in the crystal structures (and in paying the entropic cost of locking out other conformations). Second, while introducing the kink itself is energetically costly, it allows electrostatic interactions with the positively charged sides of the protein that cannot occur in straight DNA. The Δ Δ G° between loosely and tightly bent complexes is therefore a balance between the energy paid to physically bend the DNA and the energy released when additional protein-DNA interactions are made. Several lines of evidence imply that the DNA bend induced by HU is flexible, including our crystal structures, where the size of the kinks varied, and close contacts to the protein sides were missing with the smaller kinks 9; 11; 32. Since there is evidence for observably flexible bending both with and without distortions in the unbound DNA, the equilibrium constant between loosely and tightly bent complexes in both cases is probably within an order of magnitude or two from 1. Compared to the 5 orders of magnitude gained in Kd for our best distorted substrate, this implies that the energetic penalty for initially opening the kink (seen in all AHU-DNA complexes) is far greater than that for widening it to allow further contacts with the sides of the protein. This is not unexpected, since the base stacking disrupted at the initial opening step is a major stabilizing force in DNA structure.
Introducing a nick in the phosphodiester backbone at the kink in the IHF-DNA complex improved binding by 0.76 kcal/mole. The structural comparison in Figure 4 clearly shows that this nick allows relief of conformational strain. However, the stabilizing effect was much less than that of the distortions in duplex 1 and 4. Direct comparison in the IHF case is somewhat difficult because it has strong sequence specificity, but using an estimated Kd of 20μ M for nonspecific binding by IHF28, duplex 4 enhanced the stability of nonspecific IHF-DNA complexes by ~3.4 kcal/mole. These differences probably reflect the conformational dynamics of the unbound DNA: the nick is not expected to disrupt base stacking, while the single-base insertions and mismatches are.
Although IHF is undeniably a sequence-specific DNA binding protein, like HU, it binds with structural specificity. IHF’s 42nM affinity (Table 3) for 2 phased single-T insertions (duplex 4) is comparable to specific IHF sites, even though duplex 4 bears little resemblance to the IHF consensus sequence (5’-WATCARXXXXTTR)25. AHU- and IHF-bound DNA are structurally very similar whether or not a nick or T insertion is present (Figure 4), so IHF’s preference for a T insertion was predictable from a comparison of the structures. This preference is consistent with an early observation that IHF binds more tightly to its cognate H’ site when the site contains 2 adjacent T • T mismatches with 8- or 9-base pair spacing17. These preferences are also consistent with IHF’s use of indirect readout as a DNA recognition strategy.
While IHF makes a specific complex with duplex 4, its 42 nM affinity for this site is still weaker than AHU’s 0.35 nM affinity for the same site. Also, nonspecific IHF-DNA complexes are weaker than HU-DNA nonspecific complexes (Kd~20–30μ M28). Some of IHF’s specificity therefore probably reflects unfavorable interactions with incorrect sequences as well favorable interactions with correct sequences.
This work also addresses the sequence preferences of HU. Only a few such preferences have been previously demonstrated, and the structural basis for those have been unclear. The Chaconas group demonstrated a preference, in supercoiled DNA only, for E. coli HU binding to a particular sequence in Mu DNA7. E. coli HU has also been shown to have an ~10fold preference for DNA containing phased A-tracts33; 34. Through our work we have discovered a method by which previously unknown HU sequence preferences can be readily measured, and correlated with the known structure. We dictated where HU would bind by properly spacing two intercalation sites, then varied the DNA sequence at other points in the binding site. The tight binding induced by the phased T insertions also made accurate affinity measurements feasible. Using this strategy, we uncovered a 17-fold difference in Kd between a duplex containing the central sequence AAT (duplex 4) and a duplex with a central sequence CCG (duplex 8). Although others have suggested that HU’s preference for A-tracts may reflect the bending associated with phased A-tracts in naked DNA, our structural data suggests that minor groove width is actually the key feature. Helically phased A-tracts would place narrowed minor grooves not only in the center of the complex, but also at the sides, where the IHF-DNA structure also displays unusually narrow minor grooves, and on one side, a clear preference for an A-tract8; 35. The DNA duplexes in the AHU-DNA cocrystals were too short to make these all these contacts, but comparison of the protein structures suggests a similar preference for HU10. HU is highly abundant in the bacterial nucleoid. Even weak sequence preferences could lead to biologically significant differences in HU distribution on bacterial chromosomes, which could in turn affect the organization of chromosomal domains in vivo.
