

Abstract
Inactivation is a fundamental property of voltage-gated ion channels. Fast inactivation of Na+ channels involves channel block by the III–IV cytoplasmic interdomain linker. The mechanisms of nonfast types of inactivation (intermediate, slow, and ultraslow) are unclear, although the ionic environment and P-loops rearrangement appear to be involved. In this study, we employed a TTX-based P-loop domain model of a sodium channel and the MCM method to investigate a possible role of P-loop rearrangement in the nonfast inactivation. Our modeling predicts that Na+ ions can bind between neighboring domains in the outer-carboxylates ring EEDD, forming an ordered structure with interdomain contacts that stabilize the conducting conformation of the outer pore. In this model, the permeant ions can transit between the EEDD ring and the selectivity filter ring DEKA, retaining contacts with at least two carboxylates. In the absence of Na+, the electrostatic repulsion between the EEDD carboxylates disrupts the permeable configuration. In this Na+-deficient model, the region between the EEDD and DEKA rings is inaccessible for Na+ but is accessible for TMA. Taken together, these results suggest that Na+-saturated models are consistent with experimental characteristics of the open channels, whereas Na+-deficient models are consistent with experimentally defined properties of the slow-inactivated channels. Our calculations further predict that binding of LAs to the inner pore would depend on whether Na+ occupies the DEKA ring. In the absence of Na+ in the DEKA ring, the cationic group of lidocaine occurs in the focus of the pore helices' macrodipoles and would prevent occupation of the ring by Na+. Loading the DEKA ring with Na+ results in the electrostatic repulsion with lidocaine. Thus, there are antagonistic relations between a cationic ligand bound in the inner pore and Na+ in the DEKA ring.
INTRODUCTION
Inactivation is one of the fundamental properties of voltage-gated ion channels. Its physiological role is to stop conductance during prolonged depolarizing stimuli. The general term “inactivation” covers kinetically different processes that involve different regions of the channel (1). The mechanism of fast inactivation is well understood. After pore opening, the channel is blocked by a cytoplasmic segment of the protein. In K+ channels, the N-terminal domain plays the role of inactivating particle (2), whereas in Na+ channels the intracellular linker between domains III and IV is responsible for fast inactivation. Deletion of the linker and specific antibodies against the linker block fast inactivation (3–5). More precisely, mutations in a conserved IFM motif can slow or abolish fast inactivation (6). Fast inactivation is restored by the intracellular addition of the KIFMK peptide (7).
Besides fast inactivation, there are several types of nonfast inactivation, which are usually termed “intermediate”, “slow”, and “ultraslow”. Since structural features of the intermediate, slow, and ultraslow types of inactivation are unknown, we designate here these types by the general term “slow inactivation” to contrast it with fast inactivation. The molecular determinants of the slow-inactivation processes are poorly understood. However, these types of inactivation are distinct from the fast inactivation because mutations that eliminate fast inactivation do not eliminate slow inactivation (8,9). It was demonstrated that channels could enter the slow-inactivated state from the closed, open, and fast-inactivated states (10). In both K+ and Na+ channels, slow inactivation greatly depends on the P-loop residues, in particular on several positions at the selectivity filter and downstream from it (11–13). Thus, mutations of the DEKA ring (14–16) promote slow inactivation. Mutations in the outer-carboxylates ring (EEDD ring) also affect slow inactivation (17). Paired cysteine mutations demonstrate structural rearrangements of this ring during slow inactivation (18). Some residues between the DEKA and EEDD rings also affect slow inactivation (19).
Another problem is the relationships between channel inactivation, ligand binding, and ion binding and permeation. The state-dependent block of Na+ channels by LAs is usually interpreted in terms of the “guarded receptor” (20) or “modulated receptor” (21) hypotheses. The latter suggests that LAs bind with the highest affinity to the inactivated state of the channel and stabilize it. Action of LAs is antagonized by external Na+ ions (22). It was classically interpreted by the competition for the binding site inside the pore. Later it was found that external Na+ decreases the rate of slow inactivation (23). Chen et al. (24) explained coupling of these effects in a model, according to which lidocaine binding induces channels to occupy the slow-inactivated state that is inhibited by external Na+.
Characteristics of slow inactivation in Na+ and K+ channels are markedly similar. First, in both channels the slow-inactivation gates are located at the region of the selectivity filter and the outer pore (25,26). Second, in both channels, slow inactivation depends on the ionic environment (27–29). Third, in both channels, the binding of the pore ligands affects the slow inactivation (30,31). Presumably, the extracellular gate closure is linked with the ion occupancy of the pore (e.g., 32,33). X-ray structures now provide a clue for a possible mechanism of slow (C-type) inactivation in K+ channels. The key step of the channel gating at the selectivity-filter region appears to be the conformation transition that turns the backbone carbonyls out of the pore. Such conformations possibly could represent a nonconducting state of the channel (34). It seems promising to apply the emerging understanding of the slow inactivation mechanisms in K+ channels to address problems related to slow inactivation of Na+ channels.
Earlier we built a TTX receptor model of Nav1.4 using a K+ channel structure as an initial template (35). In this work, we used this model to address the problem of slow inactivation of Na+ channels. We have built Na+-saturated and Na+-deficient models of the pore domain and explored properties such as stability, ion permeability, and ligand binding. We further explored consistency of the model with experiments on disulfide cross-linking, which provide valuable distance constraints on the outer-pore geometry (18,36) and with data on accessibility of Cys mutants in the outer pore for sulfhydryl reagents (37). We found that Na+-saturated models are consistent with certain experimental characteristics of the open channels, whereas Na+-deficient models are more consistent with experimentally defined properties of the slow-inactivated channels.
METHODS
The amino acid sequence of the model corresponds to the Nav1.4 channel from rat skeletal muscle. The model consists of P-loops and S6 segments from four repeats (Table 1). In this study, we used the universal scheme of residue labeling (38). A residue label includes the domain number (I–IV), segment type (p, P-loop; i, the inner helix), and relative number of the residue in the segment (Table 1). Among available x-ray structures of the open P-loop channels (MthK, KvAP, and Kv1.2) we have chosen KvAP (39). Like Na+ channels, KvAP is a voltage-gated channel whose inner helices lack the Pro-Val-Pro motif, which affects the open-pore conformation. The KvAP template was also used for modeling the P-loop domain, but this choice is less critical because RMSDs of Cα atoms in the pore helices between the superimposed x-ray structures of MthK, KvAP, and Kv1.2 are <1 Å.
TABLE 1.
Alignment of Nav1.4 and KvAP
| Channel | Domain | First residue | 33 | 41 | 51 |
|---|---|---|---|---|---|
| KvAP | P | 193 | IKSVFDAL WWAVVTATTV GYGDVVP | ||
| Nav1.4 | IP | 383 | YDTFSWAF LALFRLMTQD YWENLFQ | ||
| IIP | 738 | MNDFFHSF LIVFRILCGE WIETMWD | |||
| IIIP | 1220 | YDNVGLGY LSLLQVATFK GWMDIMY | |||
| IVP | 1512 | FETFGNSI ICLFEITTSA GWDGLLN | |||
| 1 | 11 | 21 | |||
| KvAP | S6 | 220 | PIGKVIGIAV MLTGISALTL LIGTVSNMFQK | ||
| Nav1.4 | IS6 | 415 | KTYMIFFVVI IFLGSFYLIN LILAVVAMAYA | ||
| IIS6 | 770 | AMCLTVFLMV MVIGNLVVLN LFLALLLSSFS | |||
| IIIS6 | 1262 | LYMYLYFVIF IIFGSFFTLN LFIGVIIDNFN | |||
| IVS6 | 1565 | SIGICFFCSY IIISFLIVVN MYIAIILENFN | |||
Relative positions in the alignments are numbered according to Zhorov et al. (38). Bold type indicates ionizable residues in the selectivity filter and LA-sensing residues in the inner helices. Shading indicates residues that contribute significantly to the ligand-receptor energy in our model.
The sequences were aligned as proposed before (40). Extracellular linkers between the P-loops and S6s were not included in the model. When viewed from the extracellular side, the repeats I–IV are arranged in a clockwise direction around the central pore (41).
