

Abstract
The structures of F1-ATPase from bovine heart mitochondria inhibited with the dietary phytopolyphenol, resveratrol, and with the related polyphenols quercetin and piceatannol have been determined at 2.3-, 2.4- and 2.7-Å resolution, respectively. The inhibitors bind to a common site in the inside surface of an annulus made from loops in the three α- and three β-subunits beneath the “crown” of β-strands in their N-terminal domains. This region of F1-ATPase forms a bearing to allow the rotation of the tip of the γ-subunit inside the annulus during catalysis. The binding site is a hydrophobic pocket between the C-terminal tip of the γ-subunit and the βTP subunit, and the inhibitors are bound via H-bonds mostly to their hydroxyl moieties mediated by bound water molecules and by hydrophobic interactions. There are no equivalent sites between the γ-subunit and either the βDP or the βE subunit. The inhibitors probably prevent both the synthetic and hydrolytic activities of the enzyme by blocking both senses of rotation of the γ-subunit. The beneficial effects of dietary resveratrol may derive in part by preventing mitochondrial ATP synthesis in tumor cells, thereby inducing apoptosis.
Keywords: mitochondria, oxidative phosphorylation, rotary mechanism, crystal structure
A range of beneficial effects has been attributed to the ingestion of the phytopolyphenol resveratrol (trans3,4′,5-trihydroxystilbene) found in grapes, peanuts, berries, and various medicinal plants and to related polyphenols. They include protection against cardiovascular disease, ischemia, osteoporosis, cancer, and aging by means of mechanisms that include removal of reactive oxygen species, inhibition of mitosis and inflammation, and estrogen mimicry (1–10).
One of the many in vitro biochemical effects of resveratrol is to inhibit ATP hydrolysis and synthesis by the ATP synthase (F1Fo-ATPase) found in mitochondria (11), as do the related natural products quercetin and piceatannol (12–14). Also, they inhibit ATP hydrolysis by its separate F1 catalytic domain (15). The ATP synthase is a multisubunit assembly found in the inner membrane of the organelle. It is composed of the F1 catalytic domain (subunit composition α3β3γ1δ1ε1) attached by central (16) and peripheral stalks (17, 18) to a membrane-embedded proton-translocating domain known as Fo (19–21). The synthesis of ATP from ADP and phosphate is coupled by a mechanical rotary mechanism to a transmembrane proton-motive force generated by oxidative metabolism. This mechanism is driven by the passage of protons from the intermembrane space to the mitochondrial matrix, which impels the rotation of a ring of hydrophobic c-subunits in the Fo domain and the attached central stalk (subunits γ, δ, and ε) (22, 23). The rotating central stalk penetrates into the F1 domain through an asymmetrical α-helical coiled-coil in the γ-subunit, around which the three α- and the three β-subunits are arranged alternately (24, 25). The three catalytic sites of the enzyme, formed mainly from residues in the nucleotide-binding domains of the β-subunits, have different conformations and different affinities for nucleotides imposed by the asymmetry of the central stalk. Two catalytic subunits, known as βDP and βTP, bind either ATP (or nonhydrolyzable analogues) or ADP, but the binding to the βDP site is stronger, and it is likely that catalysis occurs at this site and not at the βTP site (25, 26). The third catalytic subunit, known as βE, is forced by the curvature of the central stalk into an “open” or “empty” conformation, which has little or no affinity for nucleotide. During ATP synthesis, the clockwise rotation of the central stalk (as viewed from the membrane) takes each catalytic site through a cycle of each of these three states, and each 360° rotation produces three ATP molecules (24). In the detergent purified F1Fo-ATPase uncoupled from the proton-motive force, or in the separate F1-ATPase domain, ATP hydrolysis energizes the rotation of the central stalk in the opposite sense to the synthetic direction of rotation (27–29).
The rotary mechanism of the mitochondrial F1-ATPase is inhibited by the binding of a range of natural products to various sites. Two molecules of the antibiotic aurovertin B bind simultaneously to equivalent sites in a cleft between the nucleotide-binding and C-terminal domains in both the βE- and βTP-subunits and appear to block catalysis by preventing closure of the catalytic interfaces (30). The efrapeptins bind in a site in the central cavity of the enzyme, thereby preventing the closure of the βE subunit during the rotary cycle (31). The natural inhibitor protein IF1 binds to a catalytic interface between the C-terminal domains of the βDP- and αDP-subunits and makes additional contacts with the γ-, βTP-, and αE-subunits (32). It blocks the rotary mechanism during ATP hydrolysis but not during ATP synthesis.
As described here, resveratrol, piceatannol, and quercetin (see Fig. 1) inhibit the rotary mechanism of F1-ATPase by binding to a fourth independent site involving the C-terminal tip of the γ-subunit, where the upper extremity of the central stalk fits into the hydrophobic annular sleeve of the “bearing” formed by loop regions below the “crown” made from β-strands in the N-terminal domains of the α- and β-subunits.
Fig. 1.

Structures of polyphenol inhibitors of bovine F1-ATPase. (I) Resveratrol. (II) Piceatannol. (III) Quercetin.
Results and Discussion
Structures of the F1-ATPase-Inhibitor Complexes.