The sequence preference described here for HU is remarkably similar to that deduced for nucleosomes that are the fundamental unit of eukaryotic chromatin25; 36; 37. Although they are also largely nonspecific, strongly preferences can be selected. As with HU, these may reflect general flexibility, but also reflect very specific DNA structural requirements that can be mapped onto the known complex structure. For instance, preferred nucleosome binding sites have similar trends of A/T rich regions where narrow minor grooves face the protein. In this case, the biological relevance of such relatively weak preferences in positioning nucleosomes has recently been verified38.
Structure-specific binding of distorted DNA is a critical feature of many DNA repair enzymes, for instance those involved in nucleotide excistion repair and mismatch repair39. For example, MutS is responsible for the initial recognition of mismatched base pairs and insertions in mismatch repair39; 40. Just as HU binds to different DNA structures, so MutS recognizes a variety of mismatches that have little structural similarity to each other. Both HU and MutS deal with this recognition challenge by using flexible DNA binding regions that undergo mutual induced fit upon DNA binding. Furthermore, both proteins seem to rely on the intrinsic duplex destabilization caused by the DNA feature that they recognize.
An important difference between MutS and HU is the degree to which we understand each protein’s involvement in DNA repair. The fact that a protein recognizes damaged DNA or repair intermediates suggests but does not prove a direct role in repair. Supporting evidence for HU’s involvement in repair includes the fact that HU-deficient cells are more sensitive to UV-induced DNA damage20. Also, genetic experiments on HU-depleted cells suggest that HU participates in homologous recombination-mediated repair19. Phenotypic pleiotropy of HU- bacteria stems from the fact that HU facilitates many different cellular processes. This multifunctionality greatly complicates the process of pinpointing HU’s role in repair. It also means that in vitro studies of HU-DNA binding such as those presented here are critical for moving towards a complete understanding of specific biological functions of HU. Although tightest-binding substrates presented here may not be physiologically relevant, they provide a detailed, structure-based understanding of the energetics and binding-site preferences of HU. This has implications not only for the dynamics of the prokaryotic nucleoid but also for the recognition mechanisms of many other DNA-binding proteins.
Materials and methods
Protein and DNA purification
AHU was cloned and purified as described in previously10. E. coli strain RJ1878 (a gift from R. Johnson), a derivative of BL21(DE3) that lacks functional chromosomal HU genes was used for protein expression. After a two-step ammonium sulfate precipitation, AHU was further purified on a heparin column (Amersham Pharmacia) followed by ion exchange on a Mono-S column (Amersham Pharmacia). The possibility of endonuclease contamination was excluded by incubating the protein with supercoiled plasmid and 10mM MgCl2 at 37° C for 2 hours. AHU has no significant absorbance at 280 nm. Protein concentration was therefore determined by quantitative amino acid analysis performed in triplicate (HHMI Biopolymer/Keck Biotechnology Research Foundation at Yale) on the stock solution (0.85mM) stored at −20° C. Protein samples were diluted freshly from this stock for each assay. IHF was a gift from S.-W. Yang and H. Nash; its concentration was determined by UV absorbance using ε 280 of 5800M−1cm−1. PAGE purified DNA was purchased from Integrated DNA Technologies.