The conformational energy expression included van der Waals, electrostatic, H-bond, hydration, and torsion components. Nonbonded interactions were calculated using the AMBER force field (42). The hydration energy was calculated by the implicit-solvent method (43). Electrostatic interactions were calculated with a distance-dependent dielectric ɛ, which is a simple way to take into account the effect of screening atomic charges by water molecules. Lidocaine docking in the inner pore was simulated with ɛ = R, where R is the distance between a pair of interacting atoms. However, this dielectric function appears too strong for systems exposed to the bulk solution with free cations and anions. Therefore, we used ɛ = 4R for modeling the outer pore. Both coefficients (as well as the intermediate ones) are widely used in molecular simulations depending on the system under consideration (44,45).
All ionizable residues were modeled in the ionized form. Nonbonded interactions were truncated at distances >8 Å. This cutoff distance allows us to speed up calculations without a noticeable decrease in the precision of energy calculations (46). The cutoff was not applied to electrostatic interactions involving Na+ ions and ionized groups; these interactions were computed at all distances. The energy was minimized in the space of generalized coordinates, which include all torsion angles, bond angles of the ligand, positions of ions, and positions and orientations of the ligand and individual segments of the protein (47). Energy minimizations were terminated when the energy gradient became <1 kcal mol−1 rad−1.
The backbone geometry of the pore helices and S6 helices was initially assigned from the KvAP template. All-trans conformations were used as starting approximations for the side chains of residues, which mismatch between KvAP and Nav1.4. The α-carbons of the pore helices and S6 helices were constrained to corresponding positions in the KvAP template with the help of pins. A pin is a flat-bottom penalty function (48) that allows a penalty-free deviation of the respective atom up to 1 Å from the template and imposes an energy penalty for larger deviations. The force constant of 10 kcal mol−1 Å−2 was used in the constraints. Conformations of the C-terminal parts of P-loops that line the outer pore were assigned from our TTX receptor model (35).
The optimal conformations of the protein models and their complexes with ions and ligands were searched by the MCM method (49). Briefly, the method is as follows. An MCM trajectory is launched from an initial point with coordinates 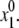 Energy minimization from
Energy minimization from 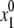 produces conformation {x1, E1} with energy E1 that is unconditionally accepted in the trajectory. Let us now consider a current energy-minimized point {xi, Ei} accepted in the trajectory. The next starting point
produces conformation {x1, E1} with energy E1 that is unconditionally accepted in the trajectory. Let us now consider a current energy-minimized point {xi, Ei} accepted in the trajectory. The next starting point 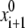 is generated by a random change of a generalized coordinate randomly selected from xi. From the starting point
is generated by a random change of a generalized coordinate randomly selected from xi. From the starting point 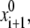 the energy is minimized to yield point {xi+1, Ei+1}. The latter is accepted in the trajectory if Ei+1 < Ei or if a random number n ∈ (0, 1) < exp[(Ei − Ei+1)/RT]. If the energy-minimized structure {xi+1, Ei+1} is rejected, another starting point
the energy is minimized to yield point {xi+1, Ei+1}. The latter is accepted in the trajectory if Ei+1 < Ei or if a random number n ∈ (0, 1) < exp[(Ei − Ei+1)/RT]. If the energy-minimized structure {xi+1, Ei+1} is rejected, another starting point 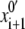 is generated by randomly modifying point xi. Trajectories were calculated at T = 600 K to increase the number of points accepted in the trajectory. A regular MCM trajectory was terminated when the last 1,000 energy minimizations did further decrease the lowest energy Em found. This stringent convergence criterion was satisfied after 3,000–30,000 energy minimizations. In addition to the lowest energy structure {xm, Em}, other conformations with energy up to 7 kcal/mol above Em were accumulated during the search.
is generated by randomly modifying point xi. Trajectories were calculated at T = 600 K to increase the number of points accepted in the trajectory. A regular MCM trajectory was terminated when the last 1,000 energy minimizations did further decrease the lowest energy Em found. This stringent convergence criterion was satisfied after 3,000–30,000 energy minimizations. In addition to the lowest energy structure {xm, Em}, other conformations with energy up to 7 kcal/mol above Em were accumulated during the search.
Due to the extremely ragged shape of the energy surface, the radius of convergence of a single MCM trajectory (the distance from the initial point  and the lowest energy structure xm) is limited. In other words, xm depends on
and the lowest energy structure xm) is limited. In other words, xm depends on 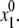 Therefore, although a single MCM trajectory is suitable for optimizing certain models (e.g., x-ray structures), a comprehensive nonlocal search for low energy structures should employ multiple MCM trajectories submitted from different initial points, which are systematically (50) or randomly (46) generated within a certain area of the conformational space. Henceforth, we use the term “multi-MCM” to refer to protocols of nonlocal optimization using multiple MCM trajectories.
Therefore, although a single MCM trajectory is suitable for optimizing certain models (e.g., x-ray structures), a comprehensive nonlocal search for low energy structures should employ multiple MCM trajectories submitted from different initial points, which are systematically (50) or randomly (46) generated within a certain area of the conformational space. Henceforth, we use the term “multi-MCM” to refer to protocols of nonlocal optimization using multiple MCM trajectories.
During a multi-MCM search, an ensemble of structures with energies up to 7 kcal/mol from the apparent global minimum was collected. Since precision of energy calculations is limited, we usually considered all structures in an ensemble as energetically possible. To reduce the size of an ensemble, geometrically similar structures were clustered. Within a cluster, matching torsions disperse by <20°, matching Cartesian coordinates of ions and root atoms in free molecules by <0.5 Å and matching Euler angles of free molecules by <10°. Each cluster was represented by its lowest energy structure, and higher energy structures were removed. Calculations were performed using the program ZMM (47; http://www.zmmsoft.com).
RESULTS
Ring EEDD in the presence and absence of TTX and Na+
The structural stability is an important feature of molecular models. P-loop channels have a ring of negatively charged residues in positions p53–p54 (Table 1). In K+ channels, these residues do not face the pore but interact with the conserved Trpp41 in the pore helices. In contrast, since Na+ channels lack Trpp41 (Table 1), the outer carboxylates can face the outer-pore lumen. The EEDD carboxylates increase the channel permeability probably by forming binding site(s) for cations (51). These carboxylates interact with TTX and STX, providing strong contributions to the ligand-binding energy (35).
To demonstrate interactions of carboxylates EEDD with the rest of the system, we partitioned their energy in different models derived from our original TTX receptor model (Table 2). In the presence of TTX, the carboxylates favorably interact with the ligand, showing negative energies. After removal of TTX followed by a single energy minimization, the energies of the carboxylates become unfavorable (positive) because of their mutual electrostatic repulsion. A multi-MCM search from 5,000 initial geometries produced an ensemble of 26 conformers (Fig. 1 A). Although these structures have a greatly variable geometry of the EEDD ring, a clear tendency is seen: the charged carboxylates repel each other and turn away from the pore lumen (Fig. 1, B and C). The average distance from the carboxylate carbon to the pore axis increased from 7.1 Å in the TTX-bound model to 8.7 Å in the ensemble of TTX-free conformations. Notably, the distribution of the distances between the carboxylate carbons and the pore axis have a major peak at 10–12 Å and an additional peak at 7 Å that corresponds to the TTX-bound model (Fig. 2). The lowest energy structure found in the multi-MCM search of the TTX-free model deviates significantly from the TTX-bound model (Fig. 1, B and C): within 20 residues in positions p49–p53, the RMSD of α-carbons, all main-chain atoms, and all heavy atoms are 2.72, 2.68, and 4.01 Å, respectively. Multi-MCM of the TTX-free model decreases the outer carboxylates' average energy by 1.6 kcal/mol compared to the respective energy-minimized model (Table 2). However, this energy is significantly more positive than in the presence of TTX, indicating that the outer carboxylates did not find good partners in the TTX-free model.
TABLE 2.
Interaction energy (kcal/mol) of outer carboxylates with the rest of the system
| TTX receptor
|
|||||
|---|---|---|---|---|---|
| Model
|
TTX-bound*
|
TTX-free†
|
Na+-deficient‡
|
Na+-saturated§
|
16-membered cycle¶
|
| Method‖ | EM | EM | Multi-MCM | Multi-MCM | MCM |
| EIp53 | −1.5 | 7.1 | 2.5 (−0.3) | −0.7 (−2.3) | −2.8 |
| EIIp53 | −0.7 | 2.2 | 0.4 (1.3) | −1.5 (−2.9) | −2.9 |
| DIIIp54 | −0.1 | −1.1 | −0.7 (−0.8) | −6.6 (−5.1) | −2.5 |
| DIVp53 | −2.1 | 0.3 | −0.1 (0.7) | −4.5 (−2.1) | −5.8 |
| Average energy | −1.1 | 2.1 | 0.5 (0.2) | −3.3 (−3.1) | −3.5 |
Numbers in parentheses are averages from the corresponding ensemble.