The structures of the F1-resveratrol, F1-quercetin and F1-piceatannol complexes were solved by molecular replacement using data to 2.3-, 2.4- and 2.7-Å resolution, respectively. The statistics for data processing and refinement are summarized in Table 1. The crystals of all three complexes belong to the space group P21, with two F1 complexes per crystallographic asymmetric unit, whereas all other crystals of bovine F1-ATPase that have been described belong to the space group P212121, with one F1 complex per asymmetric unit (16, 25, 26, 30, 31, 33–37). Crystals of yeast F1-ATPase also belong to the P21 space group, with three F1 complexes per asymmetric unit (38, 39). In the resveratrol–F1, quercetin–F1, and piceatannol–F1 structures, the two F1-ATPase complexes are virtually identical, with overall r.m.s. deviations in Cα positions of 0.09, 0.09, and 0.14 Å, respectively, and in the following text, no attempt has been made to distinguish between the two F1 complexes in each structure. Each F1 assembly in the refined resveratrol–F1 structure consists of residues αE 24–510, αTP 23–401 and 410–510, αDP 16–510, βE 9–474, βTP 9–474, βDP 9–475, γ 1–47, 67–90, 105–116, 127–148, 159–173 and 201–272. In the refined quercetin-F1 and piceatannol-F1 structures, each F1 structure contains residues αE 24–510, αTP 23–401 and 410–510, αDP 16–510, βE 9–474, βTP 9–474, βDP 9–475, γ 1–47, 72–89, 106–115, 129–140, 161–173, and 206–272. In all three structures, the electron density for the δ- and ε-subunits was too weak to allow them to be modeled.
Table 1.
Crystallographic data for bovine F1-ATPase complexed with various polyphenol inhibitors
| Resveratrol–F1 | Quercetin–F1 | Piceatannol–F1 | |
|---|---|---|---|
| Space group | P21 | P21 | P21 |
| Unit cell dimensions, Å (a, b, c) | 106.8, 277.4, 137.8 | 106.4, 282.0, 138.1 | 107.0, 281.2, 138.8 |
| Unit cell angles, ° (α, β, γ) | 90.0, 90.2, 90.0 | 90.0, 90.4, 90.0 | 90.0, 89.6, 90.0 |
| Resolution, Å | 2.30 | 2.40 | 2.70 |
| No. of unique reflections | 299,020 (40,061) | 297,401 (40,583) | 201,334 (30,153) |
| Rmerge,* % | 6.4 (23.6) | 8.2 (40.3) | 8.7 (34.9) |
| Completeness,† % | 84.7 (77.5) | 94.3 (88.1) | 90.3 (92.5) |
| Multiplicity | 1.4 (1.4) | 2.1 (2.0) | 1.4 (1.3) |
| 〈I/σ(I)〉 | 7.8 (2.1) | 7.2 (2.3) | 7.3 (1.9) |
| Wilson B factor, Å2 | 38.6 | 40.5 | 54.2 |
| Inhibitor atoms‡ | 34 | 44 | 36 |
| Water molecules | 3,847 | 2,202 | 1,113 |
| Glycerol molecules | 12 | 12 | 12 |
| R factor,§ % | 16.0 | 18.8 | 20.2 |
| Rfree,¶ % | 21.7 | 23.8 | 26.9 |
| rms deviation bonds, Å | 0.010 | 0.009 | 0.010 |
| rms deviation angles, ° | 1.2 | 1.2 | 1.3 |
Values for the highest-resolution bins (2.42–2.30, 2.53–2.40, and 2.85–2.70 Å, respectively, for the resveratrol–F1, quercetin–F1, and piceatannol–F1 complexes) are given in parentheses.
*Rmerge = Σhkl Σi|〈I(hkl)〉 − Ii(hkl)| / Σhkl Σi Ii(hkl), where 〈I(hkl)〉 is the mean weighted intensity for multiple recorded reflections i after rejection of outliers. Measurements with intensities differing >3.5 σ (I) from the weighted mean were rejected.
†The overall completeness for the resveratrol–F1 data and piceatannol–F1 data is slightly low because only 60° of data were collected.
‡Hydrogen atoms were excluded.
§The R factor is defined as Σhkl|Fo(hkl) − Fc(hkl)|/Σhkl|Fo(hkl)|, where Fo and Fc are the observed and calculated structure factor amplitudes, respectively, and was determined by using 95% of the data.
¶The free R factor is the R factor calculated for the residual 5% of the data set not included in the refinement.
The overall architectures of the three complexes are very similar to the “reference structure” of F1-ATPase (24) and to the majority of the structures of bovine F1-ATPase inhibited in various ways (25, 26, 29, 30, 31, 33–37). The reference structure superimposes well with the resveratrol–F1, quercetin–F1 and piceatannol–F1 structures, with r.m.s. deviations in Cα positions of 0.42, 0.51, and 0.44 Å, respectively. In all three inhibited complexes, an AMP-PNP molecule is bound to the βTP-subunit and to all three α-subunits, ADP and azide are bound to the βDP-subunit, and there is no nucleotide bound to the βE-subunit. There is also electron density associated with the P-loops in the βE-subunits, which was interpreted as a sulfate (or phosphate) ion as in other structures of bovine F1-ATPase.
The Inhibitor-Binding Site.