Gel mobility shift analysis
Assays were performed in binding buffer, 20mM Tris–HCl (pH 8.0), 70mM NaCl, 1 μ g/ml of salmon sperm DNA, and 5% glycerol, at 4°C by incubation of the 32P-labeled DNA (0.15 or 1.5pM) with various concentrations of protein for at least 12 hours. Diluted protein (4μ L) was added to each 20μ L reaction. Protein dilution buffer contained 25mM Hepes (pH 7.6), 50μ g/mL, BSA, 0.1M NaCl, 10% glycerol and 0.1mM EDTA. 10 μ L of the total binding reaction were loaded onto 10% polyacrylamide gels (acrylamide to bis-acrylamide 29:1, w/w) in 0.5× TBE (45mM Tris-borate, 1mM EDTA) buffer and electrophoresed at 6 V/cm for 2.0 hours at 4° C. Dried gels were visualized using PhosphorImager screens (Molecular Dynamics) scanned by a Molecular Dynamics PhosphorImager. The band intensities were quantified by using the area measurement utility in the ImageQuant V5.0 (Molecular Dynamics) software package. Because the protein concentration is in excess over the DNA concentration, free protein concentration is assumed to be approximately equal to the total protein concentration. The equilibrium dissociation constant (Kd) was therefore determined using the following relationship:
where θ is the fraction of bound DNA and Pt is the total protein concentration. Experimental data were fit using Microcal Origin 6.0. Each binding assay was repeated 2–5 times. AHU’s affinity for duplex 2 was measured under a range of conditions in order to be certain that samples reached equilibrium and that the labeled DNA concentration was sufficiently lower than the protein concentration. (shown in supplementary material Figure 1). Some substrates were bound too weakly by AHU for accurate Kd measurement. A sample polyacrylamide gel showing such weak binding is displayed in supplementary material, Figure 2. Such binding is described as above the limit of detection (ald) in Table 1.
Doubly nicked IHF-DNA complex Structure
The IHF-DNA complex structure was initially solved using crystals that contained a nick on both DNA strands8. However, when it became apparent that one nick was directly at a DNA kink, new crystals were grown with that strand intact, and data from those were used for refinement. For comparison with the AHU-DNA complex, an earlier native data set from a crystal containing doubly nicked DNA was retrieved from storage and refinement of that structure was completed. This data was collected on Fuji image plates in June 1995 at Brookhaven National Laboratory beamline x4a, and processed with denzo and scalepack41. The crystal was grown as described, but before flash-freezing was soaked in stabilizing solution containing Mg++ in place of the Cd++ in the original mother liquor. Refinement was finished using CNS_SOLVE and a modified version of rna-dna.param in which the 2’ endo sugar pucker restraints were deleted 42. The same test set indices have been reserved for Rfree in refinement of all IHF-DNA complex structures. The diffraction data are highly anisotropic, extending past 2A in c*, but no further than ~2.8 in b*. A sharp spike in the Rfactor at ~2.25A led to the realization that faint ice spots in the bad direction of diffraction were being scaled to disruptively large values by the application of an overall anisotropic B-factor. These were removed by deleting all data outside an ellipsoid with principle axes of 1/2.1, 1/2.5 and 1/1.6 A. The optimal limits were determined by monitoring the difference map signal for a known error in an intermediate version of the model. Statistics of the data and final model are shown in table 4, and they have been deposited in the protein databank (PDB ID 2HT0). Structure figures were made with Ribbons43.
Table 4.