Model of TTX receptor (35) in the presence of TTX.
Model of TTX receptor (35) in the absence of TTX.
Derived from the TTX-free model‡ by multi-MCM optimization in the absence of Na+ ions in the outer pore.
Derived from the TTX-free model‡ by multi-MCM optimization in the presence of four Na+ ions in the outer pore.
Idealized Na+-saturated model in which four outer-carboxylates and four Na+ ions form a 16-membered cycle.
EM, MCM, and multi-MCM were used to obtain a model.
FIGURE 1.

In the absence of TTX, the TTX receptor structure is unstable. (A) The extracellular view at the ensemble of the 26 lowest energy structures, which were obtained in a multi-MCM search started from the TTX receptor model. Red dots represent carbon atoms in the EEDD ring. For comparison, the outer carboxylates in the TTX receptor model are shown in the green-filled space. Roman numerals designate domains. (B and C) Top and side views of the superimposition of the TTX receptor model (green) with the lowest energy structure from the ensemble A (red). In the absence of TTX, the outer carboxylates repel each other and turn away from the pore axis.
FIGURE 2.

Distance distribution from the pore axis of Na+ ions and carboxylate carbons in the EEDD ring. The distributions are calculated for the ion-saturated and ion-deficient models. In the Na+-saturated model, Na+ ions and outer carboxylate carbons most frequently appear 6–8 Å from the pore axis, which corresponds to the TTX receptor model. In the Na+-deficient model, carboxylate carbons move away from the pore axis and show a maximum at 10 Å from the pore axis, but a significant population remains at ∼7 Å.
Charged residues facing the water-accessible pore lumen can be neutralized by counterions. In the simplest model, each carboxylate would bind a monovalent cation. To explore possible effects of metal ions in the Na+-saturated model of the outer pore, we performed a multi-MCM search from 10,000 initial points with random positions of four Na+ ions and random side-chain conformations of carboxylates' EEDD. The search yielded an ensemble of 30 structures. Unsurprisingly, in all these structures Na+ ions bind to carboxylates, whose energy becomes negative (Table 2). A nontrivial result of this simulation is that a monovalent Na+ ion binds between two carboxylates more often than to a single carboxylate. Superposition of the ensemble structures (Fig. 3 A) shows that ions are localized between neighboring carboxylates, and the region at the pore axis is poorly populated by Na+ ions. A close disposition of the outer carboxylates in the TTX receptor model (35) allows formation of two coordinating bonds per Na+ ion (Fig. 3, B and C). The distance distributions of the Na+ ions and the EEDD carboxylate carbons from the pore axis (Fig. 2) are similar and have peaks between 6 and 8 Å, which is close to the average distance between the outer carboxylates and the pore axis in the TTX-bound model.
FIGURE 3.

Stabilization of the EEDD ring by four Na+ ions. (A) Superposition of the 30 lowest energy complexes obtained by multi-MCM search. Na+ ions are shown as yellow dots. White-space-filled atoms are α-carbons of the outer carboxylates. Na+ ions are clustered between the carboxylates, whereas the pore lumen is poorly populated. (B and C) Representative examples from A that show yellow-space-filled Na+ ions interacting with neighboring carboxylates (sticks). (D) A possible conformation of the outer pore in which the outer carboxylates and four Na+ ions form a 16-membered cycle.
The idealized structure of this Na+-saturated model is a 16-membered cycle in which each Na+ ion is coordinated between two EEDD carboxylates from the neighboring domains (Fig. 3 D). The cycle is a polygon defined by four Na+ ions and four groups of −CO2. This model was obtained by a MCM of the TTX receptor model with four Na+ ions constrained between neighboring carboxylates and subsequent refinement by an unconstrained MCM trajectory. In this model, each outer carboxylate has a more negative energy than in the TTX-bound model (Table 2). Formation of the 16-membered cycle does not conflict with the TTX-bound model. Indeed, within the region p49–p53, the RMSD between the TTX-bound model and the ion-saturated 16-membering-cycle model are 0.82, 0.86, and 1.41 Å for α carbons, main-chain atoms, and all heavy atoms, respectively. Only AspIIIp54 deviates significantly between the idealized ion-saturated model and the TTX-bound model. This is not surprising because in the latter, AspIIIp54 does not interact with TTX and turns away from the pore axis to minimize electrostatic repulsion from other carboxylates. Note that interactions of AspIIIp54 are not significantly different between the TTX-bound, TTX-free, and Na+-deficient models (Table 2). This residue turns away from the pore axis in the TTX-bound model and subsequent optimization does not significantly change its energy.
A possible role of ring EEDD in Na+ permeation
In the 16-membered ring model, the distance between the DEKA ring and the EEDD ring is ∼8 Å (Fig. 4 A). In contrast, neighboring binding sites for K+ ions in K+ channels are <4 Å apart. This raises a question about the mechanism of ion permeation through Na+ channels. How would Na+ move from the EEDD ring to the DEKA ring? Strong electrostatic interactions with the outer carboxylates could cause an ionic trap, but obviously this does not happen in the permeating channel. To address this problem, we introduced an additional Na+ ion, used constraints to pull it from the extracellular space toward the DEKA ring as described (35), and collected MC-minimized structures obtained with the pulled ion at different levels of the outer pore.
FIGURE 4.

Na+ ions and EEDD and DEKA rings in the Na+-saturated (A) and Na+-excessive (B–D) models of the outer pore. A, In the Na+-saturated model, four Na+ ions form a 16-membered cycle with the outer carboxylates, and another Na+ is chelated between AspIp50 and GluIIp50 of the DEKA ring. Because of a large distance between the EEDD and DEKA rings, a single-step transition of Na+ between the rings is unlikely. (B) Insertion of an additional Na+ ion significantly perturbs coordination of Na+ ions. The 16-membered cycle at EEDD is broken and one of the Na+ ions is coordinated by two glutamates from the EEDD and DEKA rings. The model represents a possible transition state upon Na+ permeation via the outer pore. (C) Superposition of Na+ ions and the ammonium group of the DEKA Lys from 200 lowest energy structures collected in the multi-MCM search. For comparison, positions of seven K+ ions in the KcsA structure crystallized at high [K+] (11) are shown as red dots. (D) Distribution of Na+ ions and the ammonium group of the DEKA Lys along the outer pore. Vertical lines with numbers mark the outer-pore levels that correspond to K+-binding positions in KcsA (C). The average energy of Na+ ions and its standard deviations are shown.
In this Na+-excessive model, the additional Na+ ion caused significant perturbations in the outer pore. We obtained 128 low energy structures with various ion coordination patterns. The energy of the pulled Na+ varies from −7.4 to 12.5 kcal/mol. The high energy is caused by the unfavorable displacements of the resident Na+ ions by the pulled Na+. Thus, pulling a single Na+ ion does not allow us to model the ion permeation, the process that likely involves cooperative movements of several ions in a single-file pore.
Despite the limitations of the ion-pulling method, certain structures collected in the MCM trajectories provide hints that allow us to discuss structural aspects of a possible permeation mechanism. Thus, 34 structures have a Na+ ion coordinated between the EEDD and DEKA rings. Fig. 4 B shows one of the obtained structures. In this structure, the 16-membered cycle is broken by the incoming Na+ ion. Repulsion of two Na+ ions at the ends of the broken cycle leads to a spiral-like conformation in which the innermost Na+ ion approaches the DEKA ring. This, in turn, induces the rearrangement in the DEKA ring. Its resident Na+ ion moves in the intracellular direction, whereas the approaching Na+ ion becomes simultaneously coordinated by an EEDD carboxylate and GluIIp50 in the DEKA ring. Such a complex could represent a transition state during ion permeation.