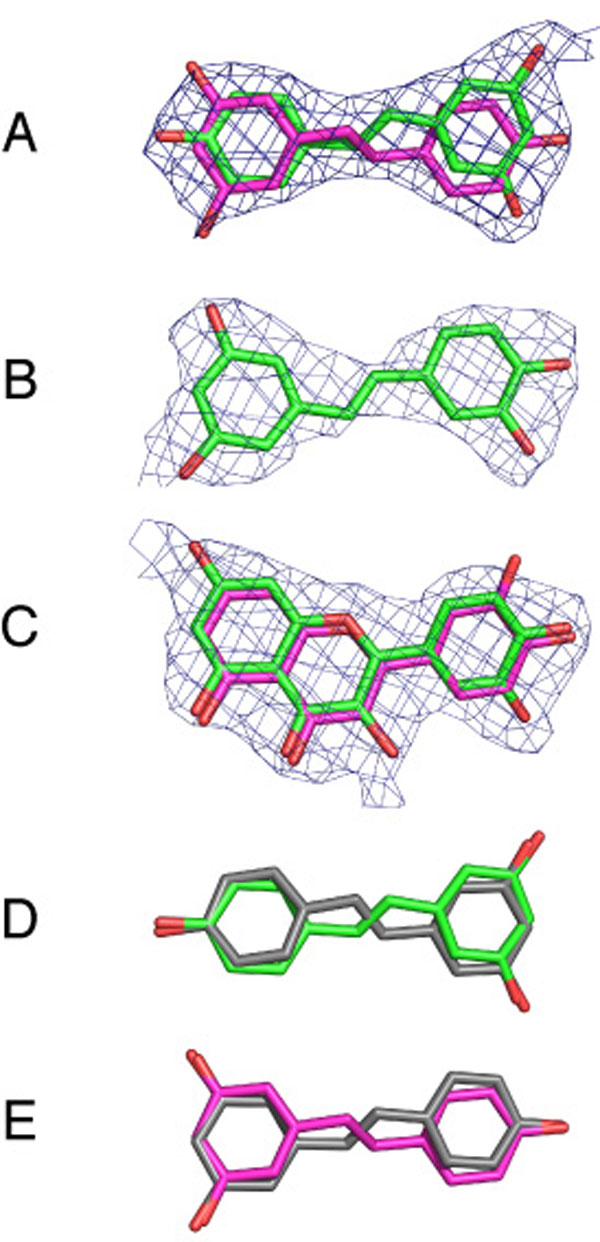
The (Fo−Fc) difference electron density maps of resveratrol–F1, piceatannol–F1, and quercetin–F1 each contained a region of positive electron density near to the C-terminal tip of the γ-subunit. The shapes of these regions of density agreed with the structures of the respective inhibitors, and they were built into the structural models [see supporting information (SI) Fig. 4]. The resveratrol and piceatannol molecules both have internal pseudosymmetry, and rotation about their long axes, and about axes orthogonal to them, produces views with similar shapes. One of these possible orientations of resveratrol fitted the initial positive (Fo−Fc) difference density map well, but after refinement with this orientation, regions of positive density were not accounted for, suggesting that other orientations of bound resveratrol were present at lower occupancy. Refinement of the map with occupancies of 75% in the first orientation and 25% for the second orientation, accounted well for the electron density. However, three other orientations, produced by rotation of resveratrol about its long axis, also fit the density, and they cannot be distinguished. Thus, it is likely that resveratrol binds to F1-ATPase in several different modes. Similarly, piceatannol could bind in four different orientations that are indistinguishable at the resolution of the electron density map. Quercetin exists as cis- and trans-rotamers produced by rotation of the exocyclic ring by 180° about the bond linking it to the benzopyranone ring. Residual density remained after refinement with either rotamer, suggesting the presence of both forms, whereas refinement with occupancies of 75% and 25% for cis and trans forms, respectively, accounted for the density.
Resveratrol, quercetin, and piceatannol are bound in a pocket between the hydrophobic tip in the C-terminal region of the γ-subunit and the hydrophobic inside, or “sleeve,” of the surrounding annulus. This annulus is made from loop regions between helices E and F and between helix G and β-strand 7 in the three β-subunits and the equivalent loops in the three α-subunits. The binding pocket lies between the βTP-subunit and the C-terminal region of the γ-subunit (Fig. 2 A and B). There is no equivalent pocket between the γ-subunit and either the βE- or the βDP-subunit (SI Fig. 5 A and B). It is distinct from the binding sites of other inhibitors and nucleotides, consistent with mixed inhibition (15), and is accessible from the solvent-filled central cavity of the enzyme, and, hence by the interfaces between the α- and β-subunits, to the external milieu.
Fig. 2.

The site of binding of resveratrol in bovine F1-ATPase. The α-, β- and γ-subunits are red, yellow, and blue, respectively, and resveratrol is green. (A) Ribbon view of F1-ATPase upwards from the mitochondrial membrane along the central axis of the γ-subunit, showing the inhibitor in solid representation bound between the γ- and βTP-subunits. (B) Side view of a solid representation of resveratrol bound in a pocket in F1-ATPase between the γ- and βTP-subunits. For clarity, the βDP and βE subunits and the three α-subunits have been removed. The pocket is in the “bearing” consisting of the sleeve provided by the N-terminal regions of α- and β-subunits in the “crown” domain of F1-ATPase and the α-helical tip of the C-terminal region of the γ-subunit. (C) Side view in stereo showing interactions of resveratrol with side chains in the binding pocket shown in stick representation with oxygen and nitrogen atoms in binding-site residues in red and dark blue, respectively. The residues shown are either within 4 Å of the inhibitor and form hydrophobic interactions, or they are linked to it via hydrogen bond networks (dotted lines) involving water molecules (light blue spheres), and by a hydrogen bond from the amido group of Val-279 to the π-electrons of the m-dihydroxyphenyl moiety of resveratrol (orange dotted line). (D and E) view of the binding pocket and bound resveratrol (with red oxygen atoms) in solid representation. D is the same view as in C, and in E, bound resveratrol and its binding pocket are viewed along the axis of γ-subunit, upwards from the mitochondrial membrane.
The resveratrol molecule is bound in the pocket in a slightly distorted planar conformation by means of hydrophobic interactions and H-bonds. There will be also entropic contributions to binding from the release of water molecules on inhibitor binding [at least five molecules relative to the [ADP–BeF3−–F1] structure (26)] and from the rigid stilbene scaffold. The hydrophobic interactions occur between the inhibitor and the γ-, βTP-, αTP-, and αDP-subunits (Fig. 2 C–E and SI Fig. 5) and involve residues γ-Lys-260, γ-Ile-263, βTP-Val-279, and βTP-Ala-278. To help accommodate the resveratrol molecule, the side chain of γ-Lys-260 has moved from a position in the cavity of the bearing between the γ-subunit and the βTP-subunit in the reference structure, to a position where its side chain interacts with γ-Glu-264. Side-chain regions of γ-Ala-256, γ-Thr-259, γ-Glu-264, αTP-Glu-292, αTP-Gly-290, and αDP-Glu-292, all within 4 Å of the resveratrol, appear to contribute additional nonpolar interactions. There are two H-bonds between the backbone amido groups of βTP-Val-279 and αTP-Glu-292, and the π-electrons of the m-dihydroxyphenyl moiety of resveratrol and its 4′-hydroxyl, respectively. A network of H-bonds between the 3-, 4′-, and 5′-hydroxyls of resveratrol and residues αTP-Arg-291, βTP-Ser-277, βTP-Gly-280, and γ-Thr-259 of the F1 domain is mediated by ordered water molecules. Quercetin and piceatannol bind in similar way to resveratrol, as shown by the superimposition of the inhibitor binding sites in the three structures (Fig. 3).