Refinement of the doubly-nicked IHF-DNA complex structure
| nominal resolution | 15 - 2Å |
| Rmerge1 | 7.7% (39.4%) |
| redundancy: >= 3fold for 73% of all data, > = 5fold for 44% | |
| resolution where <I/σ I> falls below ~2 along a*, b*, c*: 2.2, 2.8, <2.0 data were used if they lay inside an ellipsoid with principle axes of 1/2.1Å, 1/2.5Å, 1/1.6Å | |
| completeness within ellipsoid: 97.2% (99.6% in highest resolution bin, 51% of those have I/σ I > 2) | |
| R | 23.42% (31.5%) |
| Rfree2 | 27.82% (36.3%) |
| rms deviation from ideal: | |
| bond lengths | 0.006Å |
| bond angles | 1.0° |
| Ramachandran plot3: 90.7% most favorable; 9.3% additional allowed | |
values in parenthesis are for the highest resolution bin
Rmerge = ∑ ( |I − <I>| )/∑ I
4.8% of total reflections
as defined by procheck44
Supplementary Material
Acknowledgments
We thank Reid Johnson for E. coli strain RJ1878, Ying Zhang Pigli for expert help with subcloning and protein purification, Adam Conway and Arabela Grigorescu for helpful comments on this manuscript, Thomas Lynch for helpful technical advice, and John Marko for interesting discussions of kinking. We also thank Shuwei Yang, Howard Nash, Kiyoshi Mizuuchi for their contributions to the doubly nicked IHF crystal structure, and Craig Ogata for help with data collection. This study was supported by NIH grant GM66011 to PAR and an NIH predoctoral training grant NIH T32 GM07183H for KKS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Azam AT, Iwata A, Nishimura A, Ueda S, Ishihama A. Growth phase-dependent variation in protein composition of the Escherichia coli nucleoid. J Bacteriol. 1999;181:6361–70. doi: 10.1128/jb.181.20.6361-6370.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ditto MD, Roberts D, Weisberg RA. Growth phase variation of integration host factor level in Escherichia coli. J Bacteriol. 1994;176:3738–48. doi: 10.1128/jb.176.12.3738-3748.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dame RT, Goosen N. HU: promoting or counteracting DNA compaction? FEBS Lett. 2002;529:151–6. doi: 10.1016/s0014-5793(02)03363-x. [DOI] [PubMed] [Google Scholar]
- 4.Arfin SM, Long AD, Ito ET, Tolleri L, Riehle MM, Paegle ES, Hatfield GW. Global gene expression profiling in Escherichia coli K12. The effects of integration host factor. J Biol Chem. 2000;275:29672–84. doi: 10.1074/jbc.M002247200. [DOI] [PubMed] [Google Scholar]
- 5.Johnson RC, Johnson LM, Schmidt J, Garder JF. The major nucleoid proteins in the structure and function of the E. coli chromosome. In: Higgins NP, editor. Bacterial Chromosomes 1 edit. Vol. 1.1. American Society for Microbiology; 2003. [Google Scholar]
- 6.Aki T, Adhya S. Repressor induced site-specific binding of HU for transcriptional regulation. Embo J. 1997;16:3666–74. doi: 10.1093/emboj/16.12.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lavoie BD, Shaw GS, Millner A, Chaconas G. Anatomy of a flexer-DNA complex inside a higher-order transposition intermediate. Cell. 1996;85:761–71. doi: 10.1016/s0092-8674(00)81241-6. [DOI] [PubMed] [Google Scholar]
- 8.Rice PA, Yang S, Mizuuchi K, Nash HA. Crystal structure of an IHF-DNA complex: a protein-induced DNA U-turn. Cell. 1996;87:1295–306. doi: 10.1016/s0092-8674(00)81824-3. [DOI] [PubMed] [Google Scholar]
- 9.Swinger KK, Rice PA. IHF and HU: flexible architects of bent DNA. Curr Opin Struct Biol. 2004;14:28–35. doi: 10.1016/j.sbi.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 10.Swinger KK, Lemberg KM, Zhang Y, Rice PA. Flexible DNA bending in HU-DNA cocrystal structures. Embo J. 2003;22:3749–60. doi: 10.1093/emboj/cdg351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Becker NA, Kahn JD, Maher LJ., 3rd Bacterial repression loops require enhanced DNA flexibility. J Mol Biol. 2005;349:716–30. doi: 10.1016/j.jmb.2005.04.035. [DOI] [PubMed] [Google Scholar]