To explore the distribution of Na+ ions in the Na+-excessive model of the outer pore, we performed a multi-MCM search from 20,000 initial points in which six Na+ ions were randomly shifted up to 4 Å from positions shown in Fig. 4 B. Each initial point was relaxed in an MCM trajectory of 1,000 minimizations. To obtain a large ensemble, we did not cluster the low energy structures. For clarity, only 100 lowest energy structures are superimposed in Fig. 4 C to show the distribution of Na+ ions (yellow dots) and the ammonium group of LysIIIp50 (blue dots). However, the entire ensemble of ∼2000 structures was analyzed to predict a population of cations at different levels of the outer pore (Fig. 4 D). For reference, seven positions occupied by K+ ions in KcsA crystallized at high [K+] (11) are shown as red dots in Fig. 4 C; corresponding levels along the pore axis are shown as horizontal lines in Fig. 4 C and by vertical lines in Fig. 4 D with K+-binding positions numbered as in Zhou et al. (11).
Most populated are levels 6–7, where up to four Na+ ions can bind to the outer carboxylates. Levels 2, 3, and 4 in the narrow part of the outer pore are populated by Na+ ions and by the ammonium group of LysIIIp50. At these levels, cations bind, respectively, between GluIIp53 and GluIIp50, between GluIIp50 and AspIp50, and to AspIp50 (see Fig. 4 B). In our model, LysIIIp50 is permanently charged and can substitute a Na+ ion at levels 2, 3, and 4. Cooperative relocations of the LysIIIp50 ammonium group and Na+ ions between the levels of preferable occupancy could be of critical importance for permeation and selectivity.
The average energy of Na+ ions has negative values all along the outer pore with the minimum at level 4. Of course, in our coarse-grained model the Na+ energy is a scoring function rather than a free energy. It should be noted that the nature of the MCM protocol does not allow us to reveal energy barriers between closely spaced structures. To analyze such barriers, which are important for the quantitative description of permeation and selectivity, molecular dynamics simulations with more sophisticated energy functions are required.
In the proposed permeation mechanism, Na+ ions relocate from the EEDD ring to the DEKA ring, being escorted by the side chains of carboxylate residues. Such a mechanism is possible due to the flexibility of the outer pore in the Na+ channel. In contrast, the outer pore of a K+ channel has main-chain oxygens well packed, much less mobile, and unable to escort K+ ions. This dictates rather short distances between the planes of carbonyl oxygens in the outer pore of K+ channels. Thus, the spatial proximity of the EEDD carboxylates in the TTX receptor model allows different multiion configurations of the outer pore, in which Na+ ions are coordinated between several carboxylates. Flexible side chains of the acidic residues can allow Na+ permeation with transitional states in which the ion is coordinated between the EEDD and DEKA rings.
Na+ and TMA in the Na+-deficient model of the outer pore
In our Na+-deficient model of the outer pore, removal of Na+ ions induces essential conformational changes of the outer carboxylates that turn away from the pore axis (Fig. 1). How could this relocation affect permeation of Na+ via the outer pore? The ion-pulling protocol can be used to analyze the accessibility of the DEKA ring by extracellular ions because this model lacks resident Na+ ions above this ring. The structure with red side chains at Fig. 1 B was used as the initial geometry. At each level of the pore, Na+ was constrained to a plane normal to the pore axis, whereas the outer-pore geometry and positions of Na+ in the plane normal to the pore axis were sampled and MC minimized. Fig. 5 shows the energy of Na+ and its components against their position in the outer pore. Na+ has negative energy at the EEDD level due to favorable electrostatics. However, the EEDD carboxylates, which turn away from the pore axis in this model, are unable to escort the Na+ ion toward the DEKA ring. Therefore, no favorable electrostatics between the EEDD and DEKA rings compensates for the dehydration costs, which increase as the ion enters the narrow part of the pore. This results in a significant energy barrier. As the ion approaches the DEKA ring, its energy decreases due to electrostatic attraction by Asp and Glu residues. However, the total energy barrier of ∼14 kcal/mol between the EEDD and DEKA rings would preclude the Na+ access to the selectivity filter in the Na+-deficient model.
FIGURE 5.

Profiles of Na+ and TMA energy in the Na+-deficient model of the outer pore. The energy values are means from 12 independent calculations, with standard deviation shown as error bars. Vertical lines show K+ positions in KcsA (Fig. 4 C). Both Na+ and TMA experience unfavorable dehydration that increases as the ions move deeper into the outer pore (D). The electrostatic energy of Na+ has two minima at levels EEDD and DEKA (B). The weaker electrostatic energy between rings EEDD and DEKA is due to the inability of the outer carboxylates, which turn away from the pore axis, to escort Na+ toward the DEKA ring. It does not compensate the dehydration, causing an energy barrier for Na+. Unlike small Na+, a bulky TMA has favorable van der Waals interactions between the EEDD and DEKA rings that compensate the dehydration cost (C). The smooth profile of the total energy (A) evidences that TMA and compounds of similar size can reach as deep as the DEKA ring. Both Na+ and TMA profiles become highly rugged close to the DEKA ring due to competition with the resident Na+ ion there.
To further confirm that the energy barrier is not due to dimensions of the outer pore, we computed the MC-minimized energy profile of TMA using the same methodology as for Na+. In contrast to Na+, TMA experiences favorable van der Waals interactions between the rings EEDD and DEKA (Fig. 5 C). At this region, the pore dimensions fit the TMA molecule. Due to the larger size of TMA versus Na+, the electrostatic interactions of TMA with the channel are less favorable (Fig. 5 B), but the dehydration cost of TMA in the outer pore is also smaller (Fig. 5 D). Energy profiles for both Na+ and TMA show ragged energy barriers close to the DEKA ring due to competition with the resident Na+ ion there. Comparison of the total energy plots (Fig. 5 A) predicts that TMA can reach the outer-pore levels, which are not accessible by Na+ in the Na+-deficient model. Important consequences of this prediction are discussed later.
The P-loop domain models are consistent with experiments on paired Cys mutants
The TTX receptor model of Nav1.4 (35) was built using experimental constraints between TTX and the outer-pore residues involved in the TTX binding. A molecular model that is not based on a high-resolution x-ray structure needs verification by testing its consistency with experimental data, which were not used at the stage of model building. Such data are provided by paired cysteine mutations in the outer pore of Na+ channels, which revealed formation of multiple disulfide bonds (18,36,52). These data provide valuable distance constraints between the paired residues.
Based on the TTX receptor model, we generated 13 models of the P-loop domain with paired cysteine substitutions that allow disulfide bond formation according to Xiong et al. (18). Each model was MC minimized starting from coordinates of the TTX receptor model and without imposing any constraints on the geometry of the ascending limbs (residues p49–p53 in P-loops). Closing the disulfide bonds caused minor to modest deviations of the models from the TTX receptor model (Figs. 6 and 7): RMSD of 60 main-chain atoms in residues p49–p53 vary from 0.6 to 2 Å (Fig. 7 A). The largest RMSD was caused by the disulfide bonding in the double mutant EIp53C/KIIIp50C (Fig. 6), but even this deviation does not exceed 2 Å. Thus, despite a limited precision of our homology model, it is generally consistent with the data on disulfide bonding in paired Cys mutants (18). It should be noted that the interdomain disulfide bonding requires a proximity of the corresponding domains. Such proximity is an important feature of our TTX receptor model (35), which inherits a spatial disposition of the P-loop helices seen in the x-ray structures of all K+ channels. This proximity also allows Na+ ions to bind between the EEDD carboxylates in neighboring domains.
FIGURE 6.

Paired Cys mutants of the TTX receptor model. The α-carbon tracing in the selectivity filter region is shown as red rods for the original TTX receptor model (35) and as green rods for MC-minimized models of the mutants. Side chains of Cys residues are space filled. (A and B) Extracellular views; (C and D) side views in which one repeat is not shown for clarity. (A and C) The model of the EIp53C/WIVp52C double mutant that minimally deviates from the TTX receptor model. (B and D) The model of the EIp53C/KIIIp50C double mutant that maximally deviates from the TTX receptor model.
FIGURE 7.

Deformations of the TTX receptor model caused by Na+ and disulfide cross-linking. Pairs of residues cross-linked in the paired Cys mutants are indicated by their labels (see Table 1). (A) Disulfide cross-links cause up to 2 Å Cα-RMSD of the ascending limbs (positions p49–p54) versus the TTX receptor model as indicated by open bars. When compared to the TTX receptor model, distances between indicated Cα atoms usually decrease in the Na+-excessive model of the outer pore (solid bars) but increase in the Na+-deficient model (shaded bars). These predictions are consistent with the data of Xiong et al. (18) that the channel opening generally accelerates formation of disulfide cross-links, whereas slow inactivation prevents formation of certain S-S bonds. (B) Correlation between the distance change indicated by black bars at A and increase of the cross-linking rate upon the channel opening (18). For those paired Cys mutants, which deviate from the TTX receptor model up to 1 Å, the acceleration of the cross-linking reaction rate is small. For three paired Cys mutants, a big decrease of the Cα-Cα distances in the Na+-excessive model is observed. These mutants demonstrate significant acceleration of the cross-linking reaction upon the channel opening.