Fig. 3.

Comparison of the modes of binding to bovine F1-ATPase of piceatannol and quercetin with that of resveratrol. (A) Major binding modes of resveratrol (green) and piceatannol (gray). (B) Major binding mode of resveratrol (green) and cis-quercetin (gray).
The modes of binding of resveratrol and quercetin to F1-ATPase have common features with their modes of binding to other proteins. The structure of five other protein–resveratrol complexes are known [with human quinone reductase 2 (40), human transthyretin (41), stilbene synthase from Scots pine and peanuts (42, 43), and chalcone synthase from alfalfa (44)] and seven others where quercetin is bound [flavanoid glucosyl transferase from Vitis vinifera (45), phosphoinositide 3-kinase from Sus scrofa (46), quercetin 2,3-dioxygenase from Aspergillus japonicus (47), anthocyanidin synthase from Arabidopsis thalania (48), the human Src family protein kinase Hck (49), the human PIM1 kinase (50), and the multidrug-binding protein TgR from Pseudomonas putida (51)]. In most of these structures, the conformations of the bound resveratrol and quercetin molecules are slightly distorted from planar, and they are all bound in hydrophobic pockets, predominantly by means of van der Waals contacts and H-bonds involving the hydroxyl groups of the inhibitors.
All of the residues of bovine F1-ATPase that are involved in binding resveratrol are conserved in the rat enzyme (SI Fig. 6), consistent with its known inhibition by resveratrol and piceatannol (14, 15). They are conserved also in man, and so it is a reasonable assumption that the human enzyme will be affected in similar way by resveratrol and related compounds.
Mechanism of Inhibition.
The effect of resveratrol, quercetin, or piceatannol binding in the sleeve between the tip of the γ-subunit and the region of the inside surface of the annulus provided by the βTP-subunit, is to block the bearing and so to prevent the rotation of the central stalk from proceeding. Both ATP hydrolysis and ATP synthesis are inhibited by resveratrol (11), and the presence of resveratrol in the sleeve is evidently capable of preventing both senses of rotation (clockwise, as viewed from the membrane, during synthesis and counterclockwise during hydrolysis). Quercetin prevents hydrolysis of ATP, but not its synthesis (12). Given the close similarities between resveratrol- and quercetin-binding sites, this aspect of the inhibitory properties of quercetin needs to be reexamined.
One important aspect of the inhibitory mechanism of resveratrol, quercetin, and piceatannol is that it serves to emphasize the importance of the bearing in the rotary mechanism of the enzyme. The conservation of amino acid residues in the C-terminal part of the γ-subunit (52, 53) and in the residues that form the sleeve (25, 52), and the effects of mutations in this region of the enzyme in various species (54–59), provide further support for this view. However, this conclusion seems to be contradicted by experiments on the Escherichia coli, chloroplast, and Bacillus PS3 enzymes, where it has been reported that the C-terminal region α-helix of the γ-subunit can be shortened (by up to 12, 20, and 21 residues, respectively) without effect on catalytic activity (60, 61). These experiments have been interpreted as implying that this region of the γ-subunit is dispensible and that penetration of the C-terminal region of the γ-subunit into the sleeve is inessential for the rotary mechanism. However, the structural consequences of the deletions on the bacterial and chloroplast enzymes are not known, and it is possible that the structure of the shortened γ-protein is not simply the structure (modeled by homology with the known structure of the mitochondrial γ-subunit) with a shortened C-terminal α-helix of the γ-subunit no longer extending into the collar but that the shortened protein adjusts its structure, so that penetration of the central stalk into the sleeve of the bearing is maintained. One possibility that has been discussed (62) is that the C-terminal helix of the γ-subunit unwinds partially as part of this accommodation in the mutated enzyme. However, there is no experimental evidence in the wild-type enzyme that unwinding of the C-terminal tip of the γ-subunit (and rewinding) accompanies rotation, as has been suggested from molecular dynamics simulations (62). Also, irrespective of whether this explanation is correct or not, the enzyme with a truncated γ-subunit is not the wild-type enzyme. Therefore, rather than contributing to an understanding of the wild-type enzyme, studies of F1-ATPase with missing segments of sequence may reveal properties about the plasticity of the enzyme, that is, its ability to adjust to the removal of structural elements. Similar considerations apply to deletions and insertions in the peripheral stalk region of the bacterial F1-ATPase (63, 64) (where again a detailed structure is not known for either wild-type or mutated forms). These experiments have been interpreted as indicating that the peripheral stalk is a flexible structure like a rope. At our current state of knowledge, this interpretation appears to be incompatible with features of the structure of the peripheral stalk determined in the mitochondrial enzyme, which appears to be a rather rigid structure that links the top of the F1 domain to the membrane domain by the shortest route (17, 18).
Biological and Medical Implications.
The beneficial effects of dietary resveratrol and related compounds appear to derive from their interaction with one or more of a wide range of different sites in the cell, among them the mitochondrion. Mitochondrial dysfunction and energy deficiency have been linked to a number of degenerative diseases such as cardiovascular disease and neurological disorders and to aging and cancer (65, 66). Therefore, it is conceivable that the inhibition of the ATP synthase by resveratrol might play a significant role in the pathophysiology of such conditions.