- 12.Pontiggia A, Negri A, Beltrame M, Bianchi ME. Protein HU binds specifically to kinked DNA. Mol Microbiol. 1993;7:343–50. doi: 10.1111/j.1365-2958.1993.tb01126.x. [DOI] [PubMed] [Google Scholar]
- 13.Castaing B, Zelwer C, Laval J, Boiteux S. HU protein of Escherichia coli binds specifically to DNA that contains single-strand breaks or gaps. J Biol Chem. 1995;270:10291–6. doi: 10.1074/jbc.270.17.10291. [DOI] [PubMed] [Google Scholar]
- 14.Kamashev D, Balandina A, Rouviere-Yaniv J. The binding motif recognized by HU on both nicked and cruciform DNA. Embo J. 1999;18:5434–44. doi: 10.1093/emboj/18.19.5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kamashev D, Rouviere-Yaniv J. The histone-like protein HU binds specifically to DNA recombination and repair intermediates. Embo J. 2000;19:6527–35. doi: 10.1093/emboj/19.23.6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balandina A, Kamashev D, Rouviere-Yaniv J. The bacterial histone-like protein HU specifically recognizes similar structures in all nucleic acids. DNA, RNA, and their hybrids. J Biol Chem. 2002;277:27622–8. doi: 10.1074/jbc.M201978200. [DOI] [PubMed] [Google Scholar]
- 17.Grove A, Galeone A, Mayol L, Geiduschek EP. Localized DNA flexibility contributes to target site selection by DNA-bending proteins. J Mol Biol. 1996;260:120–5. doi: 10.1006/jmbi.1996.0386. [DOI] [PubMed] [Google Scholar]
- 18.Ghosh S, Grove A. Histone-like protein HU from Deinococcus radiodurans binds preferentially to four-way DNA junctions. J Mol Biol. 2004;337:561–71. doi: 10.1016/j.jmb.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 19.Li S, Waters R. Escherichia coli strains lacking protein HU are UV sensitive due to a role for HU in homologous recombination. J Bacteriol. 1998;180:3750–6. doi: 10.1128/jb.180.15.3750-3756.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boubrik F, Rouviere-Yaniv J. Increased sensitivity to gamma irradiation in bacteria lacking protein HU. Proc Natl Acad Sci U S A. 1995;92:3958–62. doi: 10.1073/pnas.92.9.3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pinson V, Takahashi M, Rouviere-Yaniv J. Differential binding of the Escherichia coli HU, homodimeric forms and heterodimeric form to linear, gapped and cruciform DNA. J Mol Biol. 1999;287:485–97. doi: 10.1006/jmbi.1999.2631. [DOI] [PubMed] [Google Scholar]
- 22.Grove A, Figueiredo ML, Galeone A, Mayol L, Geiduschek EP. Twin hydroxymethyluracil-A base pair steps define the binding site for the DNA-binding protein TF1. J Biol Chem. 1997;272:13084–7. doi: 10.1074/jbc.272.20.13084. [DOI] [PubMed] [Google Scholar]
- 23.Lynch TW, Read EK, Mattis AN, Gardner JF, Rice PA. Integration host factor: putting a twist on protein-DNA recognition. J Mol Biol. 2003;330:493–502. doi: 10.1016/s0022-2836(03)00529-1. [DOI] [PubMed] [Google Scholar]
- 24.Dickerson RE. Helix Structure and molecular recognition by B-DNA. In: Neidle S, editor. Nucleic Acid Structure. Vol. 1. Oxford University Press; Oxford: 1999. [Google Scholar]
- 25.Goodrich JA, Schwartz ML, McClure WR. Searching for and predicting the activity of sites for DNA binding proteins: compilation and analysis of the binding sites for Escherichia coli integration host factor (IHF) Nucleic Acids Res. 1990;18:4993–5000. doi: 10.1093/nar/18.17.4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu XJ, Olson WK. 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003;31:5108–21. doi: 10.1093/nar/gkg680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arnott S. Polynucleotide secondary structures: an historical perspective. In: Neidle S, editor. Nucleic Acid Structure. Vol. 1. Oxford University Press; Oxford: 1999. [Google Scholar]