Unsurprisingly, most significant deformations of the TTX receptor model are caused by disulfide bonds between nonadjacent domains I/III and II/IV (Fig. 7). These deformations may reflect an inherent flexibility of the pore-lining ascending limbs of P-loops (18,36). If a P-domain conformation necessary for disulfide bonding does not correspond to the free energy minimum, the chemical reaction should be slow and catalyst dependent. Indeed, most of the paired Cys mutants form disulfide bonds slowly (up to 20 min) and require a catalyst (36).
The RMSDs (Fig. 7 A) correlate with the distances between respective Cα atoms in the TTX receptor model (not shown). Therefore, we further explored relationships of the distances between Cα atoms in our models and the data on disulfide bonding (18). The experimental rates of disulfide bonding poorly correlate with the distances between respective Cα atoms in our models. This is not surprising because the disulfide-bonding rate depends on various factors, including local accessibility for a catalyst. However, there is a correlation between our models and the state dependence of the disulfide bonding. Xiong et al. (18) found that an increase of the stimulation frequency, which favors the channel open state, significantly accelerates the disulfide bonding in several paired Cys mutants. The maximal acceleration was observed in the following paired Cys mutants: EIp53C/EIIp53C, EIp53C/DIIIp54C, and EIp53C/MIIIp53C. It is noteworthy that respective distances Cα_EIp53/Cα_EIIp53, Cα_EIp53/Cα_DIIIp54, and Cα_EIp53/Cα_MIIIp53 decrease more significantly in the Na+-excessive model versus the TTX receptor model (Fig. 7 B). Another experimental protocol, which favors slow inactivation, prevented disulfide bonding in certain paired Cys mutants (18). This observation agrees with our finding that most of the distances between Cα atoms in the Na+-deficient model markedly increase compared to the TTX receptor model (Fig. 7 A). Thus, interresidue distances in our models correlate with the following observations of Xiong et al. (18): formation of multiple interdomain S-S bonds, accelerated formation of certain S-S bonds in the open channels, and preventing formation of some S-S bonds in the slow-inactivated channels.
State-dependent binding of local anesthetics in the inner pore
To analyze ligand binding in the inner pore, we extended the P-loop domain model of Nav1.4 (35) by adding the inner helices (S6s). The EEDD ring was saturated by four Na+ ions (see 16-membered cycle in Fig. 3 D). The DEKA ring was considered in two states: the Na+-saturated state and the Na+-deficient state. In the latter, the Na+ ion was replaced by an explicit water molecule to preserve the structural stability. The models were MC minimized before ligand docking. To avoid any bias in lidocaine docking, the multi-MCM searches were initiated from 5,000 random positions and orientations of the ligand that cover the entire inner pore. Each initial point was MC minimized by a two-stage protocol. At the first stage, a short MCM trajectory of 100 energy minimizations was used to rule out sterically clashed structures. At the second stage, 100 energetically best structures collected at the first stage were further MC minimized using the stringent criterion of convergence described in Methods.
The Na+-deficient model of lidocaine-bound Nav1.4 is represented by 28 structures in which ligand-receptor energy varies from −15.1 to −22.4 kcal/mol. The amino group of lidocaine approaches the selectivity filter in 24 structures and turns away from the filter in only 4 structures (Fig. 8 A). Mutagenesis experiments revealed several LA-sensing residues in S6s (53–60). To see whether these residues contribute to the ligand binding in our model, we partitioned ligand-receptor energy over four lowest energy structures with similarly located/oriented ligand. The P-loops and S6 segments almost equally contribute to the ligand binding energy. Electrostatic energy provides ∼90% to the interaction of P-loops with the ligand. The ligand's amino group is close to the focus of the pore helices' macrodipoles that stabilize the group. Other electrostatic contributions are provided by the DEKA residues, in agreement with an earlier finding (61). Although the KvAP template has fourfold symmetry around the pore axis, lidocaine binds tightly to domain IV (Fig. 8 E). Contributions of S6s to interaction with lidocaine are as follows: IS6 26%, IIS6 16%, IIIS6 22%, and IVS6 36%. In agreement with experiments, domains II and IV provide, respectively, the smallest and the largest contributions to the ligand-receptor energy. PheIVi15 and TyrIVi22 are the major contributors from domain IV. Thus, lidocaine strongly interacts with most of the LA-sensing residues revealed in mutational experiments (Table 1, Fig. 8). It should be emphasized that these results were obtained by the unbiased multi-MCM search.
FIGURE 8.

Binding of lidocaine in the Na+-deficient (A, C, and E) and Na+-saturated (B, D, and F) models of the inner pore. (A–D) Side views; (E and F) intracellular views. (A and B) Superposition of the 10 lowest energy structures obtained from 5,000 MCM trajectories. Lidocaine molecules are shown in a wire-frame mode with the protonated nitrogen as a red dot. (A) In the prevailing binding modes in the Na+-deficient model, the ligand's nitrogen is oriented toward the DEKA ring and occurs in the focus of macrodipoles from the pore helices. (B) Due to the electrostatic repulsion from the Na+ ion in the DEKA ring, the prevailing binding modes in the Na+-saturated model show the ligand's nitrogen oriented toward the cytoplasm. (C–F) Representative structures from A and B with a space-filled lidocaine molecule. Residues contributing to the interaction energy are shown as sticks. In the Na+-deficient model, the ligand binds tightly to IVS6 and IIIS6 and does not interact with IIS6 (C and E). In the Na+-saturated model, the ligand interacts with all four domains (D and F).
Notable exceptions are the conservative asparagines in positions i20. In our model, these residues interact with neighboring inner helices rather than face the inner pore. That is why they do not interact significantly with the ligands. These asparagines (in particular AsnIVi20) were recently shown to affect slow inactivation (62). Thus, according to our model, these residues would affect the ligand action by allosterically modulating the channel states.
The Na+-saturated model is represented by 33 structures whose ligand-receptor energy varies from −7.9 to −13.0 kcal/mol, being significantly weaker than in the Na+-deficient model. The major destabilizing factor of the Na+-saturated model is the Na+ ion in the DEKA ring that electrostatically repels cationic lidocaine. In 27 structures, lidocaine approaches the selectivity filter by its aromatic ring (Fig. 8 B) to minimize the electrostatic repulsion from the Na+ ion. In five lowest energy structures, domains I, II, III, and IV contribute, respectively, 18%, 25%, 21%, and 36% to the ligand binding energy. Segment IVS6 provides the maximal contribution but, in disagreement with experiments, IIS6 contributes more than IS6 and IIIS6. It should be noted that contributions of individual residues vary more than 20% between individual structures.
In addition to the multi-MCM search from randomly generated initial points, we also simulated lidocaine-channel interaction by systematically calculating MC-minimized energy profiles against the position of lidocaine along the pore axis. At each position, a central atom of the ligand was constrained to a plane normal to the pore axis. Another constraint limited the ligand yaw angle to 0° ± 90° or 180° ± 90°, thus imposing the cationic group orientation either toward or against the DEKA locus. From each starting position the energy was MC minimized. The ligand-protein energy was partitioned from the MC-minimized structures. To achieve statistically reliable results, each profile was averaged from 12 independent multi-MCM trajectories with different starting orientations of the ligand.
The computed plots of ligand-receptor energy are shown in Fig. 9. The lowest energy profile unambiguously corresponds to the Na+-deficient model with the ligand's amino group oriented toward the DEKA locus (Fig. 9 A). The profile has a single minimum at the position that agrees with the best structure obtained in the multi-MCM docking. Even when the cationic group of lidocaine is oriented against the DEKA locus, the ion-deficient model predicts a better interaction energy than the ion-saturated model. This difference is due to the electrostatic component, whereas profiles of van der Waals and solvation energies are similar (Fig. 9). The latter energy components are responsible for the sharp energy increase when the ligand clashes with the P-loops. Because of the clash, individual trajectories differ dramatically at the rightmost parts of the energy profiles as indicated by large error bars (Fig. 9). Thus, both the random and systematic multi-MCM docking predict that lidocaine binds most strongly with its amino group toward the DEKA locus on the Na+-deficient model. The major factors that stabilize the complex are electrostatic interactions with the macrodipoles from the pore helices and nonbonded interactions with S6s, among which IVS6 provides the largest energy contribution.