For example, during cardiac ischemia, cardioprotective benefit is thought to derive from preventing the destruction of ATP that leads to tissue damage by inhibiting the hydrolytic activity of F1Fo-ATPase (but not ATP synthesis) in mitochondria. The natural inhibitor protein IF1 acts in this way and is cardioprotective (67, 68), as do three series of synthetic cardioprotective compounds based on a 4-(N-arylimidazole)-substituted benzopyran, guanidine, and benzodiazapines (69–71). Similarly, oligomycin, which inhibits F1Fo-ATPase through its Fo domain, preserves ATP and protects against or postpones injury during ischemia (72). However, it is difficult to envisage how inhibition of mitochondrial F1Fo-ATPase by dietary resveratrol and related polyphenols could have a similar effect and so contribute to the cardiovascular protective effects associated with dietary polyphenols.
Another possible way in which inhibition of mitochondrial F1Fo-ATPase by dietary resveratrol might be beneficial is by induction of apoptosis selectively in tumor cells. Resveratrol induces cell death in tumor cells via pathways that depend on mitochondria (2, 73), and oligomycin, a specific inhibitor of mitochondrial F1Fo-ATPase, has similar effects (74), possibly by marking tumor cells for cell death by CD14, while allowing commitment to differentiation to occur in the surviving population (75). The benzodiazepine Bz-423 also inhibits the mitochondrial F1Fo-ATPase, possibly by binding to the oligomycin sensitivity-conferral protein, a component of the peripheral stalk. In mouse models of systemic lupus erythematosus, this drug suppresses autoimmunity by selective induction of apoptosis through inhibition of the F1Fo-ATPase in the disease-causing lymphocytes (76). Unlike other potent ATP synthase inhibitors such as efrapeptin and aurovertin, which are highly toxic, Bz-423 acts, not by significant depletion of ATP, but by converting the mitochondria from an actively respiring state (state 3) to resting respiration (state 4). This effect results in the production of reactive oxygen species, which triggers the apoptotic signal leading to cell death. Normal cells appear to be unaffected by the drug, but the autoimmune lymphocytes, which have altered mitochondrial bioenergetics, are sensitized to Bz-423-mediated inhibition of ATP synthase (77). Thus, it may be possible to exploit the altered bioenergetics of cancer cells in a similar way with inhibitors of ATP synthase, including resveratrol, quercetin, and piceatannol to target tumor cells selectively without affecting other cells.
Materials and Methods
Crystallization and Data Collection.
Crystals of bovine F1-ATPase with maximum dimensions of ≈0.3 mm were grown by microdialysis, as described (37). An ethanolic solution of resveratrol or piceatannol (20 mM) or a 100 mM solution of quercetin in dimethyl sulfoxide was added to the outside solution [final concentrations: 1 mM resveratrol, 5% ethanol (vol/vol); 0.2 mM piceatannol, 1% ethanol (vol/vol); 5 mM quercetin; 5% dimethyl sulfoxide (vol/vol)], and the samples were kept in the dark for 2 days at 23°C. Then the crystals were cryoprotected by adding 5% (vol/vol) glycerol to the outside buffer, which contained 14.5% (wt/vol) polyethylene glycol 6000 and 1 mM resveratrol or 0.2 mM piceatannol or 5 mM quercetin. The concentration of glycerol was increased in 5% steps to 20% and then to 22% (vol/vol) with 30 min at each concentration. Crystals were harvested with cryoloops, plunged into liquid nitrogen, and stored at 100 K. Diffraction data were collected at 100 K to 2.3-Å resolution for F1-resveratrol and 2.7-Å resolution for F1-picetannol with a charge-coupled detector (Area Detector Systems, Poway, CA) Q4 on beamline ID14–2 (λ = 0.933 Å) at the European Synchrotron Radiation Facility, Grenoble, France. Diffraction data for F1-quercetin were collected to 2.4-Å resolution under similar conditions on beamline ID14–1 (λ = 0.934 Å). They were processed with MOSFLM (78) and with programs from the Collaborative Computational Project Number 4 (CCP4) suite (79).
Structure Solution and Refinement.
The structures were solved by molecular replacement with AMoRe (80). The starting model was the structure of F1–ATPase inhibited with ADP and beryllium fluoride (BeF3−–F1) (26) with water and glycerol molecules deleted from the model, and the BeF3− groups deleted from the βTP- and βDP-subunits. After rigid-body refinement with AMoRe, the R-factor and correlation coefficient for all data from 20.0- to 4.0-Å resolution were 28.2% and 77.6%, respectively, for the resveratrol–F1 structure, 29.0% and 76.7% for the quercetin–F1 structure, and 28.8% and 74.9% for the piceatannol–F1 structure. Further refinement was carried out alternately with REFMAC5 (81) and manual rebuilding with O (82). Noncrystallographic symmetry restraints were applied during refinement of the two F1 complexes in the asymmetric unit. The coordinates for resveratrol and quercetin were taken from the crystal structures of human quinone reductase 2 (PDB ID code 1SG0) (40), quercetin 2,3-dioxygenase (PDB ID code 1H1I) (47), and flavanoid glucosyl transferase (PDB ID code 2C9Z) (45) and built into the structural models. The coordinates for piceatannol were derived from an energy-minimized model generated by PRODRG (83). The mean B-factor for resveratrol was 35 Å2, and the surrounding residues had similar B-factors. Therefore, the occupancy for the resveratrol molecule was set at 100%. The γ-phosphates for bound AMP–PNP molecules were built into the structural model by using the coordinates from the reference structure (24). An azide ion was built into the βDP-subunit.
For the calculations of the Rfree value, 5% of the diffraction data were excluded from the refinement. The stereochemistry was assessed with PROCHECK (84). For resveratrol, quercetin, and piceatannol, respectively, 92%, 91.7%, and 89.9% of the residues were assigned to the most favored region of the Ramachandran plot, 7.8%, 8.2%, and 9.8% to allowed regions, and 0.2%, 0.1%, and 0.3% to generously allowed regions. There were no residues in disallowed regions. Figures were produced with PyMOL (85).