- 28.Yang SW, Nash HA. Comparison of protein binding to DNA in vivo and in vitro: defining an effective intracellular target. Embo J. 1995;14:6292–300. doi: 10.1002/j.1460-2075.1995.tb00319.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holbrook JA, Tsodikov OV, Saecker RM, Record MT., Jr Specific and non-specific interactions of integration host factor with DNA: thermodynamic evidence for disruption of multiple IHF surface salt-bridges coupled to DNA binding. J Mol Biol. 2001;310:379–401. doi: 10.1006/jmbi.2001.4768. [DOI] [PubMed] [Google Scholar]
- 30.Yang JY, Kim K, Jayaram M, Harshey RM. A domain sharing model for active site assembly within the Mu A tetramer during transposition: the enhancer may specify domain contributions. Embo J. 1995;14:2374–84. doi: 10.1002/j.1460-2075.1995.tb07232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan J, Marko JF. Localized single-stranded bubble mechanism for cyclization of short double helix DNA. Phys Rev Lett. 2004;93:108108. doi: 10.1103/PhysRevLett.93.108108. [DOI] [PubMed] [Google Scholar]
- 32.van Noort J, Verbrugge S, Goosen N, Dekker C, Dame RT. Dual architectural roles of HU: formation of flexible hinges and rigid filaments. Proc Natl Acad Sci U S A. 2004;101:6969–74. doi: 10.1073/pnas.0308230101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wojtuszewski K, Mukerji I. HU binding to bent DNA: a fluorescence resonance energy transfer and anisotropy study. Biochemistry. 2003;42:3096–104. doi: 10.1021/bi0264014. [DOI] [PubMed] [Google Scholar]
- 34.Tanaka H, Yasuzawa K, Kohno K, Goshima N, Kano Y, Saiki T, Imamoto F. Role of HU proteins in forming and constraining supercoils of chromosomal DNA in Escherichia coli. Mol Gen Genet. 1995;248:518–26. doi: 10.1007/BF02423446. [DOI] [PubMed] [Google Scholar]
- 35.Hales LM, Gumport RI, Gardner JF. Examining the contribution of a dA+dT element to the conformation of Escherichia coli integration host factor-DNA complexes. Nucleic Acids Res. 1996;24:1780–6. doi: 10.1093/nar/24.9.1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roychoudhury M, Sitlani A, Lapham J, Crothers DM. Global structure and mechanical properties of a 10-bp nucleosome positioning motif. Proc Natl Acad Sci U S A. 2000;97:13608–13. doi: 10.1073/pnas.250476297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shrader TE, Crothers DM. Artificial nucleosome positioning sequences. Proc Natl Acad Sci U S A. 1989;86:7418–22. doi: 10.1073/pnas.86.19.7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Segal E, Fondufe-Mittendorf Y, Chen L, Thastrom A, Field Y, Moore IK, Wang JP, Widom J. A genomic code for nucleosome positioning. Nature. 2006;442:772–8. doi: 10.1038/nature04979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang W. Poor base stacking at DNA lesions may initiate recognition by many repair proteins. DNA Repair (Amst) 2006;10:654–666. doi: 10.1016/j.dnarep.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 40.Yang W, Junop MS, Ban C, Obmolova G, Hsieh P. DNA mismatch repair: from structure to mechanism. Cold Spring Harb Symp Quant Biol. 2000;65:225–32. doi: 10.1101/sqb.2000.65.225. [DOI] [PubMed] [Google Scholar]
- 41.Otwinowski Z, Minor W. Methods in Enzymology. Vol. 276. Academic Press; 1997. Processing of X-ray Data Collected in Oscillation Mode; pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 42.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54( Pt 5):905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 43.Carson M. Ribbons 2.0. J Appl Cryst. 1991;24:958–961. [Google Scholar]
- 44.Laskowski R, MacArthur M, Moss D, Thornton J. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.