FIGURE 9.

Energy profiles of lidocaine in the Na+-deficient and Na+-saturated models of the inner pore. For each model, two profiles were calculated with the ligand's ammonium group oriented either toward the DEKA ring (N-up profiles) or against the ring (N-down profiles). The energy values are means from 12 independent calculations with standard deviation shown as error bars. The vertical line marks K+ position 5 in the focus of macrodipoles of KcsA (Fig. 4 C). The ligand-receptor energy (A) has the lowest minimum in the Na+-deficient model with N-up orientation of lidocaine. This is due to electrostatic interactions (B), which are optimal when the ligand ammonium group occurs in the focus of macrodipoles from the pore helices in the absence of Na+ in the DEKA ring. This result agrees with the random search data (Fig. 8 A). Profiles of van der Waals (C) and solvation (D) energy components are rather similar. The increase of van der Waals energy close to the DEKA ring is due to the clash between the ligand and P-loops. The clash also results in significant differences between individual runs, which can be seen as large error bars.
DISCUSSION
In this work we have used the TTX receptor model of Nav1.4 (35) to build Na+-saturated and Na+-deficient models of the channel pore and considered properties of these models such as stability, ion permeability, and ligand binding. Our modeling study has several predictions. First, Na+ ions can bind between neighboring domains in the EEDD ring, forming an ordered structure with interdomain contacts that stabilize the conducting conformation of the outer pore. Second, permeant ions can transit between the EEDD and DEKA rings, retaining contacts with at least two carboxylates. Third, the Na+ deficiency can induce disordering of the EEDD ring structure. Fourth, the outer pore above the DEKA ring in the ion-deficient model is wider than in the ion-permeating model. Fifth, despite this width difference, the region between the EEDD and DEKA rings in the Na+-deficient model is inaccessible for Na+ but is accessible for TMA. Taken together, these results suggest that Na+-saturated models are consistent with experimental characteristics of the open channels, whereas Na+-deficient models are consistent with experimentally defined properties of the slow inactivated channels.
In K+ channels, disordering of the selectivity filter is coupled with significant changes in the ion disposition (11–13,63). We see the same tendency in our models of the selectivity filter region in the Na+ channel: in the absence of neutralizing Na+ ions, repulsion between the outer pore carboxylates leads to their reorientation. Such conformational changes can be associated with slow inactivation. We propose that the ion impermeability of the slow inactivated conformation is due to essential rearrangement of the outer carboxylates as compared to the conducting conformation.
Struyk and Cannon (37) demonstrated that engineered cysteines deep in the outer pore of the slow inactivated Na+ channel remain accessible for the sulfhydryl reagent MTSET, indicating that Na+ impermeability of the slow inactivated conformation is not caused by the sterical occlusion of the outer pore. This agrees with our model, in which the slow inactivated outer pore is wider than the permeating one. We demonstrated that the DEKA ring in the Na+-deficient model is accessible for TMA (which is the bulkiest fragment of MTSET) but is not accessible for Na+ (Fig. 5). Indeed, the dimensions of the trimethylammonium group of MTSET are smaller than the dimensions of hydrated Na+, whereas dehydrated Na+ is smaller than MTSET. As a result, Na+ can permeate through narrow hydrophilic pores where the energy cost of ion dehydration is low. In contrast, in the hydrophobic environment that is unfavorable for the ion dehydration, the effective dimensions of Na+ with the inner hydration shell are larger than dimensions of MTSET. Thus our study resolves a contradiction between the Na+ impermeability of the slow inactivated channel and accessibility of its outer pore for organic molecules. We also tested our models for the consistency with the experimental data on paired Cys substitutions in the outer pore region (18). Our models agree with these data and for the first time to our knowledge provide a structural explanation of the state-dependent formation of S-S bonds between cysteines as well as between cysteines and MTS reagents.
The use-dependent action of LAs is usually explained in terms of the “modulated receptor” hypothesis (21), which suggests that LAs preferably bind to the inactivated state of the channel and additionally stabilize this state. The block of the inner pore by cationic ligands, in particular LAs, may be related to slow inactivation (24). However, molecular mechanisms of the state-dependent action of LAs remain unclear (64). Our study predicts two states of the Na+ channel with the open activation gate, which significantly differ by the interaction with the cationic ligand. LAs have low affinity to the model with the Na+-bound DEKA ring but high affinity to the model with the Na+-deficient DEKA ring. Furthermore, the LA molecule that binds in the inner pore of the channel with the Na+-deficient DEKA ring would electrostatically prevent occupation of the DEKA ring by Na+. For example, the absence of Na+ in the DEKA ring explains how binding of LAs could stabilize the inactivated channel. However, relationships between occupancy of the DEKA ring by Na+ and functional states of the channel are unknown. Our slow inactivated channel model explains ion impermeability of the outer pore but does not predict the Na+ occupancy of the DEKA ring.
Lipkind and Fozzard (65) proposed a MthK-based model of the open sodium channel with LAs. The authors reported an agreement between their calculations and the experimentally obtained activities of certain LAs. Both our model and that of Lipkind and Fozzard agree on the general orientation of LAs that was earlier proposed from the site-directed mutagenesis (see, e.g., Yarov-Yarovoy et al., (56)). There are two major differences between the model of Lipkind and Fozzard and our model. First, our homology model preserves the spatial disposition of the pore helices as seen in the KvAP template, whereas Lipkind and Fozzard shifted the pore helices away from the pore axis to accommodate TTX molecules in the outer vestibule. Because of these shifts, the macrodipoles of the pore helices are unlikely to stabilize the ligand ammonium group. Besides, the ascending limbs of individual domains appear too far from each other to enable formation of disulfide bonds in the paired Cys mutants described by Xiong et al. (18). It should be noted that despite this difference, the distances between Cα atoms in the model (65) correlate with those in our TTX receptor model. In particular, in both models the largest distance is seen between α-carbons of EIp53 and DIIIp54, whereas the smallest distances are seen in the pairs EIp53/WIVp52, EIp53/DIVp53, and EIp53/GIVp51. This correlation between the two models is not surprising because both were built using the TTX binding constraints. Second, Lipkind and Fozzard employed experimental data to manually place LAs in the inner pore. These knowledge-biased models were refined in energy minimizations, which could not reveal alternative ligand-binding modes. In contrast, we did not bias the ligand binding modes but predicted them via unprecedented multi-MCM searches in sodium channel models with and without a Na+ ion in the DEKA ring. This allowed us to find alternative binding modes of lidocaine, to reveal the role of the pore helices' macrodipoles in lidocaine binding, and to demonstrate antagonism between a cationic LA in the inner pore and a Na+ ion in the selectivity filter.
We used a relatively simple method of energy calculation. In particular, we ignored lipids, employed an implicit waters approach, and used Coulomb's law for electrostatic interactions. Certainly, these approximations can raise questions about the precision of the results. On the other hand, such approximations are the essence of coarse-grained modeling, whose goal is to get a glimpse of possible structures that explain experimental data rather than analyze and animate high-resolution x-ray structures. The coarse-grained energy sampling allowed us to predict and analyze millions of energy-minimized structures. The extensive sampling is necessary because the multiple minima problem is challenging in studies not based on high-resolution structures. Taking into account these considerations, we do not discuss properties of ion channels that may be highly sensitive to “resolution” of the models. For example, we do not address the interesting question of ion selectivity because it likely depends on yet unresolved structural details. We also do not try to correlate activity of ligands with their binding energy because this requires free energy calculations with high-resolution models. Our conclusions are based on the conformational properties of the models that are not very sensitive to the particular sampling method used.
In conclusion, our models suggest that the slow inactivation gating involves disordering of the conducting structure of the outer pore rather than its sterical plugging. The structure can be disordered in different ways, which may correspond to different slow inactivated states (intermediate, slow, ultraslow). We observe disordering of the outer pore, but details of slow inactivated conformations remain unknown. A recent study further highlighted the involvement of outer carboxylates (66), but some data demonstrate that slow inactivation is sensitive to mutations beyond the P-loop domain (e.g., Chen et al. (62) and O'Reilly et al. (67)). This indicates that rearrangements during slow inactivation do not involve only the outer-pore region. In the absence of specific experimental constraints, we are unable to model such rearrangements. Our Na+-deficient models provide just a hint for possible conformations of the outer pore associated with slow inactivation. It is possible that conformational changes in S6s trigger closure of the slow inactivation gate at the level of the outer pore.