Supplementary Material
Acknowledgments
We thank the beamline staff at the European Synchrotron Radiation Facility, Grenoble, for assistance with data collection. This work was supported by the Medical Research Council, United Kingdom. J.R.G. was supported in part by a Research Studentship from Trinity College, Cambridge, U.K.
Abbreviation
- AMP-PNP
adenosine-5′-(β,γ-imino)triphosphate.
Footnotes
The authors declare no conflict of interest.
Data deposition: Coordinates and structure factors of the F1–resveratrol, F1–quercetin, and F1–piceatannol complexes were deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 2jiz, 2jj2, and 2jj1, respectively).
This article contains supporting information online at www.pnas.org/cgi/content/full/0706290104/DC1.
References
- 1.Fremont L. Life Sci. 2000;66:663–673. doi: 10.1016/s0024-3205(99)00410-5. [DOI] [PubMed] [Google Scholar]
- 2.Pervaiz S. FASEB J. 2003;17:1975–1985. doi: 10.1096/fj.03-0168rev. [DOI] [PubMed] [Google Scholar]
- 3.Ulrich S, Wolter F, Stein JM. Mol Nutr Food Res. 2005;49:452–461. doi: 10.1002/mnfr.200400081. [DOI] [PubMed] [Google Scholar]
- 4.Delmas D, Jannin B, Latruffe N. Mol Nutr Food Res. 2005;49:377–395. doi: 10.1002/mnfr.200400098. [DOI] [PubMed] [Google Scholar]
- 5.de la Lastra CA, Villegas I. Mol Nutr Food Res. 2005;49:405–430. doi: 10.1002/mnfr.200500022. [DOI] [PubMed] [Google Scholar]
- 6.Bhat KP, Pezzuto JM. Ann NY Acad Sci. 2002;957:210–229. doi: 10.1111/j.1749-6632.2002.tb02918.x. [DOI] [PubMed] [Google Scholar]
- 7.Cai YJ, Fang JG, Ma LP, Yang L, Liu ZL. Biochim Biophys Acta. 2003;1637:31–38. doi: 10.1016/s0925-4439(02)00174-6. [DOI] [PubMed] [Google Scholar]
- 8.Fan X, Mattheis JP. J Food Sci. 2001;66:200–203. [Google Scholar]
- 9.Cal C, Garban H, Jazirehi A, Yeh C, Mizutani Y, Bonavida B. Curr Med Chem Anti-Cancer Agents. 2003;3:77–93. doi: 10.2174/1568011033353443. [DOI] [PubMed] [Google Scholar]
- 10.Geahlen RL, McLaughlin JL. Biochem Biophys Res Commun. 1989;165:241–245. doi: 10.1016/0006-291x(89)91060-7. [DOI] [PubMed] [Google Scholar]
- 11.Zheng J, Ramirez VD. Br J Pharmacol. 2000;130:1115–1123. doi: 10.1038/sj.bjp.0703397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lang DR, Racker E. Biochim Biophys Acta. 1974;333:180–186. doi: 10.1016/0005-2728(74)90002-4. [DOI] [PubMed] [Google Scholar]
- 13.Di Pietro A, Godinot C, Bouillant ML, Gautheron DC. Biochimie. 1975;57:959–967. doi: 10.1016/s0300-9084(75)80218-5. [DOI] [PubMed] [Google Scholar]
- 14.Zheng J, Ramirez VD. Biochem Biophys Res Commun. 1999;261:499–503. doi: 10.1006/bbrc.1999.1063. [DOI] [PubMed] [Google Scholar]
- 15.Gledhill JR, Walker JE. Biochem J. 2005;386:591–598. doi: 10.1042/BJ20041513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gibbons C, Montgomery MG, Leslie AGW, Walker JE. Nat Struct Biol. 2000;7:1055–1061. doi: 10.1038/80981. [DOI] [PubMed] [Google Scholar]
- 17.Kane Dickson V, Silvester JA, Fearnley IM, Leslie AGW, Walker JE. EMBO J. 2006;25:2911–2918. doi: 10.1038/sj.emboj.7601177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walker JE, Kane Dickson V. Biochim Biophys Acta. 2006;1757:286–296. doi: 10.1016/j.bbabio.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 19.Walker JE. Angew Chem Int Edn. 1998;37:2309–2319. [Google Scholar]
- 20.Boyer PD. Annu Rev Biochem. 1997;66:717–749. doi: 10.1146/annurev.biochem.66.1.717. [DOI] [PubMed] [Google Scholar]
- 21.Senior AE, Nadanaciva S, Weber J. Biochim Biophys Acta. 2002;1553:188–211. doi: 10.1016/s0005-2728(02)00185-8. [DOI] [PubMed] [Google Scholar]
- 22.Stock D, Leslie AGW, Walker JE. Science. 1999;286:1700–1705. doi: 10.1126/science.286.5445.1700. [DOI] [PubMed] [Google Scholar]
- 23.Stock D, Gibbons C, Arechaga I, Leslie AGW, Walker JE. Curr Opin Struct Biol. 2000;10:672–679. doi: 10.1016/s0959-440x(00)00147-0. [DOI] [PubMed] [Google Scholar]
- 24.Abrahams JP, Leslie AGW, Lutter R, Walker JE. Nature. 1994;370:621–628. doi: 10.1038/370621a0. [DOI] [PubMed] [Google Scholar]
- 25.Bowler MW, Montgomery MG, Leslie AGW, Walker JE. J Biol Chem. 2007;282:14238–14242. doi: 10.1074/jbc.M700203200. [DOI] [PubMed] [Google Scholar]
- 26.Kagawa R, Montgomery MG, Braig K, Leslie AGW, Walker JE. EMBO J. 2004;23:2734–2744. doi: 10.1038/sj.emboj.7600293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoshida M, Muneyuki E, Hisabori T. Nat Rev Mol Cell Biol. 2001;2:669–677. doi: 10.1038/35089509. [DOI] [PubMed] [Google Scholar]