Acknowledgments
We thank Peter Backx for helpful discussions and Iva Bruhova for reading the manuscript and valuable comments. Computations were performed using the facilities of the Shared Hierarchical Academic Research Computing Network (SHARCNET: www.sharcnet.ca).
This study was supported by a grant to B.S.Z. from the Canadian Institutes of Health Research.
Abbreviations used: DEKA, the selectivity-filter ring of Asp, Glu, Lys, and Ala residues from the four P-loop domains of Na+ channels; EEDD, the ring of outer carboxylates in the outer pore comprising residues Glu, Glu, Asp, and Asp from the four P-loop domains of the Na+ channel; EM, energy minimization; KvAP, a bacterial voltage-gated potassium channel; LA, local anesthetic; MC, Monte Carlo; MCM, Monte Carlo minimization; MTSET, [2-(trimethylammonium) ethyl]methanethiosulfonate; RMSD, root mean-square deviation; STX, saxitoxin; TMA, tetramethylammonium; TTX, tetrodotoxin.
Editor: Francisco Bezanilla.
References
- 1.Ulbricht, W. 2005. Sodium channel inactivation: molecular determinants and modulation. Physiol. Rev. 85:1271–1301. [DOI] [PubMed] [Google Scholar]
- 2.Hoshi, T., W. N. Zagotta, and R. W. Aldrich. 1990. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 250:506–507. [DOI] [PubMed] [Google Scholar]
- 3.Vassilev, P. M., T. Scheuer, and W. A. Catterall. 1988. Identification of an intracellular peptide segment involved in sodium channel inactivation. Science. 241:1658–1661. [DOI] [PubMed] [Google Scholar]
- 4.Kellenberger, S., J. W. West, T. Scheuer, and W. A. Catterall. 1997. Molecular analysis of the putative inactivation particle in the inactivation gate of brain type IIA Na+ channels. J. Gen. Physiol. 109:589–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patton, D. E., J. W. West, W. A. Catterall, and A. L. Goldin. 1992. Amino acid residues required for fast sodium channel inactivation. Charge neutralizations and deletions in the III–IV linker. Proc. Natl. Acad. Sci. USA. 89:10905–10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.West, J. W., D. E. Patton, T. Scheuer, Y. Wang, A. L. Goldin, and W. A. Catterall. 1992. A cluster of hydrophobic amino acid residues required for fast Na(+)-channel inactivation. Proc. Natl. Acad. Sci. USA. 89:10910–10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eaholtz, G., T. Scheuer, and W. A. Catterall. 1994. Restoration of inactivation and block of open sodium channels by an inactivation gate. Neuron. 12:1041–1048. [DOI] [PubMed] [Google Scholar]
- 8.Featherstone, D. E., J. E. Richmond, and P. C. Ruben. 1996. Interaction between fast and slow inactivation in Skm1 sodium channels. Biophys. J. 71:3098–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vedantham, V., and S. C. Cannon. 1998. Slow inactivation does not affect movement of the fast inactivation gate in voltage-gated Na+ channels. J. Gen. Physiol. 111:83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ruff, R. L. 1996. Single-channel basis of slow inactivation of Na+ channels in rat skeletal muscle. J. Physiol. 271:971–981. [DOI] [PubMed] [Google Scholar]
- 11.Zhou, Y., J. H. Morais-Cabral, A. Kaufman, and R. MacKinnon. 2001. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature. 414:43–48. [DOI] [PubMed] [Google Scholar]
- 12.Zhou, Y., and R. MacKinnon. 2003. The occupancy of ions in the K+ selectivity filter: charge balance and coupling of ion binding to a protein conformational change underlie high conduction rates. J. Mol. Biol. 333:965–975. [DOI] [PubMed] [Google Scholar]
- 13.Lenaeus, M. J., M. Vamvouka, P. J. Focia, and A. Gross. 2005. Structural basis of TEA blockade in a model potassium channel. Nat. Struct. Mol. Biol. 12:454–459. [DOI] [PubMed] [Google Scholar]
- 14.Todt, H., S. C. Dudley, J. W. Kyle, R. J. French, and H. A. Fozzard. 1999. Ultra-slow inactivation in mu1 Na+ channels is produced by a structural rearrangement of the outer vestibule. Biophys. J. 76:1335–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hilber, K., W. Sandtner, O. Kudlacek, I. W. Glaaser, E. Weisz, J. W. Kyle, R. J. French, H. A. Fozzard, S. C. Dudley, and H. Todt. 2001. The selectivity filter of the voltage-gated sodium channel is involved in channel activation. J. Biol. Chem. 276:27831–27839. [DOI] [PubMed] [Google Scholar]
- 16.Hilber, K., W. Sandtner, T. Zarrabi, E. Zebedin, O. Kudlacek, H. A. Fozzard, and H. Todt. 2005. Selectivity filter residues contribute unequally to pore stabilization in voltage-gated sodium channels. Biochemistry. 44:13874–13882. [DOI] [PubMed] [Google Scholar]
- 17.Zhang, Z., Y. Xu, P. H. Dong, D. Sharma, and N. Chiamvimonvat. 2003. A negatively charged residue in the outer mouth of rat sodium channel determines the gating kinetics of the channel. Am. J. Physiol. Cell Physiol. 284:1247–1254. [DOI] [PubMed] [Google Scholar]
- 18.Xiong, W., R. A. Li, Y. Tian, and G. F. Tomaselli. 2003. Molecular motions of the outer ring of charge of the sodium channel: do they couple to slow inactivation? J. Gen. Physiol. 122:323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balser, J. R., H. B. Nuss, N. Chiamvimonvat, M. T. Perez-Garcia, E. Marban, and G. F. Tomaselli. 1996. External pore residue mediates slow inactivation in mu 1 rat skeletal muscle sodium channels. J. Physiol. 494:431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Starmer, C. F., A. O. Grant, and A. C. Strauss. 1984. Mechanisms of use dependent block of sodium channels in excitable membranes by local anesthetics. Biophys. J. 46:15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hille, B. 1977. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J. Gen. Physiol. 69:497–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cahalan, M. D., and W. Almers. 1979. Interactions between quaternary lidocaine, the sodium channel gates, and tetrodotoxin. Biophys. J. 27:39–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Townsend, C., and R. Horn. 1997. Effect of alkali metal cations on slow inactivation of cardiac Na+ channels. J. Gen. Physiol. 110:23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen, Z., B. H. Ong, N. G. Kambouris, E. Marban, G. F. Tomaselli, and J. R. Balser. 2000. Lidocaine induces a slow inactivated state in rat skeletal muscle sodium channels. J. Physiol. 524:37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lopez-Barneo, J., T. Hoshi, S. H. Heinemann, and R. W. Aldrich. 1993. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1:61–71. [PubMed] [Google Scholar]
- 26.Liu, Y., M. E. Jurman, and G. Yellen. 1996. Dynamic rearrangement of the outer mouth of a K+ channel during gating. Neuron. 16:859–867. [DOI] [PubMed] [Google Scholar]
- 27.Yellen, G., D. Sodickson, T. Y. Chen, and M. E. Jurman. 1994. An engineered cysteine in the external mouth of a K+ channel allows inactivation to be modulated by metal binding. Biophys. J. 66:1068–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogielska, E. M., and R. W. Aldrich. 1999. Functional consequences of a decreased potassium affinity in a potassium channel pore. Ion interactions and C-type inactivation. J. Gen. Physiol. 113:347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baukrowitz, T., and G. Yellen. 1995. Modulation of K+ current by frequency and external [K+]: a tale of two inactivation mechanisms. Neuron. 15:951–960. [DOI] [PubMed] [Google Scholar]
- 30.Grissmer, S., and M. Cahalan. 1989. TEA prevents inactivation while blocking open K+ channels in human T lymphocytes. Biophys. J. 55:203–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi, K. L., R. W. Aldrich, and G. Yellen. 1991. Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-activated K+ channels. Proc. Natl. Acad. Sci. USA. 88:5092–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Armstrong, C. M., and G. Cota. 1991. Calcium ion as a cofactor in Na channel gating. Proc. Natl. Acad. Sci. USA. 88:6528–6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Armstrong, C. M., and G. Cota. 1999. Calcium block of Na+ channels and its effect on closing rate. Proc. Natl. Acad. Sci. USA. 96:4154–4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berneche, S., and B. Roux. 2005. A gate in the selectivity filter of potassium channels. Structure. 13:591–600. [DOI] [PubMed] [Google Scholar]