- 28.Itoh H, Takahashi A, Adachi K, Noji H, Yasuda R, Yoshida M, Kinosita K. Nature. 2004;427:465–468. doi: 10.1038/nature02212. [DOI] [PubMed] [Google Scholar]
- 29.Diez M, Zimmermann B, Borsch M, Konig M, Schweinberger E, Steigmiller S, Reuter R, Felekyan S, Kudryavtsev S, Seidel CA, et al. Nat Struct Mol Biol. 2004;11:135–141. doi: 10.1038/nsmb718. [DOI] [PubMed] [Google Scholar]
- 30.van Raaij MJ, Abrahams JP, Leslie AGW, Walker JE. Proc Natl Acad Sci USA. 1996;93:6913–6917. doi: 10.1073/pnas.93.14.6913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abrahams JP, Buchanan SK, van Raaij MJ, Fearnley IM, Leslie AGW, Walker JE. Proc Natl Acad Sci USA. 1996;93:9420–9424. doi: 10.1073/pnas.93.18.9420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cabezon E, Montgomery MG, Leslie AGW, Walker JE. Nat Struct Biol. 2003;10:744–750. doi: 10.1038/nsb966. [DOI] [PubMed] [Google Scholar]
- 33.Braig K, Menz RI, Montgomery MG, Leslie AGW, Walker JE. Structure (London) 2000;8:567–573. doi: 10.1016/s0969-2126(00)00145-3. [DOI] [PubMed] [Google Scholar]
- 34.Menz RI, Leslie AGW, Walker JE. FEBS Lett. 2001;494:11–14. doi: 10.1016/s0014-5793(01)02302-x. [DOI] [PubMed] [Google Scholar]
- 35.Menz RI, Walker JE, Leslie AGW. Cell. 2001;106:331–341. doi: 10.1016/s0092-8674(01)00452-4. [DOI] [PubMed] [Google Scholar]
- 36.Orriss GL, Leslie AGW, Braig K, Walker JE. Structure (London) 1998;6:831–837. doi: 10.1016/s0969-2126(98)00085-9. [DOI] [PubMed] [Google Scholar]
- 37.Bowler MW, Montgomery MG, Leslie AGW, Walker JE. Proc Natl Acad Sci USA. 2006;103:8646–8649. doi: 10.1073/pnas.0602915103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mueller DM, Puri N, Kabaleeswaran V, Terry C, Leslie AGW, Walker JE. Acta Crystallogr D. 2004;60:1441–1444. doi: 10.1107/S0907444904012661. [DOI] [PubMed] [Google Scholar]
- 39.Kabaleeswaran V, Puri N, Walker JE, Leslie AGW, Mueller DM. EMBO J. 2006;25:5433–5442. doi: 10.1038/sj.emboj.7601410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buryanovskyy L, Fu Y, Boyd M, Ma Y, Hsieh TC, Wu JM, Zhang Z. Biochemistry. 2004;43:11417–11426. doi: 10.1021/bi049162o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klabunde T, Petrassi HM, Oza VB, Raman P, Kelly JW, Sacchettini JC. Nat Struct Biol. 2000;7:312–321. doi: 10.1038/74082. [DOI] [PubMed] [Google Scholar]
- 42.Austin MB, Bowman ME, Ferrer JL, Schroder J, Noel JP. Chem Biol. 2004;11:1179–1194. doi: 10.1016/j.chembiol.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 43.Shomura Y, Torayama I, Suh DY, Xiang T, Kita A, Sankawa U, Miki K. Proteins. 2005;60:803–806. doi: 10.1002/prot.20584. [DOI] [PubMed] [Google Scholar]
- 44.Ferrer JL, Jez JM, Bowman ME, Dixon RA, Noel JP. Nat Struct Biol. 1999;6:775–784. doi: 10.1038/11553. [DOI] [PubMed] [Google Scholar]
- 45.Offen W, Martinez-Fleites C, Yang M, Kiat-Lim E, Davis BG, Tarling CA, Ford CM, Bowles DJ, Davies GJ. EMBO J. 2006;25:1396–1405. doi: 10.1038/sj.emboj.7600970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walker EH, Pacold ME, Perisic O, Stephens L, Hawkins PT, Wymann MP, Williams RL. Mol Cell. 2000;6:909–919. doi: 10.1016/s1097-2765(05)00089-4. [DOI] [PubMed] [Google Scholar]
- 47.Steiner RA, Kalk KH, Dijkstra BW. Proc Natl Acad Sci USA. 2002;99:16625–16630. doi: 10.1073/pnas.262506299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilmouth RC, Turnbull JJ, Welford RW, Clifton IJ, Prescott AG, Schofield CJ. Structure (London) 2002;10:93–103. doi: 10.1016/s0969-2126(01)00695-5. [DOI] [PubMed] [Google Scholar]
- 49.Sicheri F, Moarefi I, Kuriyan J. Nature. 1997;385:602–609. doi: 10.1038/385602a0. [DOI] [PubMed] [Google Scholar]
- 50.Holder S, Zemskova M, Zhang C, Tabrizizad M, Bremer R, Neidigh JW, Lilly MB. Mol Cancer Ther. 2007;6:163–172. doi: 10.1158/1535-7163.MCT-06-0397. [DOI] [PubMed] [Google Scholar]
- 51.Alguel Y, Meng C, Teran W, Krell T, Ramos JL, Gallegos MT, Zhang X. J Mol Biol. 2007;369:829–840. doi: 10.1016/j.jmb.2007.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Walker JE, Fearnley IM, Gay NJ, Gibson BW, Northrop FD, Powell SJ, Runswick MJ, Saraste M, Tybulewicz VLJ. J Mol Biol. 1985;184:677–701. doi: 10.1016/0022-2836(85)90313-4. [DOI] [PubMed] [Google Scholar]
- 53.Miki J, Maeda M, Mukohata Y, Futai M. FEBS Lett. 1988;232:221–226. doi: 10.1016/0014-5793(88)80421-6. [DOI] [PubMed] [Google Scholar]
- 54.Iwamoto A, Miki J, Maeda M, Futai M. J Biol Chem. 1990;265:5043–5048. [PubMed] [Google Scholar]
- 55.Jeanteur-De Beukelaer C, Omote H, Iwamoto-Kihara A, Maeda M, Futai M. J Biol Chem. 1995;270:22850–22854. doi: 10.1074/jbc.270.39.22850. [DOI] [PubMed] [Google Scholar]
- 56.Nakamoto RK, al-Shawi MK, Futai M. J Biol Chem. 1995;270:14042–14046. doi: 10.1074/jbc.270.23.14042. [DOI] [PubMed] [Google Scholar]
- 57.Clark-Walker GD, Hansbro PM, Gibson F, Chen XJ. Biochim Biophys Acta. 2000;1478:125–137. doi: 10.1016/s0167-4838(00)00003-0. [DOI] [PubMed] [Google Scholar]
- 58.Weber ER, Rooks RS, Shafer KS, Chase JW, Thorness PE. Genetics. 1995;1240:435–442. doi: 10.1093/genetics/140.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clark-Walker GD. Mitochondrion. 2003;2:257–265. doi: 10.1016/S1567-7249(02)00107-1. [DOI] [PubMed] [Google Scholar]
- 60.Muller M, Panke O, Junge W, Engelbrecht S. J Biol Chem. 2002;277:23308–23313. doi: 10.1074/jbc.M201998200. [DOI] [PubMed] [Google Scholar]
- 61.Sokolov M, Lu L, Tucker W, Gao F, Gegenheimer PA, Richter ML. J Biol Chem. 1999;274:13824–13829. doi: 10.1074/jbc.274.20.13824. [DOI] [PubMed] [Google Scholar]
- 62.Muller M, Gumbiowski K, Cherepanov DA, Winkler S, Junge W, Engelbrecht S, Panke O. Eur J Biochem. 2004;271:3914–3922. doi: 10.1111/j.1432-1033.2004.04328.x. [DOI] [PubMed] [Google Scholar]
- 63.Sorgen PL, Bubb MR, Cain BD. J Biol Chem. 1999;274:36261–36266. doi: 10.1074/jbc.274.51.36261. [DOI] [PubMed] [Google Scholar]
- 64.Sorgen PL, Caviston TL, Perry RC, Cain BD. J Biol Chem. 1998;273:27873–27878. doi: 10.1074/jbc.273.43.27873. [DOI] [PubMed] [Google Scholar]
- 65.Wallace DC. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 66.Wallace DC. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Walker JE. Curr Opin Struct Biol. 1994;4:912–918. doi: 10.1016/0959-440x(94)90274-7. [DOI] [PubMed] [Google Scholar]
- 68.Green DW, Grover GJ. Biochim Biophys Acta. 2000;1458:343–355. doi: 10.1016/s0005-2728(00)00085-2. [DOI] [PubMed] [Google Scholar]
- 69.Atwal KS, Wang P, Rogers WL, Sleph P, Monshizadegan H, Ferrara FN, Traeger S, Green DW, Grover GJ. J Med Chem. 2004;47:1081–1084. doi: 10.1021/jm030291x. [DOI] [PubMed] [Google Scholar]
- 70.Atwal KS, Ahmad S, Ding CZ, Stein PD, Lloyd J, Hamann LG, Green DW, Ferrara FN, Wang P, Rogers WL, et al. Bioorg Med Chem Lett. 2004;14:1027–1030. doi: 10.1016/j.bmcl.2003.11.077. [DOI] [PubMed] [Google Scholar]
- 71.Hamann LG, Ding CZ, Miller AV, Madsen CS, Wang P, Stein PD, Pudzianowski AT, Green DW, Monshizadegan H, Atwal KS. Bioorg Med Chem Lett. 2004;14:1031–1034. doi: 10.1016/j.bmcl.2003.11.052. [DOI] [PubMed] [Google Scholar]
- 72.Vuorinen K, Ylitalo K, Peuhkurinen K, Raatikainen P, Ala-Rami A, Hassinen IE. Circulation. 1995;91:2810–2818. doi: 10.1161/01.cir.91.11.2810. [DOI] [PubMed] [Google Scholar]
- 73.Clement MV, Hirpara JL, Chawdhury SH, Pervaiz S. Blood. 1998;92:996–1002. [PubMed] [Google Scholar]
- 74.Wolvetang EJ, Johnson KL, Krauer K, Ralph SJ, Linnane AW. FEBS Lett. 1994;339:40–44. doi: 10.1016/0014-5793(94)80380-3. [DOI] [PubMed] [Google Scholar]
- 75.Mills KI, Woodgate LJ, Gilkes AF, Walsh V, Sweeney MC, Brown G, Burnett AK. Biochem Biophys Res Commun. 1999;263:294–300. doi: 10.1006/bbrc.1999.1356. [DOI] [PubMed] [Google Scholar]
- 76.Johnson KM, Chen X, Boitano A, Swenson L, Opipari AW, Jr, Glick GD. Chem Biol. 2005;12:485–496. doi: 10.1016/j.chembiol.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 77.Johnson KM, Cleary J, Fierke CA, Opipari AW, Jr, Glick GD. ACS Chem Biol. 2006;1:304–308. doi: 10.1021/cb600143j. [DOI] [PubMed] [Google Scholar]
- 78.Leslie AGW. Joint CCP4 and EACMB Newslett Prot Crystallogr. Vol. 26. Warrington, UK: Daresbury Laboratory; 1992. [Google Scholar]
- 79.CCP4. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 80.Navaza J. Acta Crystallogr A. 1994;50:157–163. [Google Scholar]
- 81.Murshudov GN, Vagin AA, Dodson EJ. Acta Crystallogr D. 1997;53:157–163. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 82.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 83.Schuttelkopf AW, van Alten DM. Acta Crystallogr D. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 84.Laskowski RA. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 85.DeLano WL. The Pymol Molecular Graphics System. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}