- 35.Tikhonov, D. B., and B. S. Zhorov. 2005. Modeling P-loops domain of sodium channel: homology with potassium channels and interaction with ligands. Biophys. J. 88:184–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsushima, R. G., R. A. Li, and P. H. Backx. 1997. P-loop flexibility in Na+ channel pores revealed by single- and double-cysteine replacements. J. Gen. Physiol. 110:59–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Struyk, A. F., and S. C. Cannon. 2002. Slow inactivation does not block the aqueous accessibility to the outer pore of voltage-gated Na channels. J. Gen. Physiol. 120:509–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhorov, B. S., and D. B. Tikhonov. 2004. Potassium, sodium, calcium and glutamate-gated channels: pore architecture and ligand action. J. Neurochem. 88:782–799. [DOI] [PubMed] [Google Scholar]
- 39.Jiang, Y., A. Lee, J. Chen, V. Ruta, M. Cadene, B. T. Chait, and R. MacKinnon. 2003. X-ray structure of a voltage-dependent K+ channel. Nature. 423:33–41. [DOI] [PubMed] [Google Scholar]
- 40.Yamagishi, T., R. A. Li, K. Hsu, E. Marban, and G. F. Tomaselli. 2001. Molecular architecture of the voltage-dependent Na channel: functional evidence for alpha helices in the pore. J. Gen. Physiol. 118:171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dudley Jr., S. C., N. Chang, J. Hall, G. Lipkind, H. A. Fozzard, and R. J. French. 2000. μ-conotoxin GIIIA interactions with the voltage-gated Na+ channel predict a clockwise arrangement of the domains. J. Gen. Physiol. 116:679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weiner, S. J., P. A. Kollman, D. A. Case, U. C. Singh, C. Chio, G. Alagona, S. Profeta, and P. K. Weiner. 1984. A new force field for molecular mechanical simulation of nucleic acids and proteins. J. Am. Chem. Soc. 106:765–784. [Google Scholar]
- 43.Lazaridis, T., and M. Karplus. 1999. Effective energy function for proteins in solution. Proteins. 35:133–152. [DOI] [PubMed] [Google Scholar]
- 44.Cailliez, F., and R. Lavery. 2005. Cadherin mechanics and complexation: the importance of calcium binding. Biophys. J. 89:3895–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sano, T., S. Vajda, C. L. Smith, and R. C. Cantor. 1997. Engineering subunit association of multisubunit proteins: a dimeric streptavidin. Proc. Natl. Acad. Sci. USA. 94:6153–6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bruhova, I., and B. S. Zhorov. 2007. Monte Carlo-energy minimization of correolide in the Kv1.3 channel: possible role of potassium ion in ligand-receptor interactions. BMC Struct. Biol. 7:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhorov, B. S. 1981. Vector method for calculating derivatives of energy of atom-atom interactions of complex molecules according to generalized coordinates. J. Struct. Chem. 22:4–8. [Google Scholar]
- 48.Brooks, C. L., B. M. Pettitt, and M. Karplus. 1985. Structural and energetic effects of truncating long ranged interactions in ionic polar fluids. J. Chem. Phys. 83:5897–5908. [Google Scholar]
- 49.Li, Z., and H. A. Scheraga. 1987. Monte Carlo-minimization approach to the multiple-minima problem in protein folding. Proc. Natl. Acad. Sci. USA. 84:6611–6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tikhonov, D. B., and B. S. Zhorov. 1998. Kinked-helices model of the nicotinic acetylcholine receptor ion channel and its complexes with blockers: simulation by the Monte Carlo minimization method. Biophys. J. 74:242–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chiamvimonvat, N., M. T. Perez-Garcia, G. F. Tomaselli, and E. Marban. 1996. Control of ion flux and selectivity by negatively charged residues in the outer mouth of rat sodium channels. J. Physiol. 491:51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Benitah, J. P., Z. Chen, J. R. Balser, G. F. Tomaselli, and E. Marban. 1999. Molecular dynamics of the sodium channel pore vary with gating: interactions between P-segment motions and inactivation. J. Neurosci. 19:1577–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ragsdale, D. S., J. C. McPhee, T. Scheuer, and W. A. Catterall. 1994. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science. 265:1724–1728. [DOI] [PubMed] [Google Scholar]
- 54.Ragsdale, D. S., J. C. McPhee, T. Scheuer, and W. A. Catterall. 1996. Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels. Proc. Natl. Acad. Sci. USA. 93:9270–9275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yarov-Yarovoy, V., J. Brown, E. M. Sharp, J. J. Clare, T. Scheuer, and W. A. Catterall. 2001. Molecular determinants of voltage-dependent gating and binding of pore-blocking drugs in transmembrane segment IIIS6 of the Na(+) channel alpha subunit. J. Biol. Chem. 276:20–27. [DOI] [PubMed] [Google Scholar]
- 56.Yarov-Yarovoy, V., J. C. McPhee, D. Idsvoog, C. Pate, T. Scheuer, and W. A. Catterall. 2002. Role of amino acid residues in transmembrane segments IS6 and IIS6 of the Na+ channel alpha subunit in voltage-dependent gating and drug block. J. Biol. Chem. 277:35393–35401. [DOI] [PubMed] [Google Scholar]
- 57.Wright, S. N., S. Y. Wang, and G. K. Wang. 1998. Lysine point mutations in Na+ channel D4–S6 reduce inactivated channel block by local anesthetics. Mol. Pharmacol. 54:733–739. [PubMed] [Google Scholar]
- 58.Nau, C., S. Y. Wang, and G. K. Wang. 2003. Point mutations at L1280 in Nav1.4 channel D3–S6 modulate binding affinity and stereoselectivity of bupivacaine enantiomers. Mol. Pharmacol. 63:1398–1406. [DOI] [PubMed] [Google Scholar]
- 59.Wang, G. K., C. Quan, and S. Wang. 1998. A common local anesthetic receptor for benzocaine and etidocaine in voltage-gated mu1 Na+ channels. Pflugers Arch. 435:293–302. [DOI] [PubMed] [Google Scholar]
- 60.Wang, S. Y., K. Bonner, C. Russell, and G. K. Wang. 2003. Tryptophan scanning of D1S6 and D4S6 C-termini in voltage-gated sodium channels. Biophys. J. 85:911–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sunami, A., S. C. J. Dudley, and H. A. Fozzard. 1997. Sodium channel selectivity filter regulates antiarrhythmic drug binding. Proc. Natl. Acad. Sci. USA. 94:14126–14131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen, Y., F. H. Yu, D. J. Surmeier, T. Scheuer, and W. A. Catterall. 2006. Neuromodulation of Na+ channel slow inactivation via cAMP-dependent protein kinase and protein kinase C. Neuron. 49:409–420. [DOI] [PubMed] [Google Scholar]
- 63.Cordero-Morales, J. F., L. G. Cuello, Y. Zhao, V. Jogini, D. M. Cortes, B. Roux, and E. Perozo. 2006. Molecular determinants of gating at the potassium-channel selectivity filter. Nat. Struct. Mol. Biol. 13:311–318. [DOI] [PubMed] [Google Scholar]
- 64.Nau, C., and G. K. Wang. 2004. Interactions of local anesthetics with voltage-gated Na+ channels. J. Membr. Biol. 201:1–8. [DOI] [PubMed] [Google Scholar]
- 65.Lipkind, G. M., and H. A. Fozzard. 2005. Molecular modeling of local anesthetic drug binding by voltage-gated sodium channels. Mol. Pharmacol. 68:1611–1622. [DOI] [PubMed] [Google Scholar]
- 66.Xiong, W., Y. Z. Farukhi, Y. Tian, D. Disilvestre, R. A. Li, and G. F. Tomaselli. 2006. A conserved ring of charge in mammalian Na+ channels: a molecular regulator of the outer pore conformation during slow inactivation. J. Physiol. 576:739–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.O'Reilly, J. P., S. Y. Wang, and G. K. Wang. 2001. Residue-specific effects on slow inactivation at V787 in D2–S6 of Na(v)1.4 sodium channels. Biophys. J. 81:2100–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]